
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMiddle-down approach: a choice to sequence and characterize proteins/proteomes by mass spectrometry†
P. Boomathi Pandeswari and
Varatharajan Sabareesh *
*
Advanced Centre for Bio Separation Technology (CBST), Vellore Institute of Technology (VIT), Vellore, Tamil Nadu 632014, India. E-mail: v.sabareesh@vit.ac.in; sabareesh6@gmail.com
First published on 2nd January 2019
Abstract
Owing to rapid growth in the elucidation of genome sequences of various organisms, deducing proteome sequences has become imperative, in order to have an improved understanding of biological processes. Since the traditional Edman method was unsuitable for high-throughput sequencing and also for N-terminus modified proteins, mass spectrometry (MS) based methods, mainly based on soft ionization modes: electrospray ionization and matrix-assisted laser desorption/ionization, began to gain significance. MS based methods were adaptable for high-throughput studies and applicable for sequencing N-terminus blocked proteins/peptides too. Consequently, over the last decade a new discipline called ‘proteomics’ has emerged, which encompasses the attributes necessary for high-throughput identification of proteins. ‘Proteomics’ may also be regarded as an offshoot of the classic field, ‘biochemistry’. Many protein sequencing and proteomic investigations were successfully accomplished through MS dependent sequence elucidation of ‘short proteolytic peptides (typically: 7–20 amino acid residues), which is called the ‘shotgun’ or ‘bottom-up (BU)’ approach. While the BU approach continues as a workhorse for proteomics/protein sequencing, attempts to sequence intact proteins without proteolysis, called the ‘top-down (TD)’ approach started, due to ambiguities in the BU approach, e.g., protein inference problem, identification of proteoforms and the discovery of posttranslational modifications (PTMs). The high-throughput TD approach (TD proteomics) is yet in its infancy. Nevertheless, TD characterization of purified intact proteins has been useful for detecting PTMs. With the hope to overcome the pitfalls of BU and TD strategies, another concept called the ‘middle-down (MD)’ approach was put forward. Similar to BU, the MD approach also involves proteolysis, but in a restricted manner, to produce ‘longer’ proteolytic peptides than the ones usually obtained in BU studies, thereby providing better sequence coverage. In this regard, special proteases (OmpT, Sap9, IdeS) have been used, which can cleave proteins to produce longer proteolytic peptides. By reviewing ample evidences currently existing in the literature that is predominantly on PTM characterization of histones and antibodies, herein we highlight salient features of the MD approach. Consequently, we are inclined to claim that the MD concept might have widespread applications in future for various research areas, such as clinical, biopharmaceuticals (including PTM analysis) and even for general/routine characterization of proteins including therapeutic proteins, but not just limited to analysis of histones or antibodies.
1 Introduction
1.1 Protein chemistry to proteomics
Protein sequencing, viz., elucidation of primary structure of proteins is central to any investigation related to proteins. But for the primary structure, it would not be possible to understand any biochemical or biological function of proteins, since sequence determines structure and/or conformation, which in turn regulates function or activity of proteins. Amongst several instances of sequence strongly impacting the biological function, a very popularly known case is ‘sickle cell anemia’, wherein a mutation of ‘valine’ to ‘glutamic acid’ significantly alters the structure of hemoglobin, thereby severely hampering its ability to transport oxygen (O2).1.2 Protein/peptide sequencing
For many years, proteins had been traditionally sequenced by Edman's method, popularly known as N-terminal sequencing, which is accomplished using phenylisothiocyanate (PITC).1–4 While Edman sequencing method has been successful in numerous cases, it cannot be useful to sequence proteins having blocked N-terminus, e.g., N-terminus formylated or acetylated proteins and several eukaryotic proteins are known to have modified N-terminus.3,4 Further, Edman's method can be applicable on an isolated and purified protein/peptide only, which means purity of the isolated protein/peptide is essential. Moreover, considerable time and quantity of sample would be consumed to sequence even a single protein by Edman's method.4 As a result, Edman method is not suitable for high-throughput sequencing of proteins. Nevertheless, N-terminal sequencing method indeed finds application for conventional biochemical or biophysical studies,4,5 whenever high-throughput is not necessary.However, for more than a decade or so from now, sequencing of proteins has increasingly becoming rapid and high-throughput. Such a transition can be mainly attributed to the ‘-omics’ approach to study proteins, known as ‘proteomics’ and the prime impetus for this transformation came from ‘genomics’. Rapid growth in elucidation of genome sequences from various organisms,6–8 in particular, the Human Genome Sequencing project,9,10 provided motivation to identify proteome sequences, with the main objective to identify and understand the relationship between genome and proteome. In other words, knowledge of sequences of proteome could be helpful in discerning, which genes code for proteins and which are not.11,12 Further, by comparing protein sequences with transcriptome (messenger ribonucleic acid (mRNA)), it may be possible to know the translation efficiency.11,13–15 Moreover, processes such as alternative splicing16,17 and posttranslational modifications (PTMs)18–21 expand the diversity of proteome (Scheme 1), which can be known from the deduced protein sequences, but are otherwise ambiguous to predict at the level of genome and transcriptome sequences. All these exercises enable to understand not only the normal biological processes, but also aids in knowing those factors that are responsible for abnormal or diseased conditions.19,22,23 Thus, the foremost objective of any proteomic investigation is to elucidate sequences of as many proteins as possible. As a result, there has been a radical shift from the typically followed approaches and/or methods and/or strategy to characterize and study proteins.
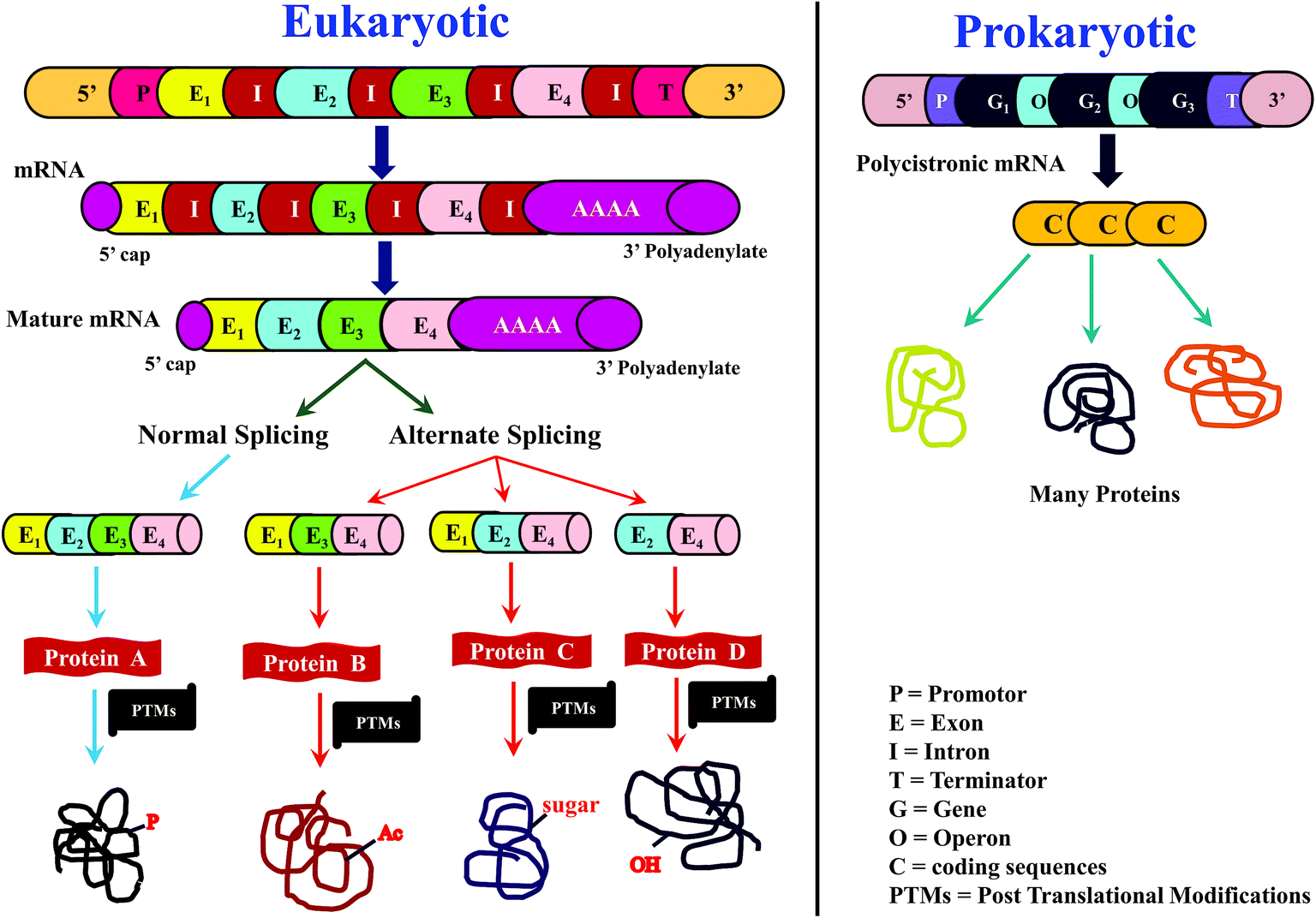 | ||
| Scheme 1 Overview of central dogma of molecular biology in (a) eukaryotic and (b) prokaryotic biological systems. This scheme highlights the importance of and the need for proteomics research, in order to correlate protein sequence information with the RNA and DNA sequence. In the case of eukaryotic system, proteomics is essential for elucidation of posttranslational modifications (PTMs), e.g., P: phosphorylation; Ac: acetylation; sugar: glycosylation; OH: hydroxylation. | ||
Mass spectrometry (MS), particularly, subsequent to the advent of electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI), enabled high-throughput determination of protein sequences at a faster rate. The key highlight of these two ionization modes is their ‘soft nature’, which enables transferring large protein molecular ions from solid or liquid phase to gas phase or vacuum, in their ‘intact’ form with little molecular fragmentations.24,25 Consequently, it became possible to determine ‘intact molecular mass’ of large proteins and peptides. Prior to the arrival of ESI and MALDI, the ionization modes such as fast atom bombardment (FAB), chemical ionization (CI), field ionization (FI) and electron impact/ionization (EI) were not capable of ionizing polar macromolecules (viz. proteins); though FAB was somewhat successful for about a decade or so, to determine intact molecular mass of certain large-sized polar compounds, for instance, polypeptides up to a mass of about 8 or 10 kilodaltons (kDa).26–29
Simultaneous innovations in the development of mass analyzers, especially ‘hybrid configuration’, wherein two or more mass analyzers are used in combination, enabled rapid sequencing of peptides and proteins.30 The basic aspect that facilitated sequencing of peptides and proteins was ‘tandem mass spectrometry’, referred as MS/MS. Through MS/MS experiments, the peptide or protein molecular ions are dissociated and the mass-to-charge (m/z) values of the resulting fragment ions are used to deduce the sequence of the peptide or protein.3,30–32 A variety of MS/MS fragmentation methods have been devised with the aim to achieve good sequence coverage.32–39 Additionally, improvements in resolution and sensitivity offered by different kinds of mass analyzers40–42 proved valuable not only for better identification of peptides/proteins, with the concomitant decrease in the false discovery rate, but also for detecting low abundant peptides/proteins with a good signal-to-noise ratio.
Not only mass spectrometry, but advancements in the field of ‘chromatography’ too contributed significantly for high-throughput identification of proteins, whereby development of various pre-fractionation and other separation methods helped in reducing the complexity of the samples, which in turn facilitated increase in the number of identifications.43 The feasibility of linking chromatography, in particular reverse phase liquid chromatography (LC) to MS (viz. LC-MS) in an online fashion, viz., analytical mode, without offline collection of eluents, proved a major step forward for realization of high-throughput identification and characterization of peptides and proteins.44 Also, there have been efforts on using two or three different chromatographic methods in tandem prior to mass spectrometric analysis, so as to reduce the complexity of the sample for better and enhanced identifications; a well known example being, MudPIT.45–48
1.3 Approaches to sequence and characterize proteins or proteomes
Sequencing of proteins and proteomes can be carried out either directly on their intact form or by truncating them. With regard to truncation, the proteins are subjected to enzymatic proteolysis (e.g. trypsin) or chemically degraded (e.g. cyanogen bromide) and the resulting peptides or polypeptides are then sequenced. Subsequently, the derived sequences of peptides/polypeptides are joined in an appropriate manner; thereby the sequence of the entire protein is elucidated. Depending on the nature of the protease and its specificity, peptide/polypeptide fragments of various sizes could be obtained from the intact protein. According to the ‘number’ and respective ‘positions’ of enzyme cleavable sites on the intact protein's sequence, the size/length of the resulting peptide or polypeptide fragments would vary. Consequently, engineering of the proteolysis step is critical, which needs to be optimized depending on the nature of proteins that are being investigated.Edman's N-terminal sequencing method is applicable to deduce the primary structure of intact proteins. Edman's method has also been shown to be useful to derive sequences of internal peptides, in the case of blocked/modified N-terminus of the protein; for which the intact protein would need to be proteolysed or chemically degraded to yield shorter peptides/polypeptides.4 In contrast, applications of mass spectrometry (MS) to elucidate sequence using only the intact form of the protein is limited, when compared to the utility of MS to derive sequences of shorter polypeptides or peptides. Although ESI and MALDI based MS has proven to be very successful to derive molecular mass of intact proteins, only few attempts of ‘directly sequencing intact protein without truncation’ by MS have yielded good results.
Altogether, the extent of usefulness and applicability of BU approach, be it to study a single purified protein or proteome, is dependent on the number of trypsin cleavable sites (viz. no. of arginines and lysines) and the sequence of the protein(s) itself, viz., average number of amino acid residues between two trypsin cleavable sites.
The major steps involved in the three approaches, as described above are summarized in Scheme 2. In this review, various strategies reported thus far by different research groups for accomplishing middle-down approach are discussed. Diverse workflows of BU and TD approaches are also briefed and compared with middle-down approach. Fundamental aspects of steps involved in the workflow such as proteolytic methods, chromatography (separation techniques), mass spectrometry and data analysis strategies are described.
 | ||
| Scheme 2 Illustration of the fundamental criteria of three different approaches for analysis of proteins or proteomes. | ||
2 Workflow of middle-down sequencing or proteomic approach
2.1 Proteolysis
As explained in the previous section, the major step that distinguishes one approach from the other is ‘proteolysis’. In the case of TD approach, proteolysis is not carried out at all, whereas the ‘extent of proteolysis’ is the key criterion or perhaps the subtlety that demarcates bottom-up and middle-down approaches. While in BU approach the proteolysis is allowed to proceed completely, the process of proteolysis in middle-down (MD) approach could be challenging, which necessitates careful optimization so as to get proteolytic peptides, whose lengths should be greater than ∼25 amino acid residues and of maximum length up to about 100 amino acid residues. Thus, MD approach entails ‘restricted digestion’, depending on the choice of protease that is employed. Certain special proteases have also been identified that could be useful to yield longer peptides specifically suited for MD approach to study proteins and proteomes.Proteases such as GluC, AspN and LysC have also been widely used for MD Proteomics (MDP).72 Of note, most of the MDP studies have been carried out particularly to characterize histones by utilizing GluC enzyme for getting longer peptides.63,73–82 AspN also has been used to study histones and their PTMs by MD approach.75,83 Additionally, MD approach has been applied to characterize phosphorylation (in cardiac myosin binding protein C) and glycosylation (in human erythropoietin, human plasma properdin, human transferrin and human α1-acid glycoprotein) by employing AspN.84–86 Further, in a study by Forbes et al., restricted digestion using LysC was shown to be useful to achieve higher sequence coverage of a mixture of proteins, whose molecular masses were greater than 70 kDa, though they have not claimed such a strategy as a MD approach.87 Microwave accelerated acid digestion experiments have been attempted on ribosomal proteins and RPMI 8226 multiple myeloma cells, which gave rise to polypeptides rich in basic amino acid residues, whose sizes varied in the range: 3–9 kDa (maximum of 12 charges).88,89
In a very recent study, Tsybin and co-workers report chemical-mediated proteolysis as an alternative to the conventional enzyme-assisted digestion, wherein they use the already well-known chemical reagents (see Table 1) to specifically effect cleavages of the peptide bonds at C-terminus to methionine, tryptophan and cysteine, on some model proteins, in order to assess the suitability of this strategy for MD proteomic applications.90 Table 1 shows list of various proteases and chemicals along with their respective specificity towards cleaving the peptide bonds.
| S. no. | Proteases | Site specificity of proteolysis |
|---|---|---|
| a E: glutamic acid (Glu); D: aspartic acid (Asp); K: lysine (Lys); M: methionine (Met); W: tryptophan (Trp); R: arginine (Arg); P: proline (Pro); A: alanine (Ala); C: cysteine (Cys).b From Staphylococcus aureus; this can proteolyze peptide bonds at C-terminus of Asp also, at a particular pH 4–6.c GluC (ref. 63, 64, 71, 73, 74–79, 81, 82, 84, 85, 91, 173, 184, and 185).d AspN (ref. 64, 75, 83, 84, 86, 92, 185).e LysC (ref. 84, 86, 172 and 259).f OmpT (ref. 94 and 98).g Sap9 (ref. 71, 95, and 96).h IdeS: (ref. 70, 97, 99, 101–105 and 133).i CNBr: cyanogen bromide; CNBr cleaves at C-terminus of tryptophan also.j BNPS: 2-(2′-nitrophenylsulfonyl)-3-methyl-3-bromoindolenine (BNPS)-skatole.k NTCB: 2-nitro-5-thiocyanobenzoic acid (NTCB). | ||
| 1. | GluCc (V8 protease)b | C-terminal side of E (C-terminal side of D) |
| 2. | AspNd | N-terminal side of D |
| 3. | LysCe | C-terminal side of K |
| 4. | OmpTf | Between two consecutive dibasic residues: R↓R, K↓K, R↓K, K↓R |
| 5. | Sap9g | C-terminal side of R, K, KR, RR, RK & KK |
| 6. | IdeSh | Between two consecutive glycine residues at the hinge region for immunoglobulin G |
| 7. | Trypsin (restricted/limited proteolysis) | C-terminal side of K & R |
| 8. | Neprosin267 | C-terminal side of P & A |
| 9. | GingisKHAN™278 | Upper hinge region of human immunoglobulin G1 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :proteins. Furthermore, it is important to understand that the extent of proteolysis also depends on the protein sequence, particularly depending on the ‘positions’ of those amino acid residues, which are the targets of a particular protease. For instance, complete LysC digestion or complete trypsinolysis of carbamidomethylated bovine pancreatic ribonuclease A (RNase A) would result in short length peptides, whereas upon digestion with GluC (V8 protease from Staphylococcus aureus), two longer peptides are obtained: residue [10–49] and residue [50–86] (see Table 2). Not only the ‘length’ of these two GluC peptides, but also their respective ESI charge state distribution (see Fig. 1 and 2; also refer Section 2.3.3, vide infra) indicate that complete GluC digestion is a better choice over complete trypsinolysis or LysC to characterize RNase A by MD approach. Thus, the workflow for the sample preparation, i.e., preparing suitable proteolytic digest for MD approach mainly depends on the sequence (viz. primary structure) of the proteins under investigation.
