
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel exchanged supported 12-tungstophosphoric acid: synthesis, characterization and base free one-pot oxidative esterification of aldehyde and alcohol†
Anish Patel and
Anjali Patel*
and
Anjali Patel*
Polyoxometalates and Catalysis Laboratory, Department of Chemistry, Faculty of Science, The Maharaja Sayajirao University of Baroda, Vadodara, India. E-mail: anjali.patel-chem@msubaroda.ac.in
First published on 11th January 2019
Abstract
The present work describes for the first time, the synthesis and characterization of a bi-functional catalyst consisting of nickel and supported 12-tungstophosphoric acid as well as its application in one pot oxidative esterification of benzaldehyde and benzyl alcohol to benzoate ester. The said reactions were operated without any base at a low temperature and atmospheric pressure. The influence of reaction parameters such as nickel concentration, molar ratio of substrate to H2O2 as well as methanol, catalyst amount, reaction temperature and reaction time were investigated to optimize the conditions for maximum conversion with good selectivity towards the desired product. The superiority of the present work lies in obtaining higher conversion as well as higher selectivity of the desired product with a high TON for both the systems under sustainable reaction conditions. Moreover, the catalyst could be recovered and reused for up to three cycles without any significant loss in its selectivity. The obtained conversion as well as selectivity were discussed with a number of control experiments and based on the obtained results, mechanisms for both the reactions were proposed. Furthermore, the difference in activity towards oxidative esterification of benzaldehyde and benzyl alcohol was also correlated with the proposed mechanism.
Introduction
Oxidative esterification of aldehydes with alcohols has received increasing attention during recent years because such raw materials are abundantly available in industry.1 Various groups have reported one step conventional methods for oxidative esterification of aldehydes, based on TS-1,2 V2O5-SPC/SPB (SPC-sodium percarbonate, SPB sodium perborate),3 supported gold nanoparticle Au/TiO2,4 Pb and Mg doping in Al2O3-supported Pd,5 palladium/NHC,6 manganese phthalocyanine immobilized on silica gel,7 supported gold–nickel oxide nanoparticles (AuNiOx),8 magnesia supported gold nanoparticles (Au/MgO),9 titanium superoxide,1 palladium complex immobilized on magnetite–graphene oxide nanocomposite,10 graphite oxide and oxone,11 iron oxalate capped iron–copper nanomaterial (Fe(ox)Fe–CuOx),12 reduced graphene oxide supported Co3O4 nanoparticles (Co3O4/rGO),13 supported iron oxide nano particles (FeNP),14 gold nanoparticles supported on Ce–Zr oxide,15 polystyrene supported rhodium nanoparticles (Rh@PS),16 PTFE supported gold nanoparticles17 and sulfate radical redox systems.18 Many methods suffer from disadvantages such as use of expensive and polluting reagents, an inert atmosphere and lengthy reaction times.7Oxidative esterification of alcohols is highly desirable from both economic and environmental points of view, as alcohols are more readily available as bulk chemicals, more stable than the carbonyl compounds, cheaper, less toxic and easier to handle.19 Hence in this direction, to improve the conversion and selectivity of the oxidative esterification of benzyl alcohol reaction, tremendous efforts have been made to develop new catalysts, including supported-monometallic catalysts such as Au, Pt, Pd, Ag, etc. and supported-bimetallic catalysts such as Au–Pd, Au–Cu, and Au–Ir catalysts.20 Various groups have reported the same, based on Co3O4–N@C,21 palladium complex immobilized on magnetite–graphene oxide nanocomposite,10 gold–palladium alloy nanoparticles on a phosphate-modified hydrotalcite support,22 supported iron oxide nanoparticles,14 Co nanoparticles embedded in nitrogen doped graphite,23 polystyrene stabilized rhodium nanoparticles16 and Pd–Bi–Te/C.24 On the other hand, gold catalysts have considerable interest due to their unique catalytic properties and selectivity in particular reactions9 such as Au/TiO2,25 Au/β-Ga2O3,26 Au nanoparticles on porous coordination polymers,27 Au/SiO2,28 nanocrystalline gold supported on Fe–Ti–Ce-modified mesoporous silica,29 Au/ZrO2,30 Au nanoparticles supported in the nanocages of SBA-16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 magnesia-supported Au nanoparticles,9 Au/Al2O332 and iron doped graphene (Fe-Gr) supported gold.20
31 magnesia-supported Au nanoparticles,9 Au/Al2O332 and iron doped graphene (Fe-Gr) supported gold.20
However, in most cases, liquid base additives (KOH and K2CO3) are required to obtain a higher yield of the ester.9 It would be interesting if the reaction could be carried out using inexpensive metal as well as without base.
A literature survey shows that even though, polyoxometalates based catalysts are found to be excellent, sustainable and have applications for number of oxidation as well as esterification reactions, very few reports are available for the oxidative esterification of the benzaldehyde. To name, anchored 12-tungstophosphoric acid and lacunary anchored phosphotungstate (PW12/ZrO2, PW12/MCM-41, PW11/ZrO2 and PW11/MCM-41),33 12-tungstophosphoric acid immobilized on the surface of silica encapsulated γ-Fe2O3 nanoparticles34 and imidazolium polyoxometalate, [bmim]3[PW12O40].35 At the same time, it is very surprising that not a single report is available on oxidative esterification of benzyl alcohol using polyoxometalates.
Recently our group has reported one pot oxidative esterification of benzaldehyde over two catalysts based on lacunary phosphotungstate (i) Cs salt of mono nickel substituted phosphotungstate (CsPW11Ni) in homogeneous medium36 (ii) supported Cs-salt of mono nickel substituted phosphotungstate catalyst (CsPW11Ni/ZrO2).37 The obtained excellent results, especially in supported nickel catalyst, encourage us to design another catalyst based on nickel and phosphotungstate and extend the work. To achieve same, new bi-functional catalyst was designed by taking the advantage of available protons of supported 12-tungstophosphoric acid (parent phosphotungstate), characterized and evaluated for one pot oxidative esterification of benzaldehyde as well as benzyl alcohol using H2O2 and methanol. Consequently, the various reaction parameters such as nickel concentration, molar ratio of substrate to H2O2 as well as methanol, catalyst amount, reaction temperature and reaction time were optimized for the maximum conversion as well as selectivity of the desired product. The catalyst was also recycled and regenerated up to three cycles. Reaction mechanisms for oxidative esterification of benzaldehyde as well as for benzyl alcohol were also proposed and based on this the difference in activity for the reactions were also discussed.
Experimental
Materials
All chemicals used were of A. R. grade. Zirconium oxychloride (ZrOCl2·8H2O) (Loba Chemie), 12-tungstophosphoric acid (H3PW12O40), nickel acetate, benzaldehyde, benzylalcohol, methanol, 30% hydrogen peroxide and dichloromethane obtained from Merck were used as received.Catalyst synthesis
The synthesis of Nickel exchanged supported 12-tungstophosphoric acid is shown in Scheme 1.
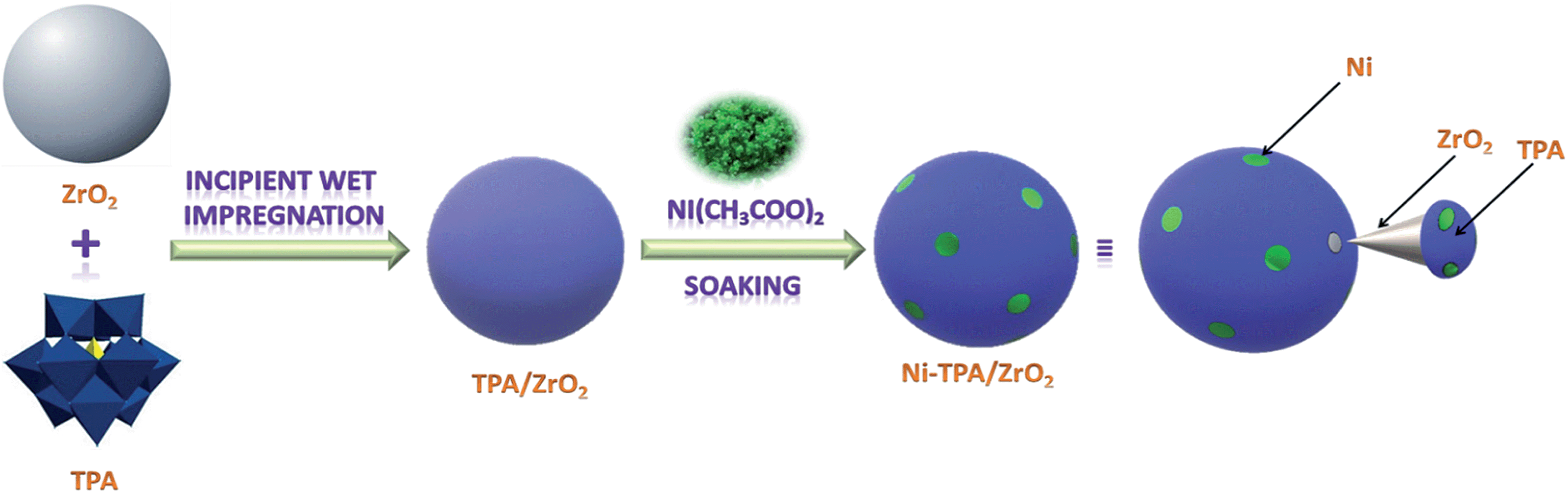 | ||
| Scheme 1 Synthesis of Ni-TPA/ZrO2. | ||
Characterization
The amount of nickel was determined by difference in the standard solution as well as sample solution by volumetric method.40 The acidity of the catalysts was determined by n-butyl amine as well as by potentiometric titration. Thermo gravimetric analysis (TGA) was performed using Mettler Toledo Star SW 7.01 up to 500 °C. Adsorption–desorption isotherms were done through Micromeritics ASAP 2010 surface area analyzer at −196 °C. Specific surface area was calculated using Brunauer–Emmett–Teller (BET method). FT-IR spectrum of the material was performed by using the KBr wafer on a Shimadzu instrument (IRAffinity-1S). The Fourier Transform Raman (FT-Raman) spectra were recorded on a FT-Raman Spectrophotometer (Model Bruker FRA 106). Electron spin resonance (ESR) spectra were recorded on a Varian E-line. Century series X-band ESR spectrometer at liquid nitrogen temperature and scanned from 2500 to 5000 gauss. X-ray photoelectron spectroscopy (XPS) measurements were performed with Auger Electron Spectroscopy (AES) Module PHI 5000 Versa Prob II. Powder X-ray Diffraction (Powder XRD) was carried out using Philips Diffractometer (Model PW-1830). TEM analysis was carried out on JEOL (JAPAN) TEM instrument (model-JEM 100CX II) with accelerating voltage of 200 kV. The samples were dispersed in ethanol and ultrasonicated for 5–10 min. A small drop of the sample was then taken in a carbon coated copper grid and dried before viewing.Acidity measurement
Catalytic evaluation
Oxidative esterification reaction of benzaldehyde/benzylalcohol (10 mmol) with H2O2 (30 mmol), methanol (5/7.5 mL respectively) and catalyst (10 mg) were charged in a 50 mL batch reactor provided with a double walled air condenser, magnetic stirrer, and a guard tube. The reaction mixture was refluxed at 80 °C for 6 h/24 h respectively. In both the cases, the obtained products were extracted with dichloromethane and analyzed on a gas chromatograph (Shimadzu-2014) using a capillary column (RTX-5).Results and discussion
Characterization of catalyst
The volumetric analysis of nickel in standard solution and filtrate show the presence of 0.24 wt%, 0.3 wt% and 0.4 wt% of Ni in (0.03 M) Ni-TPA/ZrO2, (0.04 M) Ni-TPA/ZrO2, and (0.05 M) Ni-TPA/ZrO2 respectively. Here, low exchanged amount of nickel in each catalyst, indicates that only the protons of TPA were exchanged by the Ni. EDX value of W (16.45 wt%) and Ni (0.32 wt%) in (0.04 M) Ni-TPA/ZrO2 is in good agreement with calculated one (17.17 wt% of W, 0.30 wt% of Ni). Very low % of Ni indicates that only the protons of TPA were exchanged.The types and strength of the acidic sites were determined by potentiometric titration. The strength of acidic sites in terms of initial electrode potential is shown in Table 1. The drastic increase in acidic sites as well as acidic strength of all nickel-based catalysts compare to that of TPA/ZrO2, may be due to the exchange of available protons of TPA/ZrO2 by nickel. It is interesting to note down that, as increase in nickel concentration, the total numbers of acidic sites are also increases and this may be due to the Lewis acidity of Ni. The total acidity of (0.05 M) Ni-TPA/ZrO2 is higher compare to (0.04 M) Ni-TPA/ZrO2 but the acidic strength and strong acidic sites are less, this may be due to blocking of strong acidic sites at higher exchange. Hence, (0.04 M) Ni-TPA/ZrO2 was selected for detail characterization and re-designated as Ni-TPA/ZrO2.
