
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Exosomal miR-25-3p derived from hypoxia tumor mediates IL-6 secretion and stimulates cell viability and migration in breast cancer
Zhengmin Liabc,
Fang Hed,
Zhanjia Yange,
Xueming Caof,
Shuyang Daia,
Jie Zoua,
Poshi Xua and
Zhou Zhou *g
*g
aDepartment of Medical Laboratory, Fuwai Central China Cardiovascular Hospital, Zhengzhou, Henan, China
bDepartment of Medical Laboratory, Henan Provincial People's Hospital, Zhengzhou, Henan, China
cDepartment of Medical Laboratory, Zhengzhou University People's Hospital, Zhengzhou, Henan, China
dOperating Room, Woman & Infants Hospital of Zhengzhou, Zhengzhou, Henan, China
eDepartment of Blood Transfusion, People's Hospital of Zhengzhou, Zhengzhou, Henan, China
fCardiac Care Unit, Fuwai Central China Cardiovascular Hospital, Zhengzhou, Henan, China
gDepartment of Medical Laboratory, Fuwai Hospital, Chinese Academy of Medical Sciences, No. 167, North Lishi Road, Xicheng, 100037, Beijing, China. E-mail: tiancijixiang2010@126.com; Tel: +86-010-88322030
First published on 11th January 2019
Abstract
Hypoxia is a major hallmark of solid tumors and is associated with malignant phenotypes. Exosomal miRNAs derived from hypoxia tumor cells are implicated in the modulation of cancer progression, whereas, the mechanisms underlying the association between hypoxia and exosomal miR-25-3p during breast cancer progression remain to be further clarified. The present study aimed to investigate the role of exosomal miR-25-3p in regulating breast cancer progression. Herein, we found that miR-25-3p expression was increased in hypoxia tumor-derived exosomes a HIF-1α-dependent manner. Hypoxia exosomes markedly stimulated the viability and migration of normoxia breast cancer cells, which was reversed by miR-25-3p depletion. Inhibition of exosomes miR-25-3p lowered hypoxic-induced the expression of IL-6 and NF-κB from THP-1 and RAW264.7 cells in a TLR7/8-dependent way. Treatment of macrophage supernatant (MS) initially incubated with hypoxic-responsed exosomes accelerated the viability and migration of breast cancer cells, and miR-25-3p depletion relieved the stimulatory effects of hypoxic on cell viability and migration. Moreover, miR-25-3p knockdown dramatically suppressed HIF-1α-induced tumor growth in vivo via inactivation of IL-6/STAT3 signaling pathway, reflected by the abated abundances of IL-6 and p-STAT3. These data suggested that absence of exosomal miR-25-3p rescued breast cancer aggressiveness through inhibiting cell viability and migration by regulation of IL-6 secretion from macrophages, providing a potential biomarker for breast cancer treatment.
1. Introduction
Breast cancer, one of the most commonly diagnosed female malignancies, is the prime leading cause of cancer-related death in women with an estimated 272![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 new cases and 70700 deaths in China alone in 2015.1 Despite significant advances in chemotherapy having prolonged the survival time of patients, the prognosis is also unsatisfying. Thus, further clarification of the underlying molecular mechanisms and discovery of novel biomarkers are required for breast cancer treatment.
400 new cases and 70700 deaths in China alone in 2015.1 Despite significant advances in chemotherapy having prolonged the survival time of patients, the prognosis is also unsatisfying. Thus, further clarification of the underlying molecular mechanisms and discovery of novel biomarkers are required for breast cancer treatment.
Due to the more rapid development of tumor cells than surrounding vasculature, tumor cells then subject to an avascular microenvironment with deprived of oxygen, defined as hypoxia.2 Hypoxia, appeared as a major hallmark of tumor microenvironment, is commonly served as a powerful driving force for pathological evolution of a wide range of solid tumors.3,4 Reliable studies have shown that hypoxia contributes to immunosuppression within tumors, and trigger the invasion and metastasis of cancer cells to counterbalance hypoxic microenvironment.5,6 Mechanism studies underlying physiologic responses to hypoxia confirm the prime involvement of hypoxia-inducible factors (HIF), which exists as a heterodimeric complexes composed by oxygen-dependent HIF-1α and a highly expressed HIF-1β.7,8 There are strong evidences supporting the concept that elevated expression of HIF-1α contributes to the growth, metastasis and vascularization of multiple solid tumors via activation of tumor-related genes, serving as a feature element of hypoxia-response.9–11
Exosomes, defined as membrane microvesicles with 40–100 nm in diameter, are secreted by several cell types during endocytosis, including endothelial cells, immunocytes, epithelial cells, and tumor cells.12,13 Typical exosomes play central roles in the substance and signaling communication between cells through carrying a distinct set of proteins, mRNAs, and microRNAs (miRNAs).14 Also, exosomes derived from cancer cells can function as messengers of hypoxic response and in turn contribute to tumor metastasis and angiogenesis.15 As a major element of exosomes, exosomal miRNAs are widely implicated in the progression and prognosis of various cancers, including breast cancer. For instance, exosome miR-10b and miR-155 are confirmed as pivotal regulators of tumor pathology or chemoresistance.16,17 In addition, the abundance of exosomal miR-25-3p is associated with the progression of several cancers by regulating cell–cell communication, like liposarcoma18 and kidney cancer.19 In breast cancer, miR-25-3p is reported to contribute to the proliferation of breast cancer cells through targeting BTG2.20 However, the functional role of exosomal miR-25-3p in the aggressiveness of breast cancer is still unclear.
In this study, we presented the evidence that miR-25-3p was HIF-1α-dependently upregulated in hypoxic exosomes. Absence of exosomal miR-25-3p abolished hypoxia-induced breast cancer cell viability and migration by regulation of IL-6 secretion from macrophages in a TLR-7/8 dependent way. Moreover, we also developed murine xenograft model to investigate the effect of miR-25-3p on breast cancer tumor process under hypoxic condition.