:proteins. Furthermore, it is important to understand that the extent of proteolysis also depends on the protein sequence, particularly depending on the ‘positions’ of those amino acid residues, which are the targets of a particular protease. For instance, complete LysC digestion or complete trypsinolysis of carbamidomethylated bovine pancreatic ribonuclease A (RNase A) would result in short length peptides, whereas upon digestion with GluC (V8 protease from Staphylococcus aureus), two longer peptides are obtained: residue [10–49] and residue [50–86] (see Table 2). Not only the ‘length’ of these two GluC peptides, but also their respective ESI charge state distribution (see Fig. 1 and 2; also refer Section 2.3.3, vide infra) indicate that complete GluC digestion is a better choice over complete trypsinolysis or LysC to characterize RNase A by MD approach. Thus, the workflow for the sample preparation, i.e., preparing suitable proteolytic digest for MD approach mainly depends on the sequence (viz. primary structure) of the proteins under investigation.
| LysC & Trypsin | |
|---|---|
| Lys-C digesta | Trypsin digesta |
| a Peptides of length ≤ 5 are not shown. | |
| b C* refers to carbamidomethyl cysteine | |
| 1. [2–7]: ETAAAK | 1. [2–7]: ETAAAK |
| 2. [8–31]: FERQHMDSSTSAASSSNYC*NQMMKb | 2. [11–31]: QHMDSSTSAASSSNYC*NQMMK |
| 3. [32–37]: SRNLTK | 3. [40–61]: C*KPVNTFVHESLADVQAVC*SQK |
| 4. [38–61]: DRC*KPVNTFVHESLADVQAVC*SQK | 4. [67–85]: NGQTNC*YQSYSTMSITDC*R |
| 5. [67–91]: NGQTNC*YQSYSTMSITDC*RETGSSK | 5. [86–91]: ETGSSK |
| 6. [92–98]: YPNC*AYK | 6. [92–98]: YPNC*AYK |
| 7. [99–104]: TTQANK | 7. [99–104]: TTQANK |
| 8. [105–124]: HIIVAC*EGNPYVPVHFDASV | 8. [105–124]: HIIVAC*EGNPYVPVHFDASV |
| GluC |
|---|
| Glu-C digest |
| 1. [1–9]: KETAAAKFE |
| 2. [10–49]: RQHMDSSTSAASSSNYC*NQMMKSRNLTKDRC*KPVNTFVE |
| 3. [50–86]: SLADVQAVC*SQKNVAC*KNGQTNC*YQSYSTMSITDC*RE |
| 4. [87–111]: TGSSKYPNC*AYKTTQANKHIIVAC*E |
| 5. [112–124]: GNPYVPVHFDASV |
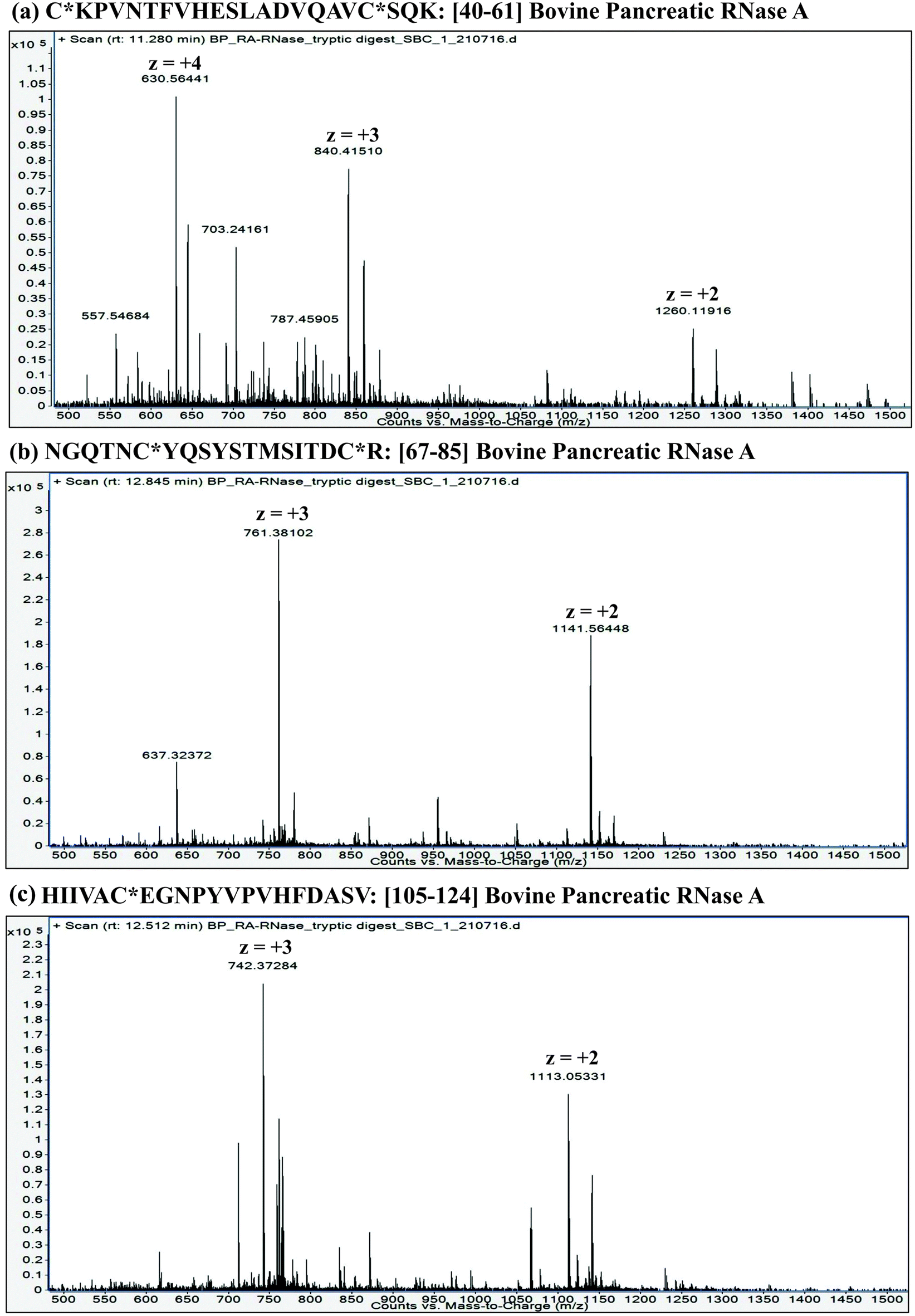 | ||
| Fig. 1 LC-ESI mass spectra of tryptic peptides: (a) Residue No. [40–61] (22 a.a. residues long); (b) Residue No. [67–85] (19 a.a. residues long) and (c) Residue No. [105–124] (20 a.a. residues long) from carbamidomethylated RNase A (Bovine pancreas). These data were acquired on an ESI-Q/TOF mass spectrometer (6540 Ultra High Definition Accurate-Mass Q-TOF LC/MS attached to 1290 Infinity LC; Agilent Technologies). Note: C* refers to carbamidomethyl cysteine. | ||
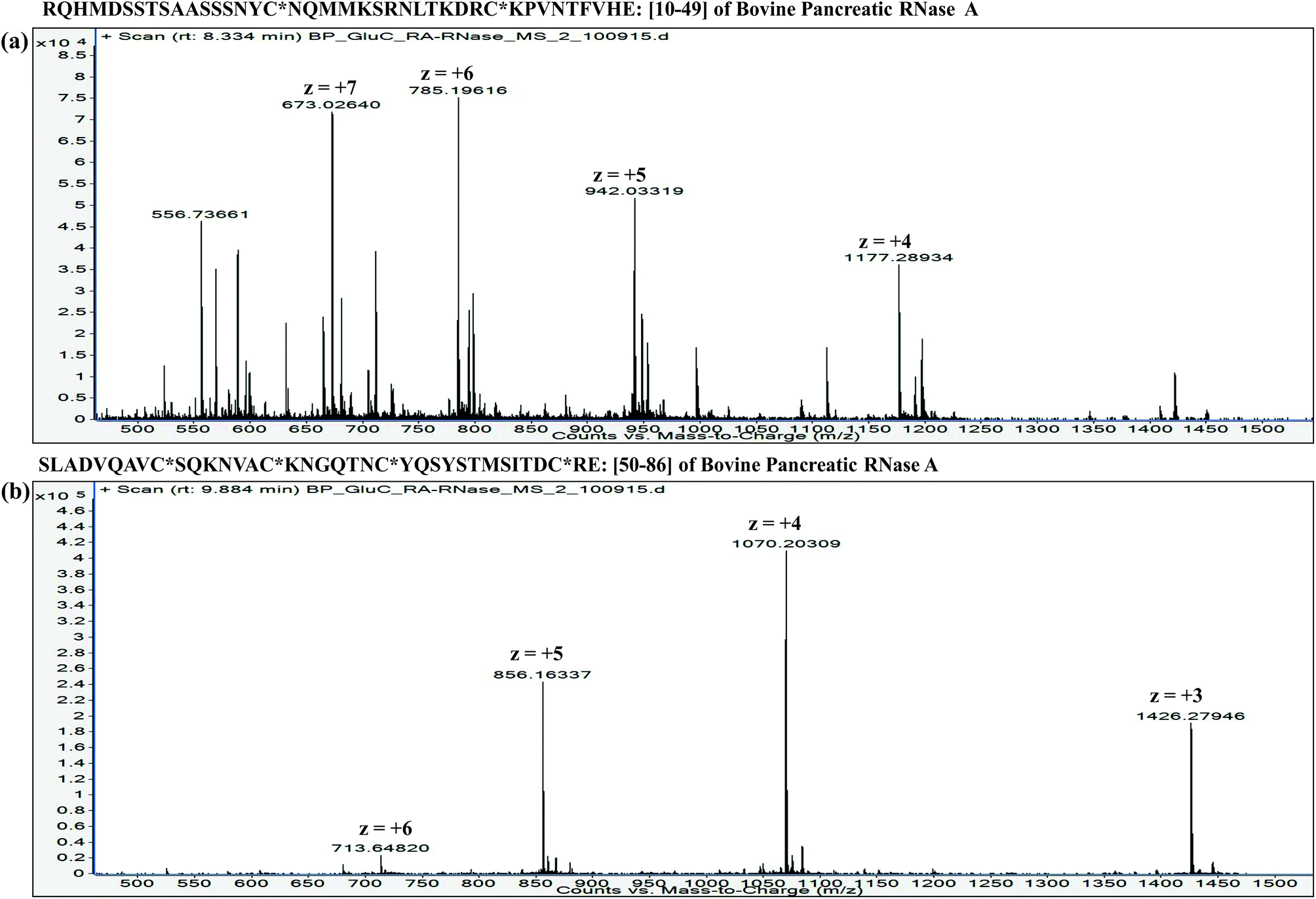 | ||
| Fig. 2 LC-ESI mass spectra of GluC digested peptides: (a) Residue No. [10–49] (40 a.a. residues long) and (b) Residue No. [50–86] (37 a.a. residues long) from carbamidomethylated RNase A (Bovine pancreas). These data were acquired on an ESI-Q/TOF mass spectrometer (6540 Ultra High Definition Accurate-Mass Q-TOF LC/MS attached to 1290 Infinity LC; Agilent Technologies). Note: C* refers to carbamidomethyl cysteine. | ||
In order to have a better understanding and obtain a clearer picture about the role of primary structure of the proteins in influencing the extent of proteolysis, in silico proteolysis was performed herein on 15 representative proteins from each of five different organisms: Homo sapiens (human), Saccharomyces cerevisiae (yeast), Escherichia coli (E. coli, bacteria), Arabidopsis thaliana (plant) and Methanococcus jannaschii (archaea). The sequences of all the representative proteins were taken from UniProt KB database and most of these are enzymes involved in glycolytic pathway and in tri-carboxylic acid cycle (see Table S1, supplementary information†). The intact molecular masses of the 15 proteins from each of these organisms are in the range: 20–100 kDa. We compared the results obtained from complete proteolysis with that of 1-missed cleavage (1-MC) proteolysis corresponding to each of the four proteases: trypsin, GluC, LysC and AspN, with the objective to find, which protease can be suitable to execute the strategy of limited proteolysis for the sake of MDP.
To simplify this analysis, the proteolytic peptides resulting from every in silico digestion were classified into five categories, based on the number of amino acid residues (a.a.r) in peptide, i.e., length of the peptide: (1) 5–15 a.a.r, (2) 16–25 a.a.r, (3) 26–35 a.a.r, (4) 36–45 a.a.r and (5) 46–55 a.a.r. The results of these in silico exercises depicting population distribution of different lengths of proteolytic peptides plotted for four different proteases can be seen in Fig. 3. An interesting aspect emerging from this in silico comparative analysis is that there is not only increase in the number of AspN peptides of lengths in the range 16–55 a.a.r due to 1-MC, when compared with the results of complete AspN digestion, but there is also a significant decrease in the number of AspN peptides of length 5–15 a.a.r because of 1-MC AspN proteolysis compared to complete AspN digestion. Likewise, reduction in the number of GluC peptides of length 5–15 a.a.r, accompanied by increase in the number of longer GluC peptides (16–55 a.a.r), due to 1-MC can be noticed. In contrast, with regard to digestion by trypsin and LysC, there is no significant decrease in the population of shorter peptides of length 5–15 a.a.r, although there is a good enhancement in the population of tryptic & LysC peptides of lengths in the range 16–55 a.a.r. It is important to note that the main purpose of restricted or limited digestion is in fact, not only to improve the yield of longer proteolytic peptides, but decrease in the population of shorter proteolytic peptides also is desirable, so that the complexity of the proteolytic peptides' concoction could be minimized. This purpose does not seem to be fulfilled by the use of trypsin and LysC over the 15 representative proteins that we have chosen from five different species. Although the population of the peptides of length 16–55 a.a.r increases due to 1-MC proteolysis in the cases of all these four proteases, the extent of decrease in the population of shorter peptides of length 5–15 a.a.r, in the cases of 1-MC AspN and 1-MC GluC proteolysis, is noteworthy.
 | ||
| Fig. 3 In silico proteolysis of 15 representative proteins (see Table S1, supplementary information†) using four different proteases: (1) trypsin, (2) LysC, (3) GluC and (4) AspN. Comparison of population of proteolytic peptides of different lengths obtained from ‘complete proteolysis (0-MC)’ and ‘limited proteolysis (1-MC)’. Based on their length (no. of a.a.r), the peptides have been classified into five different categories: (1) 5–15 a.a.r, (2) 16–25 a.a.r, (3) 26–35 a.a.r, (4) 36–45 a.a.r and (5) 46–55 a.a.r. | ||
Thus, it is apparent from this in silico analysis that restricted digestion using AspN and GluC could be more apt for performing MD studies on proteins or for proteomics, rather than performing limited trypsin or LysC digestion and to arrive at this inference, “100 proteolytic peptides” was chosen as the threshold value (Fig. 3).
Furthermore, we performed in silico proteolysis for 2-missed cleavage (2-MC) condition using all these four proteases on these seventy five proteins (Table S1, supplementary information;† also see Fig. 4). To assess the performance of 2-MC proteolysis on these seventy five proteins, it was decided to define a ratio, as shown below:
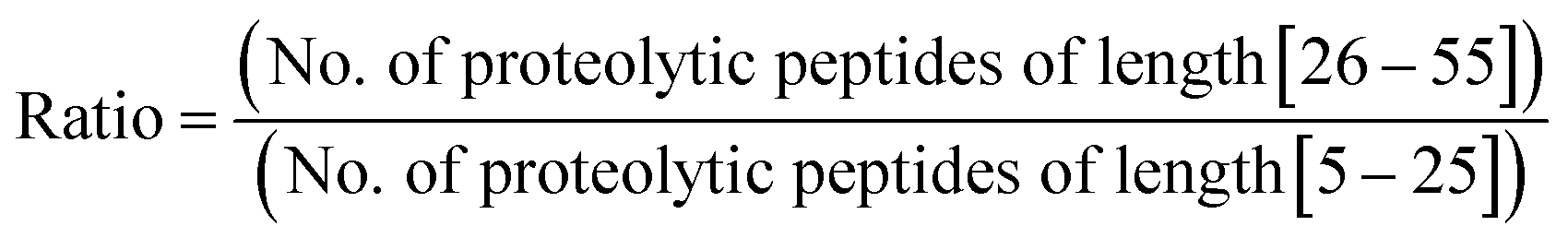
 | ||
| Fig. 4 In silico proteolysis of 15 representative proteins (see Table S1, supplementary information†) using four different proteases: (1) trypsin, (2) LysC, (3) GluC and (4) AspN. Distribution of population of proteolytic peptides of different lengths obtained from ‘2-missed cleavage (2-MC) limited proteolysis’. Based on their length (no. of a.a.r), the peptides have been classified into five different categories: (1) 5–15 a.a.r, (2) 16–25 a.a.r, (3) 26–35 a.a.r, (4) 36–45 a.a.r and (5) 46–55 a.a.r. (Compare this with Fig. 3 & also see Table 3.) | ||
The extent of proteolysis can be understood from this ratio values. Higher the value of this ratio, viz., when the value exceeds 0.5 or 1, better is that proteolytic condition suited for MD proteomic approach. We calculated this ratio for three different proteolytic conditions: 0-missed cleavage (0-MC), 1-missed cleavage (1-MC) and 2-missed cleavage (2-MC), using each of the four proteases on all of the fifteen model proteins from each of the five organisms (see Table 3). Although it is obvious to conceive that 2-MC would be better than 1-MC and 0-MC cleavage conditions for MDP, the values shown in Table 3 clearly indicate that out of these four proteases, AspN and GluC are better than trypsin and LysC, to pursue 2-MC limited proteolysis, for MDP; especially 2-MC by AspN yields better results (i.e., better ratio values) than 2-MC by GluC. Indeed, quite a number of MD investigations, particularly on histones have been accomplished by the use of AspN and GluC.63,73–79,81–83,91,92 It can also be noted in Table 3 that 2-MC by trypsin does not give favorable ratio values, excepting the case of yeast, suggesting that 2-MC by trypsin is not a useful option for MD approach.
| Comparison of results obtained from Trypsin and LysC | ||||||
|---|---|---|---|---|---|---|
| Species | Trypsin | LysC | ||||
| 0-MC | 1-MC | 2-MC | 0-MC | 1-MC | 2-MC | |
| Human | 0.0818 | 0.1681 | 0.6595 | 0.2361 | 0.6697 | 1.6434 |
| E. coli | 0.0931 | 0.2878 | 1.1659 | 0.3081 | 0.9296 | 3.1395 |
| Archaea | 0.0866 | 0.1619 | 0.3676 | 0.1501 | 0.2834 | 0.5869 |
| Plant | 0.0796 | 0.2651 | 0.7771 | 0.2051 | 0.64 | 1.8247 |
| Yeast | 0.0744 | 0.2074 | 0.4549 | 0.1648 | 0.5592 | 0.9005 |
| Comparison of results obtained from GluC and AspN | ||||||
|---|---|---|---|---|---|---|
| Species | GluC | AspN | ||||
| 0-MC | 1-MC | 2-MC | 0-MC | 1-MC | 2-MC | |
| Human | 0.25 | 0.9395 | 1.1826 | 0.4379 | 0.9006 | 2.0512 |
| E. coli | 0.2454 | 0.8209 | 2.0348 | 0.3555 | 0.8571 | 2.6415 |
| Archaea | 0.1265 | 0.3722 | 0.8299 | 0.2654 | 0.6608 | 2.4769 |
| Plant | 0.3063 | 0.8056 | 2.1647 | 0.3318 | 0.7453 | 2.7162 |
| Yeast | 0.2266 | 0.6421 | 0.9512 | 0.1895 | 0.6716 | 1.8490 |
A similar kind of analysis has been carried out by Trevisiol et al., wherein only human proteome was subjected to in silico proteolysis (using the proteases: trypsin, LysC, ArgC, LysN, AspN, GluC (D&E), GluC(E)) and the lengths of the resulting peptides, in the range 8–25 a.a.r only were analyzed; indicating the suitability of this investigation primarily on BU approach.93 Whereas, we are interested in MD approach and therefore, the analyses presented herein are focused on proteolytic peptides longer than 25 a.a.r and moreover, we have paid attention to other biological species, in addition to human.
:substrate, there have been attempts in search of special proteases, for instance OmpT, Sap9 and IdeS, that have been used to ease the process of ‘restricted proteolysis’, and to enhance the probability of consistently obtaining longer peptides suited for MD approach than those typically encountered in BU investigations.70,94–97 OmpT is a novel protease belonging to the omptin family, which has specificity to catalyze cleavage of peptide bonds between consecutive dibasic sites, K↓R, R↓R, R↓K and K↓K;94,98 whereas Sap9 (Candida albicans) is an aspartic protease derived from yapsin family; both enzymes are capable of yielding peptides of length > 2 kDa.94–96 Sap9 has been applied for extended bottom-up proteomics96 and it is also envisaged to be promiscuous for successfully carrying out MD proteomic studies.95 In the case of studies on antibodies by MD approach, immunoglobulin G-degrading enzyme (IdeS) from Streptococcus pyogenes have been employed, wherein IdeS catalyse hydrolysis of peptide bond between two consecutive glycines present specifically at the hinge region of immunoglobulin (Ig).97,99–101 For example, three larger sized proteolytic peptides were obtained by IdeS treatment on the commercially available antibodies, cetuximab, rituximab, etc.70,102–105 Proteases that have been used exclusively for MDP studies are shown in Table 1.Moreover, there is another endopeptidase, Kex2, which is specific in catalyzing the hydrolysis of peptide bonds that are C-terminal to consecutive dibasic residues, viz., C-terminus to KR or RR or PR.106 Although currently, there are no published reports about the usage of Kex2 in MD proteomic studies, it might be applicable for MD based proteomic investigations in future.