| Catalyst | Acidic strength Ei (mV) | Types of acidic sites (meq. g−1) | Total no. of acidic sites | |
|---|---|---|---|---|
| Strong | Weak | |||
| ZrO2 | 18 | 0.1 | 2.1 | 2.2 |
| TPA/ZrO2 | 45 | 1.5 | 1.2 | 2.7 |
| (0.03 M) Ni-TPA/ZrO2 | 61 | 1.9 | 1.4 | 3.3 |
| (0.04 M) Ni-TPA/ZrO2 | 87 | 2.3 | 1.7 | 4.0 |
| (0.05 M) Ni-TPA/ZrO2 | 70 | 2.1 | 2.1 | 4.2 |
TGA curve (Fig. S1†) of TPA/ZrO2 shows 12.6% weight loss in the temperature range of 70–100 °C indicating the loss of adsorbed water molecules. Further, there was no weight loss observed up to 500 °C indicating the stability of the supported catalyst. TGA of Ni-TPA/ZrO2 shows initial weight loss of 8.5% up to 180 °C indicating the loss of adsorbed water molecules. Besides this, also no significant weight loss was observed up to 500 °C, suggests higher thermal stability of synthesized catalyst.
The BET surface area of ZrO2, TPA/ZrO2 and Ni-TPA/ZrO2 were measured. Specific surface area decreased for supported catalysts TPA/ZrO2 (146 m2 g−1) as compared to that of support ZrO2 (170 m2 g−1) and in good agreement with reported fact43 that there may be decrease in surface area in case of the supported catalyst in which oxides are used as supports. This is because of strong interaction of TPA with the oxide support. The surface area of Ni-TPA/ZrO2 (221 m2 g−1) is higher as compare to that of TPA/ZrO2, indicating the exchange of available surface protons of the TPA/ZrO2 with Ni as well as high dispersion of Ni on the surface of TPA/ZrO2, which is also seen in nitrogen adsorption desorption isotherms (Fig. 1). It also confirms no change in the basic structure.
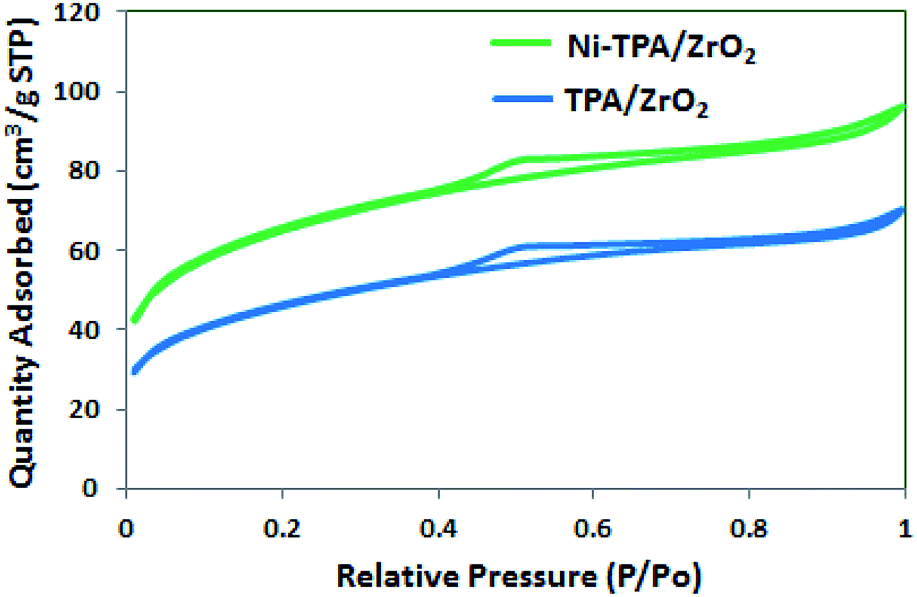 | ||
| Fig. 1 Nitrogen adsorption desorption isotherms. | ||
The FT-IR spectra of ZrO2 (Fig. 2) shows broad bands in the region of 3400, 1600 and 1370, and 600 cm−1 attributed to O–H asymmetric stretches, H–O–H and O–H–O bending, and Zr–OH bending respectively.38 FT-IR spectrum of TPA/ZrO2 (Table S1†) exhibits bands at 1070; 964; and 812 cm−1 corresponding to P–O, W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and W–O–W stretching vibration frequencies respectively. The FT-IR spectrum of Ni-TPA/ZrO2 also shows all the characteristic bands for TPA but with significant shift which may be due to the exchange of Ni with available protons of TPA, with an additional band at 495 cm−1 is observed attributing to Ni–O vibration.
O, and W–O–W stretching vibration frequencies respectively. The FT-IR spectrum of Ni-TPA/ZrO2 also shows all the characteristic bands for TPA but with significant shift which may be due to the exchange of Ni with available protons of TPA, with an additional band at 495 cm−1 is observed attributing to Ni–O vibration.
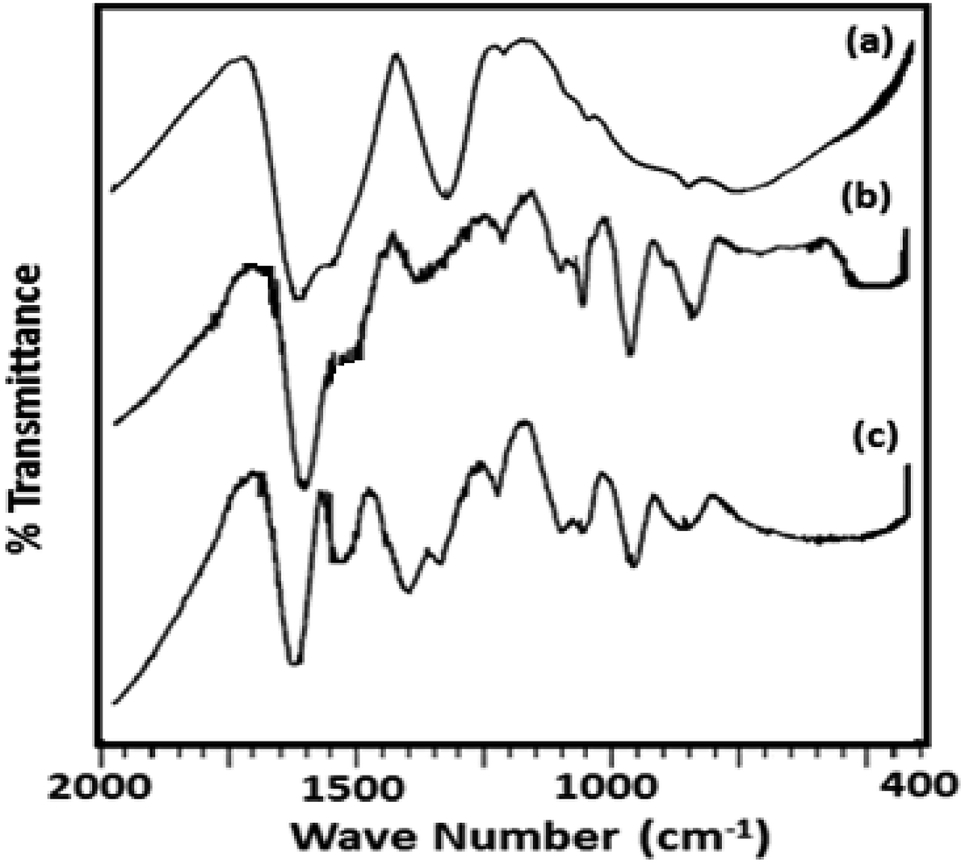 | ||
| Fig. 2 FT-IR spectra of (a) ZrO2 (b) TPA/ZrO2 and (c) Ni-TPA/ZrO2. | ||
Raman spectra of ZrO2 (Fig. S2†) shows broad peaks in the region from 200 to 800 cm−1, which are associated with long-range disordering arrangement in amorphous state.44 Raman spectra of TPA/ZrO2 shows bands at 1010, 990, 900, 550, and 217 cm−1, which are assigned to υs (W–Od), υas (W–Od), υas (W–Ob–W), υs (W–Oc–W), and υs (W–Oa), respectively where Oa, Ob, Oc, and Od correspond to the oxygen atoms linked to phosphorus, to oxygen atoms bridging two tungsten (from two different triads for Ob and from the same triad for Oc), and to the terminal oxygen WO, respectively. Raman spectra of Ni-TPA/ZrO2 shows bands at 947, 929, 859, 532 and 232, corresponding to υs (W–Od), υas (W–Od), υas (W–Ob–W), υs (W–Oc–W), and υs (W–Oa) respectively. The decrease in intensities of the bands as well as significant shifting45 in all the characteristic bands may be due to change in the environment because of the exchange of surface protons of TPA by nickel.