2. Materials and methods
2.1 Cell culture and hypoxia treatment
Human breast cancer cell lines include MDA-MB-231, monocyte cell lines THP-1 and RAW264.7 were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Mouse E0771 breast cancer cell line was obtained from American type culture collection (ATCC, Manassas, VA, USA). THP-1 (TLR8−/−) or RAW264.7 (TLR7−/−) were obtained from Biocytogen (Beijing, China). All cells were maintained in a humidified incubator of 5% CO2 at 37 °C with RPMI-1640 medium (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS, Gibco). For the hypoxia experiments, cultured cells were grown in a hypoxic condition of 1% O2, 94% N2 and 5% CO2.2.2 Plasmids and transfection
Lentivirus plasmid containing HIF-1α cDNA sequences (HIF-1α) and shRNA for HIF-1α (shHIF-1α) were constructed by Genepharma (Shanghai, China) to overexpress or inhibit HIF-1α expression. IL-6 overexpression plasmid (IL-6) was also synthesized in Genepharma by inserting IL-6 sequences into pcDNA 3.1 vector. MiR-25-3p mimics (miR-25-3p), miR-25-3p inhibitor (miR-25-3p-in) and negative control (NC) were purchased from Genepharma (Shanghai, China). Next, transfection of MDA-MB-231 and E0771 cells (1 × 105) were carried out with above oligonucleotides or plasmids using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA).2.3 Exosome isolation
After washed with PBS for three times, cells were cultured in respective medium containing exosome-depleted FBS for 48 h. Supernatants of cultured cells were collected and subjected to differential centrifugations to remove cell debris and macromolecular proteins. Then, 0.25 volume of ExoQuick-TC™ Exosome Precipitation Solution (SBI, Palo Alto, CA, USA) was mixed with the filtered supernatants and restored at 4 °C overnight. After incubation, the mixtures were centrifuged at 12000 × g for 5 min and the sediment containing exosomes was resuspended in PBS solution.
2.4 Western-blot
Total proteins derived from cells, exosomes, and tumor tissues were divided on SDS-PAGE gel and electrotransferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% non-fat milk powder overnight at 4 °C and incubated with primary antibodies against HIF-1α, CD81, CD63, anti-Tubulin, IL-6, p-STAT3, STAT3, BTG2, and anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 2 h at 37 °C. Next, horseradish peroxidase (HRP)-conjugated secondary antibodies (Cell Signaling Technology, Inc, Danvers, MA, USA) were added and incubated for another 1.5 h at 37 °C. Protein bands were visualized using Enhanced chemiluminescence kit (Biorbyt, Shanghai, China) and densitometry analysis was evaluated by Image Lab software (Bio-Rad, Hercules, CA, USA).2.5 Reverse-transcription quantitative PCR (RT-qPCR)
Total RNA was extracted from cells and exosomes using Trizol reagent (Invitrogen Carlsbad, CA, USA) referring to the manufacturer's protocol. For the detection of miR-25-3p expression, 1 μg of RNA was collected to synthesize the first strand cDNA using MicroRNA Reverse Transcription Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA) with U6 snRNA as a housekeeping gene. For IL-6 mRNA expression, cDNA was synthesized using High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) with GAPDH as a housekeeping gene. Afterwards, qPCR reactions were carried out using SYBR Green qPCR Master Mix (2×) (Thermo Fisher Scientific) mixed with special primers and ran on ABI 7500 Real-time PCR Systems (Applied Biosystems, Foster City, CA, USA). The primers were shown as below: human IL-6: 5′-TCAATGAGGAGACTTGCCTG-3′ (forward) and 5′-GATGAGTTGTCAATGTCCTGC-3′ (reverse); mouse IL-6, 5′-CTAGGAAGAACTGGCAATATG-3′ (forward) and 5′-AAACCATCTGGCTAGG TAAGA-3′ (reverse); GAPDH, 5′-TATGATGATATCAAGAGGGTAGT-3′ (forward) and 5′-TGTATCCAAACTCATTGTCATAC-3′ (reverse). Primers for miR-25-3p and U6 snRNA were obtained from Genepharma (Shanghai, China). All samples were detected in triplicate and relative expression levels were calculated by 2−ΔΔCt method.2.6 Cell viability assay
Cell viability was evaluated using MTT Assay Kit (Solarbio, Beijing, China). Briefly, MDA-MD-231 and E0771 cells were seeded into 96-well plates with a density of 5 × 103 cells per well. About 48 h after incubation with exosomes or macrophage supernatant, 0.5 mg mL−1 MTT was introduced and further incubated for 4 h. Then, the supernatant of medium was carefully removed and 150 μL dimethyl sulfoxide (DMSO) was added to dissolve the generated formazan. The absorbance at 490 nm was detected by a microplate reader (Bio-Rad). All experiments were performed for three repetitions.2.7 Cell migration assay
Cell migration ability was assessed using Transwell chambers (BD Biosciences, San Jose, CA, USA). Briefly, MDA-MD-231 and E0771 cells treated with respective exosomes or macrophage solution were harvested, resuspended in serum-free medium, and then added to the upper chambers. The bottom chambers were treated with complete medium containing 10% FBS. After incubation under a definite condition for 24 h, the cells on the upper membrane were carefully removed, and those migrated to the basal side of the membrane were fixed and stained with crystal violet (Sigma-Aldrich, St. Louis, MO, USA). Finally, three visual fields were selected and photographed, followed by the counted of migrated cells by a microscope (Olympus, Tokyo, Japan).2.8 Enzyme-linked immunosorbent assay (ELISA)
IL-6 and NF-κB secretion were determined using IL-6 ELISA kit (Abcam, Cambridge, UK) or NF-κB ELISA Detection kit (Jianglaibio, Shanghai, China) according to manufacturers' instructions.2.9 Tumor xenograft
All experimental procedures were carried out in accordance with the Guidelines of Animal Care and Use with the approval of Animal Research committee of Sanquan College of Xinxiang Medical University. C57/BL6 and non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice (females, 6 weeks of age) were purchased from Hufukang (Beijing, China). E0771 cells (2 × 106) initially transfected with miR-25-3p-in, HIF-1α, HIF-1α + miR-25-3p-in, or corresponding controls were administered into mammary fat pad of C57/BL6 mice. Cells transfected with miR-25-3p-in and negative control were inoculated orthotopically to NOD/SCID mice in the same manner. Tumors size was measured using Vernier caliper weekly and volume was calculated as the cubature formula of (length × width2)/2. About 6 weeks after tansplantation, mice were sacrificed, and tumors were collected to detect the protein levels of IL-6, p-STAT3, STAT3, and β-actin.2.10 Statistical analysis