2.2 Separation technology
Proteomic studies are relatively more complicated, when compared to genomic investigations because proteomic studies involve characterization of protein isoforms, PTMs and various kinds of analyses to monitor expression levels at different stages of growth of cells/tissues. Therefore, different types of separation strategies are essential for proteomics, so as to decrease the complexity of proteome. Application of various separation or chromatographic methods can significantly influence the detection and analysis of even the low abundance level of proteins/proteolytic peptides by MS.107–110 Separation of proteome can be carried out by either of the two approaches: gel-based or gel-free.111–1132.2.3.1 Reverse phase liquid chromatography (RPLC). Most of the proteomic studies are successfully done by reverse phase-high performance liquid chromatography (HPLC) or RPLC.107 The mobile phase or solvents of RPLC are water, acetonitrile and methanol which are compatible to ESI-MS and consequently, it was possible to successfully couple RP-HPLC with ESI-MS, which is denoted as LC-MS or LC-ESI-MS.134,135 In general, online separation of peptides by LC-MS can save time, sample and also can be more sensitive, when compared to the offline direct infusion method. Moreover, with the introduction of ultra performance or ultra high performance LC (UPLC/UHPLC), there was further improvement in the speed, sensitivity and resolution of separation of complex samples containing peptides for LC-MS in proteomics, which can be attributed to the use of sub 2 μm particle size containing reverse phase columns.136,137
Silica based C18, C8 and C4 are some well known column chemistries that have been more widely used for RP-HPLC of peptides and proteins3 and thus, these column chemistries are also extensively applied for several online LC-MS experiments in proteomics. For BUP, C18 columns are predominantly used for LC-MS as it involves characterization of short length peptides.45,46 In the case of MD approach, since longer or larger proteolytic peptides are encountered, C4, C5 and C8 columns have been utilized for online LC-MS.65,68,70,71,90,99,104,105,138,139 Nevertheless, C18 column (in the form of Nano LC) also has been utilized for efficient separation of proteolytic peptides, in certain MD studies.64,75,85,89,139
2.2.3.2 Other liquid chromatographic methods. With regard to application of other LC methods for MD approach based studies, there are perhaps no reports showing the utility of affinity chromatography for MDP. However, for BUP, IMAC has been proven to be of immense utility.140 A major application of IMAC in BUP is enrichment of phosphorylated peptides by Ti4+ and Zr4+ based IMAC.141,142 Recently, IMAC using monolithic columns for proteomics is on the rise and in this connection, IMAC has been shown to be a pre-fractionation method with the use of monolith disks.143,144 Furthermore, lectin affinity chromatography has been well utilized for the study of glycated peptides and glycans in BUP.145–147 Boronate affinity chromatography also has been of utility for enrichment of glycoproteins and glycopeptides, prior to mass spectrometric analysis.148,149 With regard to size exclusion chromatography (SEC), excepting an investigation, no reports are available on its use for MD approach; Hummel et al. have applied SEC to extract 2S albumin from the mustard.138 Ion exchange chromatography, particularly weak cation exchange (WCX) chromatography in conjunction with hydrophilic interaction chromatography (HILIC) has been successfully applied for MD approach based studies (cf. vide infra).
2.2.3.3 Multi-dimensional LC. The introduction of multi-dimensional protein identification technology (MudPIT) revolutionized the field of proteomics, in particular, for BUP.45–47 MudPIT involves combining the use of more than one separation method or technology for achieving better separation of proteins/proteolytic peptides, thereby decreasing the complexity of the sample for further analysis. Coupling strong cation exchange (SCX) chromatography with RPLC, viz., SCX-RPLC has been the predominantly followed MudPIT approach,45,47,64 which has proven to be successful in several BUP investigations.48 Other examples of MudPIT strategies applied for proteomics are: LC with CE, SEC with RPLC, RP-RPLC and IMAC coupled to CE.47,150,151 In the case of MD approach, WCX in conjunction with HILIC, termed as WCX-HILIC has been widely followed, particularly for the investigations focused on histones.76–79,82,92,152 Young et al. reported the first use of “saltless-pH gradient” to carry out WCX-HILIC (online coupled to ESI-MS/MS), which became widely applicable to several other MD based studies on histones.92 In contrast, in a recent report by Shabaz Mohammed, Heck and co-workers, traditional MudPIT approach encompassing SCX-RPLC has been shown to be useful for MDP to separate longer proteolytic peptides.64 Scheme 3 summarizes different types of gel-based and non-gel-based (gel-free) separation techniques that have been applied for different kinds of proteomic analyses including that of MD approach based investigations. In addition to gel-based and gel-free approaches, separation has been attempted in gas phase also, accomplished by means of ion-mobility.153,154
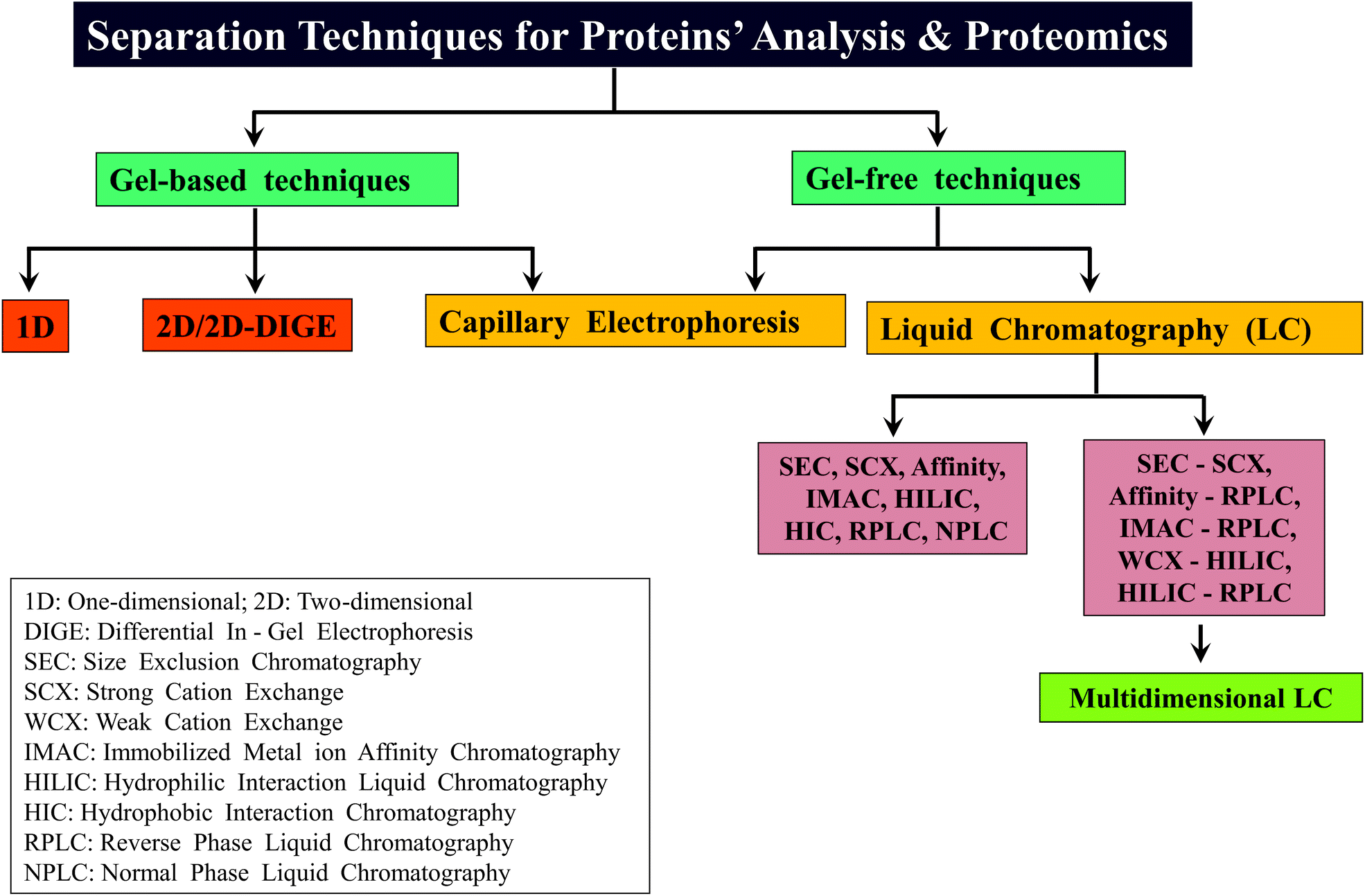 | ||
| Scheme 3 An overview of different types of separation techniques that have been widely followed for the analysis of proteins as well as for various kinds of proteomic analyses including MD approach based studies. | ||
2.3 Conventional MS and tandem MS (MS/MS)
Mass spectrometers basically have three modules namely ion source or ionization source, mass analyzer and detector. Because of the emergence of soft-ionization methods such as ESI and MALDI,24,25 it became possible to successfully apply these two ionization modes in various fields, mainly for the purpose of intact molecular mass measurements. In fact, the rapid progress in the field of proteomics was primarily driven by applying these two ionization modes. MALDI generates predominantly singly protonated ions of intact peptide or protein molecules, whereas multiply protonated ionic species of proteins or peptides are detected/generated by means of ESI.(2) The charged droplets would then begin to disperse from the Taylor cone (atmospheric pressure) towards mass analyzer, which is housed in high vacuum region. Consequently, the charged droplets move under the influence of pressure as well as electric potential gradient. During the course of this movement, the sizes of the droplets begin to shrink, attributed to the processes of ‘Coulomb explosion’ and ‘desolvation’. When the population of like-charged analyte ions contained within a drop exceeds a certain limit (Rayleigh limit), Coulomb explosion happens, because under that circumstance the electrostatic repulsive forces between the like-charged analyte ions exceed the surface tension of that drop, leading to division of those drops into smaller-sized droplets. Desolvation is accomplished by supplying heated dry nitrogen gas (about 200–300 °C), opposite to the direction of the flow of the charged droplets, which facilitates evaporation of solvent molecules, e.g., water, methanol, leading to reduction in the size of the droplets. The processes of ‘Coulomb explosion’ and ‘desolvation’ continue to take place until the analyte ions are completely devoid of any solvation. Thus, the ions are released in the gas phase or vacuum and then directed to the mass analyzer.
It is important to realize that MALDI is a pulsed ionization method, as it is accomplished by use of ‘pulsed’ lasers, typically of nanosecond or picosecond pulse width, whereas ions are generated continuously by ESI.
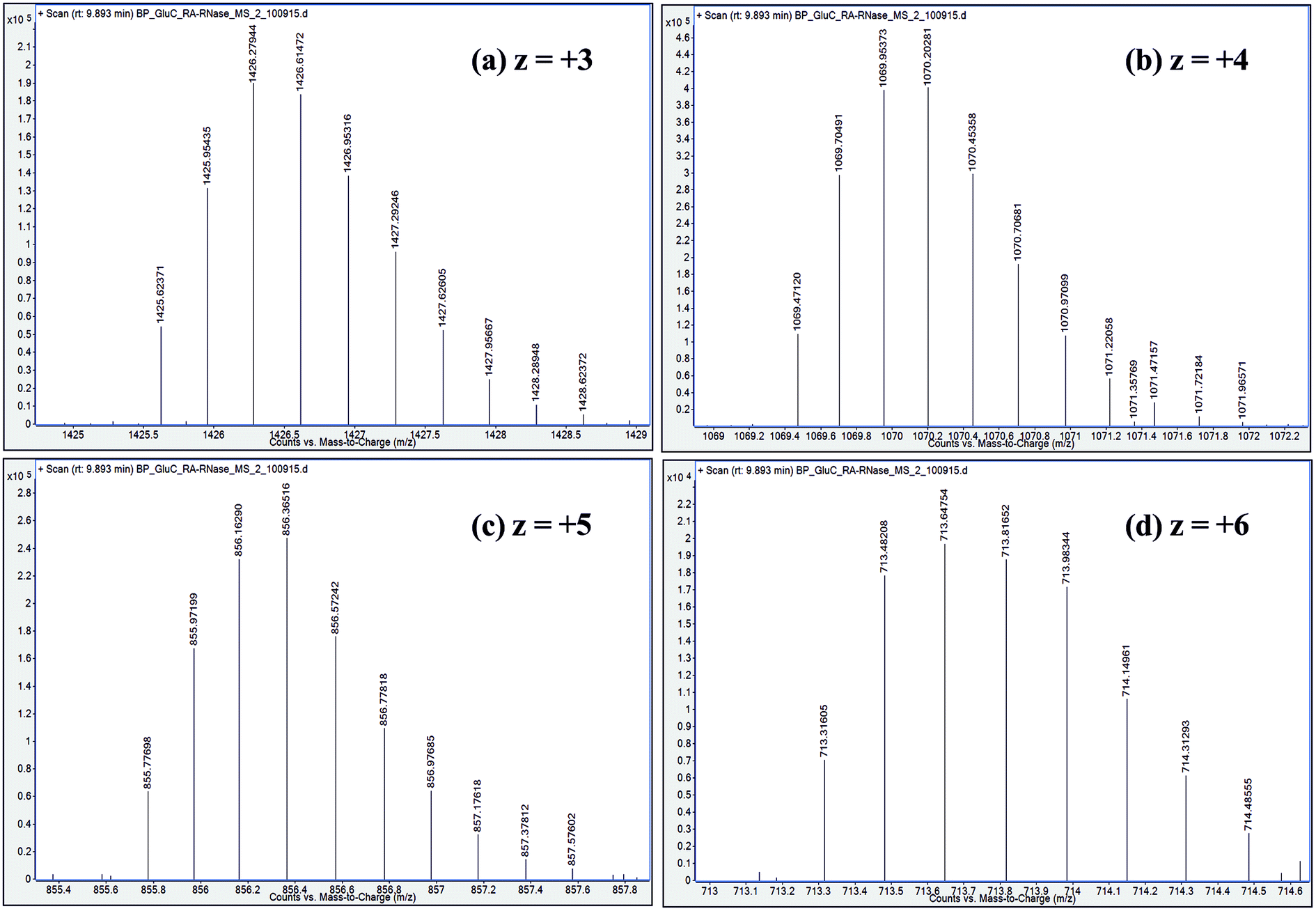 | ||
| Fig. 5 Distribution of isotope peaks corresponding to charge states: z = +3, z = +4, z = +5 and z = +6, observed in ESI mass spectrum of a GluC proteolytic peptide (Residues No. [50–86], 37 a.a. residues long), zoomed-in from the spectrum shown in Fig. 2b. | ||
2.3.4.1 Quadrupole. Quadrupole is the one of the earliest and widely used mass analyzers consisting of four cylindrical rods, which are arranged in a parallel manner that would have hyperbolic cross-section.162 A combination of direct current (DC) and radio frequency (RF) voltages are applied to opposing pair of rods. The ions are trapped and guided from one end to the other end of the quadrupole by suitably varying the RF and DC voltages. The stability of the ions' trajectories can be understood and manipulated through the Mathieu's stability diagram, from which it is possible to determine the optimum values of RF and DC voltages meant for a particular configuration of quadrupole.162 Thus, under certain conditions of RF and DC potentials, those ions that have ‘stable’ trajectories would be detected and their respective m/z values would be measured. Those ions traversing with unstable trajectories would go undetected.
By applying apt RF and DC voltages, ions of a particular m/z value can be trapped within the quadrupole and the ions of other m/z values would not be let into the quadrupole. Therefore, quadrupole can also function as a ‘mass filter’, in addition to being used for conventional mass analysis or mass measurements. The virtue of the quadrupole as a ‘mass filter’ enables it to be applied for tandem mass spectrometry (MS/MS), whereby it is used for the purpose of precursor ion selection (cf. vide infra).
Quadrupole mass analyzer mostly offers unit mass resolution. Most of the BUP studies have been carried out in quadrupole based mass spectrometers quite successfully. Thus far, only a few MD studies have been conducted in quadrupole containing spectrometers, where the quadrupole works as mass filter for precursor ion selection, while the fragment or product ions' analysis is done by high resolution mass analyzers, such as ion cyclotron resonance (ICR) and orbitrap, viz., Fourier Transform MS (FTMS).63,73,101,163
2.3.4.2 Ion trap. Ion trap is another most commonly used mass analyzer, which consists of three electrodes, where a ring electrode is placed between two end cap electrodes.162 A quadrupolar-like field can be produced within the ion trap by applying RF voltage to the central ring electrode only (and no potentials to the two end-cap electrodes), and this field can capture and store the ions (in millisecond timescale) within the trap. It is possible to formulate Mathieu's stability diagram for this three-electrode design of ion trap also, thereby optimum RF and DC voltages to achieve stable ion trajectories can be known. Therefore, an ion trap can function as both mass filter as well as conventional mass analyzer.162,164 A fascinating aspect about the ion trap is that multi-dimensional tandem MS experiments, viz., MS2, MS3, MS4, etc. can be performed within this ‘single’ device,162,164–167 which is not possible by a single quadrupole. In other words, tandem MS experiments in an ion trap are performed as a function of time (millisecond timescale).30
While this configuration of three electrodes (Paul type) is called as ‘three-dimensional (3D) ion trap’, there have been attempts to build two-dimensional ion traps also based on the quadrupole (four electrode system), for example, linear ion trap (LIT) and linear trap quadrupole (LTQ).168–170 LIT or LTQ has certain advantages over the 3D ion trap, such as, better ion trapping efficiency, greater ion storage capacity and lesser space-charge effects.158,169 Further, multi-dimensional MS/MS experiments are possible to be effected in LTQ instruments also.169,171 Several MD approach based studies have been accomplished using LTQ for MS/MS experiments, which are then analyzed in mass analyzers used in FTMS: ICR172 as well as orbitrap.68,70,83,88,89,139 Among these MD studies, Fenselau's group reported about the utility of LTQ coupled to orbitrap for collision induced dissociation (CID) MS/MS,88,89 whereas it may be interesting to note that Jennifer Brodbelt and co-workers implemented ultraviolet photodissociation (UVPD) method in a dual linear ion trap for MS/MS experiments, by adopting a MD proteomic workflow.139 In addition, there are several MD based investigations that report on the use of electron transfer dissociation (ETD) MS/MS carried out in LTQ-Orbitrap.70,71,78,81–83,99,173
2.3.4.3 Time-of-flight (TOF). In TOF, mass analysis is based on the time taken by the ions to reach the detector. The ions are pushed or accelerated into an empty hollow tube of known length, which is maintained at a high vacuum. The acceleration of ions is achieved by applying a very high voltage, e.g., about 15–20 kilovolts (kV) and the usual length of the TOF tube would be about 1 metre. There are two different ways of operating a TOF analyzer: (1) linear mode and (2) reflectron mode.30,158
In the linear mode of operation, ions traverse in the field-free region. All the ions in the analyte receive same electrical energy due to the acceleration potential and hence, those ions of lighter mass would travel at a higher velocity than the ions of heavier mass; which can be understood from the following equations:
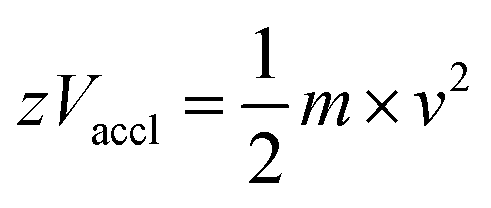 | (1) |
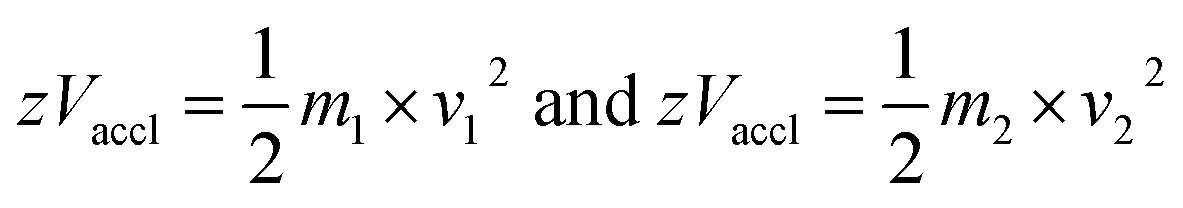 | (2) |
If m1 < m2 and z of m1 = z of m2, then v1 > v2, because Vaccl is same (or fixed) for all the ions.