The full range (5000–2500 G) X-band liquid nitrogen temperature ESR spectrum for Ni-TPA/ZrO2 (Fig. S3†) was recorded. The calculated g value (g ∼ 2.05) confirms the presence of Ni(II) species, in good agreement with reported one.46 Further, the presence of Ni(II) is confirmed by XPS.
The XPS of Ni-TPA/ZrO2 is displayed in Fig. 3. A low intense peak at 854.5 eV (2p3/2) is in good agreement with reported one47 confirming the presence of Ni(II) on the surface. However, the obtained poor-quality spectra with low peak intensity of 2p3/2 may be due to the presence of very low concentration of Ni (0.3 %wt) in the synthesized catalyst.
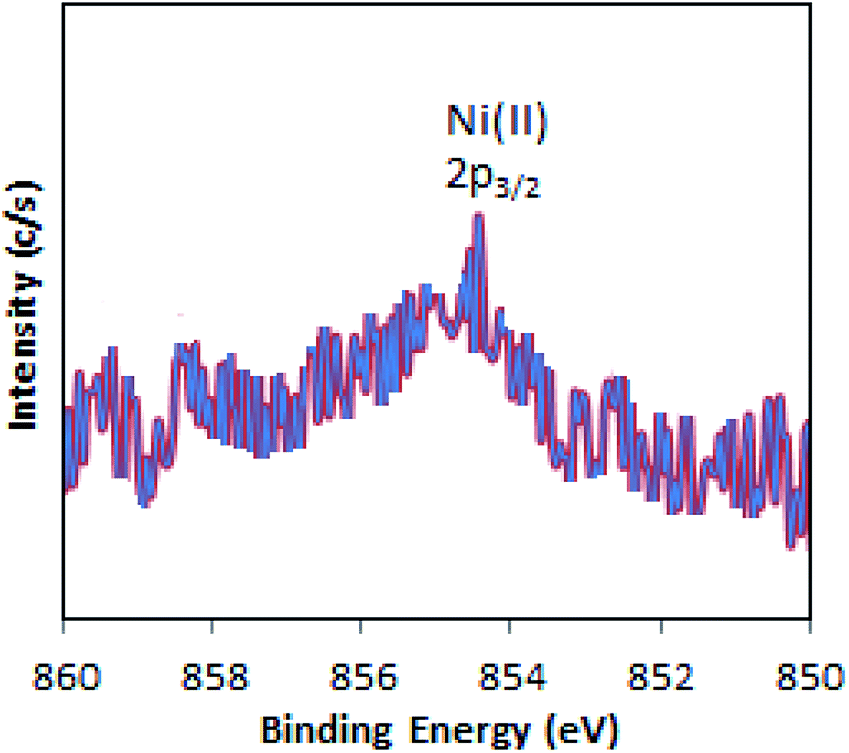 | ||
| Fig. 3 X-ray photoelectron spectrum of Ni-TPA/ZrO2. | ||
The XRD patterns of ZrO2, TPA, TPA/ZrO2 and Ni-TPA/ZrO2 catalysts are shown in Fig. 4. The XRD patterns of ZrO2 (Fig. 4a) shows the amorphous nature of the support. Absence of characteristic diffraction lines corresponding to TPA, in the XRD pattern of TPA/ZrO2 (Fig. 4c), indicates a high dispersion of TPA on the surface of ZrO2. The XRD patterns of Ni-TPA/ZrO2 (Fig. 4d) are very similar to TPA/ZrO2, suggesting very high dispersion of nickel on the non-crystalline surface of TPA/ZrO2, which is also observed in TEM.
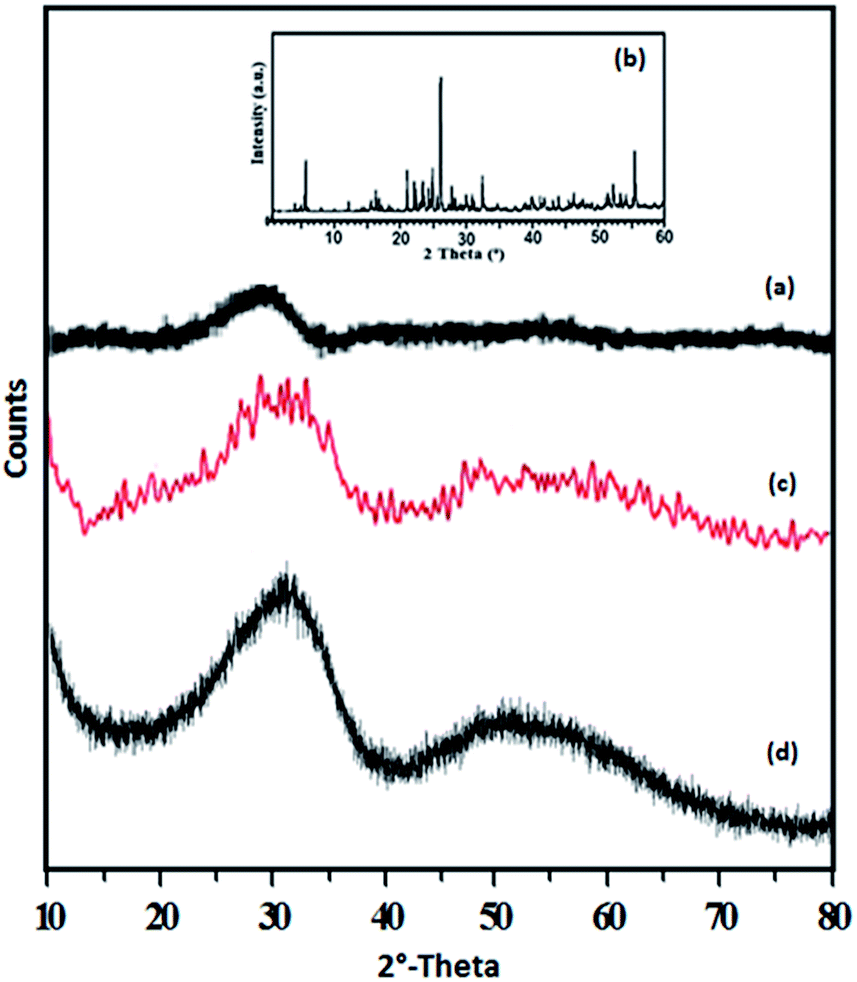 | ||
| Fig. 4 XRD spectra of (a) ZrO2, (b) TPA (c) TPA/ZrO2 and (d) Ni-TPA/ZrO2. | ||
TEM images of the fresh catalyst Ni-TPA/ZrO2 are presented in (Fig. S4†) at various magnifications. SAED image (a) indicates the non-crystalline and uniform distribution of nickel in the synthesized catalyst. Images (b and c) show the uniform dispersion of Ni on the surface of TPA/ZrO2.
In summary, FT-IR and Raman spectrum indicate the retention of TPA structure in the catalyst as well as exchange of nickel with available surface protons of TPA/ZrO2. ESR and XPS studies confirm the presence of Ni(II) in the catalyst. While XRD and TEM reveals the uniform dispersion of Ni over TPA/ZrO2.
Catalytic activity
 | ||
| Scheme 2 Oxidative esterification of benzaldehyde. | ||
The reaction was carried out in presence of different concentration of nickel and the results are presented in Table S2.† It can be seen that initially, % conversion increases with increase in nickel concentration without change in % selectivity of the desired product. This can be explained on the bases of the acidity of the catalyst (Table 1). With increase in concentration of nickel, the % conversion also increases due to the increase in total number of acidic sites of the catalyst. With further increase in nickel concentration (0.05 M) the % conversion decreases with increase in % selectivity of the desired product. This may be due to the lower acidic strength of the (0.05 M) Ni-TPA/ZrO2 compare to Ni-TPA/ZrO2 (Table 1). Conversion was achieved highest in case of Ni-TPA/ZrO2 compared to (0.03 M) Ni-TPA/ZrO2 and (0.05 M) Ni-TPA/ZrO2. Hence, further optimization was carried out using Ni-TPA/ZrO2.
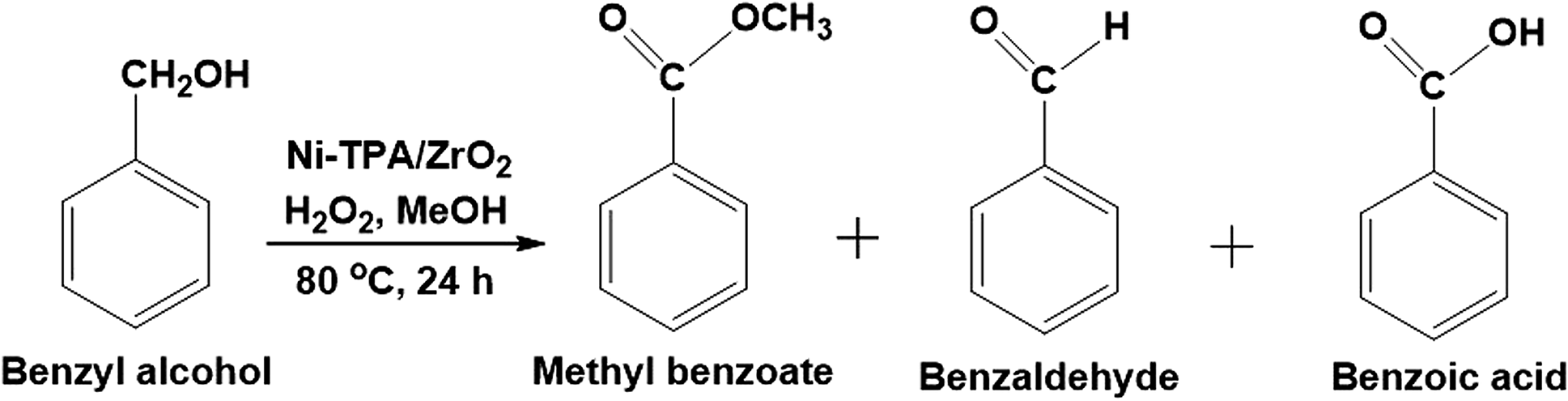
The effect of the catalyst amount was evaluated by varying it from 5 to 25 mg (Fig. 5A). By increasing the catalyst amount up to 10 mg conversion was decreases with increase in % selectivity of desired product, with further increase in catalyst amount, % conversion as well as selectivity decreases. This is due to unproductive decomposition of H2O2 which generates additional water and results in decrease in selectivity of ester, is in good agreement with the reported one.37 Maximum % conversion with highest % selectivity of ester was obtained for 10 mg of catalyst. The temperature effect screened from 60 to 90 °C, with increase in temperature conversion also increases with decrease in ester selectivity (Fig. 5B), may be due to the thermal decomposition of H2O2.37 80 °C temperature was optimized to achieve maximum conversion as well as ester selectivity. Then, effect of methanol volume was carried out from 1 to 6 mL (Fig. 5C). Results show that with enlarging methanol amount, conversion decreases with increase in ester selectivity. Here, decrease in conversion is because of dilution of the substrate concentration.37 From obtained results, 5 mL of methanol was optimized. Finally, the time effect was performed with in 4 h to 12 h range (Fig. 5D). Results indicate that with rise in time the conversion increases whereas selectivity of ester decreases. This is because of decomposition of H2O2 which results in the formation of the water molecule to hydrolyze the ester to aldehyde. From the results, 6 h time was optimized for the reaction. The obtained results are summarized in Fig. 5.
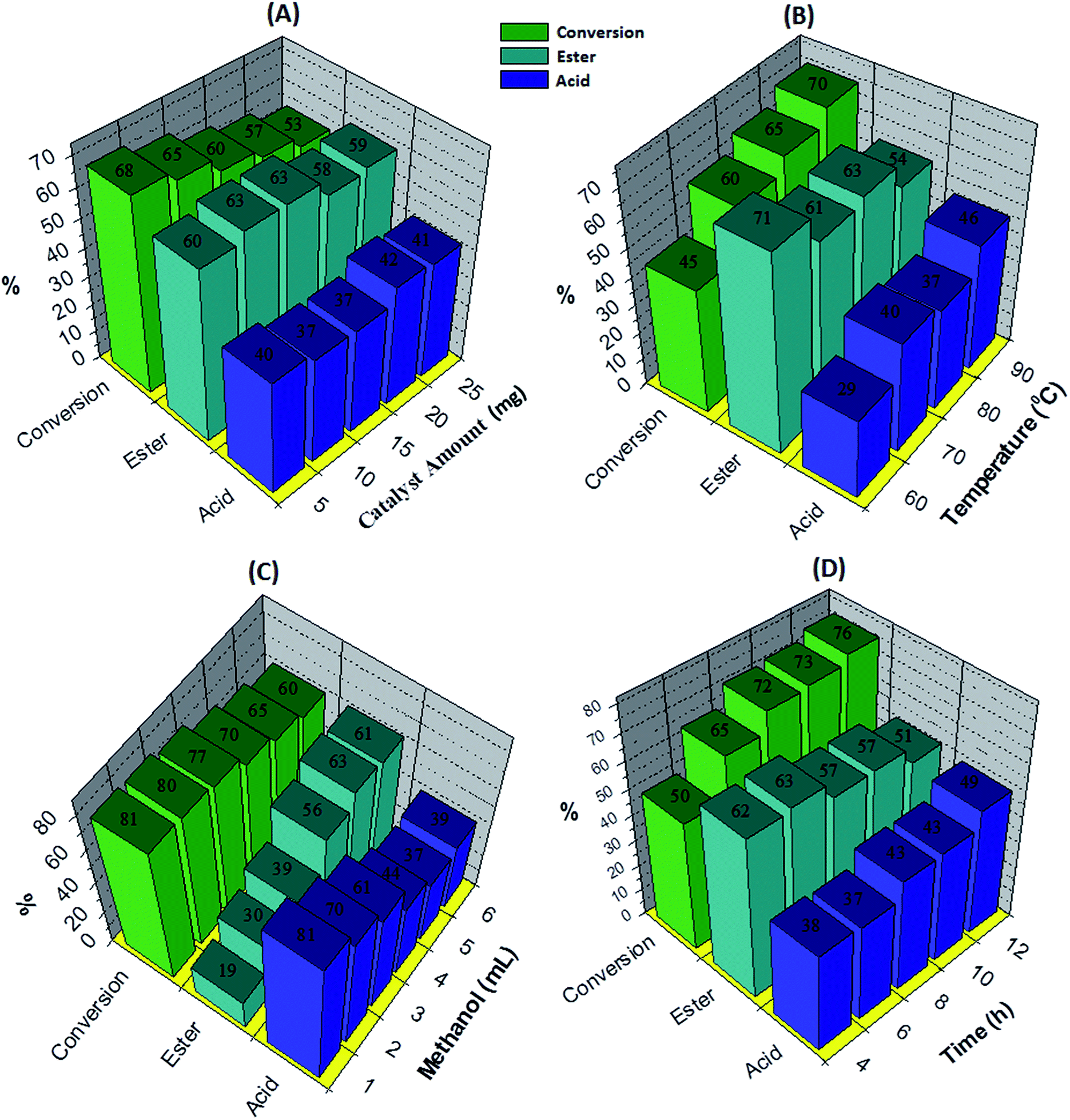 | ||
| Fig. 5 Optimization of parameters for oxidative esterification of benzaldehyde. Optimization of parameters for oxidative esterification of benzaldehyde. Reaction conditions: (A) effect of the catalyst amount: benzaldehyde (10 mmol), H2O2 (30 mmol), methanol (5 mL), catalyst/substrate ratio (5.11 × 10−5), time (6 h), temp. (80 °C); (B) effect of the temperature: benzaldehyde (10 mmol), H2O2 (30 mmol), methanol (5 mL), catalyst (10 mg), catalyst/substrate ratio (5.11 × 10−5), time (6 h); (C) effect of the methanol amount: benzaldehyde (10 mmol), H2O2 (30 mmol), catalyst (10 mg), catalyst/substrate ratio (5.11 × 10−5), time (6 h), temp. (80 °C) and (D) effect of the time: benzaldehyde (10 mmol), H2O2 (30 mmol), methanol (5 mL), catalyst/substrate ratio (5.11 × 10−5), temp. (80 °C). | ||
Further to see the effect of H2O2, the reaction was carried out with different mole ratio of substrate:H2O2 keeping all other parameters constant (Table 2). From the results it can be seen that the conversion increases with increasing oxidant amount up to mole ratio (1:3) and is in good agreement with the chemical dynamics34 according to which the oxidative esterification could be improved by increasing the amount of H2O2. Simultaneously, the selectivity of the desired product, ester decreases while that of acid increases because higher amount of oxidant will tolerate more benzaldehyde to oxidize in benzoic acid. Further with increase in oxidant amount (1:4) the drastic decrease in conversion was found and hence substrate to oxidant ratio (1:3) was optimized.
| Substrate:H2O2 |
% Conversion | % Selectivity | |
|---|---|---|---|
| Ester | Acid | ||
| a Reaction conditions: benzaldehyde (10 mmol), oxidant (H2O2), methanol (5 mL), catalyst (10 mg), time (6 h), temp. (80 °C). | |||
| 1:1 |
34 | 74 | 26 |
| 1:2 |
54 | 70 | 30 |
| 1:3 |
65 | 63 | 37 |
| 1:4 |
51 | 56 | 44 |
From the above study, the conditions optimized for the maximum conversion are: benzaldehyde (10 mmol), H2O2 (30 mmol), methanol (5 mL), conc. of Ni (5.11 × 10−4 mmol), catalyst/substrate ratio (5.11 × 10−5), time (6 h), temp. (80 °C). Conversion = 65% with TON = 12712.
The control experiments were also carried out with Ni(CH3COO)2, ZrO2, TPA/ZrO2 and Ni-TPA/ZrO2 in identical experimental conditions and results are presented in Table 3. ZrO2 and TPA/ZrO2 both were moderately active with low selectivity of the desired product benzoate ester. However, Ni(CH3COO)2 gives 54% conversion and 52% ester selectivity as expected. The increase in the conversion as well as selectivity in case of Ni-TPA/ZrO2 shows the synergic effect due to the presence of nickel, which is also confirmed by acidity measurement. Total number of acidic sites as well as acidic strength (Table 1) is almost double and responsible for increase in activity.
| Catalyst | % Conversion | % Selectivity | |
|---|---|---|---|
| Ester | Acid | ||
| a Reaction conditions: benzaldehyde (10 mmol), H2O2 (30 mmol), methanol (5 mL), catalyst (10 mg), catalyst/substrate ratio (5.11 × 10−5), time (6 h), temp. (80 °C). aActive amount of nickel (5.11 × 10−4 mmol). | |||
| aNi(CH3COO)2 | 54 | 52 | 48 |
| ZrO2 | 27 | 41 | 59 |
| TPA/ZrO2 | 52 | 43 | 57 |
| aNi-TPA/ZrO2 | 65 | 63 | 37 |
For oxidative esterification of benzaldehyde, we are expecting the same mechanism as reported by us earlier,36 contradictory to general mechanism. The general mechanism demonstrates the formation of acetal as intermediate.48 To explain the contradiction, following set of experiments was carried out. (1) When benzoic acid is used as a substrate instead of benzaldehyde under identical conditions (in absence of H2O2), 34% conversion with 100% selectivity of methyl benzoate was obtained. (2) In another reaction, benzaldehyde was reacted with methanol in presence of catalyst and absence of H2O2 gives 11% conversion with 100% selectivity for benzoic acid in 6 h. On prolonging the reaction for 3 h after addition of H2O2, 46% conversion was achieved with 60% ester selectivity. The study shows that the oxidation of benzaldehyde does not proceed through an acetal intermediate, but through in situ formation of benzoic acid. The proposed reaction mechanism is shown in Scheme 3. Benzaldehyde is first converted into benzoic acid in presence of H2O2 and the formed benzoic acid is esterified in presence of methanol to desire ester. It must be noted that both the steps take place in situ. Here, it might be possible that in the beginning, the formation of metal-peroxo active intermediate (here, metal can be both, W as well as Ni) takes place.49 However, as reported by us earlier, the detail mechanism of the synthesis of esters involving POM and transition metal is not clear.49
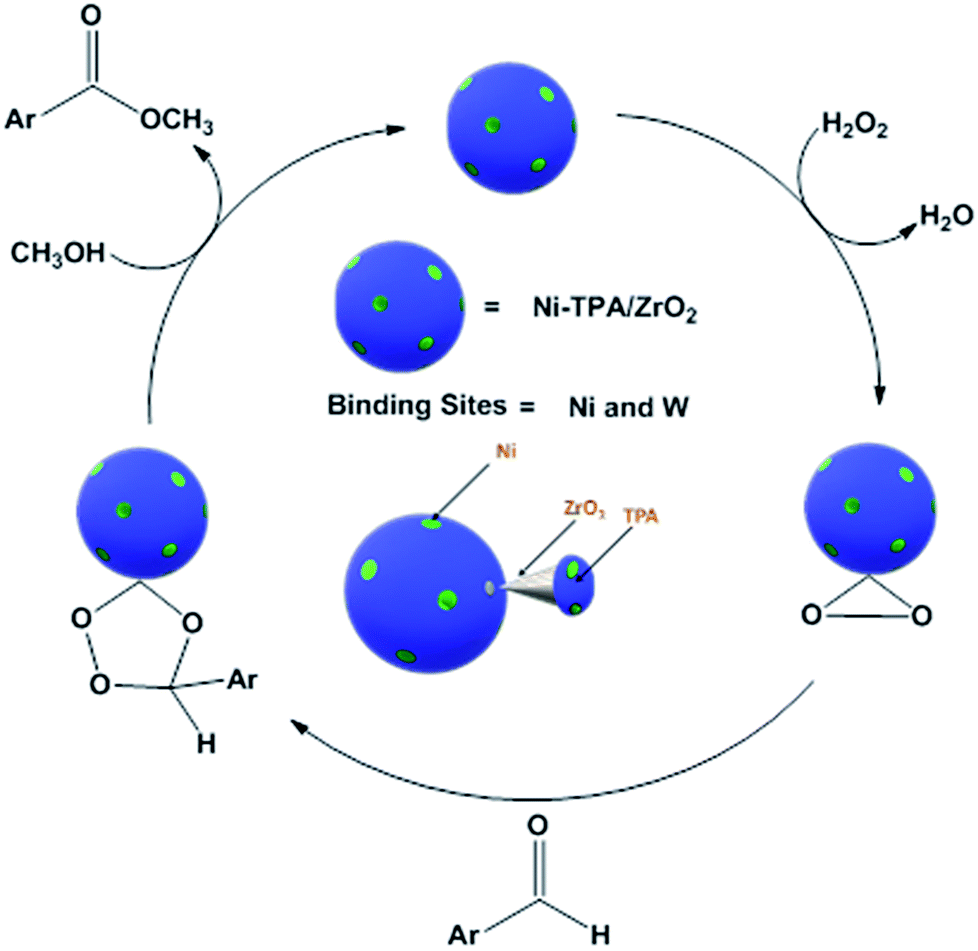 | ||
| Scheme 3 Proposed reaction mechanism for oxidative esterification of benzaldehyde. | ||
 | ||
| Scheme 4 Oxidative esterification of benzyl alcohol. | ||
To determine the effect of nickel concentration, the reaction was carried using (0.03 M) Ni-TPA/ZrO2, Ni-TPA/ZrO2 and (0.05 M) Ni-TPA/ZrO2. The obtained results are shown in Table S4.† The trend in the % conversion as well as % selectivity was found to be the same as earlier (Table S2†). And hence, further optimization was carried out using Ni-TPA/ZrO2.
To optimize the catalyst amount, the reaction was performed by varying amount (i.e. from 5 to 25 mg) by keeping other parameters constant. Oxidative esterification is significantly affected by acidity as well as oxidizing property of the catalyst. The increase in the conversion can be attributed to an increase in the number of available catalytically active sites. From Fig. 6 it can be seen that initially the conversion increases with increasing the amount from 5 to 10 mg. Further, with increasing the amount, the conversion as well as selectivity of ester decreases as observed earlier (Fig. 5A). This may be due to the rapid catalytic decomposition of H2O2 in presence of excess amount of catalyst which generates the water molecules.37 As esterification is reversible process in presence of water, the selectivity of the ester product decreases with increase in catalyst amount.
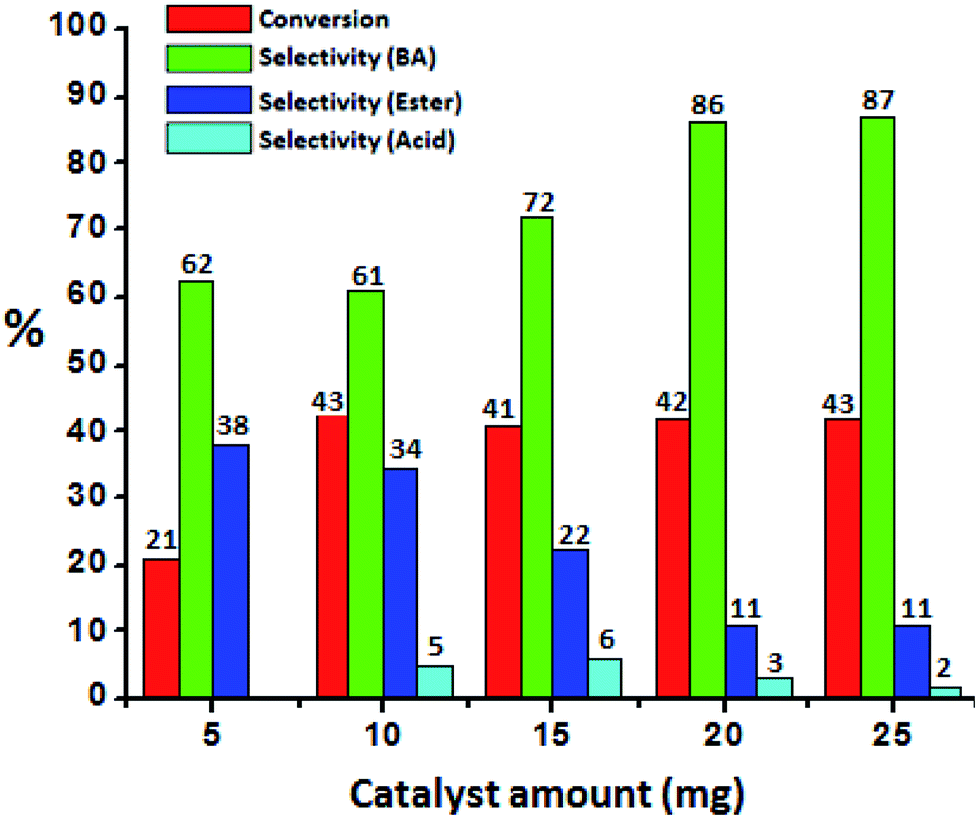 | ||
| Fig. 6 Effect of catalyst amount. Reaction conditions: benzyl alcohol (10 mmol), H2O2 (30 mmol), methanol (7.5 mL), time (24 h), temp. (80 °C). | ||
Effect of methanol amount was studied in the range of 1 mL to 8 mL and obtained results are presented in Table 4. Initially, the amount of methanol increased up to 7.5 mL, the % conversion remains almost same with increase in the % selectivity of the desired product. With further increase in methanol quantity (8 mL), % conversion decreases; this is because of higher the volume of methanol, which dilute the concentration of the substrates. Hence, 7.5 mL volume of methanol was optimized for the reaction for further study.
| Methanol (mL) | % Conversion | % Selectivity | ||
|---|---|---|---|---|
| Benzaldehyde | Ester | Acid | ||
| a Reaction condition: benzyl alcohol (10 mmol), H2O2 (30 mmol), catalyst (10 mg), catalyst/substrate ratio (5.11 × 10−5), temp. (80 °C), time (24 h). | ||||
| 1 | 38 | 61 | 13 | 26 |
| 2 | 45 | 77 | 11 | 12 |
| 3 | 49 | 75 | 15 | 10 |
| 4 | 48 | 75 | 16 | 9 |
| 5 | 47 | 72 | 21 | 7 |
| 6 | 46 | 69 | 27 | 4 |
| 7 | 45 | 65 | 31 | 4 |
| 7.5 | 43 | 61 | 34 | 5 |
| 8 | 35 | 55 | 40 | 5 |
The effect of temperature was carried out and obtained results were presented in Fig. 7, it shows that conversion as well selectivity of ester increases with increase in the temperature from 50 °C to 80 °C. Further, with increase in temperature to 90 °C, conversion also increases but ester selectivity decreases. This may be due to the thermal decomposition of the H2O2. Decomposition of H2O2 produces water molecule and hence ester hydrolysed to benzaldehyde.37
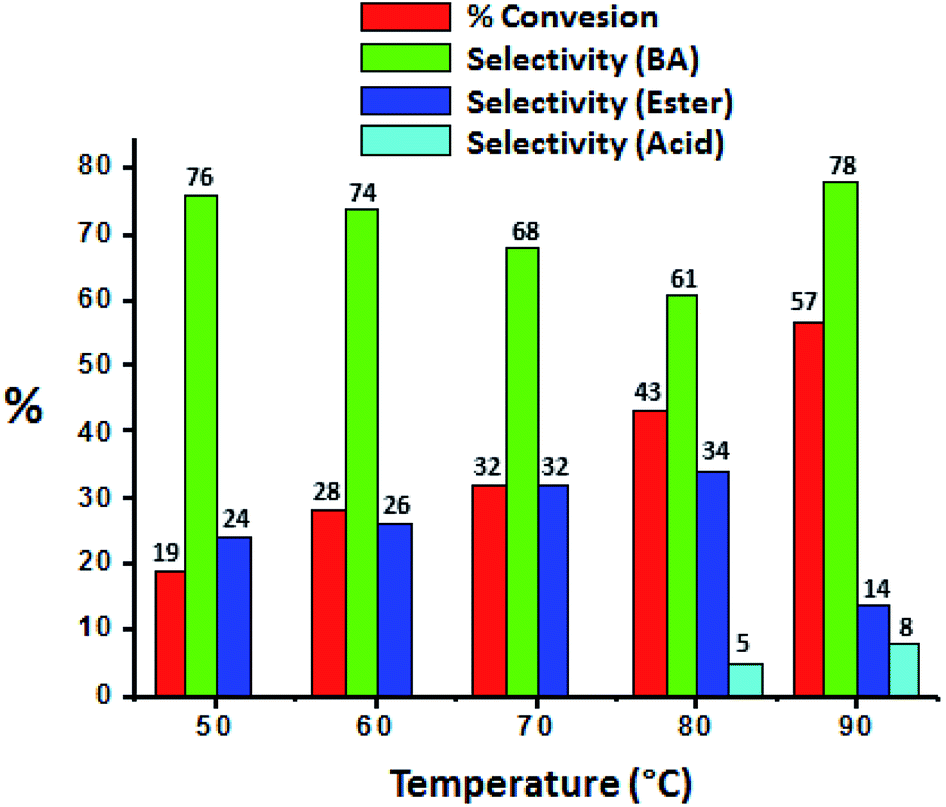 | ||
| Fig. 7 Effect of temperature. Reaction condition: benzyl alcohol (10 mmol), H2O2 (30 mmol), methanol (7.5 mL), catalyst (10 mg), catalyst/substrate ratio (5.11 × 10−5), time (24 h). | ||
The effect of time was investigated by varying the reaction time. As shown in Fig. S5,† the reaction was performed at three different time intervals. Initially, increase in the reaction time from 18 h to 24 h, conversion as well as ester selectivity increases. Further, increasing the reaction time shows no notable change in the results because of attainment of reaction equilibrium.37 So, 24 h is optimized for the maximum reaction conversion and ester selectivity.
Finally, to see the effect of H2O2, the reaction was carried out with different mole ratio of substrate:H2O2 keeping all other parameters constant (Table 5). From the results it can be seen that the conversion increases with increasing the oxidant amount with the selectivity of desired product ester up to mole ratio (1:3). Because it is well known that the oxidative esterification could be improved by increasing the amount of H2O2, according to the chemical dynamics.34 Further with increase in oxidant amount (1:4) the slight decrease in conversion was found. Hence, (1:3) substrate to oxidant ratio was optimized for further catalytic optimization.
| Substrate:H2O2 |
% Conversion | % Selectivity | ||
|---|---|---|---|---|
| Aldehyde | Ester | Acid | ||
| a Reaction conditions: benzyl alcohol (10 mmol), oxidant (H2O2), methanol (7.5 mL), catalyst (10 mg), time (24 h), temp. (80 °C). | ||||
| 1:1 |
10 | 89 | 11 | — |
| 1:2 |
34 | 80 | 20 | — |
| 1:3 |
43 | 61 | 34 | 5 |
| 1:4 |
41 | 41 | 33 | 4 |
From the above study, the conditions optimized for the maximum conversion (43%) with TON = 8415 are: benzyl alcohol (10 mmol), H2O2 (30 mmol), methanol (7.5 mL), conc. of Ni (5.11 × 10−4 mmol), catalyst/substrate ratio (5.11 × 10−5), time (24 h) and temp. (80 °C).
Control experiments and investigation of mechanism. Number of experiments (Table S5†) were carried out in order to study the role of each substrate as well as catalyst: (i) without catalyst (benzyl alcohol + methanol + H2O2), the reaction did not progress significantly (ii) without oxidant (benzyl alcohol + methanol + catalyst) reaction did not occur, (iii) without methanol (benzyl alcohol + H2O2 + catalyst), oxidation of benzyl alcohol to benzaldehyde takes place. Two things can be concluded from the above studies (i) H2O2, methanol as well as catalyst are essential for the feasibility of the oxidative esterification reaction and (ii) W and Ni both are responsible (due to the synergic effect) for the oxidation as in later case the found conversion was almost double.
The control experiments were also carried out with Ni(CH3COO)2, ZrO2, TPA/ZrO2 and Ni-TPA/ZrO2 in identical experimental conditions and results are presented in Table 6. ZrO2 and TPA/ZrO2 both were moderately active with low selectivity of the desired product, benzoate ester. In presence of nickel salt Ni(CH3COO)2, only 5% conversion with 32% ester selectivity was obtained. The increase in the conversion as well as selectivity in case of Ni-TPA/ZrO2 shows the synergic effect due to the presence of nickel, which is also confirmed by acidity measurement. Total number of acidic sites as well as acidic strength (Table 1) is almost double and responsible for increase in activity.| Catalyst | % Conversion | % Selectivity | ||
|---|---|---|---|---|
| Benzaldehyde | Ester | Acid | ||
| a Reaction conditions: benzyl alcohol (10 mmol), H2O2 (30 mmol), methanol (7.5 mL), catalyst (10 mg), catalyst/substrate ratio (5.11 × 10−5), time (24 h), temp. (80 °C). aActive amount of nickel (5.11 × 10−4 mmol). | ||||
| aNi(CH3COO)2 | 5 | 68 | 32 | — |
| ZrO2 | 10 | 77 | 23 | — |
| TPA/ZrO2 | 22 | 61 | 27 | 12 |
| aNi-TPA/ZrO2 | 43 | 61 | 34 | 5 |
For oxidative esterification of benzyl alcohol, two possible mechanisms are known (1) two step mechanism involving oxidation of alcohol to benzaldehyde and further oxidative esterification of benzaldehyde to ester via benzoic acid formation and (2) formation of hemiacetal as an intermediate,25,26 whose further dehydration gives the ester. In order to know about the possible mechanism, the following experiments have been carried out. (1) Benzyl alcohol was reacted with H2O2 in presence of catalyst, without adding methanol, under the identical conditions, 23% conversion with 98% selectivity of benzaldehyde was obtained. Benzoic acid was obtained with poor selectivity (2%). (2) Benzyl alcohol was reacted with methanol in presence of catalyst, without adding H2O2, no conversion was found. However, on prolonging the same reaction with addition of H2O2 up to 24 h, 41% conversion was achieved with % selectivity 68, 26 and 8 for benzaldehyde, ester and acid respectively. The obtained results are consistent with two step mechanism i.e. oxidation of benzyl alcohol to benzaldehyde (also confirmed from Table S5†) and further oxidative esterification of benzaldehyde to ester formation. Hence, we are assuming same mechanism, as proposed for oxidative esterification of aldehyde. Only the difference is that in present case, first alcohol is converted in to aldehyde and then it follows the same mechanism. Based on the above study, the following reaction mechanism is proposed (Scheme 5).
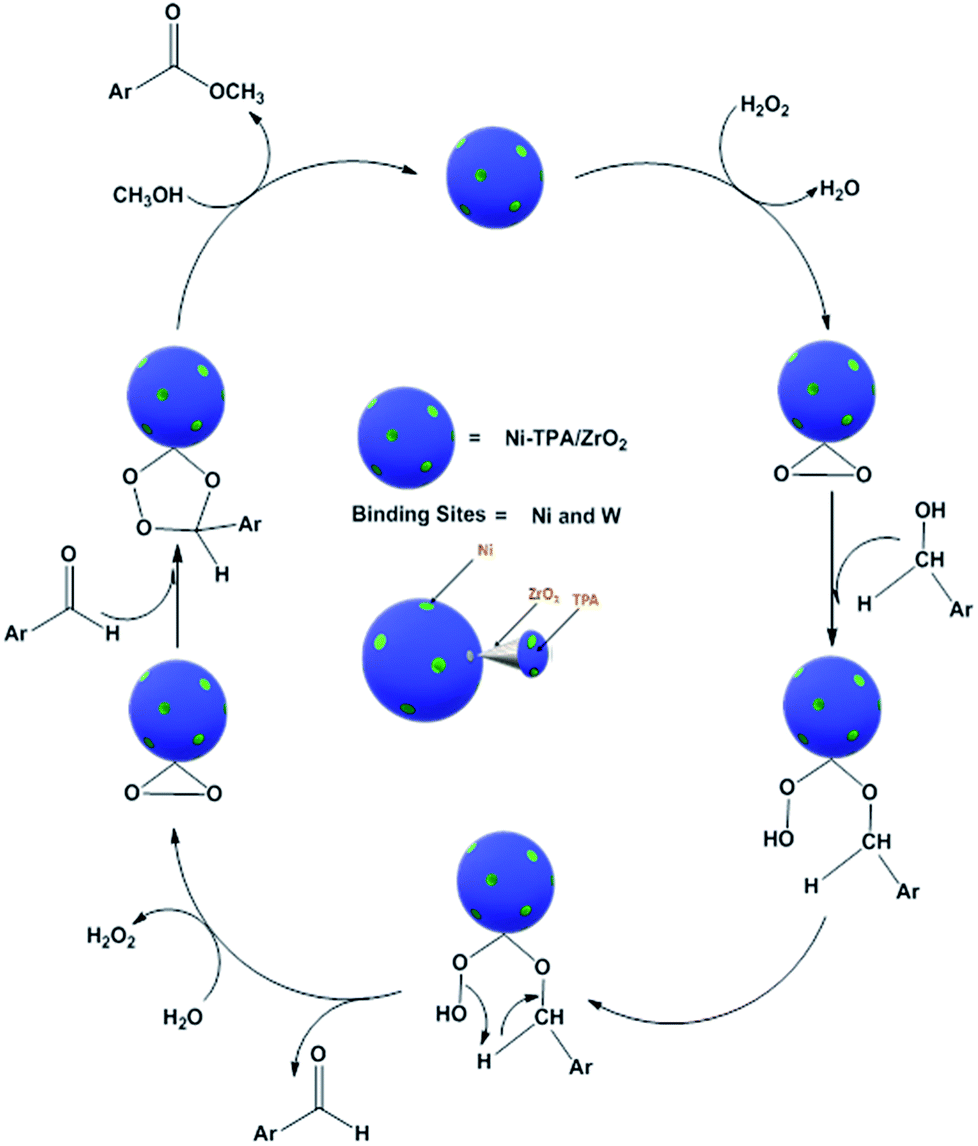 | ||
| Scheme 5 Proposed reaction mechanism for oxidative esterification of benzyl alcohol. | ||
It is interesting to note that activity of the catalyst for oxidative esterification of benzaldehyde is greater than benzyl alcohol. This is in good agreement with the proposed mechanism, which shows that the latter involves one more additional step. i.e. oxidation of benzyl alcohol to benzaldehyde.
Heterogeneity test
In both the cases, heterogeneity test was carried out by centrifuging the catalyst from the reaction mixture. In the case of oxidative esterification of benzaldehyde (Table 7) reaction was carried out up to 4 h, then reaction mixture was centrifuged and filtrate was allowed to react up to 6 h. Similarly, oxidative esterification of benzyl alcohol (Table 8), reaction was carried out for 12 h, centrifuged to remove the catalyst and then filtrate was allowed to react up to 24 h. After that obtained reaction mixtures were extracted by using dichloromethane and analyzed by Gas chromatogram. Results showed that there was no significant change in conversion of the reaction after removing the catalyst, indicates no leaching of Ni during the reaction. This study indicates that TPA plays an important role to bind Ni very strongly and thus does not allow the leaching of Ni into the reaction mixture, making it a true heterogeneous catalyst to recycle and reuse. Here, heterogeneous nature of the catalyst and withholding of the active species on the support during the reaction shows that present catalyst is of category C.50| Catalyst | % Conversion | % Selectivity | |
|---|---|---|---|
| Ester | Acid | ||
| a Reaction conditions: benzaldehyde (10 mmol), H2O2 (30 mmol), methanol (5 mL), catalyst (10 mg), catalyst/substrate ratio (5.11 × 10−5), temp. (80 °C). | |||
| Ni-TPA/ZrO2 | 50 (after 4 h) | 62 | 38 |
| 50 (after 6 h) | 61 | 39 | |
| Catalyst | % Conversion | % Selectivity | ||
|---|---|---|---|---|
| Benzaldehyde | Ester | Acid | ||
| a Reaction conditions: benzyl alcohol (10 mmol), H2O2 (30 mmol), catalyst (10 mg), methanol (7.5 mL), catalyst/substrate ratio (5.11 × 10−5), temp. (80 °C). | ||||
| Ni-TPA/ZrO2 | 20 (after 12 h) | 71 | 25 | 4 |
| 19 (after 24 h) | 72 | 24 | 4 | |
The recovery and reusability of the catalyst is a key issue for the sustainability of any catalytic process. Results of recyclability for oxidative esterification of benzaldehyde and oxidative esterification of benzyl alcohol are shown in Fig. 8 (a and b) respectively for Ni-TPA/ZrO2. After completion of reaction, the organic layer was extracted by dichloromethane. The catalyst was recovered by simple centrifugation, washed with dichloromethane followed by water and then dried in oven at 100 °C for an hour and finally used for the next cycle. Present catalyst was examined up to three cycles only, due to the small scale of reaction with very low concentration of catalyst. In both the cases obtained results show that there was no significant change in conversion as well as in desired product selectivity. The constant results indicate the activity of the catalyst towards its repetitive use and its true heterogeneity nature during the reaction.