The data from three independent experiments were expressed as mean ± standard deviation (SD). Significant group differences were assessed using SPSS 20.0 software (SPSS, Chicago, IL, USA) with Student t-test or one-way ANOVA. P value less than 0.05 (P < 0.05) was considered to be statistically significant.3. Result
3.1 Hypoxia stimulates miR-25-3p expression in tumor-derived exosomes in a HIF-1α dependent manner
In present study, we first confirmed the elevated abundance of HIF-1α protein following hypoxia treatment and HIF-1α transfection in MDA-MB-231 and E0771 cells by western-blot assay (Fig. 1A and B). RT-qPCR showed that miR-25-3p was markedly upregulated in hypoxia tumor cells (Fig. 1C) and hypoxia tumor-derived exosomes (Fig. 1E) compared to normoxia group. Moreover, we further confirmed the presence of the known exosome markers CD63 and CD81 in exosomes and cells by western-blot analysis. Results revealed that hypoxia and HIF-1α induced the release of exosomes compared with normoxia group (Fig. 1D). In addition, miR-25-3p level was obviously higher in hypoxia tumor-derived exosomes than in hypoxia tumor cells, however, the introduction of shHIF-1α inhibited the expression of miR-25-3p in MDA-MB-231 and E0771 cells or exosomes under the oxygen deficit condition (Fig. 1F and G), hinting that hypoxia induced miR-25-3p expression both in tumor cells and exosomes in a HIF-1α dependent manner.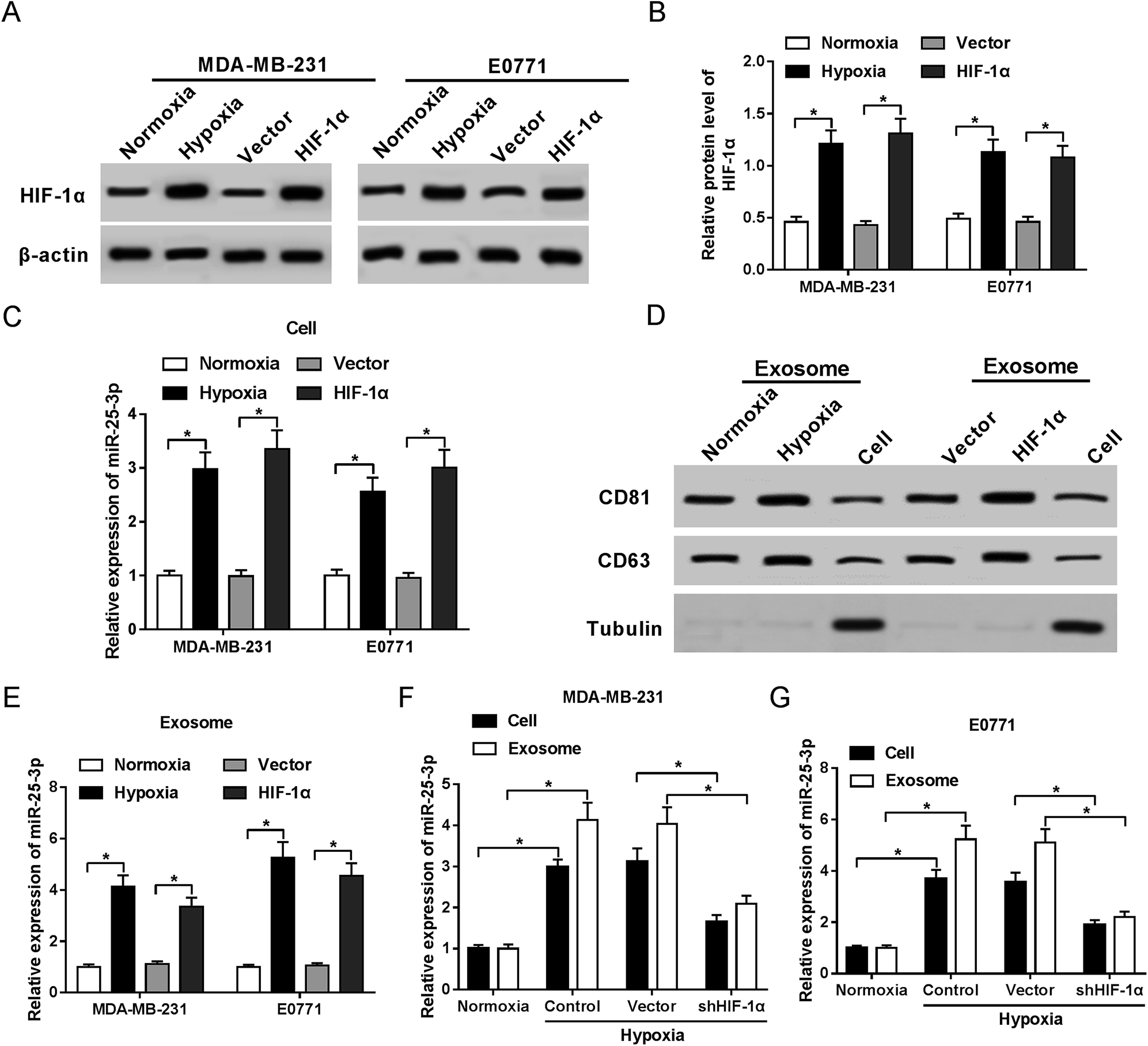 | ||
| Fig. 1 Hypoxia stimulated exosomal miR-25-3p expression in a HIF-1α dependent manner. The protein expression of HIF-1α in normoxia, hypoxia, or HIF-1α-transfected MDA-MB-231 (A) and E0771 cells (B) was measured by western-blot assay. (C) miR-25-3p expression in normoxia, hypoxia, or HIF-1α-transfected MDA-MB-231 and E0771 cells was detected by RT-qPCR. (D) Exosome-related proteins CD81 and CD63 in normoxia or hypoxia-derived exosomes, as well as in MDA-MB-231 cells were determined by western-blot assay. (E) miR-25-3p expression in normoxia, hypoxia, or HIF-1α-derived exosome was detected by RT-qPCR. (F and G) Knockdown of HIF-1α inhibited miR-25-3p expression in hypoxia-responsive breast cancer cells and exosomes. | ||
3.2 Depletion of exosomal miR-25-3p inhibits hypoxic exosome-induced cell viability and migration in normoxic breast cancer cells
Exosomes have the potential to deliver biological information by interacting with adjacent or distant cells.21 In this study, we aimed to investigate whether hypoxic tumor-derived exosomes modulate normoxic tumor cell phenotypes via depending on miR-25-3p. MTT and Transwell assays revealed that hypoxic exosomes evidently induced the viability and migration of normoxic MDA-MB-231 and E0771 cells, while exosomes derived from miR-25-3p-antagonized tumor cells inhibited hypoxic exosome-induced target cell viability and migration (Fig. 2A–D).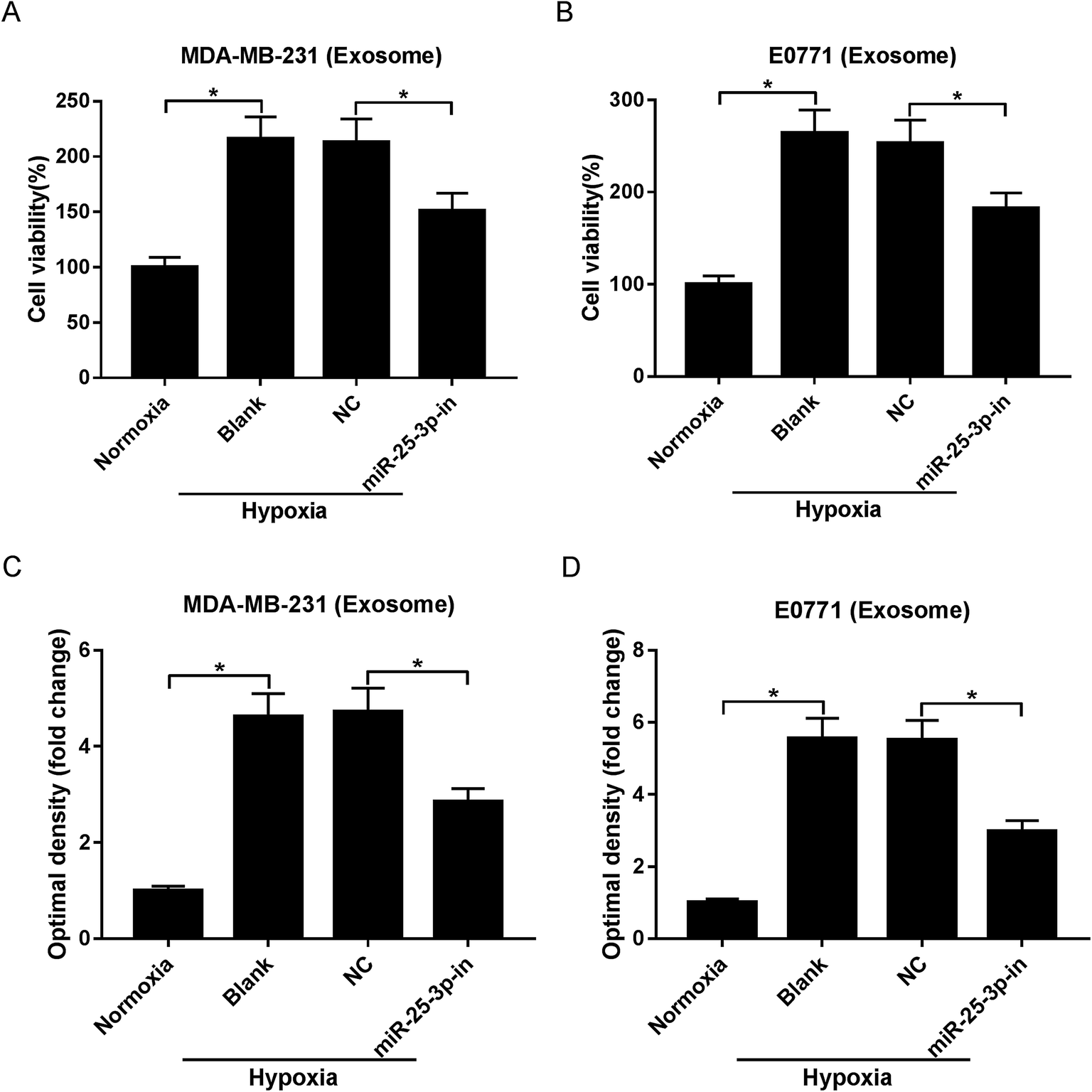 | ||
| Fig. 2 Exosomes derived from hypoxic breast cancer cells contributed to target cell viability and migration through delivering miR-25-3p. Normoxia, hypoxia, or hypoxia + miR-25-3p-derived exosomes were added into normoxia MDA-MB-231 and E0771 cells. About 48 h after incubation, the viability (A and B) and migration (C and D) of tumor cells were assessed by MTT and Transwell assays. | ||
3.3 Exosomal miR-25-3p stimulates IL-6 secretion from macrophages via NF-κB pathway and promotes breast cancer cell proliferation and migration