Since length of the tube is known (L) and times of flight of every ion are measured experimentally, the m/z value of every ion can be determined from the following equation:
| m/z = 2 × Vaccl × (L/t)2 | (3) |
But, operating TOF in linear mode may not offer good resolution, since the entire population of the ions of ‘same m/z value’ may not reach the detector at the same time, leading to peak broadening. Consequently, masses may not be determined accurately. This problem can be overcome by following reflectron mode, where the ions are not allowed to move in field-free region. In reflector TOF, suitable electric potentials are applied to a set of electrodes that are appropriately positioned within the hollow tube and this helps to alter the trajectories of the ions of the same m/z value; whereby the path of the slowly moving ions is adjusted to a shorter distance and the path of the faster moving ions is altered to take up a longer distance. Thus, by following reflectron mode, the ‘entire population’ of the ‘ions of same m/z value’ could be ‘focused’ appropriately to reach the detector at the ‘same time’, thereby enabling accurate mass determination. Another method to improve mass resolution is ‘delayed extraction’, where the accelerating potential would be momentarily not applied for a very short duration, viz., 100 nanosecond or 500 nanosecond, just after the application of laser pulse (for desorption/ionization), which helps in better or enhanced focusing of the ‘whole population’ of the ‘ions of same m/z value’ to reach the detector at the ‘same time’.30,158
Not many MD investigations report the involvement of TOF based mass spectrometer, excepting a few, which have been carried out on antibodies to verify their sequences and to probe their extent of heterogeneity.104,105,133
2.3.4.4 Fourier transform mass spectrometry (FTMS). Applying ‘Fourier Transform (FT)’ mathematical operation for mass spectrometric data processing leads to tremendous improvement in resolution, accuracy and sensitivity.158 The two mass analyzers: ICR and orbitrap, incorporate FT for data processing and consequently, these two analyzers are capable of offering high resolution, better mass accuracy and enhanced sensitivity.41 In both ICR and orbitrap, a broad range of frequencies due to ionic motions are measured and by applying FT to these measured frequencies, m/z values of the ions are determined. Since MD approach involves characterization of the longer or medium-sized peptides (or polypeptides), high resolution mass analyzers are preferred and FTMS can be a technique of choice (cf. vide infra).
2.3.4.5 Fourier transform-ion cyclotron resonance (FT-ICR). Ion cyclotron resonance (ICR) is the first mass spectrometric technique to which FT was applied and therefore, it is customary to refer this as FT-ICR-MS.174,175 In FT-ICR, ions are analyzed by applying both magnetic field and RF (alternating current: AC) voltage. The measurement of m/z values of ions is based on their rotational frequencies in the presence of magnetic field, i.e., cyclotron frequencies. Each ion would have its own characteristic cyclotron frequency depending on its m/z value (see eqn (4)). In other words, ions of higher m/z values would traverse the circular path with lower cyclotron frequencies and vice versa.
| m/z = B/2πf | (4) |
It is important to note that the determination of m/z value is dependent only on the cyclotron frequency (angular velocity) and independent of the linear velocity of the ion.
When the frequency of RF or AC voltage (electromagnetic wave; which would be applied orthogonal to the direction of magnetic field), suitably matches with the cyclotron frequency of the ions, a condition of ‘resonance excitation’ is achieved. Such resonantly excited ions would then move in a circular path of a larger radius. Thus, by resonant excitation, the circular trajectories corresponding to ions of every m/z value are brought closer to the detection plates and the signals are detected in the form of ‘image currents’. These signals encompass a broad range of various cyclotron frequencies and hence, such a dataset would need to be deconvoluted by applying FT. Subsequently, the mass spectrum is plotted utilizing the cyclotron frequency values.
There are certain technical difficulties for online coupling of LC to FT-ICR-MS.176 Although, a few studies on the application of online LC-FT-ICR-MS are reported, there are not many investigations on its utility to proteomics. Nevertheless, FT-ICR-MS has been well applied for TD investigations of proteins, where the samples have been introduced into the ICR by means of direct infusion (viz., offline nanoESI).177,178
2.3.4.6 Orbitrap179. Orbitrap is another high resolution mass analyzer that utilizes FT for data processing. It consists of two outer barrel-like electrodes, which are co-axial with an inner spindle shaped electrode.40,180 A special aspect about orbitrap is that it utilizes only electrostatic fields to trap ions for m/z measurements. The ions not only orbit around the central spindle shaped electrode, but also oscillate in the axial direction. The frequencies of such axial oscillatory motions are recorded as image currents and these frequencies are then deconvoluted by applying FT, in a manner similar to FT-ICR. The frequency of the oscillation depends on the m/z value of the ion and it is proportional to (m/z)−1/2.
Orbitrap was first commercialized by coupling it with LTQ (Thermo) and subsequently, different ionization modes such as ESI and MALDI have been combined with orbitrap for high resolution mass analysis,41 thereby orbitrap found numerous applications in proteomics.181–183 And there are a good number of MD proteomic approach based studies reporting the utility of orbitrap, since MD investigations demand high mass resolution (cf. vide infra).
2.4 Tandem mass spectrometry (MS/MS)
MS/MS occupies a very important position not only in proteomics, but in several other studies that involve structure elucidation process of different types of molecules. In fact, MS/MS is now indispensable for any kind of proteomic studies. The three key steps in any MS/MS experiment are: (1) selection or isolation of precursor ion, (2) activation or fragmentation of the isolated precursor ion, leading to the formation of fragment ions or product ions and (3) detection and plotting mass spectrum of product ions, which is called as MS/MS spectrum. The first step, which is the isolation of precursor ion is mostly accomplished by quadrupole, which is capable of functioning as a mass filter; ion traps are also used for precursor ion isolation. Various methods have been developed to carry out the second step, viz., for effecting fragmentation of molecular ions (i.e. precursor ion), among which collision induced dissociation (CID) method has found tremendous applications.31,32,187 CID can be carried out in a quadrupole or a hexapole or an ion trap (linear as well as three-dimensional) or in an ICR cell.The process of CID involves kinetic excitation of the selected precursor molecular ions that are allowed to collide with neutral inert gas, referred as ‘collision gas’, such as helium (He), nitrogen (N2) and argon (Ar). Zero grade air is also used for CID, but in selected instruments only. As a result of inelastic collisions with the neutral inert gas (atoms or molecules), the precursor ions begin to dissociate, which may become observable beyond a certain threshold value of the kinetic energy. In other words, when the amount of energy that is applied for kinetic excitation exceeds the internal energy of the precursor ion, the precursor ion starts to fragment. Thus, by suitably altering the kinetic energy that is deposited on the precursor ion for its excitation, which can also be referred as ‘collision energy’, it would be possible to effect complete dissociation of the entire structure of the precursor ion, giving rise to fragment ions or product ions. The degree of dissociation of the isolated precursor ion is also dependent on the property of its molecular structure, meaning certain chemical bonds of the precursor molecular ion would readily dissociate, whereas certain bonds would be refractory towards dissociation, at a particular value of collision energy. In other words, a specific value of collision energy would cause vibrational excitation of only a few selected chemical bonds in the precursor molecular ion, while some other chemical bonds would be vibrationally excited at a different or higher collision energy values. Thus, when the energy of the collision (that takes place between the neutral gas atoms/molecules and the precursor ion) exceeds the energy of the chemical bond(s) (viz., bond energy) in the precursor molecular ion, fragment ions or product ions are produced. Dissociation of different chemical bonds results in the generation of fragment ions of different sizes, i.e., different m/z values. Consequently, the m/z values of the product ion peaks in CID MS/MS spectrum are useful to elucidate or to ascertain the molecular structure. Not only collision energy, the extent of fragmentation can also be varied or optimized by adjusting the collision gas pressure, whereby the number of collisional events occurring between the neutral gas atoms/molecules and the precursor ions can be controlled. But, in some instruments, the option of changing the collision gas pressure may not be available, as the manufacturers preset or fix the pressure of the collision gas at a particular value and would permit the end-users for optimization of collision energy only. Another vital factor that severely impacts the extent of the precursor ion's fragmentation is, the charge state or protonation state, viz., the number of protons (or charges) appended on the precursor ion and this is true in many cases, when CID is carried out with ESI. In this regard, precursor ion of higher charge state would tend to undergo better fragmentation than the lower charge state of the precursor ion.
Further, MS/MS data can be acquired by two different modes: (1) data dependent acquisition (DDA)71,75,82 and (2) data independent acquisition (DIA).79 DDA relies on the ionic intensities or abundances of the precursor ions for MS/MS data acquisitions, whereby a few ‘topmost intense’ precursor ions are only selected for activation or fragmentation. In contrast, when all the ions, irrespective of their relative abundances are subjected to fragmentation, it is called as data independent acquisition, which means MS/MS data are acquired independent of the intensity or abundance of the precursor ions. So, in DIA process, all those ions that successfully reach the mass analyzer from the source would be precursor ions, whereas by DDA mode, MS/MS fragmentation process is allowed to take place solely on a selected number of ions and the criterion to choose a narrow range of precursor ions for MS/MS is based on their ionic intensities.
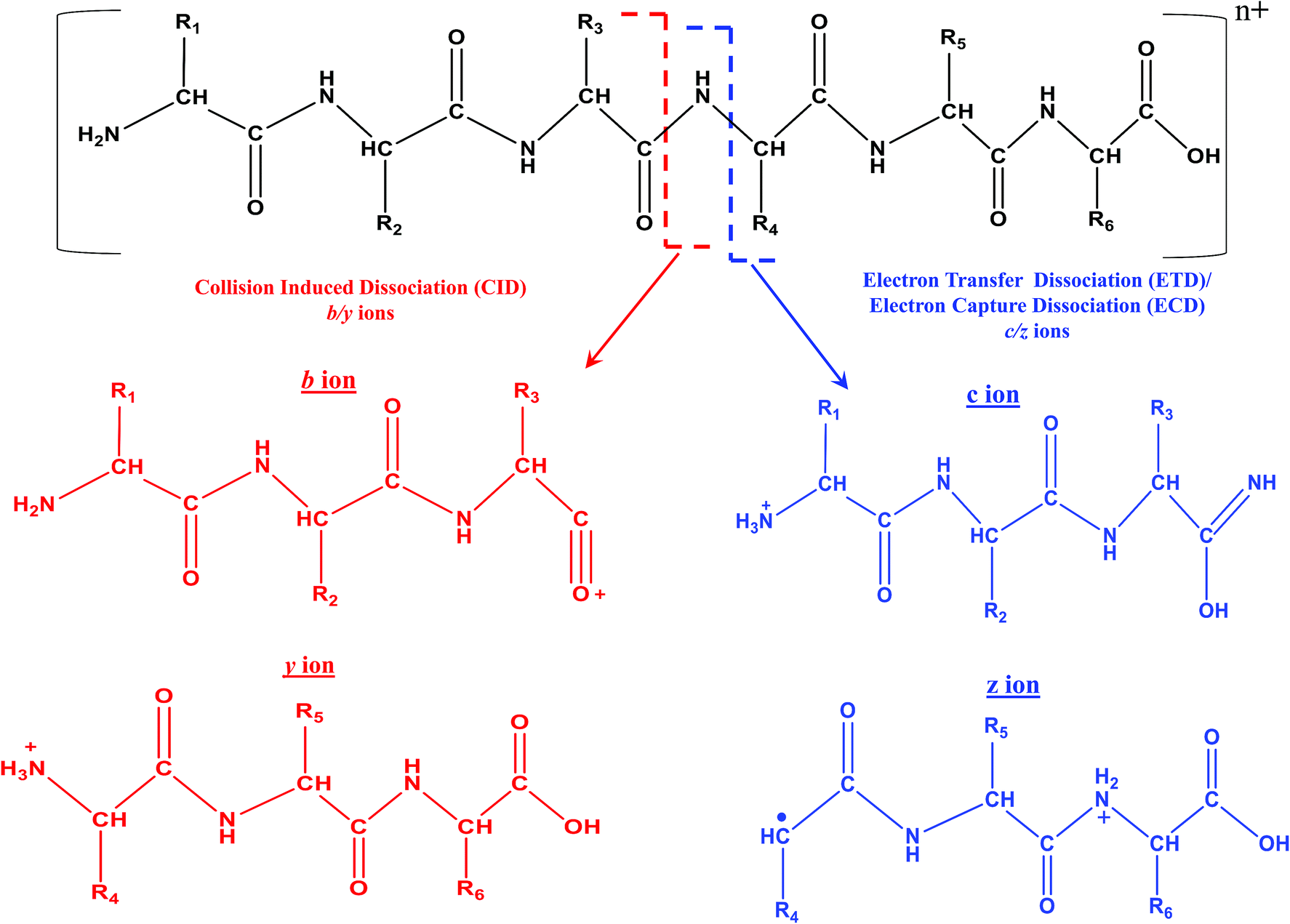 | ||
| Scheme 4 Representative fragmentation pattern of a hexapeptide due to CID and ECD/ETD MS/MS. Molecular structure of different types of product/fragment ions resulting from CID and ECD/ETD MS/MS. In the case of MALDI, the charge state (n+) on the peptide is n = 1 and for ESI, n ≥ 1. In the case of ESI, higher values of n are possible, depending on the length and nature of amino acid residues constituting the peptide/protein. ECD and ETD MS/MS are not possible with MALDI, when n = 1. | ||
In fact, ECD was observed serendipitously for the first time by Zubarev and Kelleher in McLafferty's research lab.33 An interesting aspect is that ECD of multiply protonated proteins or peptides would predominantly yield c- and z-type ions, viz., c′ and z˙ ions (see Scheme 4), which arise from radical mediated cleavage process of the backbone N–Cα bonds.33 Hence, the information obtained from ECD is complementary to that of CID. Despite its suitability for TD analysis of proteins, ECD could be performed in ICR mass spectrometer only.35 As a consequence, research attempts began in order to accomplish ECD like process in ion trap or quadrupole, which are not suitable analyzers to trap or store electrons. Eventually, such attempts paved way for the creation of a new MS/MS method called ‘electron transfer dissociation (ETD)’, which is actually an ion–ion reaction that is allowed to take place between an anion or radical anion (anthracene or fluoranthene) and the protonated protein/peptide precursor, within a quadrupole or two-dimensional or three-dimensional ion trap.35,200–202 Under such reaction conditions, the anion or radical anion is the source of electron, which upon transferring to the protonated peptide/protein precursor results in the formation of ‘unstable’ radical cationic species of peptide/protein precursor that undergo dissociation. Intriguingly, the process of ETD too yields c- and z-type of ions, again attributed to the radical mediated cleavage process of backbone N–Cα bonds of peptides/proteins (see Scheme 4).35 It is important to realize that ECD and ETD can be carried out on multiply charged precursors only, viz., minimum prerequisite to carry out ECD or ETD is doubly protonated precursors: [M + nH]n+, n ≥ 2. Since, ETD could be accomplished in ion traps itself there was a rapid progress in applying ETD MS/MS for proteomics. Prior to applying for proteomics, the efficacy of ETD was evaluated in certain BU approach based model cases and it was observed that significantly greater number of tryptic or LysC or LysN peptides were identified by ETD MS/MS than CID MS/MS, whereby a larger proportion of these identifications were from the precursor charge state, +3 ≤ z ≤ +5.203,204 Moreover, unlike CID MS/MS, the primary structure (viz. sequence) of peptides would not drastically influence the extent of fragmentation by ETD (BU approach) and hence, a better sequence coverage was obtainable from ETD of peptides than CID MS/MS, in BU approach based studies.35,203,205 Another virtue of ETD MS/MS was that it could be utilized to identify PTMs and consequently, the application of ETD particularly for phosphoproteomics proved to be beneficial.35,206–213 Additionally, ETD has been of use to elucidate glycosylated modified sites on proteins and peptides.214–216 Because MD approach principally focuses on the longer proteolytic peptides, ETD (or ECD) based MS/MS were the most sought method(s) of choice, which not only aided in yielding better sequence coverage, but also useful for PTM identifications (see Table 4).
| S. no | Chromatographya (for online LC-MS/MS) | LTQ-Orbitrap: MS/MS methods | Samples | References | |
|---|---|---|---|---|---|
| CID or HCD | ETD | ||||
| a Offline chromatographic methods used for fraction collection prior to LC-MS.b RPLC (C18) used as smaller trap column prior to WCX-HILIC.c UVPD.d Studies on histones.e Supplemented by collision induced decay (CID) using a collision energy of 10%.f Q-FTICR. | |||||
| 1 | RPLC (C8) | CID | — | Ubiquitin | 65 |
| 2 | RPLCa (C8) & WCX-HILIC | — | ETD | HeLa S3 cellsd | 92 |
| 3 | RPLC (C18) | CID | — | MCF 7 breast cells | 88 |
| 4 | Gel-filtrationa | — | ETD | Apomyoglobin, BSA, RHD3 | 69 |
| 5 | RPLC (C18) | CID | — | RPMI 8226 myeloma cells | 89 |
| 6 | RPLC (Phenyl) | — | — | Monoclonal antibody | 102 |
| 7 | RPLCa (C18) & WCX-HILIC | — | ETD | Mouse embryonic stem cells (ESC)d | 184 |
| 8 | RPLC (C4) | — | ETD | Monoclonal antibodies | 70 |
| 9 | RPLC (C18)-WCX-HILICb | — | ETD | Mouse ESC d | 76 |
| 10 | RPLCa (C18) & WCX-HILIC | — | ETD | Histonesd | 81 |
| 11 | SECa & RPLC (C4) | — | — | Mustard allergen | 138 |
| 12 | RPLC (C18)-WCX-HILICb | — | ETD | Caenorhabditis elegansd | 173 |
| 13 | RPLC (C18) | CID | ETD | HeLa S3 cellsd | 83 |
| 14 | WCX-HILIC | — | ETD | Human hepatocytesd | 82 |
| 15 | WCX-HILIC | — | ETD | C. elegansd | 78 |
| 16 | RPLC (C5) | — | ETD | Monoclonal antibodies | 99 |
| 17 | RPLC (C8) | CID/HCD | ETD | Monoclonal antibodies | 71 |
| 18 | RPLC (C8) | HCD | — | Model proteins (e.g. enolase, RNase A, lysozyme, etc.) | 90 |
| S. no | Chromatographya (for online LC-MS/MS) | Orbitrap fusion: MS/MS methods | Samples | References | |
|---|---|---|---|---|---|
| CID or HCD | ETD | ||||
| 1 | WCX-HILIC | HCD | EThcD | Murine erythroleukemia cellsd | 91 |
| 2 | RPLC (C18)-WCX-HILICb | — | ETD | HeLa S3 cellsd | 77 |
| 3 | RPLC (C18) | HCD | EThcD | Monoclonal antibodies | 85 |
| 4 | RPLC (C4) | — | ETD | Monoclonal antibodies | 68 |
| 5 | RPLC (C18) | HCD | EThcD | HeLa cells | 64 |
| 6 | RPLC (polymer) | — | ETD | Ubiquitin | 67e |
| 7 | RPLC (polymer) | HCD | — | Ubiquitin, myoglobin, carbonic anhydrase | 139c |
| 8 | RPLC (C18)-WCX-HILICb | — | ETD | HeLa cellsd | 79 |
| S. no | Chromatographya (for online LC-MS/MS) | LTQ-FT-ICR: MS/MS methods | Samples | References |
|---|---|---|---|---|
| 1 | RPLCa (C18) | ECD | Rat brain tissuesd | 63 |
| 2 | RPLCa(C18) | ECD | Ten rat tissuesd | 73 |
| 3 | RPLCa | ECD & CAD | Cardiac myosin binding protein C | 84 |
| 4 | RPLC (C18) | CID & ETDf | Human serum peptides | 260 |
| 5 | RPLC (C18) | ECD | Calf thymus, HeLa, Jurkat, MCF-7d | 75 |
| 6 | Chip based nano-ESI | ECD | Ubiquitin | 66 |
| 7 | RPLCa (C3) & WCX-HILIC | ECD | Murine erythroleukemia cellsd | 186 |
| 8 | SECa & RPLC (polymer) | ECD | Cardiac myosin heavy chain | 185 |
Another important aspect about ETD is the application of supplemental collisional activation concurrent with ETD, which was referred as ETcaD.217 ETcaD method was developed to specifically fragment the intact or undissociated ‘electron transferred (ET)’ product (radical) species produced from the doubly protonated [M + 2H]2+ tryptic peptide precursor ions. Although, ETcaD enabled in enhancing the sequence coverage better than that was observed from ETD alone or CID, it also triggered the process of hydrogen atom (radical), viz., H˙ migration, which was observed preponderantly with the [M + 2H]2+ tryptic peptide precursors, during the course of ETcaD. Such a kind of H˙ migration resulted in the formation of odd-electron c˙ (c−1) and even-electron z′ (z+1) fragment ions, which were often detected besides the usually observed even-electron c′ and odd-electron z˙ ions in the ETD MS/MS spectrum of [M + 2H]2+ tryptic peptide precursors.217 However, the extent of H˙ migration was observed to be less pronounced, when ETcaD was carried out over the triply and other higher charge states of the proteolytic peptide ions.204,218 Consequently, most of the ETD MS/MS experiments are performed by applying supplemental collisional activation on an appropriate charge state of the precursor ion. It is important to note that such a ETcaD process, when carried out in Paul-type (3-dimensional) ion trap (Bruker Daltonics), it is called as ‘Smart Decomposition’,218,219 whereas when the supplemental activation is applied by means of higher energy collision dissociation (HCD; Thermo Scientific), then it is referred as EThcD.210,216 Over a period of time, instruments became available that had the option to vary even the collision energy corresponding to the supplemental activation, which is applied during the process of ETD; for instance, Yang et al. had applied 20% of supplemental activation energy for EThcD, whereas Cristobal et al. used 40% supplemental activation energy for their EThcD experiments (also see Table 4).64,85,91
In the case of ECD also, supplemental activation has been applied through different procedures such as increasing temperature of the ICR cell,220 by infrared (IR) irradiation221 or by in-beam collision activation with a background gas (N2).222 These activation methods were devised in order to aid in disrupting the probable non-covalent interactions that may not allow the release of the backbone dissociated c-type and z-type ions.220,222 Such activation processes have helped in improving the yield of c-type and z-type of ions, thereby facilitating enhancement of the sequence coverage,221,222 but additionally, even-electron z′ (z+1) and odd-electron c˙ (c−1) fragment ions too are observed under the activated conditions, indicative of hydrogen atom or H˙ migration.220,221 In fact, hydrogen atom (H˙) transfer has been a commonly occurring process giving rise to even-electron z′ (z+1) and odd-electron c˙ (c−1) fragment ions, even under normal conditions of ECD without application of any kind of activation, as evidenced from the ECD MS/MS data of 15000 tryptic peptides, acquired from their respective [M + 2H]2+ precursors.223 Tsybin et al., have reported decreased ratio of population of (c˙/c′) and increased ratio of abundance of (z˙/z′), by applying IR radiation (vibrational excitation) prior to the ECD process.224
Overall, the above described details pertaining to ECD and ETD illustrate the significance of charge state of the precursor ion, in impacting the extent of fragmentation by ECD or ETD, so as to obtain better sequence coverage unambiguously. Table 4 summarizes various methods of chromatography and MS/MS (viz., online LC-MS/MS) that have been carried out on different instruments for MD investigations on different biological samples. Extensive applications of ETD and ECD MS/MS for MD approach based studies on a variety of biological systems are evident from Table 4.