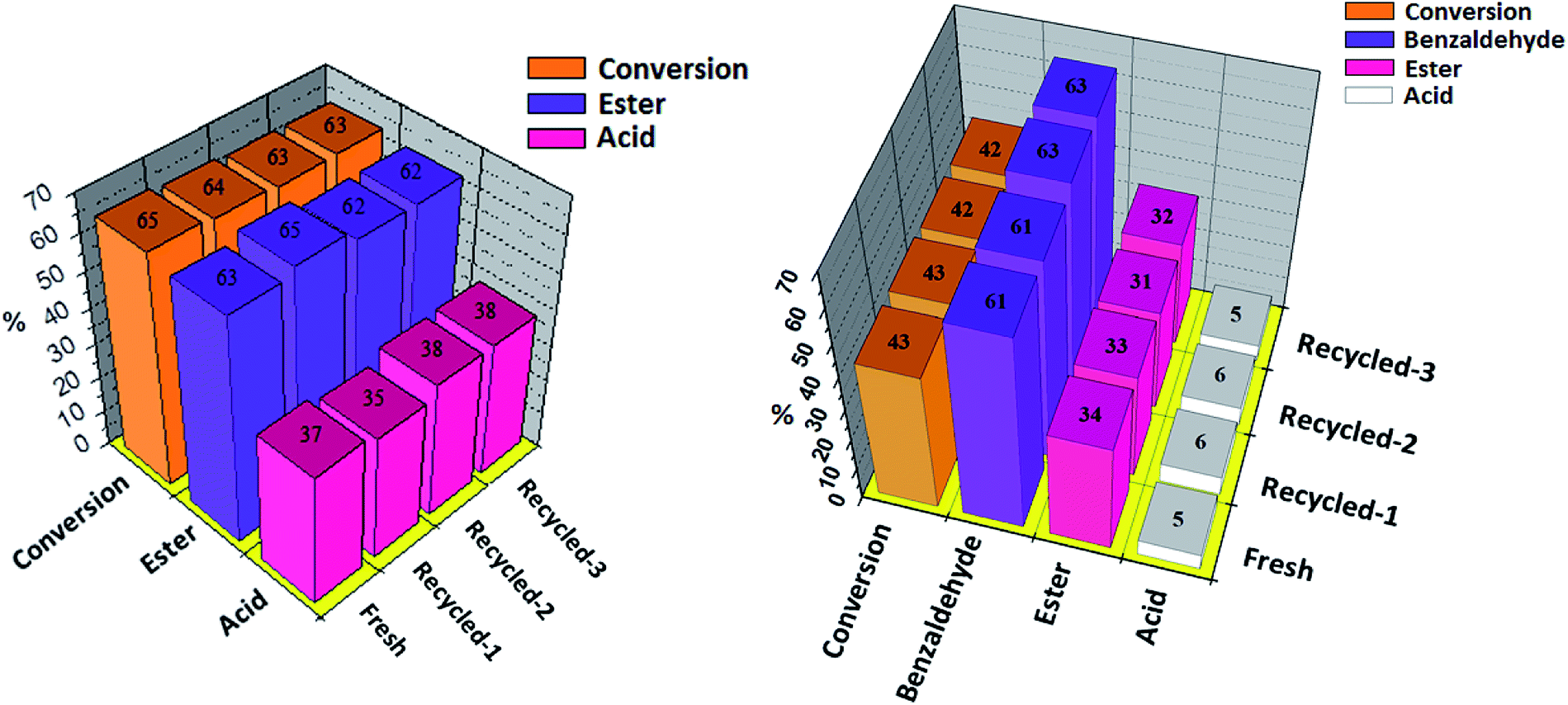 | ||
| Fig. 8 Oxidative esterification of (a) benzaldehyde and (b) benzyl alcohol with fresh and regenerated catalyst. Reaction conditions: benzaldehyde (10 mmol), H2O2 (30 mmol), methanol (5 mL), time (6 h). Reaction conditions: benzyl alcohol (10 mmol), H2O2 (30 mmol), methanol (7.5 mL), time (24 h). Catalyst (10 mg), temp. (80 °C), catalyst/substrate ratio (5.11 × 10−5). | ||
Characterization of regenerated catalyst
Regenerated catalyst was characterized by elemental analysis (EDX), n-butyl amine acidity, potentiometric acidity, FT-IR and TEM analysis for the confirmation of the catalyst structure retention.EDX values of Ni (0.34 wt%) and W (16.52 wt%) of regenerated Ni-TPA/ZrO2 is in good agreement with values of fresh catalyst (16.45 wt% of Ni, 0.32 wt% of Ni) confirming no emission of Ni as well as TPA from ZrO2 during the reaction.
The acidity and total number of acidic sites of the fresh and regenerated catalyst were determined by volumetric titration with n-butyl amine as well as by potentiometric titration, as shown in Table S6† respectively. From the tables it can be seen that acidic sites of regenerated catalyst slightly decrease compared to fresh catalyst whereas acidic strength in terms of initial electrode potential also decreases. This obtained result indicates that few strong acidic sites get blocked during the reaction. Here, blocking of strong acidic sites did not alter the activity of the regenerated catalyst, which can be seen from the catalytic activity of the regenerated catalyst as shown in Fig. 8.
The FTIR spectra of the fresh catalyst and regenerated catalyst are shown in Fig. 9. From the figure it can be seen that almost identical spectrum was obtained without any significant shift in the bands of regenerated catalyst (R-Ni-TPA/ZrO2) compare to the fresh catalyst (Ni-TPA/ZrO2), indicates that catalyst structure remains unaltered even after the regeneration. However, the spectrum is slightly different from the fresh one in terms of intensity. This might be due to the sticking of the substrates on the surface, although this might not be significant in the reutilization of the catalyst.51
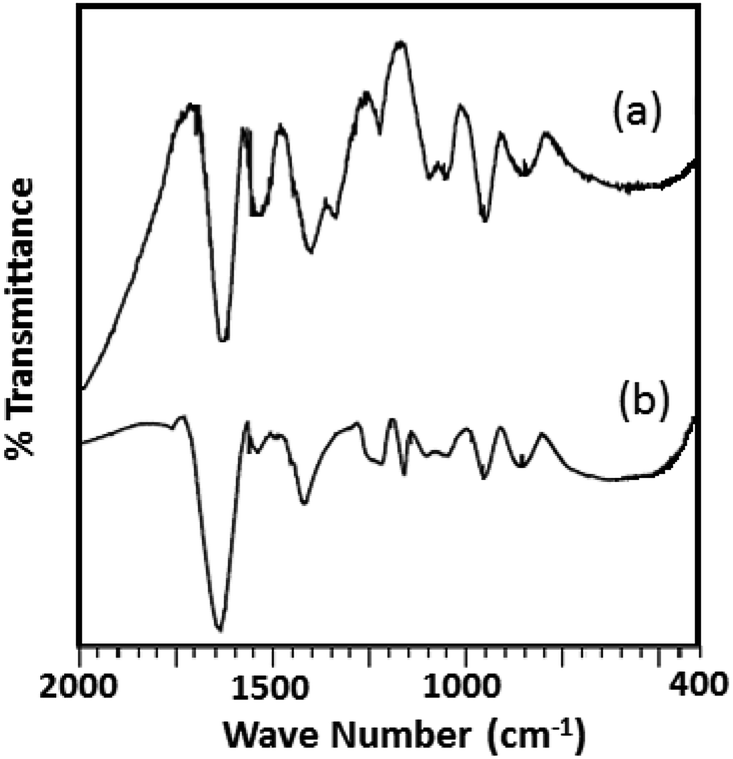 | ||
| Fig. 9 FTIR spectra of (a) Ni-TPA/ZrO2 and (b) R-Ni-TPA/ZrO2. | ||
The X-ray photoelectron spectrum of regenerated Ni-TPA/ZrO2 is displayed in Fig. 10. A low intense peak at 854.6 eV (2p3/2) is in agreement with reported one47 and confirms the presence of Ni(II) on the surface. Obtained spectrum is identical with the fresh catalyst Ni-TPA/ZrO2, confirms that catalyst is sustainable during the reaction.
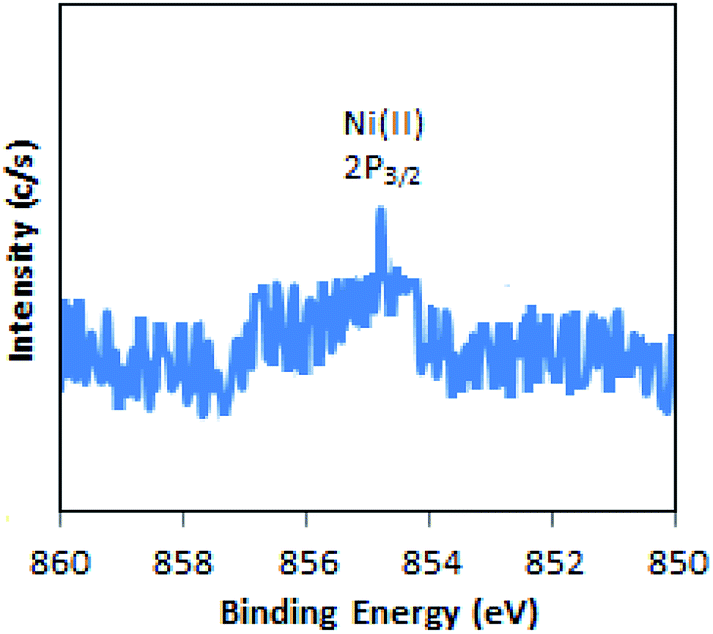 | ||
| Fig. 10 XPS of regenerated Ni-TPA/ZrO2. | ||
TEM images of regenerated catalyst Ni-TPA/ZrO2 are displayed in Fig. S6† at various magnifications. SAED image (a) indicates the non-crystalline and uniform distribution of nickel in the regenerated catalyst. Images (b and c) show the dark uniform suspension in the amorphous nature of the catalyst. This indicates the uniform dispersion of the catalyst over the surface of the support. Obtained images were identical with the fresh catalyst, show the stability and sustainability of the catalyst during the reaction.
Comparison with reported catalyst
Catalytic activity of the present catalyst is also compared with reported one (Table 9). Literature survey shows that only two reports are available, and that are by our group only, in which polyoxometalate and nickel based catalysts are used for the oxidative esterification of benzaldehyde: (i) Cs salt of mono nickel substituted phosphotungstate (CsPW11Ni) in homogeneous medium36 and (ii) heterogeneous catalyst, supported Cs salt of mono nickel substituted phosphotungstate (CsPW11Ni/ZrO2).37 Comparison shows that present catalyst gives higher % conversion compared to CsPW11Ni/ZrO2, which may be due to presence of higher total number of acidic sites whereas selectivity of ester was low due to lower acidic strength. However, homogeneous catalyst CsPW11Ni gives the identical % conversion compare to present catalyst, but it must be noted that in case of homogeneous catalyst the active amount of nickel was 3 times more compared to present catalyst.| Catalyst | Acidic strength Ei (mV) | Types of acidic sites (meq. g−1) | Total no. of acidic sites | % Conversion | % Selectivity | |||
|---|---|---|---|---|---|---|---|---|
| Very Strong | Strong | Weak | Ester | Acid | ||||
| a Reaction conditions: benzaldehyde (10 mmol), H2O2 (30 mmol), methanol (5 mL), catalyst (10 mg), catalyst/substrate ratio (5.11 × 10−5), time (6 h), temp. (80 °C). aActive amount of nickel (5.11 × 10−4 mmol). bActive amount of nickel (1.5 × 10−3 mmol). cActive amount of nickel (5 × 10−4 mmol). | ||||||||
| a Ni-TPA/ZrO2 | 87 | 0 | 2.3 | 1.7 | 4.0 | 65 | 63 | 37 |
| b CsPW11Ni36 | 50 | 0 | 0.7 | 1.7 | 2.4 | 65 | 55 | 45 |
| c CsPW11Ni/ZrO237 |
110 | 0.2 | 1.3 | 2.2 | 3.7 | 40 | 75 | 25 |
At the same time, not a single report is available on oxidative esterification of benzyl alcohol using polyoxometalate.
Conclusion
New versatile bi-functional heterogeneous catalyst comprising nickel and supported 12-tungstophosphoric acid was introduced for base free one pot oxidative esterification of benzaldehyde as well as benzyl alcohol. The catalyst proved to be a bi-functional one by combining their acidic and redox properties. The superiority of present work lies in obtaining higher conversion as well as higher selectivity of the desired product with high TON (>8000 in both the cases) under sustainable reaction conditions. The activity of the catalyst for oxidative esterification of benzyl alcohol was lower than benzaldehyde because the former reaction involves one more additional step. i.e. oxidation of benzyl alcohol to benzaldehyde, which undergoes oxidative esterification. The catalyst can be recycled up to three cycles without any degradation. In addition, the advantages of using Ni as an alternative of more expensive metals as heterogeneous catalyst makes this methodology interesting from an economic as well as an ecological point of view.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to Department of Atomic Energy (DAE) and Board of Research in Nuclear Science (BRNS), Project No. 37(2)/14/34/2014-BRNS, Mumbai, for the financial support. One of the authors Mr Anish Patel is thankful to the same for the grant of JRF. We are also thankful to Department of Chemistry, The Maharaja Sayajirao University of Baroda for BET surface area analysis.References
- S. Dey, S. K. Gadakh and A. Sudalai, Org. Biomol. Chem., 2015, 13, 10631 RSC.
- S. P. Chavan, S. W. Dantale, C. A. Govande, M. S. Venkatraman and C. Praveen, Synlett, 2002, 267 CrossRef.
- R. Gopinath, B. Barkakaty, B. Talukdar and B. K. Patel, J. Org. Chem., 2003, 68, 2944 CrossRef CAS PubMed.
- C. Marsden, E. Taarning, D. Hansen, L. Johansen, S. K. Klitgaard, K. Egeblad and C. H. Christensen, Green Chem., 2008, 10, 168 RSC.
- Y. Diao, R. Yan, S. Zhang, P. Yang, Z. Li, L. Wang and H. Dong, J. Mol. Catal. A: Chem., 2009, 303, 35 CrossRef CAS.
- M. Zhang, S. Zhang, G. Zhang, F. Chen and J. Cheng, Tetrahedron Lett., 2011, 52, 2480 CrossRef CAS.
- R. K. Sharma and S. Gulati, J. Mol. Catal. A: Chem., 2012, 363–364, 291 CrossRef CAS.
- K. Suzuki, T. Yamaguchi, K. Matsushita, C. Iitsuka, J. Miura, T. Akaogi and H. Ishida, ACS Catal., 2013, 3, 1845 CrossRef CAS.
- X. Wan, W. Deng, Q. Zhang and Y. Wang, Catal. Today, 2014, 233, 147 CrossRef CAS.
- S. Verma, D. Verma, A. K. Sinha and S. L. Jain, Appl. Catal., A, 2015, 489, 17 CrossRef CAS.
- M. Mirza-Aghayan, S. Zonoubi, M. Molaee Tavana and R. Boukherroub, Ultrason. Sonochem., 2015, 22, 359 CrossRef CAS PubMed.
- R. Kashyap, D. J. Talukdar and S. Pratihar, New J. Chem., 2015, 39, 1430 RSC.
- V. Panwar, A. Al-Nafiey, A. Addad, B. Sieber, P. Roussel, R. Boukherroub and S. L. Jain, RSC Adv., 2015, 5, 88567 RSC.
- F. Rajabi, R. A. D. Arancon and R. Luque, Catal. Commun., 2015, 59, 101 CrossRef CAS.
- Y. Li, L. Wang, R. Yan, J. Han and S. Zhang, Catal. Sci. Technol., 2015, 5, 3682 RSC.
- N. R. Guha, S. Sharma, D. Bhattacherjee, V. Thakur, R. Bharti, C. B. Reddy and P. Das, Green Chem., 2016, 18, 1206 RSC.
- M. Penhoat, T. Vanbésien, A. Cocud, A. Addad, H. Vezin and C. Rolando, New J. Chem., 2016, 40, 9460 RSC.
- Y.-F. Guo, S. Mahmood, B.-H. Xu, X.-Q. Yao, H.-Y. He and S.-J. Zhang, J. Org. Chem., 2017, 82, 1591 CrossRef CAS PubMed.
- L. Wang, J. Li, W. Dai, Y. Lv, Y. Zhang and S. Gao, Green Chem., 2014, 16, 2164 RSC.
- J. Sun, X. Tong, Z. Liu, S. Liao, X. Zhuang and S. Xue, Catal. Commun., 2016, 85, 70 CrossRef CAS.
- R. V. Jagadeesh, H. Junge, M.-M. Pohl, J. Radnik, A. Brückner and M. Beller, J. Am. Chem. Soc., 2013, 135, 10776 CrossRef CAS PubMed.
- Q. Xiao, Z. Liu, A. Bo, S. Zavahir, S. Sarina, S. Bottle, J. D. Riches and H. Zhu, J. Am. Chem. Soc., 2015, 137, 1956 CrossRef CAS PubMed.
- W. Zhong, H. Liu, C. Bai, S. Liao and Y. Li, ACS Catal., 2015, 5, 1850 CrossRef CAS.
- D. S. Mannel, M. S. Ahmed, T. W. Root and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 1690 CrossRef CAS PubMed.
- I. S. Nielsen, E. Taarning, K. Egeblad, R. Madsen and C. H. Christensen, Catal. Lett., 2007, 116, 35 CrossRef CAS.
- F.-Z. Su, J. Ni, H. Sun, Y. Cao, H.-Y. He and K.-N. Fan, Chem.–Eur. J., 2008, 14, 7131 CrossRef CAS PubMed.
- T. Ishida, M. Nagaoka, T. Akita and M. Haruta, Chem.–Eur. J., 2008, 14, 8456 CrossRef CAS PubMed.
- R. L. Oliveira, P. K. Kiyohara and L. M. Rossi, Green Chem., 2009, 11, 1366 RSC.
- L. A. Parreira, N. Bogdanchikova, A. Pestryakov, T. A. Zepeda, I. Tuzovskaya, M. H. Farías and E. V. Gusevskaya, Appl. Catal., A, 2011, 397, 145 CrossRef CAS.
- W. Cui, M. Jia, W. Ao and B. Zhaorigetu, React. Kinet., Mech. Catal., 2013, 110, 437 CrossRef CAS.
- Y. Hao, Y. Chong, S. Li and H. Yang, J. Phys. Chem. C, 2012, 116, 6512 CrossRef CAS.
- S. Rautiainen, O. Simakova, H. Guo, A.-R. Leino, K. Kordás, D. Murzin, M. Leskelä and T. Repo, Appl. Catal., A, 2014, 485, 202 CrossRef CAS.
- S. Singh and A. Patel, Catal. Lett., 2014, 144, 1557 CrossRef CAS.
- E. Rafiee and S. Eavani, J. Mol. Catal. A: Chem., 2013, 373, 30 CrossRef CAS.
- H. Li, Y. Qiao, L. Hua, Z. Hou, B. Feng, Z. Pan, Y. Hu, X. Wang, X. Zhao and Y. Yu, ChemCatChem, 2010, 2, 1165 CrossRef CAS.
- S. Singh, A. Patel and P. Prakashan, Appl. Catal., A, 2015, 505, 131 CrossRef CAS.
- A. Patel, S. Pathan and P. Prakashan, RSC Adv., 2016, 6, 51394 RSC.
- S. Patel, N. Purohit and A. Patel, J. Mol. Catal. A: Chem., 2003, 192, 195 CrossRef CAS.
- N. Bhatt, C. Shah and A. Patel, Catal. Lett., 2007, 117, 146 CrossRef CAS.
- A. I. Vogel and G. H. Jeffery, Vogel's textbook of quantitative chemical analysis, Longman Scientific & Technical, 1989 Search PubMed.
- H. Ranjan Sahu and G. Ranga Rao, Bull. Mater. Sci., 2000, 23, 349 CrossRef.
- P. Vázquez, L. Pizzio, C. Cáceres, M. Blanco, H. Thomas, E. Alesso, L. Finkielsztein, B. Lantaño, G. Moltrasio and J. Aguirre, J. Mol. Catal. A: Chem., 2000, 161, 223 CrossRef.
- I. Yermakov Yu, Stud. Surf. Sci. Catal., 1980, 52, 2075 Search PubMed.
- H. Wang, G. Li, Y. Xue and L. Li, J. Solid State Chem., 2007, 180, 2790 CrossRef CAS.
- S. Rana and K. M. Parida, Catal. Sci. Technol., 2012, 2, 979 RSC.
- W. L. Feng, T. H. Chen, L. J. Wang and J. J. Chen, Optik-Int. J. Light Electron Opt., 2010, 121, 362 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. S. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771 CrossRef CAS.
- W.-J. Yoo and C.-J. Li, Tetrahedron Lett., 2007, 48, 1033 CrossRef CAS.
- J. Wang, L. Yan, G. Li, X. Wang, Y. Ding and J. Suo, Tetrahedron Lett., 2005, 46, 7023 CrossRef CAS.
- R. A. Sheldon, M. Wallau, I. W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485 CrossRef CAS.
- S. Singh and A. Patel, J. Cleaner Prod., 2014, 72, 46 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08419j |
| This journal is © The Royal Society of Chemistry 2019 |