We next asked whether miR-25a-3p in tumor-derived exosomes involved in the modulation of IL-6 levels from macrophages. RT-qPCR and ELISA assays showed that miR-25-3p or hypoxia-derived exosomes notably increased macrophage-secreted IL-6 mRNA (Fig. 3A and B) and protein levels (Fig. 3C and D) both in THP-1 and RAW264.7 cells. However, miR-25-3p-deprived exosomes weakened hypoxia exosomes-induced IL-6 mRNA and protein expression (Fig. 3A–D), indicating that hypoxia exosomes promoted IL-6 secretion in macrophages via delivering miR-25-3p.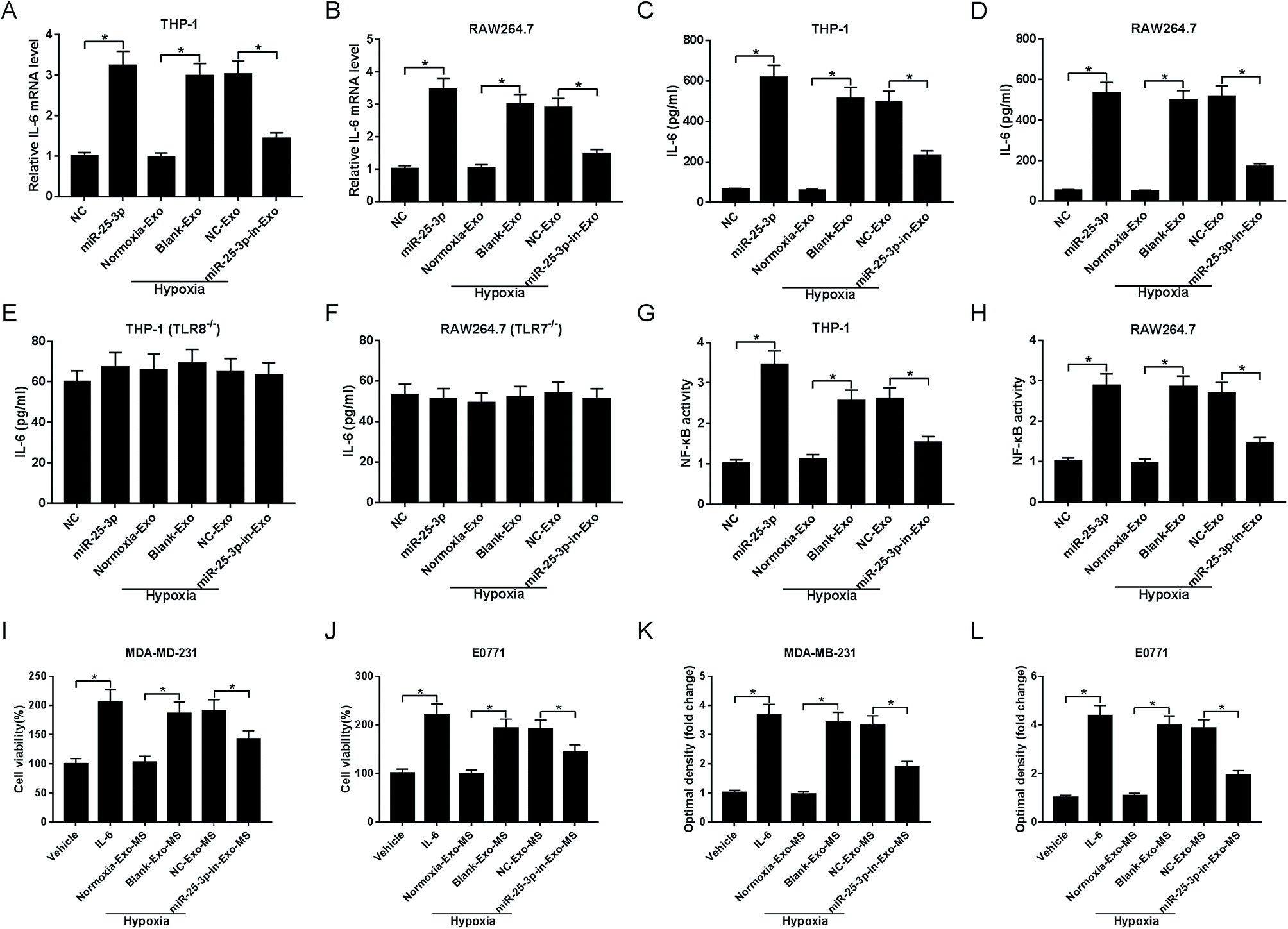 | ||
| Fig. 3 Absence of exosomal miR-25-3p inhibited IL-6 secretion from macrophages via NF-κB in a TLR7/8-dependent manner and then receded breast cancer cell viability and migration. Exosomes derived from tumor cells were introduced into THP-1, RAW264.7, THP-1 (TLR8−/−), or RAW264.7 (TLR7−/−) cells. About 48 h after incubation, RT-qPCR and ELISA assays were carried out to detect the mRNA (A and B) and protein levels (C–F) of IL-6. NF-κB activity was measured by ELISA assay (G and H). The viability (I and J) and migration (K and L) of MDA-MD-231 and E0771 cells treated with IL-6, vehicle, or MS previously incubated with hypoxia or miR-25-3p-lacked hypoxia exosomes were determined by MTT and Transwell assays. | ||
To verify whether IL-6 secretion induced by exosomal miR-25-3p occurred in a TLR7/8-dependent manner, THP-1 and RAW264.7 cells with TLR8 or TLR7 knockout were constructed, followed by the detection of IL-6 secretion. As our conjecture, knockout of TLR8 or TLR7 in human or murine macrophages abrogated the inhibitory effect of exosomal miR-25-3p depletion on IL-6 expression (Fig. 3E and F), suggesting that murine TLR7 and human TLR8 receptors were needed for this process. On the basis of these findings, we probed whether exosomal miR-25-3p-mediated IL-6 secretion from macrophages through NF-κB pathway. In line with IL-6 expression, NF-κB activity in THP-1 and RAW264.7 cells was prominently accelerated following the treatment of miR-25-3p or hypoxia-derived exosomes compared to respective controls. However, miR-25-3p inhibition weakened hypoxia-induced NF-κB activity of macrophages (Fig. 3G and H).
Next, we further investigated the effect of macrophage-secreted IL-6 on cell viability and migration of normoxia MDA-MD-231 and E0771 cells. Human and murine macrophages previously incubated with normoxia exosomes (normoxia-exo), hypoxia exosomes (hypoxia + blank-exo), NC-derived hypoxia exosomes (hypoxia + NC-exo), or miR-25-3p-in-derived hypoxia exosomes (hypoxia + miR-25-3p-in-exo) were incubated for 48 h, followed by the extraction of macrophage supernatant (MS). Then, normoxic MDA-MD-231 and E0771 cells were treated with vehicle, IL-6, or respective macrophage solution for 48 h to evaluate cell viability and migration ability. IL-6-overexpression or incubation with MS previously incubated with hypoxia exosomes induced the viability (Fig. 3I and J) and migration (Fig. 3K and L) of normoxia breast cancer cells. However, cell viability and migration was notably suppressed following the introduction of MS, which was previously treated with hypoxia + miR-25-3p-in-exo (Fig. 3I–L). These findings indicated that miR-25-3p knockdown decreased the release of macrophage-secreted IL-6 and then negatively regulated tumor growth.