2.5 Data analysis & database search engines: computational methods
 | ||
| Scheme 5 Venn diagram representation of various computational tools used for each of the three different proteomic approaches. The tools shown within the category of TD approach are available at https://www.topdownproteomics.org/resources/software/. | ||
Additionally, certain special computational software packages have been developed for the purpose of ‘quantification’ of peptides in BU proteomics, such as, MaxQuant,239,240 Skyline,241,242 Census,243 etc. While several of these software programs meant for quantification are commercial that are license-protected, a good number of open-sourced software programs are also freely available.244
Peptide-based protein identification by BU approach can also been performed by means of matching with the MS/MS spectral library that have been catalogued from the experimental data.245 This approach can be more reliable for identifying peptides and proteins than the typical search method based on sequence databases, since the intensities of peptide fragment ions (i.e., b-ions and y-ions, obtained by CID MS/MS) in the MS/MS spectrum are also taken into account during the process of matching with the MS/MS spectrum library.246,247
When TD proteomic experiments are carried out by involving offline direct infusion mode of sample introduction, mixture of proteins would be simultaneously introduced into FT-ICR mass spectrometer, which would result in spectra containing isotope peaks of many proteins overlapping with one another. Hence, both deisotoping as well as deconvolution processes are required to analyze such convoluted data. In order to decrease the complexity of the mixture and introduce only simple mixtures (that would contain three or four proteins) into FT-ICR mass spectrometer, pre-fractionation or chromatographic steps are necessary prior to performing offline direct infusion61 or by LC-MS mode, so that deisotoping can be performed on the data acquired from a simple mixture in a relatively easier manner. For example, anion exchange chromatography has been applied preceding the online reverse phase LC-FT-ICR-MS for the TD proteomic analysis of Shewanella oneidensis MR-1 and Saccharomyces cerevisiae.58,248 In the case of Shewanella oneidensis MR-1, THRASH algorithm249 was used to process and analyze the TD mass spectral data, whereas ProSightPC and ProSight PTM250 were utilized for analyzing the TD data acquired on yeast. The deconvolution and deisotoping processes are incorporated within the THRASH algorithm and ProSight PTM. Another key aspect about TD approach is identification of PTMs, which are given importance in database search engines and in the relevant software.251 Not only PTMs, but also proteoforms (isoforms) can be detected by TD approach and suitable software tools have been developed to analyze TD mass spectral data for the purpose of identifying proteoforms.251–253 LC-MS based TD proteomics has been performed with the orbitrap as well62,254,255 and different computational strategies mentioned above have been adapted to analyze the TD mass spectral or proteomic data arising from orbitrap too. In a recently published study, a new standalone software called VisioProt-MS was reported, which provides a 2D LC-MS map representation by taking-up LC-MS data of TD studies.256 Further, VisioProt-MS can take-up data from different types of instrument configurations, e.g., FT-ICR, orbitrap, Q-TOF and therefore, aids in better comparison of the TD LC-MS data.
Since, most of the MD proteomic investigations have been performed over histones, efforts have been focused to create software tool to analyze data recorded on histones, namely ‘isoScale’ and ‘Histone Coder’ that are available in the site https://middle-down.github.io.76–80,82,173 Skyline software also has been utilized for quantifying histones, which were characterized by MD approach.186 Other software, such as ProsightPC and MASH-Suite, which were primarily developed for analysis of TD proteomic data, have also been applied well for the purpose of analyzing MD proteomic data.66,67,70,88,89,99,172,185 Algorithms to process the raw data of MDP for the purpose of deconvolution are, Xtract, THRASH249 and MS-Deconv.75,91,257 Among these, Xtract and THRASH algorithms have been used mostly to analyze FT-MS data of longer proteolytic peptides.63,70,73,77,79,82,84,102,172 Further, Xtract has been used for the deconvolution of mass spectra obtained from N-terminal tails of histones, which were extracted from HeLa cells and Caenorhabditis elegans embryos for the detection of PTMs.79,173 Based on Xtract algorithm, an application software, called cRAWler 2.0 (Thermo Fisher Scientific, San Jose, CA) was useful to process LC-MS/MS data (Thermo Scientific raw data files).172 cRAWler has also been used using THRASH algorithm for the purpose of deconvolution.88 YADA is another software tool developed for deisotoping and decharging (deconvolution), which was applied on ProLuCID and DTASelect to analyze large-scale MDP data.258 XDIA is a computational strategy meant to exclusively analyze ETD MS/MS spectra for MDP, where it helps in enhancing the sequence coverage and increase the number of identified peptides; this was demonstrated by conducting experiments on complex sample, such as crude yeast cell lysate digested using LysC.259 Furthermore, BioTools of Bruker Daltonics has been useful to process and analyze data acquired on middle-down-sized peptides from human serum samples, which involved ESI-CID and ETD MS/MS, carried out using FT-ICR-MS.260 With regard to the application of MALDI-MS for MDP, a few studies have reported the utility of MALDI-in source decay (ISD) method to analyze middle-down sized peptides obtained from biosimilar monoclonal antibodies (mAbs) and in those studies, BioTools was used to process and analyze the data.104,105 Various kinds of software applied for BU, TD and MD approaches for analysis of proteins and proteome, have been summarized in the form of a Venn diagram in Scheme 5.
It should to be realized that the deconvolution or decharging and deisotoping are not required for the analysis of ESI-MS/MS spectra acquired in BU proteomic studies. This is because quite often, the charge state ([M + nH]n+) of the proteolytic peptide precursor ions encountered in BU approach, does not exceed 3 or 4, i.e., 1 or 2 ≤ n ≤ 3 or 4. In fact, majority of the ESI-MS/MS spectral data in BU proteomic studies are from [M + 2H]2+ (doubly protonated) or [M + 3H]3+ (triply protonated) of the proteolytic (typically tryptic) peptide precursors.203 Consequently, in majority of the cases, singly charged fragment ions are observed and rarely doubly and triply charged fragment ions are detected in BU studies. Moreover, there is no need for deconvolution for the analysis of MALDI-MS/MS data, be it BU or MD approach (MALDI-ISD studies in the case of MD approach), since MALDI-MS/MS experiments are always/mostly carried out on singly charged precursors only.
3 Applications of MD approach
Since, MD approach is relatively a new strategy, which was introduced about 5–10 years ago from now, thus far, there have not been many studies reporting about its application. A major motivation to put forth the concept of MD approach came from the investigations conducted on histones and consequently, MD strategy has been successfully applied predominantly on histones related studies, particularly for identification of PTMs on histones (see Table 4). This is because, PTMs on histones strongly influence epigenetic regulation of gene expression.261,262 Further, posttranslationally modified histones alter the structure of chromatin of eukaryotes, thereby modulate the chromatins' activities, such as gene transcription, DNA repair and DNA replication.263 Hence, studying variants of histones and their PTMs could be useful to understand the dynamics of variations occurring on chromatin; which in turn can be helpful to comprehend the causal factors of diseases.264,265 In this connection, MD strategy has been shown to be useful for better identification of novel isoforms of histones and their PTMs.73,76–79,833.1 PTMs on histones
With regard to identifying PTMs on histones, both MD and TD approaches have been followed, though there are relatively more studies reported on the application of MD workflow, in comparison to TD approach. For example, studies carried out on Caenorhabditis elegans have revealed different levels of methylated lysine (me-Lys), methylated arginine (me-Arg) and acetylated lysine (ac-Lys) in histone H3, whose relative abundances have been quantified at various developmental stages of this worm, through MD proteomic approach.78,173 N-terminal proteolytic processing of histones has an important role in nucleosome dynamics, and such proteolytic clippings have been noted in a study, wherein differences in PTM patterns (ac-Lys, me-Lys and me-Arg) were detected between intact and N-terminal tail clipped H2B and H3 histones in human hepatocytes by following MD in conjunction with TD approach.82 Additionally, MD approach also has been fruitful to successfully identify even ‘phosphorylation’ of histones (viz., H4) in HeLa S3 cells, whereby the phosphorylation status of serine was monitored along with the other PTMs, such as ac-Lys, me-Lys and me-Arg, at different stages of cell cycle.83 Additionally, in a very recently published study, phosphorylation of histones (H4) have been identified even in breast cancer cells (MDA-MB-231 and MCF-10A) at different cell cycle stages.266 Furthermore, tissue-specific variations in PTMs of histones H3 in a rat model was reported by Garcia et al., for which tissues from kidney, spleen, brain, bladder, lung, liver, heart, ovary, testes and pancreas were analyzed by MD proteomic approach.73 PTMs on histones have also been investigated and quantitated in mouse embryonic stem cells by MD based workflow, which involved the use of protease GluC and WCX-HILIC chromatography coupled online to ETD MS/MS.76,184 Very recently, Schräder et al. have reported MD strategy by using neprosin, a novel propyl-endoprotease for getting the middle-range sized peptides to characterize the H3 and H4 histones of HeLa S3 cells.267 It needs to be noted that BU approach may not be suitable to study histones, especially their PTMs.77,268 This is because N-terminal tails of histones are rich in lysine and arginine residues and therefore, very short length peptides would be produced upon carrying out trypsin digestion. Such short length peptides are not useful to identify the primary structures of the histones due to the ambiguities posed by the presence of paralogs and variants of histones. Also, these short length peptides are not suitable for identification of PTMs, in particular of co-occurring combinatorial PTMs.3.2 Therapeutic antibodies
In addition to its comprehensive application for the study on histones, MD approach has been proven to be successful for characterization of therapeutic antibodies too, particularly, during the last few years (see Table 4). For example, MD based mass spectrometric approach was shown to be useful to evaluate the quality of two anti-CD20 monoclonal antibody (mAb) drug products, whose structural analyses were carried out by using IdeS enzyme and LC-ESI-MS.102 In an investigation performed on a single immunoglobulin (IgG) and mixture of IgGs, Tsybin and co-workers demonstrated that extended BU approach by employing Sap9 protease was helpful to obtain up to 99% and 100% sequence coverage of heavy chain (Hc) and light chain (Lc) of the respective IgG.71 However, in another study the same group applied MD strategy by using IdeS enzyme on a sample containing mixture of commercial IgGs, and showed that this workflow yielded better sequence coverage than the TD approach.70It may be interesting to note that MD based approach has been applied along with hydrogen/deuterium exchange (HDX) for the structural characterization of a recombinant therapeutic antibody Herceptin (restricted pepsin digestion in conjunction with online ETD MS/MS) to probe the alterations to the three-dimensional structure of the antibody that could happen due to deglycosylation.68 In fact, this is the first study reporting about the applicability of MD approach together with HDX method on the antibody, which may be extended for structural characterization of other therapeutic antibodies and other proteins as well.
In addition to MD approach, in recent times, ‘middle-up’ approach is gaining importance, particularly for characterization of therapeutic antibodies. Unlike MD strategy, middle-up approach does not involve the application of MS/MS at all. Nevertheless, similar to MD strategy, in middle-up approach also, limited proteolysis is carried out. Therefore, in the case of middle-up investigations on therapeutic antibodies, the IdeS enzyme is often employed and the resulting three large polypeptide fragments from the immunoglobulins are analyzed in their intact form by high-resolution MS. For instance, Sokolowska et al. adopted middle-up strategy to analyze the subunits of certain therapeutic antibodies, which were digested by IdeS enzyme and the resulting polypeptide fragments were characterized by Q-TOF mass spectrometer (Waters Xevo).269 Bruker maxis Q-TOF instrument was used for middle-up characterization of a therapeutic antibody, Cetuximab.270 In another study, middle-up approach in combination with BU approach was applied to characterize antibody drug conjugate (Brentuximab vedotin) by utilizing Triple TOF mass spectrometer (Sciex).271
In addition to a wealth of studies carried out by ESI-MS/MS based MD approach, there have been efforts to apply MALDI as well for characterization of certain important commercial antibodies, which were analyzed by MD approach involving ‘In-Source decay (ISD)’ (Bruker); see Table 4.104,105,133
3.3 Other applications of MD approach
Other studies involving MD approach are characterization of ubiquitin,65–67 ribosomal proteins,88 mustard 2s albumin,138 leukocytes,163 nuclear proteins,172 serum peptides,260 and recombinant proteins;84 all these examples are included in Table 4. Among other biological effects of ubiquitin, a popularly known role is ubiquitin-mediated degradation of proteins in impacting the cell cycle events.272,273 Perturbation to such proteolytic process may lead to adverse effects such as uncontrolled proliferation, genomic instability and even cancer.272 Ubiquitin can covalently bind to the target protein in several modes, which involves modification of more than one site (i.e., ε-amino group of lysine) of the target proteins giving rise of different chain lengths, various kinds of linkages and configuration, for example, linear and branched.67 Consequently, it has become important to investigate different chain linkages and topological configurations of ubiquitin modified proteins, for which MD proteomic strategy has been shown to be quite successful.65,67,274 Furthermore, the utility of MD approach along with intact protein MS to analyze microheterogeneity due to PTM features (e.g., glycosylation) in recombinant human erythropoietin and human plasma properdin was shown.85 Additionally, MD approach also has been successfully applied for identification of many proteins, (i.e., typical proteomic analysis), e.g., several nuclear proteins from HeLa-S3 cells were identified by following LysC digestion172 and in another study, acid-hydrolysis in combination with microwave treatment was performed on ribosomal proteins in human MCF7 cancer cells.883.4 Combination of MD and TD approaches
Furthermore, MD approach has been applied in tandem with TD strategy, which involves MS/MS experiments done on intact proteins as well as middle-down sized proteolytic peptides.275 For example, the extent of micro-heterogeneity in glycosylated proteins, which include biopharmaceutical products, e.g., monoclonal antibodies, recombinant erythropoietin, etc. was investigated by combining MD with TD approach.85,99,100,103,276 Similarly, by integrating MD and TD methods, it was possible to obtain full sequence coverage of a 142 kDa cardiac myosin binding protein C (cMyBP-C), where MD was helpful to acquire details of the middle part of the sequence.84 Moreover, considerable number of investigations on histones by adopting both MD and TD workflows has been reported.63,81,82,268,277 Studies conducted on human leukocytes, mustard 2S albumin allergen and monoclonal antibody Fc/2 are other examples, in which both MD and TD approaches have been adopted.133,138,163 In the case of studies on mustard albumin allergen, combination of MD with TD workflow aided in identification of some isoforms of Sin a 1, which included a few novel isoforms too.138 In a very recently published study, Tsybin and co-workers have reported about multiplexed TD/MD MS workflow, which combines both TD and MD approaches, particularly for characterization of monoclonal antibodies.2784 Summary & conclusion
It is thus apparent from the previous sections that majority of MD based investigations have been performed on histones, a key reason being that the intact histone N-terminal tail (∼50–60 a.a. residues) has most PTMs.80 MD approach has also been shown to be very beneficial for the characterization of biopharmaceutically relevant antibodies and therapeutic proteins, since identifying PTMs is very important in these cases.68,70,71,85,99,102,104,105,133 In addition, certain applications of MD approach to determine the topological configuration of ubiquitylated proteins have been reported.65–67 Thus, it seems that excepting the studies conducted on histones, biopharmaceutical products and ubiquitinated proteins, MD approach has not been completely explored or not appropriately applied for research in other domains or fields yet, despite the availability of the knowhow and the necessary technological facilities to implement the MD workflow, in the current era. For instance, MD strategy can be useful for ‘elucidation of the primary structure’ of novel proteins or newly discovered enzymes that are of significance in translational research and/or of relevance to biotechnological research or industries. In other words, MD approach can be promising for the purpose of ‘de novo protein sequencing’, which can be pursued either on fully purified or even on partially purified chromatographic samples.Further, MD approach can be performed without the need for FTMS, meaning, MD approach based experiments can be done using Q-TOF (e.g. Sciex or Bruker or Waters or Agilent) or ion trap-TOF (e.g. Shimadzu), wherein the requirement for high resolution can be satisfactorily achieved with the help of TOF mass analyzer. Additionally, MD strategy can be extended for high-throughput work, viz., MD proteomics. Because of the development of new proteases such as OmpT, Sap9 and the already known enzymes, e.g., AspN and GluC, it is indeed feasible to carry out MD proteomics for microbial or plant investigations and this can be achieved in a manner similar to the workflow of BU approach, whereby MD proteomics may yield better results than BU proteomics. However, it is somewhat surprising to note that only a few studies report about applying MD approach for proteomics.88,89,172
Another domain, where MD strategy can be efficacious is ‘proteogenomics’, which is also emerging.279 The results obtained by MD approach can be integrated with the transcriptome and genomic data, not only to identify unannotated missing proteins and/or to confirm annotated coding regions in the genome, but also to correct either the erroneously annotated gene sequences or to correct the proteome sequence databases.280–282 In this connection, there is a huge scope for MD proteomic approach to contribute immensely for the development of this upcoming field, since investigations thus far involving proteogenomics have mostly followed the BU proteomic approach only.281,283,284
Additionally, it needs to be noted that there has been no accurate definition for MD approach, which could clearly demarcate the boundary between BU and MD approaches. For instance, Tsybin and co-workers defined or proposed another approach called ‘Extended Bottom-Up’, which may be regarded as an offshoot of MD approach.72,96 According to their definition, studies on proteolytic peptides of molecular masses in the range 3–7 kDa would be considered as ‘Extended Bottom-Up approach’, whereas investigations on 7–12 kDa proteolytic peptides were categorized under ‘MD approach’.72 In this regard, Tsybin's research group applied a novel protease, Sap9 (secreted aspartic protease 9) to evaluate its utility for ‘extended BUP’96 and they have also demonstrated the usefulness of this approach to characterize monoclonal antibodies by utilizing Sap9.71 But, according to our view, if a particular study on proteins is carried out with the help of ‘a protease’, whose usage results in the formation of (longer) proteolytic peptides of molecular masses > 2.5 kDa or > 3 kDa, then such an investigation can be called as a ‘middle-down’ based study of proteins or proteome. In other words, those studies that follow restricted or limited proteolysis using any protease that specifically produces peptides of molecular masses > 2.5 kDa or > 3 kDa can be regarded as an investigation adopting MD approach, e.g., Forbes et al. and Jung et al., have indeed adopted MD approach in their respective studies (as per our definition proposed herein), though they have not explicitly claimed or mentioned specifically about the approach.87,184
Overall, from our (re)view on the published studies reporting the utility of MD strategy that we have presented herein, it is clear that MD approach indeed has an immense potential in future to successfully implement the objectives in both low- as well as high-throughput works, i.e., for ‘sequencing’ purified protein(s) (one-by-one) as well as for ‘proteomics’. Indeed, 100% sequence coverage by MD approach is achievable, when it is applied on a highly purified and homogeneous protein sample, which can be (realistically) obtained by adept application of suitable chromatographic methods. And therefore, in the present scenario, MD based mass spectrometry does not seem to be surreal, though for proteomic scale, it may now be cumbersome.284 Perhaps, the current trend could undergo transformation in future paving way for brighter prospects for MD proteomics. Altogether, MD approach could be a better choice for identification and characterization of the protein or proteome, where it could serve to ‘bridge the gaps’285 observed in BU and TD studies and hence, it can become indispensable for fulfilling the objectives in various biological, biomedical, biochemical and biotechnological domains of research. The flow chart depicting various details and methods followed at different stages corresponding to each of the three approaches (BU, MD and TD) are illustrated in Scheme 6.
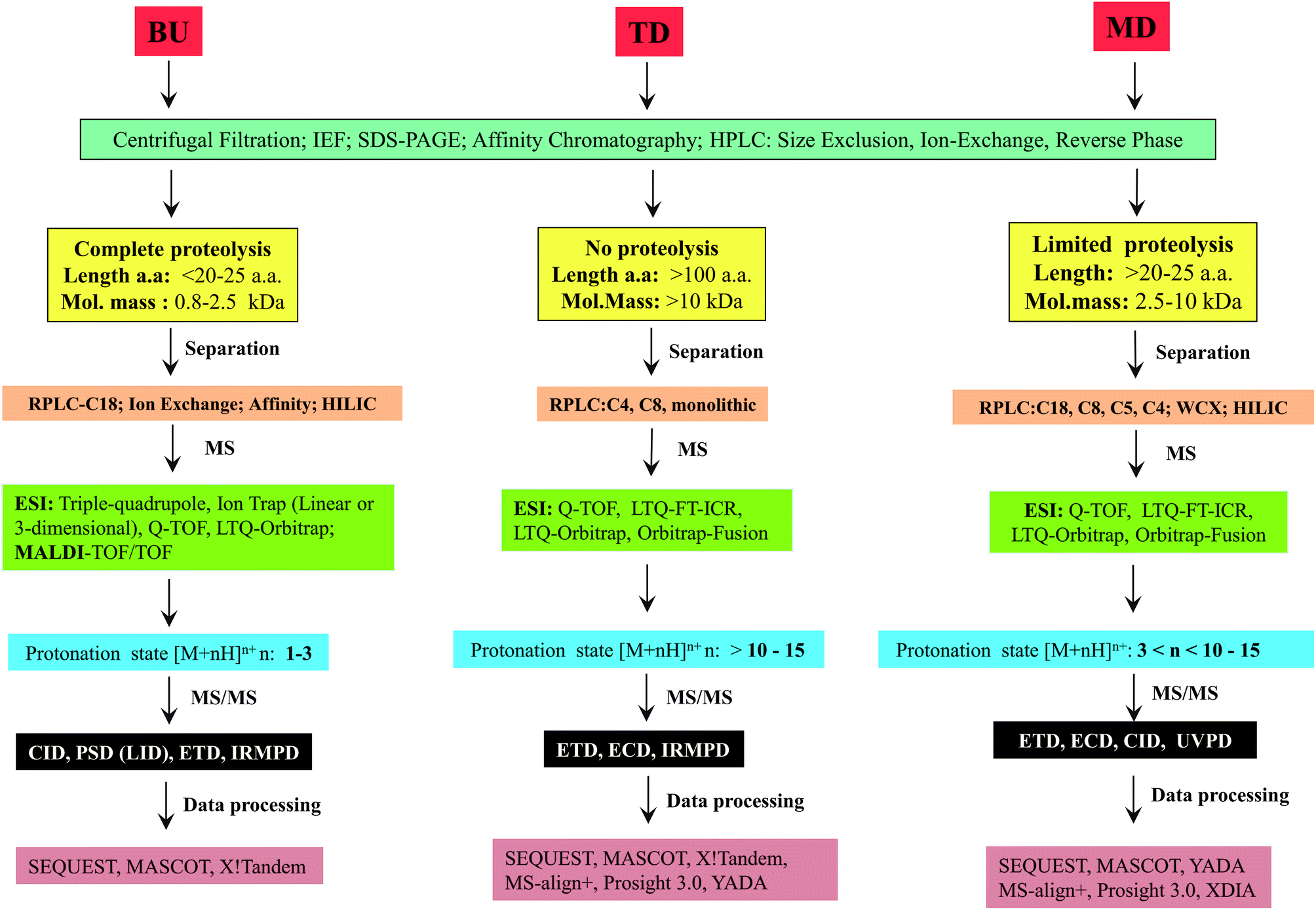 | ||
| Scheme 6 Flow chart depicting various details and methods followed for each of the three approaches shown in Scheme 2, for analysis of proteins or proteomes. | ||
Before concluding, we wish to highlight salient features of two published reports that describe the efforts put forward for de novo sequencing. For de novo sequencing of a 72-residue polypeptide, both BU and TD approaches had to be executed involving application of MALDI-FT-ICR, nano-LC-ESI-Q-TOF and nano-ESI-FT-ICR, through which various types of MS/MS experiments, e.g., CID, ECD and IRMPD were conducted.286 This example shows that multiple sophisticated mass spectrometric techniques were sought after to elucidate the sequence of a polypeptide of molecular mass less than 10 kDa. In another study, sequence of a protein of molecular mass greater than 10 kDa (i.e., 13.6 kDa) deduced de novo by TD MALDI-ISD, was confirmed by BU approach by use of LC-ESI-MS/MS.287 These two examples indicate that a single approach cannot be full-proof and may not answer the problem completely. Depending on the question that is being addressed, it may be imperative to integrate two or more methods for designing a strategy. But, in doing so, the pros and cons of those methods and the designed strategy also must be borne in mind. Accordingly, the results need to be interpreted. Based on the nature of the results obtained by implementing a particular strategy, it may be essential to redesign the strategy and the efficacy of the newly redesigned strategy may have to be evaluated. On a final note, the choice of approach or strategy must be based on the case or the problem or the question that is undertaken for investigation.288
Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. Boomathi Pandeswari thanks Council of Scientific and Industrial Research (CSIR), Govt. of India, for Senior Research Fellowship. V. Sabareesh wishes to acknowledge the completed Project No. SB/YS/LS-370/2013, ‘Start-Up Research Grant (Young Scientists)’, which was funded by Science and Engineering Research Board (SERB), Govt. of India.References
- P. Edman, Arch. Biochem. Biophys., 1949, 22, 475–480 CAS.
- P. Edman and G. Begg, Eur. J. Biochem., 1967, 1, 80–91 CrossRef CAS PubMed.
- R. J. Simpson, Proteins and Proteomics. A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 2003, I.K. International Pvt. Ltd., New Delhi, India Search PubMed.
- K. D. Speicher, N. C. Gorman and D. W. Speicher, Curr. Protoc. Protein Sci., 2009, 57, 11.10.1–11.10.31 Search PubMed.
- Y. Nishiyama, T. Fukamizo, K. Yoneda and T. Araki, Protein J., 2017, 36(2), 98–107 CrossRef CAS PubMed.
- A. Goffeau, B. G. Barrell, H. Bussey, R. W. Davis, B. Dujon, H. Feldmann, F. Galibert, J. D. Hoheisel, C. Jacq and M. Johnston, et al., Science, 1996, 274(5287), 563–567 CrossRef.
- F. R. Blattner, G. Plunkett III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau and Y. Shao, Science, 1997, 277, 1453–1462 CrossRef CAS PubMed.
- S. T. Cole, R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas and C. E. Barry 3rd, et al., Nature, 1998, 393(6685), 537–544 CrossRef CAS PubMed.
- E. S. Lander, L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K. Devon, K. Dewar, M. Doyle and W. FitzHugh, et al., Nature, 2001, 409(6822), 860–921 CrossRef CAS PubMed.
- J. C. Venter, M. D. Adams, E. W. Myers, P. W. Li, R. J. Mural, G. G. Sutton, H. O. Smith, M. Yandell, C. A. Evans and R. A. Holt, et al., Science, 2001, 291(5507), 1304–1351 CrossRef CAS PubMed.
- M. S. Kim, S. M. Pinto, D. Getnet, R. S. Nirujogi, S. S. Manda, R. Chaerkady, A. K. Madugundu, D. S. Kelkar, R. Isserlin and S. Jain, et al., Nature, 2014, 509, 575–581 CrossRef CAS PubMed.
- G. S. Omenn, L. Lane, E. K. Lundberg, R. C. Beavis, C. M. Overall and E. W. Deutsch, J. Proteome Res., 2016, 15(11), 3951–3960 CrossRef CAS PubMed.
- T. Maier, M. Güell and L. Serrano, FEBS Lett., 2009, 583, 3966–3973 CrossRef CAS PubMed.
- S. Haider and R. Pal, Curr. Genomics, 2013, 14, 91–110 CrossRef CAS PubMed.
- Y. Bai, S. Wang, H. Zhong, Q. Yang, F. Zhang, Z. Zhuang, J. Yuan, X. Nie and S. Wang, Sci. Rep., 2015, 5, 14582, DOI:10.1038/srep14582.
- H. D. Li, R. Menon, G. S. Omenn and Y. Guan, Trends Genet., 2014, 30, 340–347 CrossRef CAS PubMed.
- C. L. Barrett, C. DeBoever, K. Jepsen, C. C. Saenz, D. A. Carson and K. A. Frazer, Proc. Natl. Acad. Sci. U. S. A., 2015, E3050–E3057 CrossRef CAS PubMed.
- C. T. Walsh, S. Garneau-Tsodikova and G. J. Gatto Jr., Angew. Chem., Int. Ed., 2005, 44(45), 7342–7372 CrossRef CAS PubMed.
- T. Bartke, J. Borgel and P. A. DiMaggio, Briefings Funct. Genomics, 2013, 12, 205–218 CrossRef CAS PubMed.
- C. Choudhary, B. T. Weinert, Y. Nishida, E. Verdin and M. Mann, Nat. Rev. Mol. Cell Biol., 2014, 15(8), 536–550 CrossRef CAS PubMed.
- S. Doll and A. L. Burlingame, ACS Chem. Biol., 2015, 10(1), 63–71 CrossRef CAS PubMed.
- R. Aebersold and M. Mann, Nature, 2016, 537, 347–355 CrossRef CAS PubMed.
- J. E. Van Eyk, F. J. Corrales, R. Aebersold, F. Cerciello, E. W. Deutsch, P. Roncada, J.-C. Sanchez, T. Yamamoto, P. Yang, H. Zhang and G. S. Omenn, J. Proteome Res., 2016, 15(11), 3979–3987 CrossRef CAS PubMed.
- K. Tanaka, Angew. Chem., Int. Ed., 2003, 42, 3861–3870 CrossRef CAS PubMed.
- J. B. Fenn, Angew. Chem., Int. Ed., 2003, 42, 3871–3894 CrossRef CAS PubMed.
- M. Barber, R. S. Bordoli, R. D. Sedgwick and A. N. Tyler, J. Chem. Soc., Chem. Commun., 1981a, 325–327 Search PubMed.
- M. Barber, R. S. Bordoli, R. D. Sedgwick and A. N. Tyler, Nature, 1981b, 293, 270–275 Search PubMed.
- J. W. Crabb, L. G. Armes, S. A. Carr, C. M. Johnson, G. D. Roberts, R. S. Bordoli and W. L. McKeehan, Biochemistry, 1986, 25, 4988–4993 CrossRef CAS PubMed.
- R. S. Johnson and K. Biemann, Biochemistry, 1987, 26, 1209–1214 CrossRef CAS PubMed.
- M. Kinter and N. E. Sherman, Protein sequencing and identification using tandem mass spectrometry, John Wiley & Sons, New York, 2000 Search PubMed.
- L. Sleno and D. A. Volmer, J. Mass Spectrom., 2004, 39, 1091–1112 CrossRef CAS PubMed.
- J. S. Brodbelt, Anal. Chem., 2016, 88, 30–51 CrossRef CAS PubMed.
- R. A. Zubarev, N. L. Kelleher and F. W. McLafferty, J. Am. Chem. Soc., 1998, 120, 3265–3266 CrossRef CAS.
- R. A. Zubarev, D. M. Horn, E. K. Fridriksson, N. L. Kelleher, N. A. Kruger, M. A. Lewis, B. K. Carpenter and F. W. McLafferty, Anal. Chem., 2000, 72, 563–573 CrossRef CAS PubMed.
- J. E. P. Syka, J. J. Coon, M. J. Schroeder, J. Shabanowitz and D. F. Hunt, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9528–9533 CrossRef CAS PubMed.
- M. C. Crowe and J. S. Brodbelt, J. Am. Soc. Mass Spectrom., 2004, 15, 1581–1592 CrossRef CAS PubMed.
- R. Bakhtiar and Z. Guan, Biotechnol. Lett., 2006, 28, 1047–1059 CrossRef CAS PubMed.
- J. A. Madsen and J. S. Brodbelt, J. Am. Soc. Mass Spectrom., 2009, 20, 349–358 CrossRef CAS PubMed.
- M. S. Kim and A. Pandey, Proteomics, 2012, 12, 530–542 CrossRef CAS PubMed.
- Q. Hu, R. J. Noll, H. Li, A. Makarov, M. Hardman and R. G. Cooks, J. Mass Spectrom., 2005, 40, 430–443 CrossRef CAS PubMed.
- R. A. Zubarev and A. Makarov, Anal. Chem., 2013, 85, 5288–5296 CrossRef CAS PubMed.
- S. Eliuk and A. Makarov, Annu. Rev. Anal. Chem., 2015, 8, 61–80 CrossRef PubMed.
- P. G. Righetti, A. Castagna, P. Antonioli and E. Boschetti, Electrophoresis, 2005, 26, 297–319 CrossRef CAS PubMed.
- S. L. Wu, A. F. R. Huhmer, Z. Hao and B. L. Karger, J. Proteome Res., 2007, 6, 4230–4244 CrossRef CAS PubMed.
- M. P. Washburn, D. Wolters and J. R. Yates III, Nat. Biotechnol., 2001, 19(3), 242–247 CrossRef CAS PubMed.
- J. Peng and S. P. Gygi, J. Mass Spectrom., 2001, 36, 1083–1091 CrossRef CAS PubMed.
- J. R. Yates, C. I. Ruse and A. Nakorchevsky, Annu. Rev. Biomed. Eng., 2009, 11, 49–79 CrossRef CAS PubMed.
- X. Zhang, A. Fang, C. P. Riley, M. Wang, F. E. Regnier and C. Buck, Anal. Chim. Acta, 2010, 664(2), 101–113 CrossRef CAS PubMed.
- R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CrossRef CAS PubMed.
- E. Vandermarliere, M. Mueller and L. Martens, Mass Spectrom. Rev., 2013, 32(6), 453–465 CAS.
- L. C. Gillet, A. Leitner and R. Aebersold, Annu. Rev. Anal. Chem., 2016, 9, 449–472 CrossRef PubMed.
- F. Xie, R. D. Smith and Y. Shen, J. Chromatogr. A, 2012, 1261, 78–90 CrossRef CAS PubMed.
- N. Siuti and N. L. Kelleher, Nat. Methods, 2007, 4, 817–821 CrossRef CAS PubMed.
- B. A. Garcia, J. Am. Soc. Mass Spectrom., 2010, 21, 193–202 CrossRef CAS PubMed.
- H. Zhou, Z. Ning, A. E. Starr, M. Abu-Farha and D. Figeys, Anal. Chem., 2012, 84, 720–734 CrossRef CAS PubMed.
- Y. Ge, B. G. Lawhorn, M. ElNaggar, E. Strauss, J. H. Park, T. P. Begley and F. W. McLafferty, J. Am. Chem. Soc., 2002, 124(4), 672–678 CrossRef CAS PubMed.
- S. M. Patrie, J. P. Charlebois, D. Whipple, N. L. Kelleher, C. L. Hendrickson, J. P. Quinn, A. G. Marshall and B. Mukhopadhyay, J. Am. Soc. Mass Spectrom., 2004, 15, 1099–1108 CrossRef CAS PubMed.
- B. A. Parks, L. Jiang, P. M. Thomas, C. D. Wenger, M. J. Roth, M. T. Boyne II, P. V. Burke, K. E. Kwast and N. L. Kelleher, Anal. Chem., 2007, 79(21), 7984–7991 CrossRef CAS PubMed.
- H. Zhang, W. Cui, J. Wen, R. E. Blankenship and M. L. Gross, J. Am. Soc. Mass Spectrom., 2010, 21(12), 1966–1968 CrossRef CAS PubMed.
- W. Cui, H. W. Rohrs and M. L. Gross, Analyst, 2011, 136, 3854–3864 RSC.
- A. D. Catherman, O. S. Skinner and N. L. Kelleher, Biochem. Biophys. Res. Commun., 2014, 445(4), 683–693 CrossRef CAS PubMed.
- T. K. Toby, L. Fornelli and N. L. Kelleher, Annu. Rev. Anal. Chem., 2016, 9(1), 499–519 CrossRef CAS PubMed.
- B. A. Garcia, N. Siuti, C. E. Thomas, C. A. Mizzen and N. L. Kelleher, Int. J. Mass Spectrom., 2007, 259, 184–196 CrossRef CAS.
- A. Cristobal, F. Marino, H. Post, H. W. P. van den Toorn, S. Mohammed and A. J. R. Heck, Anal. Chem., 2017, 89, 3318–3325 CrossRef CAS PubMed.
- P. Xu and J. Peng, Anal. Chem., 2008, 80, 3438–3444 CrossRef CAS PubMed.
- E. M. Valkevich, N. A. Sanchez, Y. Ge and E. R. Strieter, Biochemistry, 2014, 53, 4979–4989 CrossRef CAS.
- S. O. Crowe, A. S. J. B. Rana, K. K. Deol, Y. Ge and E. R. Strieter, Anal. Chem., 2017, 89, 4428–4434 CrossRef CAS.
- J. Pan, S. Zhang, A. Chou and C. H. Borchers, Chem. Sci., 2016, 7, 1480–1486 RSC.
- Y. J. Tan, W. H. Wang, Y. Zheng, J. Dong, G. Stefano, F. Brandizzi, R. M. Gatavito, G. E. Reid and M. L. Brueining, Anal. Chem., 2012, 84(19), 8357–8363 CrossRef CAS PubMed.
- L. Fornelli, D. Ayoub, K. Aizikov, A. Beck and Y. O. Tsybin, Anal. Chem., 2014, 86, 3005–3012 CrossRef CAS PubMed.
- K. Srzentic, L. Fornelli, U. A. Laskay, M. Monod, A. Beck, D. Ayoub and Y. O. Tsybin, Anal. Chem., 2014, 86, 9945–9953 CrossRef CAS PubMed.
- U. A. Laskay, A. A. Lobas, K. Srzentic, M. V. Gorhkov and Y. O. Tsybin, J. Proteome Res., 2013, 12, 5558–5569 CrossRef CAS.
- B. A. Garcia, C. E. Thomas, N. L. Kelleher and C. A. Mizzen, J. Proteome Res., 2008, 7(10), 4225–4236 CrossRef CAS.
- P. Voigt, G. LeRoy, W. J. Drury III, B. M. Zee, J. Son, D. Beck, N. L. Young, B. A. Garcia and D. Reinberg, Cell, 2012, 151(1), 181–193 CrossRef CAS.
- A. Kalli, M. J. Sweredoski and S. Hess, Anal. Chem., 2013, 85, 3501–3507 CrossRef CAS PubMed.
- S. Sidoli, V. Schwammle, C. Ruminowicz, T. A. Hansen, X. Wu, K. Helin and O. N. Jensen, Proteomics, 2014, 14, 2200–2211 CrossRef CAS.
- S. Sidoli, S. Lin, K. R. Karch and B. A. Garcia, Anal. Chem., 2015, 87, 3129–3133 CrossRef CAS PubMed.
- S. Sidoli, J. Vandamme, A. E. Salcini and O. N. Jensen, Proteomics, 2016, 16(3), 459–464 CrossRef CAS PubMed.
- S. Sidoli, C. Lu, M. Coradin, X. Wang, K. R. Karch, C. Ruminowicz and B. A. Garcia, Epigenet. Chromatin, 2017, 10, 34 CrossRef.
- S. Sidoli and B. A. Garcia, Expert Rev. Proteomics, 2017, 14(7), 617–626 CrossRef CAS PubMed.
- R. C. Molden and B. A. Garcia, Curr. Protoc. Protein Sci., 2014, 77, 1–28 Search PubMed.
- A. Tvardovskiy, K. Wrzesinski, S. Sidoli, S. J. Fey, A. Rogowska-Wrzesinska and O. N. Jensen, Mol. Cell. Proteomics, 2015, 14, 3142–3153 CrossRef CAS PubMed.
- K. Yamamoto, Y. Chikaoka, G. Hayashi, R. Sakamoto, R. Yamamoto, A. Sugiyam, T. Kodama, A. Okamoto and T. Kawamura, Mass Spectrom., 2015, 4(1), A0039 CrossRef PubMed.
- Y. Ge, I. N. Rybakova, Q. Xu and R. L. Moss, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(31), 12658–12663 CrossRef CAS PubMed.
- Y. Yang, F. Liu, V. Franc, L. A. Halim, H. Schellekens and A. J. R. Heck, Nat. Commun., 2016, 7, 13397 CrossRef CAS PubMed.
- K. Khatri, Y. Pu, J. A. Klein, J. Wei, C. E. Costello, C. Lin and J. Zaia, J. Am. Soc. Mass Spectrom., 2018, 29(6), 1075–1085 CrossRef CAS PubMed.
- A. J. Forbes, M. T. Mazur, H. M. Patel, C. T. Wals and N. L. Kelleher, Proteomics, 2001, 1, 927–933 CrossRef CAS PubMed.
- J. Cannon, K. Lohnes, C. Wyne, Y. Wang, N. Edwards and C. Fenselau, J. Proteome Res., 2010, 9(8), 3886–3890 CrossRef CAS PubMed.
- J. R. Cannon, N. J. Edwards and C. Fenselau, J. Mass Spectrom., 2013, 48(3), 340–343 CrossRef CAS PubMed.
- K. Srzentic, K. O. Zhurov, A. A. Lobas, G. Nikitin, L. Fornelli, M. V. Gorshkov and Y. O. Tsybin, J. Proteome Res., 2018, 17(6), 2005–2016 CrossRef CAS PubMed.
- M. J. Sweredoski, A. Moradian, M. Raedle, C. Franco and S. Hess, Anal. Chem., 2015, 87, 8360–8366 CrossRef CAS PubMed.
- N. L. Young, P. A. DiMaggio, M. D. Plazas-Mayorca, R. C. Baliban, C. A. Floudas and B. A. Garcia, Mol. Cell. Proteomics, 2009, 8, 2266–2284 CrossRef CAS PubMed.
- S. Trevisiol, D. Ayoub, A. Lesur, L. Ancheva, S. Gallien and B. Domon, Proteomics, 2016, 16(5), 715–728 CrossRef CAS PubMed.
- C. Wu, J. C. Tran, L. Zamdborg, K. R. Durbin, M. Li, D. R. Ahlf, B. P. Early, P. M. Thomas, J. V. Sweedler and N. L. Kelleher, Nat. Methods, 2012, 9(8), 822–824 CrossRef CAS.
- U. A. Laskay, K. Srzentic, L. Fornelli, O. Upir, A. N. Kozhinov, M. Monod and Y. O. Tsybin, Chimia, 2013, 67, 244–249 CrossRef CAS.
- U. A. Laskay, K. Srzentic, M. Monod and Y. O. Tsybin, J. Proteomics, 2014, 110, 20–31 CrossRef CAS PubMed.
- M. H. Ryan, D. Petrone, J. F. Nemeth, E. Barnathan, L. Bjorck and R. E. Jordan, Mol. Immunol., 2008, 45, 1837–1846 CrossRef CAS PubMed.
- K. Okuno, M. Yabuta, K. Kawanishi, K. Ohsuye, T. Ooi and S. Kinoshita, Biosci., Biotechnol., Biochem., 2002, 66, 127–134 CrossRef CAS PubMed.
- B. Q. Tran, C. Barton, J. Feng, A. Sandjong, S. H. Yoon, S. Aswasti, T. Liang, M. M. Khan, D. P. A. Kilgour, D. R. Goodlett and Y. A. Goo, Data Brief, 2016, 6, 68–76 CrossRef PubMed.
- L. He, L. C. Anderson, D. R. Barnidge, D. L. Murray, C. L. Hendrickson and A. G. Marshall, J. Am. Soc. Mass Spectrom., 2017, 28(5), 827–838 CrossRef CAS.
- A. M. Belov, L. Zang, R. Sebastiano, M. R. Santos, D. R. Bush, B. L. Karger and A. R. Ivanov, Electrophoresis, 2018, 39(16), 2069–2082 CrossRef CAS.
- B. Wang, A. C. Gucinski, D. A. Keire, L. F. Buhse and M. T. Boyne II, Analyst, 2013, 138, 3058–3065 RSC.
- L. Fornelli, K. Srzentić, R. Huguet, C. Mullen, S. Sharma, V. Zabrouskov, R. T. Fellers, K. R. Durbin, P. D. Compton and N. L. Kelleher, Anal. Chem., 2018, 90(14), 8421–8429 CrossRef CAS PubMed.
- D. Ayoub, W. Jabs, A. Resemann, W. Evers, C. Evans, C. Baessmamm, E. Wagner-Rousset, D. Suckau and A. Beck, mAbs, 2013, 5(5), 699–710 CrossRef.
- A. Resemann, W. Jabs, A. Wiechmann, E. Wagner, O. Colas, W. Evers, E. Belau, L. Vorwerg, C. Evans, A. Beck and D. Suckau, mAbs, 2016, 8(2), 318–330 CrossRef CAS.
- K. Mizuno, T. Nakamura, T. Ohshima, S. Tanaka and H. Matsuo, Biochem. Biophys. Res. Commun., 1989, 159(1), 305–311 CrossRef CAS.
- Y. Shi, R. Xiang, C. Horvath and J. A. Wilkins, J. Chromatogr. A, 2004, 1053, 27–36 CrossRef CAS PubMed.
- B. R. Fonslow and J. R. Yates III, J. Sep. Sci., 2009, 32(8), 1175–1188 CrossRef CAS PubMed.
- C. Desiderio, D. V. Rossetti, F. Iavarone, I. Messana and M. Casagnola, J. Pharm. Biomed. Anal., 2010, 53(5), 1161–1169 CrossRef CAS.
- G. Lin, C. Gao, Q. Zheng, Z. Lei, H. Geng, Z. Lin, H. Yang and Z. Cai, Chem. Commun., 2017, 53(26), 3649–3652 RSC.
- L. Monteoliva and J. P. Albar, Briefings Funct. Genomics Proteomics, 2004, 3(3), 220–239 CrossRef CAS.
- G. Baggerman, E. Vierstraete, A. De Loof and L. Schoofs, Comb. Chem. High Throughput Screening, 2005, 8, 669–677 CrossRef CAS.
- J. P. Lambert, M. Ethier, J. C. Smith and D. Figeys, Anal. Chem., 2005, 77, 3771–3788 CrossRef CAS PubMed.
- M. Unlü, M. E. Morgan and J. S. Minden, Electrophoresis, 1997, 18(11), 2071–2077 CrossRef.
- G. Zhou, H. Li, D. DeCamp, S. Chen, H. Shu, Y. Gong, M. Flaig, J. W. Gillespie, N. Hu, P. R. Taylor, M. R. Emmert-Buck, L. A. Liotta, E. F. Petricoin III and Y. Zhao, Mol. Cell. Proteomics, 2002, 1, 117–124 CrossRef CAS.
- R. Westermeier and B. Scheibe, Methods Mol. Biol., 2008, 424, 73–85 CrossRef CAS.
- J. S. Minden, S. R. Dowd, H. E. Meyer and K. Stühler, Electrophoresis, 2009,(Suppl 1), S156–S161 CrossRef.
- A. Venkatesan, K. Palaniyandi, D. Sharma, D. Bisht and S. Narayanan, Can. J. Microbiol., 2018, 64(4), 243–251 CrossRef CAS PubMed.
- S. Ananthi, C. N. P. Lakshmi, P. Atmika, K. Anbarasu and S. Mahalingam, Sci. Rep., 2018, 8(1), 1567, DOI:10.1038/s41598-018-19937-3.
- L. Sun, G. Zhu, X. Yan, Z. Zhang, R. Wojcik, M. M. Champion and N. J. Dovichi, Proteomics, 2016, 16, 188–196 CrossRef CAS PubMed.
- J. W. Cooper, Y. Wang and C. S. Lee, Electrophoresis, 2004, 25, 3913–3926 CrossRef CAS.
- D. C. Simpson and R. D. Smith, Electrophoresis, 2005, 26, 1291–1305 CrossRef CAS.
- Z. Zhu, J. J. Lu and S. Liu, Anal. Chim. Acta, 2012, 709, 21–31 CrossRef CAS PubMed.
- V. Brocke, G. Nicholson and E. Bayer, Electrophoresis, 2001, 22, 1251–1266 CrossRef.
- T. Manabe, Electrophoresis, 1999, 20, 3116–3121 CrossRef CAS.
- H. Mischak, J. J. Coon, J. Novak, E. M. Weissinger, J. Schanstra and A. F. Dominiczak, Mass Spectrom. Rev., 2009, 28(5), 703–724 CrossRef CAS PubMed.
- Y. Li, M. M. Champion, L. Sun, P. A. Champion, R. Wojcik and N. J. Dovichi, Anal. Chem., 2012, 84(3), 1617–1622 CrossRef CAS PubMed.
- G. Zhu, L. Sun, X. Yan and N. J. Dovichi, Anal. Chem., 2013, 85, 2569–2573 CrossRef CAS.
- K. Faserl, B. Sarg, L. Kremser and H. Lindner, Anal. Chem., 2011, 83(19), 7297–7305 CrossRef CAS.
- Y. Li, P. D. Compton, J. C. Tran, I. Ntai and N. L. Kelleher, Proteomics, 2014, 14(10), 1158–1164 CrossRef PubMed.
- R. Wojcik, G. Zhu, Z. Zhang, X. Yan, Y. Zhao, L. Sun, M. M. Champion and N. J. Dovichi, Bioanalysis, 2016, 8(2), 89–92 CrossRef CAS PubMed.
- Y. Zhao, N. M. Riley, L. Sun, A. S. Hebert, X. Yan, M. S. Westphall, M. J. P. Rush, G. Zhu, M. M. Champion, F. M. Medie, P. A. D. Champion, J. J. Coon and N. J. Dovichi, Anal. Chem., 2015, 87, 5422–5429 CrossRef CAS PubMed.
- M. Biacchi, N. Said, A. Beck, E. Leize-Wagner and Y. N. Francois, J. Chromatogr. A, 2017, 1498, 120–127 CrossRef CAS PubMed.
- C. M. Whitehouse, R. N. Dreyer, M. Yamashita and J. B. Fenn, Anal. Chem., 1985, 57(3), 675–679 CrossRef CAS PubMed.
- Y. Shen and R. D. Smith, Electrophoresis, 2002, 23, 3106–3124 CrossRef CAS.
- G. Mitulovic and K. Mechtler, Briefings Funct. Genomics Proteomics, 2006, 5(4), 249–260 CrossRef CAS PubMed.
- Y. Y. Zhao and R. C. Lin, Chem.-Biol. Interact., 2014, 215, 7–16 CrossRef CAS PubMed.
- M. Hummel, T. Wigger and J. Brockmeyer, J. Proteome Res., 2015, 14, 1547–1556 CrossRef CAS PubMed.
- J. D. Sanders, S. M. Greer and J. S. Brodbelt, Anal. Chem., 2017, 89(21), 11772–11778 CrossRef CAS PubMed.
- X. Sun, J. F. Chiu and Q. Y. He, Expert Rev. Proteomics, 2005, 2(5), 649–657 CrossRef CAS PubMed.
- J. Fila and D. Honys, Amino Acids, 2012, 43, 1025–1047 CrossRef CAS PubMed.
- Y. Zhang, B. R. Fonslow, B. Shan, M. C. Baek and J. R. Yates III, Chem. Rev., 2013, 113, 2343–2394 CrossRef CAS.
- R. R. Prasanna, S. Sidhik, A. S. Kamalanathan, K. Bhagavatula and M. A. Vijayalakshmi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 955–956, 42–49 CrossRef CAS PubMed.
- K. Karkra, K. K. R. Tetala and M. A. Vijayalakshmi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1052, 1–9 CrossRef CAS PubMed.
- X. Wei and L. Li, Briefings Funct. Genomics Proteomics, 2008, 8(2), 104–113 CrossRef PubMed.
- S. Pan, R. Chen, R. Aebersold and T. A. Brentnall, Mol. Cell. Proteomics, 2011, 10, 1–14 Search PubMed.
- J. Nilsson, A. Halim, A. Grahn and G. Larson, Glycoconjugate J., 2013, 30, 119–136 CrossRef CAS PubMed.
- Q. Zhang, N. Tang, J. W. C. Brock, H. M. Mottaz, J. M. Ames, J. W. Baynes, R. D. Smith and T. O. Metz, J. Proteome Res., 2007, 6(6), 2323–2330 CrossRef CAS PubMed.
- C. Zhou, X. Chen, Z. Du, G. Li, X. Xia and Z. Cai, J. Chromatogr. A, 2017, 1498, 90–98 CrossRef CAS PubMed.
- H. J. Issaq, Electrophoresis, 2001, 22, 3629–3638 CrossRef CAS PubMed.
- H. Liu, D. Lin and J. R. Yates III, BioTechniques, 2002, 32, 898–911 CrossRef CAS PubMed.
- Y. Zheng, X. Huang and N. L. Kelleher, Curr. Opin. Chem. Biol., 2016, 33, 142–150 CrossRef CAS PubMed.
- J. A. McLean, B. T. Ruotolo, K. J. Gillig and D. H. Russell, Int. J. Mass Spectrom., 2005, 240, 301–315 CrossRef CAS.
- D. Helm, J. P. C. Vissers, C. J. Hughes, H. Hahnet, B. Ruprecht, F. Pachl, A. Grzyb, K. Richardson, J. Wildgoose, S. K. Maier, H. Marx, M. Wilhelm, I. Becher, S. Lemeer, M. Bantscheff, J. I. Langridge and B. Kuster, Mol. Cell. Proteomics, 2014, 13(12), 3709–3715 CrossRef CAS PubMed.
- A. A. Shvartsburg, Y. Zheng, R. D. Smith and N. L. Kelleher, Anal. Chem., 2012, 84, 4271–4276 CrossRef CAS PubMed.
- A. Beck, F. Debaene, H. Diemer, E. Wagner-Rousset, O. Colas, A. Van Dorsselaer and S. Cianférani, J. Mass Spectrom., 2015, 50(2), 285–297 CrossRef CAS PubMed.
- A. Garabedian, M. A. Baird, J. Porter, K. Jeanne Dit Fouque, P. V. Shliaha, O. N. Jensen, T. D. Williams, F. Fernandez-Lima and A. A. Shvartsburg, Anal. Chem., 2018, 90(4), 2918–2925 CrossRef CAS PubMed.
- E. de Hoffmann and V. Stroobant, Mass spectrometry - Principles and Applications, John Wiley & Sons Ltd, Chichester, West Sussex, England, 3rd edn, 2007 Search PubMed.
- M. Karas, D. Bachmann and F. Hillenkamp, Anal. Chem., 1985, 57, 2935–2939 CrossRef CAS.
- M. Karas, D. Bachmann, U. Bahr and F. Hillenkamp, Int. J. Mass Spectrom. Ion Processes, 1987, 78, 53–68 CrossRef CAS.
- J. Yergey, D. Heller, G. Hansen, R. J. Cotter and C. Fenselau, Anal. Chem., 1983, 55(2), 353–356 CrossRef CAS.
- W. Paul, Angew. Chem. Int. Ed., 1990, 29, 739–748 CrossRef.
- M. J. Roth, B. A. Parks, J. T. Ferguson, M. T. Boyne II and N. L. Kelleher, Anal. Chem., 2008, 80, 2857–2866 CrossRef CAS PubMed.
- R. E. March, J. Mass Spectrom., 1997, 32, 351–369 CrossRef CAS.
- K. R. Jonscher and J. R. Yates III, Anal. Biochem., 1997, 244, 1–15 CrossRef CAS PubMed.
- M. Mohamed-Benkada, M. Montagu, J. F. Biard, F. Mondeguer, P. Verite, M. Dalgalarrondo, J. Bissett and Y. F. Pouchus, Rapid Commun. Mass Spectrom., 2006, 20(8), 1176–1180 CrossRef CAS PubMed.
- V. Sabareesh, Mass spectrometric sequencing of acyclic and cyclic peptides, PhD thesis, Indian Institute of Science, Bengaluru, Karnataka, India, 2007.
- J. W. Hager, Rapid Commun. Mass Spectrom., 2002, 16(6), 512–526 CrossRef CAS.
- J. C. Schwartz, M. W. Senko and J. E. P. Syka, J. Am. Soc. Mass Spectrom., 2002, 13, 659–669 CrossRef CAS PubMed.
- J. W. Hager and J. C. Yves Le Blanc, Rapid Commun. Mass Spectrom., 2003, 17(10), 1056–1064 CrossRef CAS.
- B. Macek, L. F. Waanders, J. V. Olsen and M. Mann, Mol. Cell. Proteomics, 2006, 5(5), 949–958 CrossRef CAS PubMed.
- M. T. Boyne II, B. A. Garcia, M. Li, L. Zamdborg, C. D. Wenger, S. Babai and N. L. Kelleher, J. Proteome Res., 2009, 8(1), 374–379 CrossRef PubMed.
- J. Vandamme, S. Sidoli, L. Mariani, C. Friis, J. Christensen, K. Helin, O. N. Jensen and A. E. Salcini, Nucleic Acids Res., 2015, 43(20), 9694–9710 CAS.
- M. B. Comisarow and A. G. Marshall, Chem. Phys. Lett., 1974, 25(2), 282–283 CrossRef CAS.
- A. G. Marshall, C. L. Hendrickson and G. S. Jackson, Mass Spectrom. Rev., 1998, 17, 1–35 CrossRef CAS.
- W. Schrader and H. W. Klein, Anal. Bioanal. Chem., 2004, 379, 1013–1024 CrossRef CAS PubMed.
- F. Meng, B. J. Cargile, S. M. Patrie, J. R. Johnson, S. M. McLoughlin and N. L. Kelleher, Anal. Chem., 2002, 74, 2923–2929 CrossRef CAS.
- V. Zabrouskov, L. Giacomelli, K. J. van Wijk and F. W. McLafferty, Mol. Cell. Proteomics, 2003, 2, 1253–1260 CrossRef CAS PubMed.
- A. Makarov, Anal. Chem., 2000, 72, 1156–1162 CrossRef CAS PubMed.
- R. H. Perry, R. G. Cooks and R. J. Noll, Mass Spectrom. Rev., 2008, 27(6), 661–699 CrossRef CAS.
- K. J. Webb, T. Xu, S. K. Park and J. R. Yates III, J. Proteome Res., 2013, 12, 2177–2184 CrossRef CAS PubMed.
- F. Meier, P. E. Geyer, S. Virreira Winter, J. Cox and M. Mann, Nat. Methods, 2018, 15(6), 440–448 CrossRef CAS.
- R. Danda, K. Ganapathy, G. Sathe, A. K. Madugundu, U. M. Krishnan, V. Khetan, P. Rishi, H. Gowda, A. Pandey, K. Subramanian, T. S. K. Prasad and S. V. Elchuri, Proteomics: Clin. Appl., 2018, e1700101, DOI:10.1002/prca.201700101.
- H. R. Jung, S. Sidoli, S. Haldbo, R. R. Sprenger, V. Schwämmle, D. Pasini, K. Helin and O. N. Jensen, Anal. Chem., 2013, 85(17), 8232–8239 CrossRef CAS PubMed.
- Y. Jin, L. Wei, W. Cai, Z. Lin, Z. Wu, Y. Peng, T. Kohmoto, R. L. Moss and Y. Ge, Anal. Chem., 2017, 89, 4922–4930 CrossRef CAS PubMed.
- A. Moradian, C. Franco, M. J. Sweredoski and S. Hess, J. Anal. Sci. Technol., 2015, 6, 21 CrossRef.
- A. W. Jones and H. J. Cooper, Analyst, 2011, 136, 3419–3429 RSC.
- K. Biemann, Methods Enzymol., 1990, 193, 886–887 CAS.
- V. H. Wysocki, G. Tsaprailis, L. L. Smith and L. A. Breci, J. Mass Spectrom., 2000, 35, 1399–1406 CrossRef CAS.
- B. Paizs and S. Suhai, Mass Spectrom. Rev., 2005, 24, 508–548 CrossRef CAS PubMed.
- K. F. Medzirahradszky, Methods Enzymol., 2005, 402, 209–244 Search PubMed.
- P. L. Ross, Y. N. Huang, J. N. Marchese, B. Williamson, K. Parker, S. Hattan, N. Khainovski, S. Pillai, S. Dey, S. Daniels, S. Purkayastha, P. Juhasz, S. Martin, M. Bartlet-Jones, F. He, A. Jacobson and D. J. Pappin, Mol. Cell. Proteomics, 2004, 3, 1154–1169 CrossRef CAS.
- S. Wiese, K. A. Reidegeld, H. E. Meyer and B. Warscheid, Proteomics, 2007, 7(3), 340–350 CrossRef CAS.
- M. Bantscheff, M. Schirle, G. Sweetman, J. Rick and B. Kuster, Anal. Bioanal. Chem., 2007, 389, 1017–1103 CrossRef CAS.
- E. Sachon, S. Mohammed, N. Bache and O. N. Jensen, Rapid Commun. Mass Spectrom., 2006, 20(7), 1127–1134 CrossRef CAS.
- B. McDonagh, P. Martínez-Acedo, J. Vázquez, C. A. Padilla, D. Sheehan and J. A. Bárcena, Int. J. Proteomics, 2012, 2012, 514847, DOI:10.1155/2012/514847.
- H. R. Fuller and G. E. Morris, Quantitative proteomics using iTRAQ labeling and mass spectrometry, Integrative Proteomics, ed. H.-C. Leung, InTech, London, United Kingdom, 2012, pp. 347–362 Search PubMed.
- F. W. McLafferty, D. M. Horn, K. Breuker, Y. Ge, M. A. Lewis, B. Cerda, R. A. Zubarev and B. K. Carpenter, J. Am. Soc. Mass Spectrom., 2001, 12, 245–249 CrossRef CAS PubMed.
- A. Pamreddy and N. R. Panyala, J. Appl. Bioanal., 2016, 2(2), 52–75 CrossRef CAS.
- Y. Xia, P. A. Chrisman, D. E. Erickson, J. Liu, X. Liang, F. A. Londry, M. J. Yang and S. A. McLuckey, Anal. Chem., 2006, 78(12), 4146–4154 CrossRef CAS PubMed.
- G. C. McAlister, D. Phanstiel, D. M. Good, W. T. Berggren and J. J. Coon, Anal. Chem., 2007, 79(10), 3525–3534 CrossRef CAS PubMed.
- R. Hartmer, D. A. Kaplan, C. R. Gebhardt, T. Ledertheil and A. Brekenfeld, Int. J. Mass Spectrom., 2008, 276, 82–90 CrossRef CAS.
- D. M. Good, M. Wirtala, G. C. McAlister and J. J. Coon, Mol. Cell. Proteomics, 2007, 6(11), 1942–1951 CrossRef CAS PubMed.
- R. J. Chalkley, K. F. Medzihradszky, A. J. Lynn, P. R. Baker and A. L. Burlingame, Anal. Chem., 2010, 82(2), 579–584 CrossRef CAS PubMed.
- H. Molina, R. Matthiesen, K. Kandasamy and A. Pandey, Anal. Chem., 2008, 80, 4825–4835 CrossRef CAS PubMed.
- L. M. Mikesh, B. Ueberheide, A. Chi, J. J. Coon, J. E Syka, J. Shabanowitz and D. F. Hunt, Biochim. Biophys. Acta, 2006, 1764(12), 1811–1822 CrossRef CAS PubMed.
- H. Molina, D. M. Horn, N. Tang, S. Mathivanan and A. Pandey, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(7), 2199–2204 CrossRef CAS.
- D. L. Swaney, C. D. Wenger, J. A. Thomson and J. J. Coon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(4), 995–1000 CrossRef CAS.
- Y. Zhou, J. Dong and R. W. Vachet, Curr. Pharm. Biotechnol., 2011, 12(10), 1558–1567 CAS.
- C. K. Frese, H. Zhou, T. Taus, A. F. Altelaar, K. Mechtler, A. J. Heck and S. Mohammed, J. Proteome Res., 2013, 12(3), 1520–1525 CrossRef CAS PubMed.
- A. Schmidt, D. B. Trentini, S. Spiess, J. Fuhrmann, G. Ammerer, K. Mechtler and T. Clausen, Mol. Cell. Proteomics, 2014, 13, 537–550 CrossRef CAS PubMed.
- E. L. de Graaf, P. Giansanti, A. F. M. Altelaar and A. J. R. Heck, Mol. Cell. Proteomics, 2014, 13, 2426–2434 CrossRef CAS PubMed.
- L. von Stechow, C. Francavilla and J. V. Olsen, Expert Rev. Proteomics, 2015, 12(5), 469–487 CrossRef CAS PubMed.
- I. Perdivara, R. Petrovich, B. Alliquant, L. J. Deterding, K. B. Tomer and M. Przybylski, J. Proteome Res., 2009, 8(2), 631–642 CrossRef CAS PubMed.
- Y. Mechref, Curr. Protoc. Protein Sci., 2012, 12.11.1–12.11.11 Search PubMed.
- Q. Yu, B. Wang, Z. Chen, G. Urabe, M. S. Glover, X. Shi, L. W. Guo, K. C. Kent and L. Li, J. Am. Soc. Mass Spectrom., 2017, 28(9), 1751–1764 CrossRef CAS.
- D. L. Swaney, G. C. McAlister, M. Wirtala, J. C. Schwartz, J. E. Syka and J. J. Coon, Anal. Chem., 2007, 79(2), 477–485 CrossRef CAS PubMed.
- V. Sabareesh, P. Sarkar, A. A. Sardesai and D. Chatterji, Analyst, 2010, 135, 2723–2729 RSC.
- K. Gupta, M. Kumar, K. Chandrashekara, K. S. Krishnan and P. Balaram, J. Proteome Res., 2012, 11(2), 515–522 CrossRef CAS PubMed.
- V. A. Mikhailov and H. J. Cooper, J. Am. Soc. Mass Spectrom., 2009, 20(5), 763–771 CrossRef CAS PubMed.
- C. Lin, J. J. Cournoyer and P. B. O'Connor, J. Am. Soc. Mass Spectrom., 2008, 19(6), 780–789 CrossRef CAS PubMed.
- D. M. Horn, Y. Ge and F. W. McLafferty, Anal. Chem., 2000, 72, 4778–4784 CrossRef CAS PubMed.
- M. M. Savitski, F. Kjeldsen, M. L. Nielsen and R. A. Zubarev, J. Am. Soc. Mass Spectrom., 2007, 18, 113–120 CrossRef CAS PubMed.
- Y. Tsybin, H. He, M. R. Emmett, C. L. Hendrickson and A. G. Marshall, Anal. Chem., 2007, 79(20), 7596–7602 CrossRef CAS PubMed.
- J. S. Brodbelt, Chem. Soc. Rev., 2014, 43(8), 2757–2783 RSC.
- V. C. Cotham, A. P. Horton, J. Lee, G. Georgiou and J. S. Brodbelt, Anal. Chem., 2017, 89, 6498–6504 CrossRef CAS PubMed.
- T. Chen, J. Zhao, J. Ma and Y. Zhu, Genomics, Proteomics Bioinf., 2015, 13, 36–39 CrossRef PubMed.
- K. Verheggen, H. Ræder, F. S. Berven, L. Martens, H. Barsnes and M. Vaudel, Mass Spectrom. Rev., 2017, 1–15, DOI:10.1002/mas.21543.
- V. Nanjappa, J. K. Thomas, A. Marimuthu, B. Muthusamy, A. Radhakrishnan, R. Sharma, A. Ahmad Khan, L. Balakrishnan, N. A. Sahasrabuddhe, S. Kumar, B. N. Jhaveri, K. V. Sheth, R. Kumar Khatana, P. G. Shaw, S. M. Srikanth, P. P. Mathur, S. Shankar, D. Nagaraja, R. Christopher, S. Mathivanan, R. Raju, R. Sirdeshmukh, A. Chatterjee, R. J. Simpson, H. C. Harsha, A. Pandey and T. S. Prasad, Nucleic Acids Res., 2014, 42, D959–D965 CrossRef CAS PubMed.
- B. Muthusamy, G. Hanumanthu, S. Suresh, B. Rekha, D. Srinivas, L. Karthick, B. M. Vrushabendra, S. Sharma, G. Mishra and P. Chatterjee, et al., Proteomics, 2005, 5, 3531–3536 CrossRef CAS PubMed.
- D. N. Perkins, D. J. Pappin, D. M. Creasy and J. S. Cottrell, Electrophoresis, 1999, 20(18), 3551–3567 CrossRef CAS PubMed.
- J. K. Eng, A. L. McCormack and J. R. Yates III, J. Am. Soc. Mass Spectrom., 1994, 5, 976–989 CrossRef CAS.
- B. Ma, K. Zhang, C. Hendrie, C. Liang, M. Li, A. Doherty-Kirby and G. Lajoie, Rapid Commun. Mass Spectrom., 2003, 17(20), 2337–2342 CrossRef CAS PubMed.
- L. Y. Geer, S. P. Markey, J. A. Kowalak, L. Wagner, M. Xu, D. M. Maynard, X. Yang, W. Shi and S. H. Bryant, J. Proteome Res., 2004, 3, 958–964 CrossRef CAS PubMed.
- L. Martens, H. Hermjakob, P. Jones, M. Adamski, C. Taylor, D. States, K. Gevaert, J. Vandekerckhove and R. Apweiler, Proteomics, 2005, 5(13), 3537–3545 CrossRef CAS PubMed.
- R. Craig and R. Beavis, Bioinformatics, 2004, 20, 1466–1467 CrossRef CAS PubMed.
- A. Bertsch, C. Gröpl, K. Reinert and O. Kohlbacher, Methods Mol. Biol., 2011, 696, 353–367 CrossRef CAS PubMed.
- E. W. Deutsch, L. Mendoza, D. Shteynberg, T. Farrah, H. Lam, N. Tasman, Z. Sun, E. Nilsson, B. Pratt, B. Prazen, J. K. Eng, D. B. Martin, A. Nesvizhskii and R. Aebersold, Proteomics, 2010, 10(6), 1150–1159 CrossRef CAS PubMed.
- J. Cox and M. Mann, Nat. Biotechnol., 2008, 26(12), 1367–1372 CrossRef CAS PubMed.
- S. Tyanova, T. Temu and J. Cox, Nat. Protoc., 2016, 11, 2301–2319 CrossRef CAS PubMed.
- B. MacLean, D. M. Tomazela, N. Shulman, M. Chambers, G. L. Finney, B. Frewen, R. Kern, D. L. Tabb, D. C. Liebler and M. J. MacCoss, Bioinformatics, 2010, 26(7), 966–968 CrossRef CAS PubMed.
- L. K. Pino, B. C. Searle, J. G. Bollinger, B. Nunn, B. MacLean and M. J. MacCoss, Mass Spectrom. Rev., 2017 DOI:10.1002/mas.21540.
- S. K. Park, J. D. Venable, T. Xu and J. R. Yates III, Nat. Methods, 2008, 5(4), 319–322 CrossRef CAS PubMed.
- F. F. Gonzalez-Galarza, C. Lawless, S. J. Hubbard, J. Fan, C. Bessant, H. Hermjakob and A. R. Jones, OMICS, 2012, 16(9), 431–442 CrossRef CAS PubMed.
- R. Craig, J. C. Cortens, D. Fenyo and R. C. Beavis, J. Proteome Res., 2006, 5(8), 1843–1849 CrossRef CAS PubMed.
- B. E. Frewen, G. E. Merrihew, C. C. Wu, W. S. Noble and M. J. MacCoss, Anal. Chem., 2006, 78, 5678–5684 CrossRef CAS PubMed.
- S. Dasari, M. C. Chambers, M. A. Martinez, K. L. Carpenter, A.-J. L. Ham, L. J. Vega-Montoto and D. L. Tabb, J. Proteome Res., 2012, 11(3), 1686–1695 CrossRef CAS PubMed.
- S. Sharma, D. C. Simpson, N. Tolic, N. Jaitly, A. M. Mayampurath, R. D. Smith and L. Pasa-Tolic, J. Proteome Res., 2007, 6, 602–610 CrossRef CAS PubMed.
- D. M. Horn, R. A. Zubarev and F. W. McLafferty, J. Am. Soc. Mass Spectrom., 2000, 11, 320–332 CrossRef CAS PubMed.
- G. K. Taylor, Y.-B. Kim, A. J. Forbes, F. Meng, R. McCarthy and N. L. Kelleher, Anal. Chem., 2003, 75(16), 4081–4086 CrossRef CAS PubMed.
- R. T. Fellers, J. B. Greer, B. P. Early, X. Yu, R. D. LeDuc, N. L. Kelleher and P. M. Thomas, Proteomics, 2015, 15(7), 1235–1238 CrossRef CAS PubMed.
- Q. Kou, L. Xun and X. Liu, Bioinformatics, 2016, 32(22), 3495–3497 CAS.
- W. Cai, H. Guner, Z. R. Gregorich, A. J. Chen, S. Ayaz-Guner, Y. Peng, S. G. Valeja, X. Liu and Y. Ge, Mol. Cell. Proteomics, 2016, 15(2), 703–714 CrossRef CAS PubMed.
- C. Wynne, N. J. Edwards and C. Fenselau, Proteomics, 2010, 10, 3631–3643 CrossRef CAS PubMed.
- K. Scheffler, Methods Mol. Biol., 2014, 1156, 465–487 CrossRef CAS.
- M. Locard-Paulet, J. Parra, R. Albigot, E. Mouton-Barbosa, L. Bardi, O. Burlet-Schiltz and J. Marcoux, Bioinformatics, 2018 DOI:10.1093/bioinformatics/bty680.
- X. Liu, Y. Inbar, P. C. Dorrestein, C. Wynne, N. Edwards, P. Souda, J. P. Whitelegge, V. Bafnat and P. Pevzner, Mol. Cell. Proteomics, 2010, 9, 2772–2782 CrossRef CAS PubMed.
- P. C. Carvalho, T. Xu, X. Han, D. Cociorva, V. C. Barbosa and J. R. Yates III, Bioinformatics, 2009, 25(20), 2734–2736 CrossRef CAS PubMed.
- P. C. Carvalho, X. Han, T. Xu, D. Cociorva, M. da Gloria Carvalho, V. C. Barbosa and J. R. Yates III, Bioinformatics, 2010, 26(6), 847–848 CrossRef CAS PubMed.
- S. Nicolardi, H. Dalebout, M. R. Bladergroen, W. E. Mesker, R. A. E. M. Tollenaar, A. M. Deelder and Y. E. M. van der Burgt, Int. J. Proteomics, 2012, 2012, 804036 CrossRef PubMed.
- E. Bártová, J. Krejčí, A. Harničarová, G. Galiová and S. Kozubek, J. Histochem. Cytochem., 2008, 56(8), 711–721 CrossRef PubMed.
- P. Chi, C. D. Allis and G. G. Wang, Nat. Rev. Cancer, 2010, 10(7), 457–469 CrossRef CAS.
- E. S. Chrun, F. Modolo and F. I. Daniel, Pathol., Res. Pract., 2017, 213, 1329–1339 CrossRef CAS PubMed.
- A. Portela and M. Esteller, Nat. Biotechnol., 2010, 28(10), 1057–1068 CrossRef CAS PubMed.
- E. L. Greer and Y. Shi, Nat. Rev. Genet., 2012, 13(5), 343–357 CrossRef CAS PubMed.
- T. Jiang, M. E. Hoover, M. V. Holt, M. A. Freitas, A. G. Marshall and N. L. Young, Proteomics, 2018, 18(11), e1700442 CrossRef PubMed.
- C. U. Schräder, D. S. Ziemianowicz, K. Merx and D. C. Schriemer, Anal. Chem., 2018, 90(5), 3083–3090 CrossRef PubMed.
- A. Moradian, A. Kalli, M. J. Sweredoski and S. Hess, Proteomics, 2014, 14, 489–497 CrossRef CAS PubMed.
- I. Sokolowska, J. Mo, J. Dong, M. J. Lewis and P. Hu, mAbs, 2017, 9(3), 498–505 CrossRef CAS PubMed.
- M. Biacchi, R. Gahoual, N. Said, A. Beck, E. Leize-Wagner and Y.-N. François, Anal. Chem., 2015, 87(12), 6240–6250 CrossRef CAS PubMed.
- N. Said, R. Gahouala, L. Kuhn, A. Beck, Y.-N. Françoisa and E. Leize-Wagner, Anal. Chim. Acta, 2016, 918, 50–59 CrossRef CAS PubMed.
- K. Nakayama and K. Nakayama, Nat. Rev. Cancer, 2006, 6(5), 369–381 CrossRef CAS PubMed.
- D. M. Koepp, Methods Mol. Biol., 2014, 1170, 61–73 CrossRef.
- J. Peng, BMB Rep., 2008, 41(3), 177–183 CrossRef CAS.
- C. D. Wenger, M. T. Boyne, J. T. Ferguson, D. E. Robinson and N. L. Kelleher, Anal. Chem., 2008, 80, 8055–8063 CrossRef CAS.
- Y. Jin, Z. Lin, Q. Xu, C. Fu, Z. Zhang, Q. Zhang, W. A. Pritts and Y. Ge, mAbs, 2018 DOI:10.1080/19420862.2018.1525253.
- Z. Tian, N. Tolic, R. Zhao, R. J. Moore, S. M. Hengel, E. W. Robinson, D. L. Stenoien, S. Wu, R. D. Smith and L. Pasa-Tolic, Genome Biol., 2012, 13, R86 CrossRef CAS PubMed.
- K. Srzentić, K. O. Nagornov, L. Fornelli, A. A. Lobas, D. Ayoub, A. N. Kozhinov, N. Gasilova, L. Menin, A. Beck, M. V. Gorshkov, K. Aizikov and Y. O. Tsybin, Anal. Chem., 2018, 90(21), 12527–12535 CrossRef PubMed.
- A. Nesvizhskii, Nat. Methods, 2014, 11(11), 1114–1125 CrossRef CAS PubMed.
- D. S. Kelkar, D. Kumar, P. Kumar, L. Balakrishnan, B. Muthusamy, A. K. Yadav, P. Shrivastava, A. Marimuthu, S. Anand, H. Sundaram, R. Kingsbury, H. C. Harsha, B. Nair, T. S. Prasad, D. S. Chauhan, K. Katoch, V. M. Katoch, P. Kumar, R. Chaerkady, S. Ramachandran, D. Dash and A. Pandey, Mol. Cell. Proteomics, 2011, 10(12), M111.011627 CrossRef.
- S. E. Lee, J. Song, K. Bösl, A. C. Müller, D. Vitko, K. L. Bennett, G. Superti-Furga, A. Pandey, R. K. Kandasamy and M. S. Kim, Proteomics, 2018, 18(8), e1700386 CrossRef PubMed.
- G. M. Sheynkman, M. R. Shortreed, A. J. Cesnik and L. M. Smith, Annu. Rev. Anal. Chem., 2016, 9(1), 521–545 CrossRef.
- N. E. Castellana, S. H. Payne, Z. Shen, M. Stanke, V. Bafna and S. P. Briggs, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(52), 21034–21038 CrossRef CAS PubMed.
- B. Meyer, D. G. Papasotiriou and M. Karas, Amino Acids, 2011, 41, 291–310 CrossRef CAS.
- L. He, C. R. Weisbrod and A. G. Marshall, Int. J. Mass Spectrom., 2018, 427, 107–113 CrossRef CAS.
- M. Ma, R. Chen, Y. Ge, H. He, A. G. Marshall and L. Li, Anal. Chem., 2009, 81(1), 240–247 CrossRef CAS PubMed.
- A. Resemann, D. Wunderlich, U. Rothbauer, B. Warscheid, H. Leonhardt, J. Fuchser, K. Kuhlmann and D. Suckau, Anal. Chem., 2010, 82, 3283–3292 CrossRef CAS PubMed.
- J. J. Thelen and J. A. Miernyk, Biochem. J., 2012, 444(2), 169–181 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information available. See DOI: 10.1039/c8ra07200k |
| This journal is © The Royal Society of Chemistry 2019 |