3.4 Depletion of miR-25-3p suppressed HIF-1α-induced tumorigenesis via serving as a pro-inflammatory factor in breast cancer
To investigate the effects of miR-25-3p on tumor growth in vivo, E0771 cells transfected with miR-25-3p-in or NC were injected into the mammary fat pad of C57/BL6 or NOD/SCID mice. Compared to NC group, reduction of miR-25-3p resulted in an obvious reduction of tumor volume in C57/BL6 mice (Fig. 4A), but for NOD/SCID mice, the tumor volume had less change following miR-25-3p inhibition compared to C57/BL6 mice (Fig. 4B), suggesting that miR-25-3p stimulated tumorigenesis in an immune-dependent way. To further confirmed HIF-1α-derived hypoxia could mediate tumor growth via miR-25-3p in vivo, E0771 cells transfected with vector, HIF-1α, or HIF-1α + miR-25-3p-in were injected into C57/BL6 mice. Result showed that knockdown of miR-25-3p abrogated HIF-1α-induced tumor growth (Fig. 4C). Reliable evidences have verified the activation of IL-6/STAT3 pathway in tumor progression.22 In this study, miR-25-3p knockdown inhibited the protein expressions of IL-6 and p-STAT3 induced by HIF-1α (Fig. 4D and E), indicating that lack of miR-25-3p inhibited tumorigenesis induced by oxygen deprivation through inactivation of IL-6/STAT3 pathway.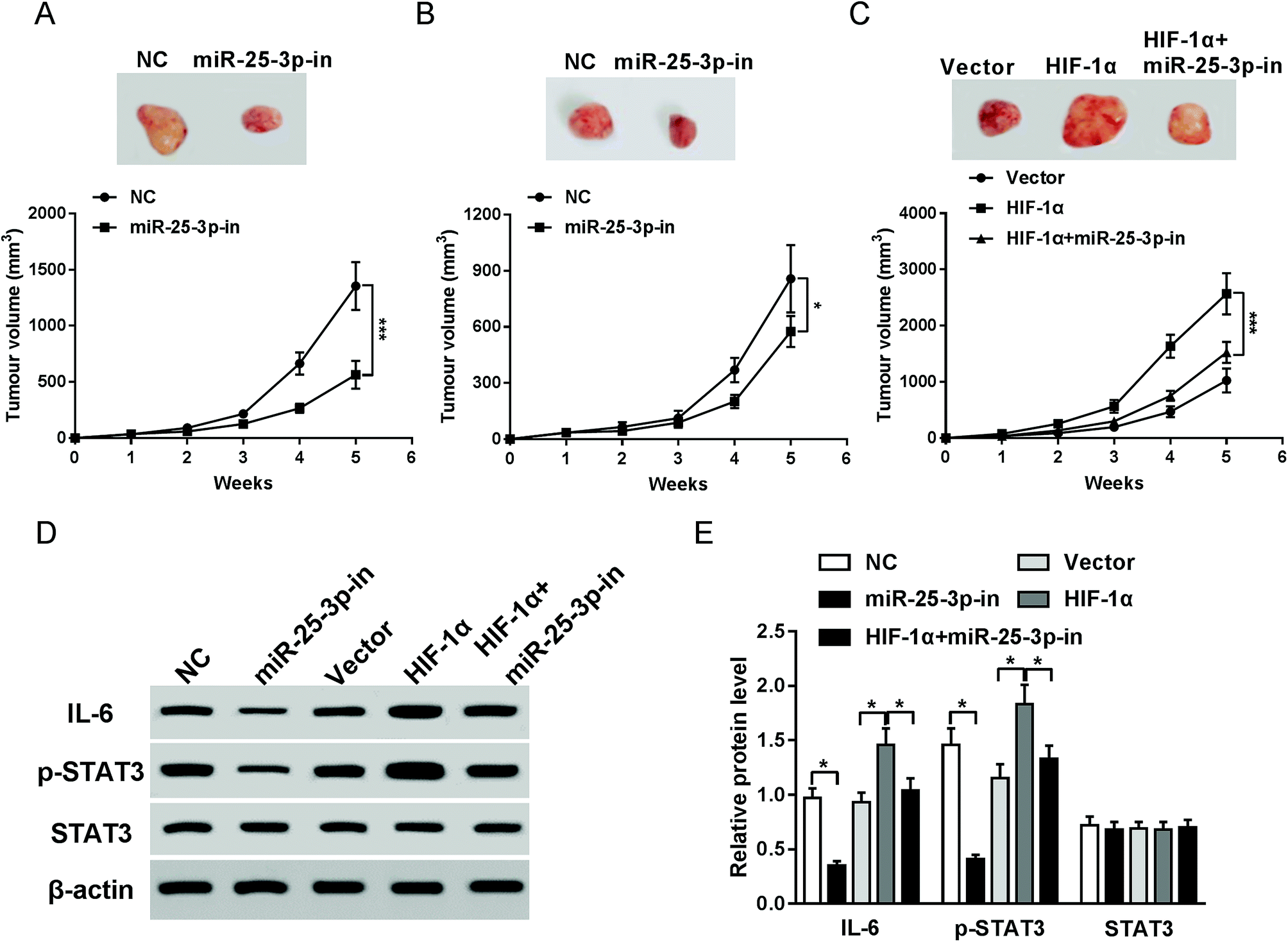 | ||
| Fig. 4 Lack of miR-25-3p suppressed HIF-1α-induced tumorigenesis via regulation of IL-6/STAT3 signaling pathway. Tumor volumes of C57/BL6 (A and C) and E0771 mice (B) injected with NC, miR-25-3p-in, HIF-1α, or HIF-1α + miR-25-3p-in-transfected E0771 cells were calculated every week. (D and E) The protein expressions of IL-6, p-STAT3, and STAT3 in tumor tissues of C57/BL6 mice were detected by western-blot assay. | ||
4. Discussion
Breast cancer is a serious threat for women's health and life with relatively poor prognosis and lacks effective targeted therapy. In this study, we provided a new insight into the mechanism of hypoxia exosome-induced tumor growth via delivering miR-25-3p by stimulating the release of IL-6 from macrophages in a TLR7/8 dependent way (Fig. 5).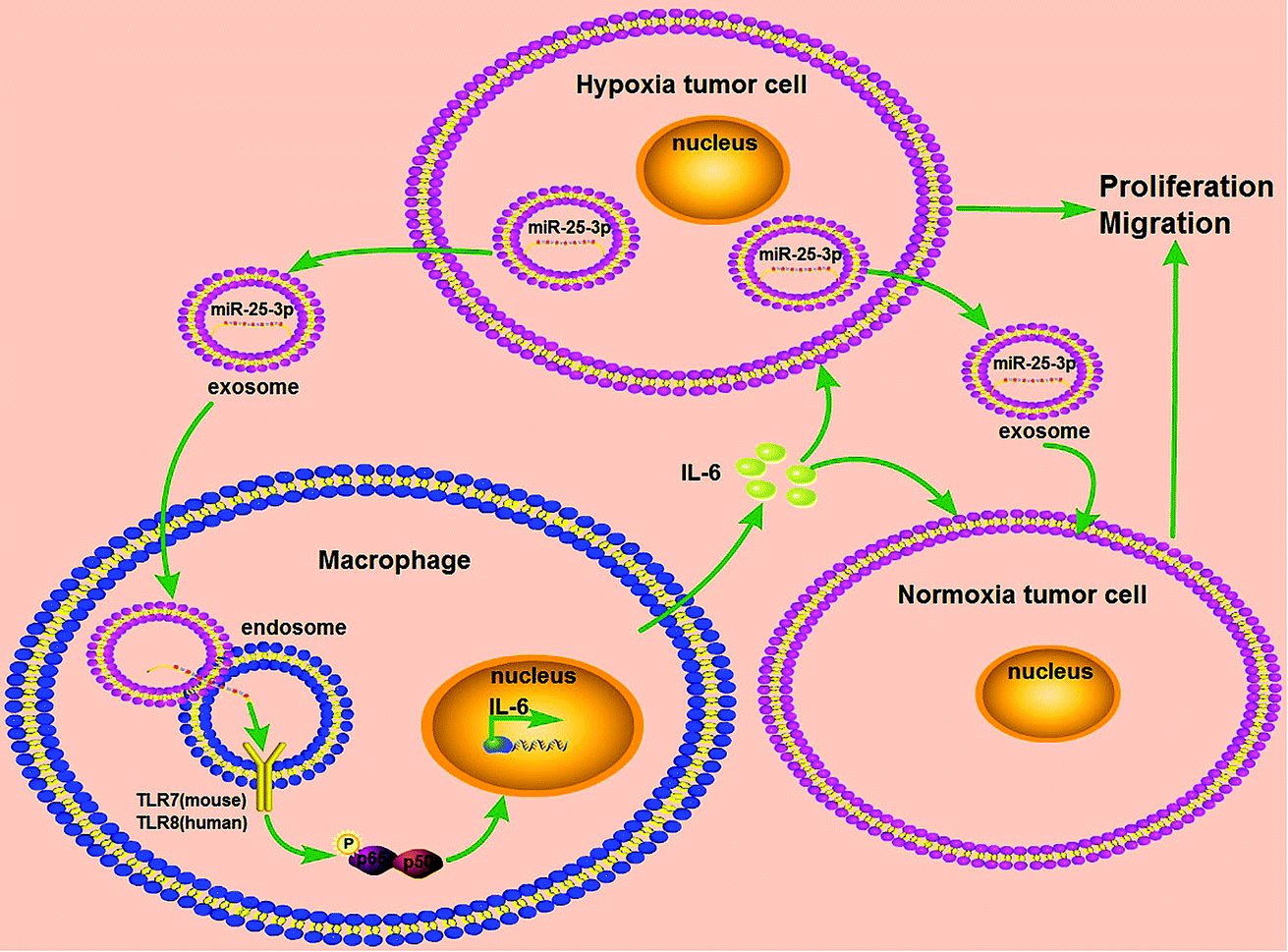 | ||
| Fig. 5 Schematic representation of exosomal miR-25-3p in the modulation of breast cancer progression. Hypoxia stimulated the expression of miR-25-3p in breast cancer cells and induced the release of this miRNA into exosomes. Exosomal miR-25-3p in turn promoted the proliferation and migration of breast cancer cells through induced IL-6 secretion from macrophages via NF-κB pathway in a TLR7/8-dependent manner. | ||
Mounting evidences suggests that hypoxia is a major promoter for tumor progression by modulation of multistage tissue metastasis, angiogenesis, growth factor signaling, and poor prognosis.23 HIF-1α can be modulated via oxygen-independent mechanism in a set of cell types. In hypoxia condition, HIF-1α protein degradation is blocked, leading to the accumulation of HIF-1α.24 In line with previous viewpoint, we observed an elevated level of HIF-1α in hypoxia-treated or HIF-1α-transfected breast cancer cells. Recently, the vital roles of tumor-derived exosomes in the malignant processes of cancers have been confirmed and hypoxia is speculated to stimulate the secretion of exosomes derived from tumor cells.25 In present study, we first purified exosomes from breast cancer cell supernatant and observed the enrichment of exosome-related protein CD81 and CD63 in normoxia or hypoxia exosomes, especially in hypoxia exosomes. This is also agreement with those evidences in support of hypoxia stimulating the release of exosomes derived from breast cancer cells.26
As lipid-bilayer-enclosed extracellular vesicles, exosomes can mediate the interaction between tumor cells and microenvironment by carrying a wild series of complex components, including proteins, ribonucleotides, and miRNA.27,28 MiRNAs have been considered as key players of cancer progression through regulating cancer-related gene expression.29 A specific set of exosomal microRNAs, including miR-25-3p, are reported to involve in the malignant evolution of several cancers. However, the detail roles of exosomal miR-25-3p in breast cancer have not been uncovered. In this study, we observed an elevated expression of exosomal miR-25-3p in hypoxia exosomes in a HIF-1α-dependent fashion. Further loss-of-function assay revealed that deprivation of exosomal miR-25-3p inhibited hypoxia-induced cell viability and migration in normoxia breast cancer cells, prompting that exosomal miR-25-3p may function as an oncogene in breast cancer.
A growth number of evidences suggest that chronic inflammation is wildly involved in the multi-stages of tumorigenesis. Interleukin-6 (IL-6) is demonstrated to be a pivotal inflammatory cytokine predominantly produced by macrophages.30,31 Elevated IL-6 is relevant to the poor outcome of cancer patients, and overproduction of IL-6 promotes cell growth, metastasis, angiogenesis in a series of malignancies. For example, overexpressed IL-6 was significantly associated with advance clinical stage, higher recurrence rate, and lower survival rate in bladder cancer.32 In addition, IL-6 combined with TNF-α and Th17-type cytokines contributed to the growth of colorectal cancer cells via activating NF-κB and STAT3.33 Also, IL-6 strikingly induced the proliferation and metastasis of osteosarcoma.34 Recent literature highlights the vital role of NF-κB/IL-6/STAT3 feedback loop in human cancers. In most cases, NF-κB pathway mediates the releases of pro-inflammatory cytokines, noteworthy IL-6. Enhanced abundance of IL-6 in turn results in the activation of STAT3 pathway,35 indicating that IL-6 is implicated in the malignant progression of various tumors mostly via NF-κB/IL-6/STAT3 pathway.
In the following study, we attempted to explore whether miR-25-3p promoted breast cancer tumor growth via modulation of IL-6. Our results exhibited abated abundances of IL-6 and NF-κB in macrophages after incubating with miR-25-3p-depleted hypoxia exosomes. However, there was no significant change of IL-6 levels in TLR7/8-knockout macrophages, which indicated that miR-25-3p might promote the release of pro-inflammatory cytokine IL-6 from macrophages via NF-κB in a TLR7/8-dependent way. Further study showed that the viability and migration ability of normoxia breast cancer cells incubated with macrophages previously treated with miR-25-3p-lacked exosomes were obviously inhibited via downregulating IL-6. In vivo experiments also indicated that knockdown of miR-25-3p suppressed HIF-1α-induced tumor growth, possibly through inactivation of IL-6/STAT3 signaling pathway.
5. Conclusions
Taken together, our data showed that hypoxia induced the expression of exosomal miR-25-3p HIF-1α dependently. In addition, miR-25-3p-lacked exosomes promoted the transformation of tumor cells toward an anti-metastasis phenotype via suppressing the secretion of IL-6 from macrophages in a TLR7/8-dependent manner. These findings also raise the possibility that exosomal miR-25-3p may be employed as potential therapeutic target for breast cancer.Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant No. U1704167).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Not applicable.References
- W. Chen, R. Zheng, P. D. Baade, S. Zhang, H. Zeng, F. Bray, A. Jemal, X. Q. Yu and J. He, Ca-Cancer J. Clin., 2016, 66, 115–132 CrossRef PubMed.
- G. Babu, Wilderness Environ. Med., 2015, 26, e3–e4 CrossRef.
- E. B. Rankin and A. J. Giaccia, Science, 2016, 352, 175–180 CrossRef CAS PubMed.
- X. Lu and Y. Kang, Clin. Cancer Res., 2010, 16, 5928–5935 CrossRef CAS PubMed.
- P. Asis, A. Julián, M. K. Aizea, D. L. Manuel Ortiz and M. Ignacio, Clin. Cancer Res., 2012, 18, 1207–1213 CrossRef PubMed.
- V. Kumar and D. I. Gabrilovich, Immunology, 2014, 143, 512–519 CrossRef CAS PubMed.
- G. L. Semenza, Oncogene, 2010, 29, 625–634 CrossRef CAS PubMed.
- R. H. Wenger, M. Gassmann, M. Kurowska, A. Hirth, S. Gay and O. Distler, Arthritis Rheumatol., 2004, 50, 10–23 CrossRef PubMed.
- G. Guan, Y. Zhang, Y. Lu, L. Liu, D. Shi, Y. Wen, L. Yang, Q. Ma, T. Liu and X. Zhu, Cancer Lett., 2015, 357, 254–264 CrossRef CAS PubMed.
- T. Zhao, H. Ren, J. Li, J. Chen, H. Zhang, W. Xin, Y. Sun, L. Sun, Y. Yang and J. Sun, Cancer Res., 2015, 75, 111–119 CrossRef CAS PubMed.
- S. W. Wang, S. C. Liu, H. L. Sun, T. Y. Huang, C. H. Chan, C. Y. Yang, H. I. Yeh, Y. L. Huang, W. Y. Chou and Y. M. Lin, Carcinogenesis, 2015, 36, 104–114 CrossRef CAS PubMed.
- J. Wahlgren, T. D. L. Karlson, M. Brisslert, F. V. Sani, E. Telemo, P. Sunnerhagen and H. Valadi, Nucleic Acids Res., 2012, 40, e130 CrossRef CAS PubMed.
- J. Zhang, S. Li, L. Li, M. Li, C. Guo, J. Yao and S. Mi, Genomics, Proteomics Bioinf., 2015, 13, 17–24 CrossRef CAS PubMed.
- X. Huang, T. Yuan, M. Tschannen, Z. Sun, H. Jacob, M. Du, M. Liang, R. L. Dittmar, L. Yong and M. Liang, BMC Genomics, 2013, 14, 319 CrossRef CAS PubMed.
- Y. Yang, X. Yang, Y. Yang, H. Zhu, X. Chen, H. Zhang, F. Wang, Q. Qin, H. Cheng and X. Sun, Curr. Med. Chem., 2015, 22, 4189–4195 CrossRef CAS PubMed.
- R. Singh, R. Pochampally, K. Watabe, Z. Lu and Y. Y. Mo, Mol. Cancer, 2014, 13, 256 CrossRef PubMed.
- J. C. Santos, N. D. S. Lima, L. O. Sarian, A. Matheu and M. L. Ribeiro, Sci. Rep., 2018, 8, 829 CrossRef PubMed.
- L. Casadei, F. Calore, C. J. Creighton, M. Guescini, K. Batte, O. H. Iwenofu, A. Zewdu, D. Braggio, K. L. Bill and P. Fadda, Cancer Res., 2017, 77, 3846–3856 CrossRef CAS PubMed.
- H. Butz, R. Nofechmozes, Q. Ding, K. Hwz, P. M. Szabó, M. Jewett, A. Finelli, J. Lee, M. Ordon and R. Stewart, Eur. Urol. Focus, 2016, 2, 210–218 CrossRef.
- H. Chen, H. Pan, Y. Qian, W. Zhou and X. Liu, Mol. Cancer, 2018, 17, 4 CrossRef PubMed.
- D. Sun, X. Zhuang, S. Zhang, Z. B. Deng, W. Grizzle, D. Miller and H. G. Zhang, Adv. Drug Delivery Rev., 2013, 65, 342–347 CrossRef CAS PubMed.
- H. Kitamura, Y. Ohno, Y. Toyoshima, J. Ohtake, S. Homma, H. Kawamura, N. Takahashi and A. Taketomi, Cancer Sci., 2017, 108, 1947–1952 CrossRef CAS PubMed.
- A. L. Harris, Nat. Rev. Cancer, 2002, 2, 38–47 CrossRef CAS.
- B. Bedessem and A. Stéphanou, PLoS One, 2014, 9, e110495 CrossRef.
- G. K. Panigrahi, P. P. Praharaj, T. C. Peak, J. Long, R. Singh, J. S. Rhim, Z. Elmageed and G. Deep, Sci. Rep., 2018, 8, 6645 CrossRef PubMed.
- H. W. King, M. Z. Michael and J. M. Gleadle, BMC Cancer, 2012, 12, 421 CrossRef CAS PubMed.
- R. J. Simpson, J. Proteomics, 2010, 73, 1907–1920 CrossRef PubMed.
- S. Rana, K. Malinowska and M. Zöller, Neoplasia, 2013, 15, 281–295 CrossRef CAS.
- M. V. Iorio and C. M. Croce, Carcinogenesis, 2012, 33, 1126–1133 CrossRef CAS PubMed.
- R. Bharti, G. Dey and M. Mandal, Cancer Lett., 2016, 375, 51–61 CrossRef CAS PubMed.
- M. J. Waldner and M. F. Neurath, Semin. Immunol., 2014, 26, 75–79 CrossRef CAS.
- M. F. Chen, P. Y. Lin, C. F. Wu, W. C. Chen and C. T. Wu, PLoS One, 2013, 8, e61901 CrossRef CAS.
- S. V. De, E. Franzè, G. Ronchetti, A. Colantoni, M. C. Fantini, F. D. Di, G. S. Sica, P. Sileri, T. T. Macdonald and F. Pallone, Oncogene, 2015, 34, 3493–3503 CrossRef PubMed.
- B. Tu, L. Du, Q. M. Fan, Z. Tang and T. T. Tang, Cancer Lett., 2012, 325, 80–88 CrossRef CAS PubMed.
- B. C. Mcfarland, S. W. Hong, R. Rajbhandari, G. B. T. Jr, G. K. Gray, H. Yu, E. N. Benveniste and S. E. Nozell, PLoS One, 2013, 8, e78728 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |