
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering O-glycosylation in modified N-linked oligosaccharide (Man12GlcNAc2∼Man16GlcNAc2) Pichia pastoris strains†
Siqiang Li ab,
Peng Suna,
Xin Gonga,
Shaohong Changa,
Enzhong Lib,
Yuanhong Xub,
Jun Wu*a and
Bo Liu*a
ab,
Peng Suna,
Xin Gonga,
Shaohong Changa,
Enzhong Lib,
Yuanhong Xub,
Jun Wu*a and
Bo Liu*a
aBeijing Institute of Biotechnology, Beijing 100071, China. E-mail: junwu1969@163.com; liubo7095173@163.com; Fax: +861063833521
bSchool of Biological and Food Engineering, Huanghuai University, Zhumadian 463000, China
First published on 12th March 2019
Abstract
Yeast have been engineered for the production of therapeutic glycoproteins with humanized N-linked oligosaccharides. Both N- and O-linked oligosaccharides engineered yeast have been attractive prospects, since yeast-specific O-mannosylated proteins were reported to induce an aberrant immune response and alter pharmacokinetics in vivo. In the present study, we genetically manipulated O-glycosylation by disrupting O-mannosyltransferase PMT1 and PMT5 in a low-mannose type N-linked oligosaccharide (Man12GlcNAc2∼Man16GlcNAc2) engineered Pichia pastoris strain to produce therapeutic glycoproteins. The O-mannosyltransferase PMT1 mutant produces anti-Her-2 antibodies with reduced O-linked oligosaccharides and protein degradation, but this strain exhibited growth defects. However, the deletion of O-mannosyltransferase PMT5 individually has a minimal effect on O-glycosylation, degradation of the anti-Her-2 antibody, and strain growth. Thus, by disrupting O-mannosyltransferase PMT1 in an N-glycosylation engineered Pichia pastoris strain, we generated an effective glycoengineered Pichia pastoris strain to effectively produce therapeutic glycoproteins with both engineered N- and O-linked oligosaccharides.
1 Introduction
Glycoproteins including antibodies, interferons and cytokines have become a rapidly growing class of therapeutic agents.1,2 Today, therapeutic glycoproteins are produced by the mammalian expression system (e.g., Chinese hamster ovary cell lines, CHO), which has been the only process known to yield human-like glycoproteins.3 To meet the increasing demand, a low-cost and efficient expression system is urgently required. Yeast is an excellent candidate for the production of therapeutic glycoproteins because they can perform many posttranslational modification reactions, including N-, and O-glycosylation; besides, yeast have high proliferation rates and high yields of the secreted protein, etc.4,5 However, the N- and O-linked oligosaccharide structures of glycoproteins produced from yeast significantly differ from those of mammalian cells and humans,6,7 which induce aberrant immune response and alter in vivo pharmacokinetics.8 Therefore, genetic manipulation in yeast to produce therapeutic glycoproteins with oligosaccharides of the low-mannose type, or that closely relates to those of mammalian cells and humans, is urgently needed.To solve these problems, several yeasts have been engineered to produce glycoproteins with low-mannose type N-linked oligosaccharides,9 hybrid-type10 and complex sialylated-type.11 In addition to N-glycosylation, yeast O-glycosylation is also different from that in mammalian cells and humans. Yeast O-glycosylation is initiated in the endoplasmic reticulum (ER); the O-mannosyltransferase family (Pmtps) catalyzes dolichol phosphate mannose (Dol-P-Man) and Ser/Thr to form the O-mannose protein, which is significantly different from in mammalian cells, where it is initiated in the Golgi apparatus.12,13 The extension of yeast O-glycans occurred in the Golgi apparatus; additional mannoses are added to form full O-linked oligosaccharides via related enzymes that are located in the Golgi membrane.13 This yeast-type O-mannosylation leads to poor pharmacokinetic behavior. Furthermore, this yeast-type O-mannosylation may influence the folding, function and yield of glycoproteins. For example, the O-mannosylated antibody cannot assemble efficiently.14 Besides, glycoproteins with complex O-glycosylation from mammal cells and humans are related to specific functions.15 Therefore, glycoproteins with O-linked oligosaccharides produced from yeast should be engineered for the development of therapeutic agents.
In our lab, we have developed a glycoengineered P. pastoris strain GJK01 (Δoch1) that produces glycoproteins with Man12GlcNAc2∼Man16GlcNAc2 N-linked oligosaccharides.16 As one of the most popular eukaryotic expression systems, P. pastoris has many advantages; besides, glycoengineered P. pastoris has been applied to produce glycoproteins with N-linked oligosaccharides similar to those of mammalian cells and humans.17,18 We report herein the genetic manipulation of O-mannosylation in GJK01 (Δoch1). A yeast strain that produces the anti-Her-2 antibody with decreased O-linked oligosaccharides and protein degradation was isolated by disrupting PMT1, but a growth defect was exhibited. However, the PMT5 mutant had little effect upon O-glycosylation, the degradation of the anti-Her-2 antibody, and strain growth.
2 Results and discussion
Although N- and O-glycosylation humanized in yeast have severally received significant achievements,19 to our best knowledge, there are few reports of the engineering of both N- and O-linked oligo-saccharides in yeast cells,20 due partly to mannoproteins being crucial in the biosynthesis of the cell wall.21,22 Due to the Pmtps initial O-mannosylation at the ER, we developed both N- and O-linked oligosaccharide engineered P. pastoris strains via inactivated Pmtps in GJK01 (Δoch1). All seven Pmtps family members (Pmt1p to Pmt7p) are integral ER membrane proteins and can be subdivided into the Pmt1p (Pmt1p and Pmt5p), Pmt2p (Pmt2p and Pmt3p), and Pmt4p (Pmt4p and Pmt6p) subfamilies to form distinct functional subclusters.12 Although several O-mannosylated acceptor proteins were reported,23 O-mannosylated signals and determinants of the different substrate specificities by Pmtps subclusters were unknown. Thus, which Pmtps member catalyzes O-mannosylation of the target protein (anti-Her-2 antibody) is unknown. Pmt1p and Pmt5p were selected in the present study mainly because: (1) the Pmt1p and Pmt2p subfamilies have always been considered to act on many more acceptor proteins than other subfamilies;24 (2) the Pmt1p/Pmt2p complex could in part compensate for Pmt3p defects;24 (3) we found that PMT2 mutant in low-mannose type N-linked oligosaccharide engineered P. pastoris strains may be lethal (data not shown).2.1. Two-step recombination deletion of GJK01 (Δoch1) PMT5 gene
The two-step homologous recombination method for knocking out yeast genes provides several advantages, such as the high efficiency of the mutant strain (approximately 50%), well-studied and applicable to knock out several yeast genes in our lab.25–27 Here, the GJK01 (Δoch1) PMT5 gene was knocked out via the two-step homologous recombination method. The 1.7 kb PMT5 gene upstream (Fig. S1A†) and 2.1 kb downstream (Fig. S1B†) flanking fragments of the PMT5 gene were amplified from P. pastoris GS115 genomic DNA and fused via PCR (3.8 kb, Fig. S1C†). The 3.8 kb of the PCR products was patched in the pYES-URA3 vector and were then transformed into E. coli DH5α. The transformants were selected on LB/Amp plates, and DNA sequencing was used to confirm the presence of the desired plasmid.The deletion of the GJK01 (Δoch1) PMT5 gene was performed as previously reported;25 besides, an overview of the PMT5 gene deletion strategy using the two-step recombinant was shown in Fig. S2.† The △pmt5-pYES2 vector was linearized with the restriction enzyme Bstx I in the upstream flanking fragment of PMT5 and transformed into GJK01 (Δoch1) by electroporation. After incubation and the selection of transformants at 25 °C for 5 days on MD + RH plates, they were picked up and identified by PCR. The desired transformants were incubated in YPD+U medium at 25 °C with shaking at 200 rpm. After 12 h of cultivation, the cultures were then spread onto an MD-URHF plate at 25 °C for 6 days. 5-Fluoroorotic acid was employed for the selection of cells that had lost the △pmt5-pYES2 vector by excisional recombination between the homologous upstream or downstream flanking fragment repeats of the gene. Correct disruption was confirmed by PCR analysis with primers (Fig. 1A and B) based on genomic DNA.
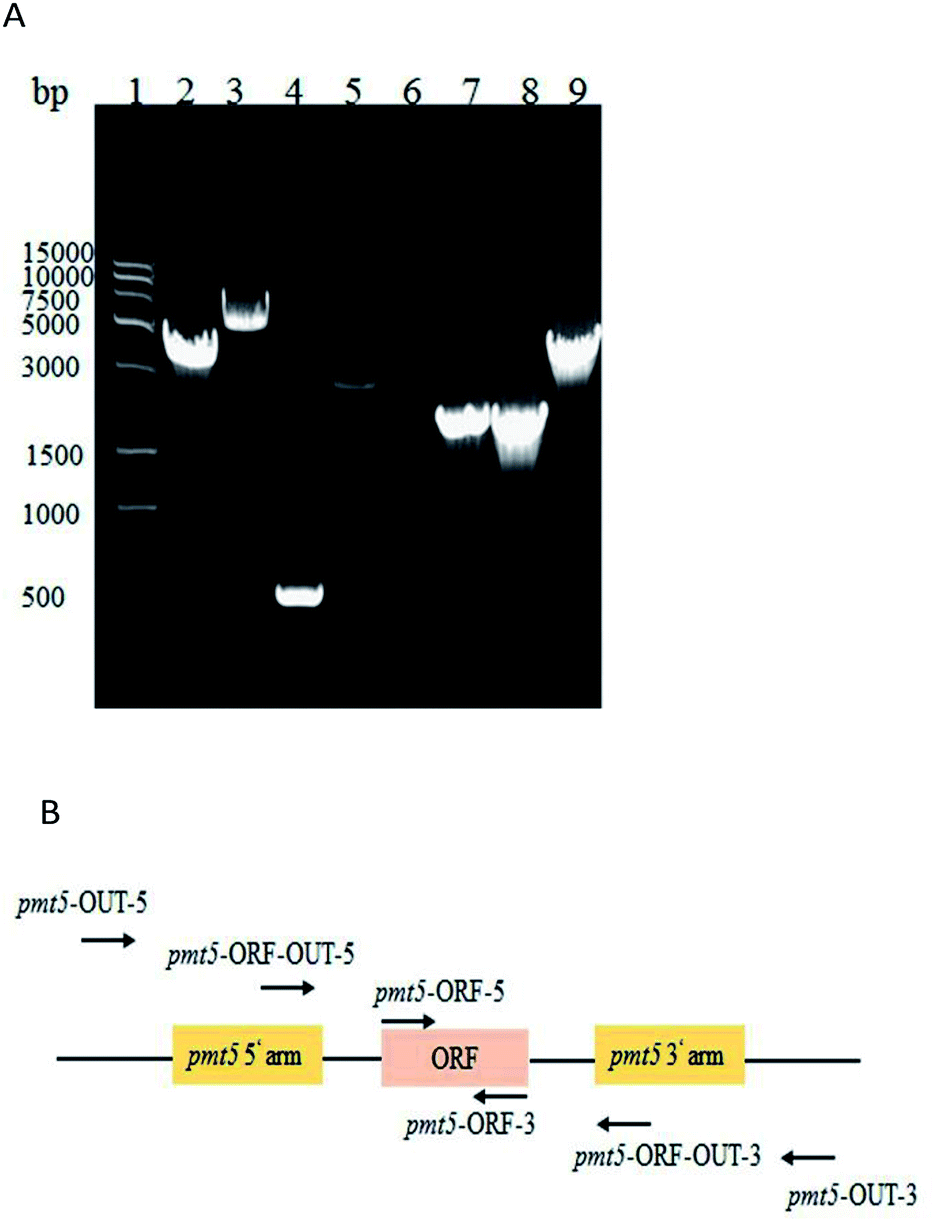 | ||
| Fig. 1 (A) PCR identification of PMT5 gene knockout; molecular marker (lane 1); PCR identified GJK15(Δoch1, Δpmt5) via the primers of pmt5-OUT-5 and pmt5-OUT-3 (4.1 kb, lane 2); pmt5-ORF-OUT-5 and pmt5-ORF-OUT-3 (2.9 kb, lane 4); pmt5-ORF-5 and pmt5-ORF-3 (0, lane 6); pmt5-ORF-OUT-5 and pmt5-OUT-3 (1.9 kb, lane 8); and GJK01 (Δoch1) (6.5 kb, lane 3), (0.5 kb, lane 5); (1.8 kb, lane 7), (4.5 kb, lane 9) as contrasts, respectively; and (B) PMT5 gene locus. | ||
2.2. Insertional inactivation of the GJK01-HL (Δoch1) PMT1 gene
PMT1 was not knocked out via the two-step homologous recombination method. This was likely because PMT1 was crucial in low-mannose type N-linked oligosaccharide engineered P. pastoris, and the wildtype had overlapped the defective type on MD + URH plates during the two-step homologous recombination. Finally, inactive PMT1 was carried out via insertional inactivation.The AOX1TT terminator was designed to further terminate the expression of URA3 (Fig. S3†); the pYES2-URA3-AOX1TT vector containing AOX1TT was transformed into E. coli DH5α, selected on LB/Amp plates, and identified via DNA sequencing. PMT1 (63–900 bp) was amplified via a routine PCR procedure (Fig. S4A†) to directional integrate plasmid at the PMT1 gene region locus. To terminate yeast PMT1 gene expression, termination codons were added at both ends of the amplified PMT1 gene (63–900 bp); thus, the CYC1TT terminator was amplified (Fig. S4B†) and fused with the PMT1 gene (63–900 bp) (Fig. S4C†) to further inhibit the expression of the yeast PMT1 gene (Fig. S3†). The △pmt1-pYES2 vector was transformed into E. coli DH5α and selected on LB/Amp plates; DNA sequencing was performed to confirm the presence of the desired vector. The △pmt1-pYES2 vector was linearized by EcoR V, electrotransformed into the GJK01-HL (Δoch1) competent cell, and then transformants were selected on the MD + RH plates. Transformants contain URA3 would be picked up on MD + RH plates. Further identification was performed via PCR using primers of pmt1-ORF-OUT-5 and pmt1-ORF-OUT-3. PCR products of 8.6 kb fragments showed △pmt1-pYES2 integrated into the PMT1 gene region locus, while the contrast was 3 kb (Fig. 2). Due to variations of the termination codons and terminators designed in △pmt1-pYES2, corrected integration of GJK01-HL (Δoch1) strains would not express PMT1. The mutant was named GJK11-HL (Δoch1, Δpmt1).
 | ||
| Fig. 2 (A) PCR identification of PMT1 gene disruption; molecular marker (lane 1); GJK11-HL (Δoch1, Δpmt1) (lane 2 and 3); GJK01-HL (Δoch1) (lane 4); and (B) PMT1 gene locus. | ||
2.3. Comparison growth rate of GJK01-HL (Δoch1), GJK11-HL (Δoch1, Δpmt1) and GJK15-HL (Δoch1, Δpmt5)
To identify whether the deletion of PMT1 or PMT5 influenced the growth rate of P. pastoris strains, we compared the growth rates of GJK01-HL (Δoch1), GJK11-HL (Δoch1, Δpmt1) and GJK15-HL (Δoch1, Δpmt5). To ensure the uniformity of the initial concentration of these strains, GJK01-HL (Δoch1), GJK11-HL (Δoch1, Δpmt1) and GJK15-HL (Δoch1, Δpmt5) were incubated for 5 days in 2.5 mL of YPD+U medium and diluted to OD580 = 30 and were then diluted at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 with fresh BMGY+U medium, typically to a total volume of 5 mL, simultaneously. The culture was collected and the OD580 was measured after 24, 60, 96, 120, 144, 168 and 192 h, respectively. Fig. 3 showed that the OD580 of GJK11-HL (Δoch1, Δpmt1) was lower than for GJK01-HL (Δoch1), indicating that the inactivity of the PMT1 gene in the low-mannose type N-linked oligosaccharide engineered P. pastoris strain caused the growth defect. However, the deletion of PMT5 had little effect on the P. pastoris growth rate (Fig. 3).
:20 with fresh BMGY+U medium, typically to a total volume of 5 mL, simultaneously. The culture was collected and the OD580 was measured after 24, 60, 96, 120, 144, 168 and 192 h, respectively. Fig. 3 showed that the OD580 of GJK11-HL (Δoch1, Δpmt1) was lower than for GJK01-HL (Δoch1), indicating that the inactivity of the PMT1 gene in the low-mannose type N-linked oligosaccharide engineered P. pastoris strain caused the growth defect. However, the deletion of PMT5 had little effect on the P. pastoris growth rate (Fig. 3).
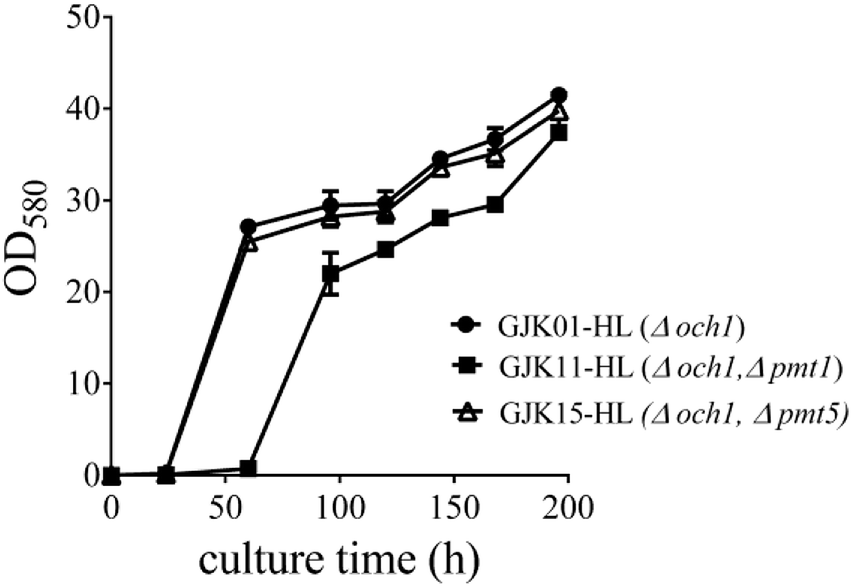 | ||
| Fig. 3 The influence of PMT1 (GJK11-HL (Δoch1, Δpmt1)) or PMT5 (GJK15-HL (Δoch1, Δpmt5)) gene disruption on the growth rate of yeast; GJK01-HL (Δoch1) was utilized as a control. Error bars represent the standard deviations of the three experiments. | ||
2.4. Expression, purification and identification of anti-Her-2 antibodies
GJK15-HL (Δoch1, Δpmt5) was constructed via conventional protocols to express the anti-Her-2 antibody. SDS-PAGE and western blot studies (Fig. S5A and S5B†) of the purified proteins show that their molecular weights (55 kDa for the heavy chain and 25 kDa for the light chain, respectively) agreed with the predicted molecular weights of the anti-Her-2 antibody. Fig. S5A† also revealed that the proteins were essentially homogeneous. The purified protein was also analyzed using a BCA protein quantification kit to determine that the protein expression level of the anti-Her-2 antibody was 66.71 ± 2.87 mg L−1. The protein expression levels of the anti-Her-2 antibody were respectively 74.63 ± 1.42 mg L−1 and 67.39 ± 13.79 mg L−1 from GJK01-HL (Δoch1) and GJK11-HL (Δoch1, Δpmt1). All these data indicated that the protein expression levels from all three mutants were higher than for the wildtype strain GS115, which was 53.7 ± 2.9 mg L−1.28 The procedures for the purification and identification of anti-Her-2 antibodies were the same as for GJK15-HL (Δoch1, Δpmt5), which will be discussed next.2.5. Detection of O-glycosylation of the anti-Her-2 antibody
Next, we examined O-glycosylation of the anti-Her-2 antibody via western blot combined with lectin Con A.29 Compared to other O-glycosylation analysis methods (e.g., matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) and peptide mass fingerprint (PMF)), western blot combined with lectin Con A could provide directive evidence for the content of mannose without any need for the consideration of releasing O-chains from proteins, the determination of the molecular weights of proteins, complex data-processing operations or expensive equipment.29 To eliminate the effects of mannose from N-linked oligosaccharides and to block buffer proteins, PNGase F was performed to remove N-linked oligosaccharides, and Q- and Con A Sepharose 4B columns were utilized to purify bovine serum albumin (BSA). Human IgG and RNB served as perfect contracts because only N-linked but O-linked oligosaccharides existed.30 Using this strategy, we have reported that the anti-Her-2 antibody heavy chain from GJK01 (Δoch1) have O-glycans.29 In addition, since there are no O-glycosylation in the light chains of antibodies expressed via GJK01 (Δoch1),29 only the heavy chains of antibodies were analyzed in the present research.After incubation with PNGase F, molecular RNB (Fig. 4A and B, lane 1 and 2; Fig. 5A, lane 2 and 3) and human IgG (Fig. 5A, lane 4 and 5) decreased, indicated that N-linked oligosaccharides were removed; besides, there was no mannose detected in RNB (Fig. 5C, lane 2 and 3) and human IgG (Fig. 4B, lane 1 and 2; Fig. 5C, lane 4 and 5), suggesting that all N-glycans were removed thoroughly. To avoid the potential influences of PNGase F concentration on the assays, PNGase F was utilized in large excess. In addition, the concentration of proteins between pre- or post-incubation with PNGase F was the same (Fig. 4A, lane 1 and 2; Fig. 5A, lane 2 and 3, and lane 4 and 5; Fig. 5B, lane 4 and 5), and PNGase F contains no mannose (Fig. 5A and C, lane 1), indicating that Con A stained bands were only caused by O-linked oligosaccharides.
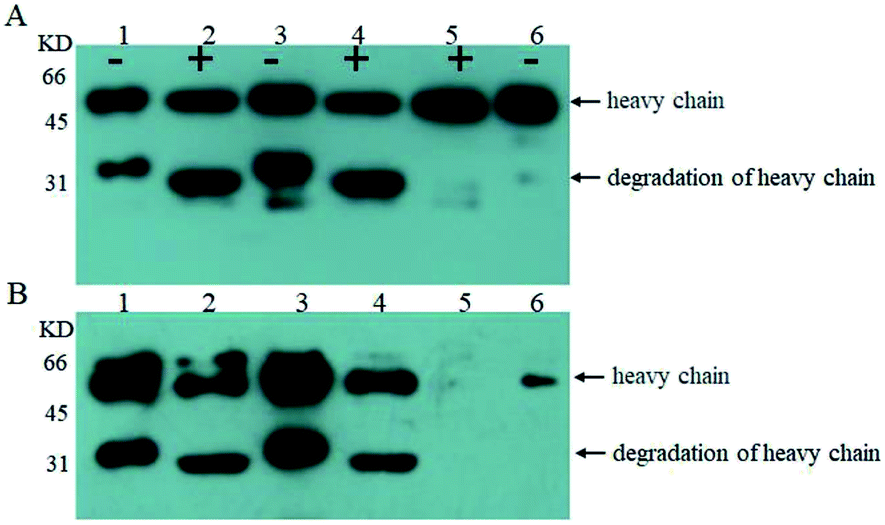 | ||
| Fig. 4 Western blot stained with goat anti-human IgG Fc polyclonal antibody (A) and western blot stained with Con A (B); analysis of the anti-Her-2 antibody expressed by GJK01-HL (Δoch1) (lane 1), PNGase F digestion of the anti-Her-2 antibody expressed by GJK01-HL (Δoch1) (lane 2), the anti-Her-2 antibody expressed by GJK15-HL (Δoch1, Δpmt5) (lane 3), PNGase F digestion of the anti-Her-2 antibody expressed by GJK15-HL (Δoch1, Δpmt5) (lane 4), PNGase F digestion of human IgG (lane 5), human IgG standard (lane 6); “+” indicate proteins that were treated with PNGase F, while “−” indicates those that were not. | ||
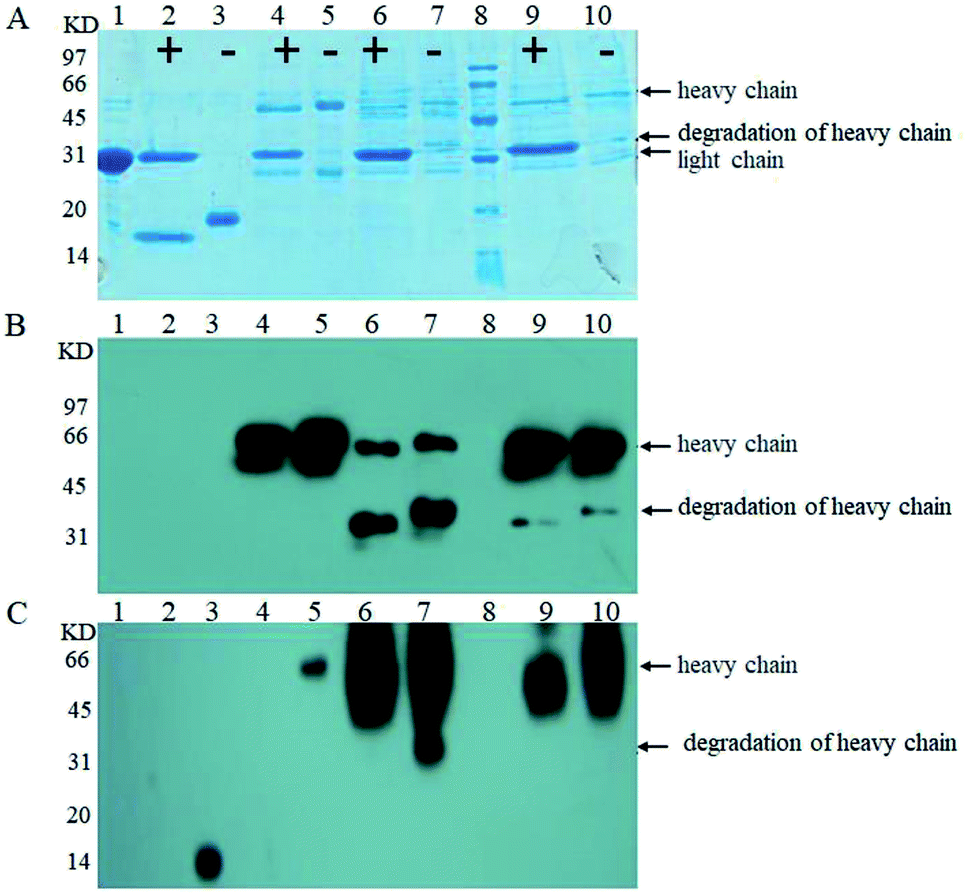 | ||
| Fig. 5 SDS-PAGE (A); western blot stained with goat anti-human IgG Fc polyclonal antibody (B); and western blot stained with Con A (C); analysis of PNGase F standard (lane 1), PNGase F digestion of RNB (lane 2), RNB standard (lane 3), PNGase F digestion of the human IgG standard (lane 4), the human IgG standard (lane 5), PNGase F digestion of anti-Her-2 antibody expressed by GJK01-HL (Δoch1) (lane 6), the anti-Her-2 antibody expressed by GJK01-HL (Δoch1) (lane 7), molecular marker (lane 8), PNGase F digestion of the anti-Her-2 antibody expressed by GJK11-HL (Δoch1, Δpmt1) (lane 9), and the anti-Her-2 antibody expressed by GJK11-HL (Δoch1, Δpmt1) (lane 10), “+” indicates proteins that were treated with PNGase F, while “−” indicates those that were not. | ||
Firstly, we compared O-linked oligosaccharides of the anti-Her-2 antibody from GJK01-HL (Δoch1) and GJK15-HL (Δoch1, Δpmt5). The anti-Her-2 antibody from GJK01-HL (Δoch1) was utilized as the control here. It seemed that the deletion of PMT5 had little effect on the O-mannosylation of the anti-Her-2 antibody (Fig. 4B, lane 2 and 4) and the degradation of the anti-Her-2 antibody (Fig. 4A and B, lane 1 and 3). Due to the Pmtps family mannosylated different acceptor proteins23 and how the deletion of PMT5 had little effect on the growth rate, we would not be surprised. The deletion of PMT5 did not influence the O-mannosylation of the anti-Her-2 antibody, mainly because: (1) the anti-Her-2 antibody was not an acceptor protein of Pmt5p; (2) the Pmt1p/Pmt2p complex compensates for the deletion of Pmt5p.31
We then compared O-linked oligosaccharides of the anti-Her-2 antibody from GJK01-HL (Δoch1) and GJK11-HL (Δoch1, Δpmt1). The result showed that O-linked oligosaccharides from GJK11-HL (Δoch1, Δpmt1) significantly decreased (Fig. 5C, lane 9), even though the concentration of the anti-Her-2 antibody from GJK11-HL (Δoch1, Δpmt1) (Fig. 5A and B, lane 9) was 2-fold higher than from GJK01-HL (Δoch1) (Fig. 5A and B, lane 6). Besides, after internal calibration, the IOD values determined from the PNGase F digestion of the anti-Her-2 antibody expressed by GJK01-HL (Δoch1) and GJK11-HL (Δoch1, Δpmt1) were 4.43 ± 0.43 and 3.09 ± 0.34, respectively (P < 0.05, Table SIII†). Due to the N-glycans of the human IgG standard containing 3 mannoses but no O-glycans,32 it can be utilized as an internal reference protein to calculate the mannose content of proteins, which is caused by O-glycosylation. Using this strategy, we preliminary determined the amounts of the O-linked oligosaccharides of the anti-Her-2 antibody to be 14 and 5 O-mannose per molecule expressed by GJK01-HL (Δoch1) and GJK11-HL (Δoch1, Δpmt1), respectively (Table SIII†). All these data indicated that the inactivity of PMT1 significantly decreased O-linked oligosaccharides. Considering the amount of protein from GJK11-HL (Δoch1, Δpmt1), which after analysis by lectin blot was determined to be two-fold higher than from GJK01-HL (Δoch1), this paper provided incontrovertible evidence to suggest that Pmt1p was responsible for the initial O-mannosylation of the anti-Her-2 antibody. Furthermore, other Pmtps members, which will be further studied next, also catalyze the initial O-glycosylation of the anti-Her-2 antibody, because O-linked oligosaccharides from GJK11-HL (Δoch1, Δpmt1) were not removed thoroughly (Fig. 5C, lane 9). Since Pmtps subfamilies have different substrate specificities,12 Pmt4p, Pmt6p and Pmt7p probably are not responsible for the initial O-mannosylation of the anti-Her-2 antibody. However, further studies are needed to confirm the reference. Thus, we have succeeded in decreasing the relative amounts of O-linked oligosaccharides in N-linked oligosaccharide engineered strains. Although this is not the first Pichia pastoris strain with both engineered N- and O-linked oligosaccharides, our research also has some positive significance.33 For example, this article utilized the anti-Her-2 antibody as reporter proteins to verify the decrease of both N- and O-linked oligosaccharides. Considering the high specificity of O-mannosyltransferases (Pmtps), studies on different reporter proteins are significant. Furthermore, this article provided direct evidence that O-glycosylation was decreased via western blot with lectin. In addition, more remarkably, a widely used analytical method, high-performance anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD), requires beta-elimination to separate oligosaccharides from proteins and allow for filtration via the filtrate membrane to purify oligosaccharides, both of which are difficult to quantitatively detect. We constructed the PMT1 gene mutant strain via an insertion inactivation but two-step homologous recombination method, which would provide an idea for PMTs gene mutation in N-glycosylation engineered yeast. In addition to Pichia pastoris strains, the Hiroko Abe group developed N- and O-linked oligosaccharide engineered Saccharomyces cerevisiae strains; the strain had a limited capacity to decrease O-mannosylation, and the double defectivity of PMT1 and PMT2 result in severe growth defects. Besides, the mutagenesis technique probably causes reverse mutation during microorganism cultivation.20 To verify whether reverse mutation existed, GJK11-HL (Δoch1, Δpmt1) was incubated in uracil defective medium and showed stability in at least ten consecutive batches (data not shown). Besides, uracil defective medium caused overwhelming growth of PMT1 mutant strains during incubation. Thus, GJK11-HL (Δoch1, Δpmt1) is a genetically stable strain to produce therapeutic glycoproteins with both N- and O-linked modified oligosaccharides. Compared to other approaches for decreasing O-glycosylation, such as treatment stains with rhodanine-3-acetic acid derivative34 and treating glycoproteins with mannosidase,35 we believe that the construction of both N- and O-glycosylation engineered P. pastoris strains as described in this study is a simple and effective method for suppressing O-mannosylation of recombinant glycoproteins with low-mannose type N-linked oligosaccharide, because no extra procedures are required.
Furthermore, degradation of the heavy chains of the anti-Her-2 antibody from GJK11-HL (Δoch1, Δpmt1) was significantly decreased (Fig. 5A and B, lane 7 and 10), suggesting that the suppression of O-mannosylation significantly decreased degradation of proteins expressed from P. pastoris. Protein degradation is one of the challenges in the production and purification of heterologous proteins from yeast.36 Traditionally, defective protease include protease A (PEP4), protease B (PRB1) and yapsin1, and optimal incubation conditions were established to reduce protein degradation; however, a series of new problems arose, such as the growth defects of host strains and the substrate specificity of proteases.37 The decrease in the O-glycosylation of the anti-Her-2 antibody reduced the degradation of the protein in the present study, providing a new strategy for the reduction of recombinant protein degradation from yeast. It was proposed that hyper-O-glycosylated proteins in the ER of yeast caused ER stress and protein degradation.38 Further study will combine O-glycosylation defects in defective proteases and/or the up-regulation of ER transcription factors to decrease protein degradation and increase the production of expressed proteins.
3 Experimental
3.1. Bacterial strains, yeast strains, plasmids and other materials
The strains and plasmids used in the present study are listed in Table SI.† Yeast extract, agar and tryptone from OXOID; yeast nitrogen base without amino acids (YNB) from Difco; T4 DNA ligase, DNA ladder (100–5000 bp), restriction endonuclease, and LA DNA polymerase from New England Biolabs (NEB); low molecular weight protein markers (17-0446-01) from Amersham; DNA extraction kits and yeast genomic DNA kits from Cwbiotech; as well as BSA, human IgG and goat anti-human IgG from Sigma were purchased, respectively. Other reagents were obtained from Sangon biotech unless stated otherwise.3.2. Construction of PMT1 insertional inactivation plasmid (△pmt1-pYES2)
△pmt1-pYES2 is listed in Table SI† and further details are provided in the ESI.†3.3. Insertional inactivation of GJK01-HL (Δoch1) PMT1 gene
The △pmt1-pYES2 recombinant plasmids were linearized by EcoRV and then electrotransformed into a GJK01-HL (Δoch1) competent cell; the transformants were selected on MD + RH plates (1.5% agar, 1% YNB, 1% glucose, 4 × 10−5% biotin, 0.01% histidine and 0.01% arginine) and identified via PCR using primers of pmt1-ORF-OUT-5, pmt1-ORF-OUT-3.3.4. Construction of PMT5 gene deletion plasmid (△pmt5-pYES2)
△pmt5-pYES2 is listed in Table SI† and further details are provided in the ESI.†3.5. Deletion of the GJK01 (Δoch1) PMT5 gene
The △pmt5-pYES2 recombinant plasmids were linearized by Bstx I and then electro-transformed into the GJK01 (Δoch1) competent cell; the transformants were selected on MD + RH plates. The first recombinants were identified via PCR using primers of pmt5-ORF-OUT-5 and pmt5-ORF-OUT-3. The desired recombinant was incubated in 2.5 mL of YPD+U (2% tryptone, 1% yeast extract, 2% glucose and 0.1% uracil) at 25 °C. After 12 h of cultivation, the cultures were then spread onto an MD-URHF plate (1.5% agar, 1%YNB, 1% glucose, 4 × 10−5% biotin, 0.1% uracil, 0.01% histidine, 0.01% arginine and 0.1% 5-fluoroorotic acid) at 25 °C for 6 days. Correct disruption was confirmed by PCR analysis with primers (Table SII†) based on genomic DNA.3.6. Expression of the anti-Her-2 antibody
For the expression of the anti-Her-2 antibody, GJK01-HL (Δoch1), GJK11-HL (Δoch1, Δpmt1), GJK15-HL (Δoch1, Δpmt5) were constructed and incubated; further details are provided in the ESI.†3.7. Purification of the anti-Her-2 antibody
Anti-Her-2 antibodies were purified with a HiTrap rProtein A FF column, and further details are provided in the ESI.†3.8. Comparison of the growth rates of GJK01-HL (Δoch1), GJK11-HL (Δoch1, Δpmt1) and GJK15-HL (Δoch1, Δpmt5)
Growth rates of target strains were determined using colorimetry at OD580, and further details are provided in the ESI.†3.9. Detection of O-glycosylation of anti-Her-2 antibody
O-Glycosylation of the anti-Her-2 antibody was detected by previously reported methods.29 For the deletion of N-glycans, 550 μL of target proteins (100 μg) containing 5% SDS and 4 M DTT were incubated in boiling water for 10 min. After cooling to room temperature, NP-40, PBS (pH 7.5) and PNGase F were added at final concentrations of 1%, 50 mM and 30 U mL−1, respectively, and were then incubated at 37 °C for 18 h.For western blot analysis, 20 μg of proteins from GJK11-HL (Δoch1, Δpmt1) and 10 μg of other proteins from SDS-PAGE gels were transferred to a PVDF membrane overnight at 45 V in a transfer buffer. The membranes were then blocked in 0.1 mg mL−1 of BSA (BSA was purified contains no mannose in two steps with a Q-column and a Con A Sephadex 4B-column, respectively) for 2 h at room temperature. This was followed by incubation for an additional 1 h in a 1:5000 dilution of horseradish peroxidase conjugated goat anti-human IgG antibody; the parallel membranes were incubated for 16 h at room temperature in 3 μg L−1 of Con A. The membranes were then washed five times for 5 min in PBST. Chemiluminescent detection was carried out according to instructions supplied by Immobilon™ Western. The integral optic density (IOD) of each ConA band was measured via an Image pro plus 6.0. With the Con A IOD ratios of the target protein to the internal reference protein (human IgG standard), the relative quantitative of the O-glycans was calculated.
4 Conclusions
We generated an effective glycoengineered Pichia pastoris strain to effectively produce therapeutic glycoproteins with both engineered N- and O-linked oligosaccharides. For example, the anti-Her-2 antibody with Man12GlcNAc2∼Man16GlcNAc2 N-linked oligosaccharides and reduced O-linked oligosaccharides was produced by GJK11-HL (Δoch1, Δpmt1) in our lab. These yeast proteins modified with low-mannose type N- and O-linked oligosaccharides probably have limitless applications in the discovery and generalization of therapeutic glycoproteins. On the other hand, this study clarified that O-glycosylation of the anti-Her-2 antibody is partly catalysed by not Pmt1p but Pmt5p. Encouraged by this, we will further decrease O-glycosylation in GJK11-HL (Δoch1, Δpmt1) in future studies. However, the insertional inactivation of PMT2, and both PMT1 and PMT5 failed, indicating that the disruption of these genes was lethal in low-mannose type N-linked oligosaccharide engineered P. pastoris and that more work is needed to produce an effective strain.Conflicts of interest
There are no conflicts to declare.Abbreviations
| Man | Mannose |
| GlcNAc | N-Acetylglucosamine |
| rpm | Revolutions per minute |
| IgG | Immunoglobulin G |
| RNB | Ribonuclease B |
| YNB | Yeast nitrogen base without amino acids |
| PCR | Polymerase chain reaction |
| LB | Luria–Bertani medium |
| MD | Minimal dextrose medium |
| YPD | Yeast extract peptone dextrose medium |
| BMGY | Buffered minimal glycerol-complex medium |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| PBS | Phosphate buffered saline |
| Con A | Concanavalin A |
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (No. 81673339 & No. 81773619), the Beijing Natural Science Foundation (No. 7152112), the Scientific Research Foundation for Advanced Talents of Huanghuai University (No. 1000.12.01.1678), the Preparation for the National Natural Science Foundation of Huanghuai University (No. 1000. 11.07.1678.1002) and the Project of Education Department of Henan Province (19A180022).References
- S. Li, H. Wang, J. Ma, G. Gu, Z. Chen and Z. Guo, Chem. Commun., 2016, 52, 13995–13998 RSC.
- P. Umana, J. Jean-Mairet, R. Moudry, H. Amstutz and J. E. Bailey, Nat. Biotechnol., 1999, 17, 176–180 CrossRef CAS.
- N. Pristovsek, H. G. Hansen, D. Sergeeva, N. Borth, G. M. Lee, M. R. Andersen and H. F. Kildegaard, Biotechnol. J., 2018, 13, e1700216 CrossRef PubMed.
- J. M. Cregg, J. L. Cereghino, J. Shi and D. R. Higgins, Mol. Biotechnol., 2000, 16, 23–52 CrossRef CAS.
- J. Wu and L. Shi, Lett. Biotechnol., 2005, 60–63 Search PubMed.
- S. Wildt and T. U. Gerngross, Nat. Rev. Microbiol., 2005, 3, 119–128 CrossRef CAS PubMed.
- R. Kornfeld and S. Kornfeld, Annu. Rev. Biochem., 1985, 54, 631–664 CrossRef CAS PubMed.
- B. Laukens, C. De Visscher and N. Callewaert, Future Microbiol., 2015, 10, 21–34 CrossRef CAS PubMed.
- X. Yang, B. Liu, M. Song, X. Gong, S. Chang, K. Xue and J. Wu, Chin. J. Biotechnol., 2011, 27, 108–117 CAS.
- B. K. Choi, P. Bobrowicz, R. C. Davidson, S. R. Hamilton, D. H. Kung, H. Li, R. G. Miele, J. H. Nett, S. Wildt and T. U. Gerngross, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5022–5027 CrossRef CAS PubMed.
- K. Tomimoto, Y. Fujita, T. Iwaki, Y. Chiba, Y. Jigami, K. Nakayama, Y. Nakajima and H. Abe, Biosci., Biotechnol., Biochem., 2013, 77, 2461–2466 CrossRef CAS PubMed.
- V. Girrbach, T. Zeller, M. Priesmeier and S. Strahl-Bolsinger, J. Biol. Chem., 2000, 275, 19288–19296 CrossRef CAS PubMed.
- T. Willer, W. Amselgruber, R. Deutzmann and S. Strahl, Glycobiology, 2002, 12, 771–783 CrossRef CAS PubMed.
- K. Kuroda, K. Kobayashi, Y. Kitagawa, T. Nakagawa, H. Tsumura, T. Komeda, D. Shinmi, E. Mori, K. Motoki, K. Fuju, T. Sakai, K. Nonaka, T. Suzuki, K. Ichikawa, Y. Chiba and Y. Jigami, Appl. Environ. Microbiol., 2008, 74, 446–453 CrossRef CAS PubMed.
- A. Bondt, S. Nicolardi, B. C. Jansen, T. M. Kuijper, J. Hazes, Y. van der Burgt, M. Wuhrer and R. Dolhain, Arthritis Res. Ther., 2017, 19, 160 CrossRef PubMed.
- X. Yang, B. Liu, M. Song, X. Gong, S. Chang, K. Xue and J. Wu, Chin. J. Biotechnol., 2011, 27, 108–117 CAS.
- D. Zha, Methods Mol. Biol., 2013, 988, 31–43 CrossRef CAS.
- T. I. Potgieter, M. Cukan, J. E. Drummond, N. R. Houston-Cummings, Y. Jiang, F. Li, H. Lynaugh, M. Mallem, T. W. McKelvey, T. Mitchell, A. Nylen, A. Rittenhour, T. A. Stadheim, D. Zha and M. D'Anjou, J. Biotechnol., 2009, 139, 318–325 CrossRef CAS PubMed.
- Y. Jigami, Biosci., Biotechnol., Biochem., 2008, 72, 637–648 CrossRef CAS PubMed.
- H. Abe, K. Tomimoto, Y. Fujita, T. Iwaki, Y. Chiba, K. I. Nakayama and Y. Nakajima, Glycobiology, 2016, 26, 1248–1256 CAS.
- F. M. Klis, A. Boorsma and P. W. De Groot, Yeast, 2006, 23, 185–202 CrossRef CAS PubMed.
- G. Lesage and H. Bussey, Microbiol. Mol. Biol. Rev., 2006, 70, 317–343 CrossRef CAS PubMed.
- J. Hutzler, M. Schmid, T. Bernard, B. Henrissat and S. Strahl, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7827–7832 CrossRef CAS PubMed.
- M. Gentzsch and W. Tanner, Glycobiology, 1997, 7, 481–486 CrossRef CAS PubMed.
- B. Liu, X. Gong, S. Chang, Y. Yang, M. Song, D. Duan, L. Wang, Q. Ma and J. Wu, J. Biotechnol., 2009, 143, 95–102 CrossRef CAS PubMed.
- Y. Wang, X. Gong, S. Chang, B. Liu, M. Song, H. Huang and J. Wu, Chin. J. Biotechnol., 2007, 907–914 CrossRef.
- K. Xue, B. Liu, S. Chang, X. Gong, W. Xu and J. Wu, Lett. Biotechnol., 2010, 650–654 CAS.
- L. Wang, W. Zhang and J. Wu, Lett. Biotechnol., 2008, 843–847 CAS.
- S. Li, X. Gong, S. Chang, J. Wu, B. Liu and G. Gao, Lett. Biotechnol., 2011, 834–837 CAS.
- S. R. Hamilton, R. C. Davidson, N. Sethuraman, J. H. Nett, Y. Jiang, S. Rios, P. Bobrowicz, T. A. Stadheim, H. Li, B. K. Choi, D. Hopkins, H. Wischnewski, J. Roser, T. Mitchell, R. R. Strawbridge, J. Hoopes, S. Wildt and T. U. Gerngross, Science, 2006, 313, 1441–1443 CrossRef CAS PubMed.
- V. Girrbach and S. Strahl, J. Biol. Chem., 2003, 278, 12554–12562 CrossRef CAS.
- F. Clerc, K. R. Reiding, B. C. Jansen, G. S. M. Kammeijer, A. Bondt and M. Wuhrer, Glycoconjugate J., 2016, 33, 309–343 CrossRef CAS PubMed.
- J. H. Nett, W. J. Cook, M. T. Chen, R. C. Davidson, P. Bobrowicz, W. Kett, E. Brevnova, T. I. Potgieter, M. T. Mellon, B. Prinz, B. K. Choi, D. Zha, I. Burnina, J. T. Bukowski, M. Du, S. Wildt and S. R. Hamilton, PLoS One, 2013, 8, e68325 CrossRef CAS PubMed.
- M. G. Orchard, J. C. Neuss, C. M. Galley, A. Carr, D. W. Porter, P. Smith, D. I. Scopes, D. Haydon, K. Vousden, C. R. Stubberfield, K. Young and M. Page, Bioorg. Med. Chem. Lett., 2004, 14, 3975–3978 CrossRef CAS PubMed.
- S. Gomathinayagam and S. R. Hamilton, Appl. Microbiol. Biotechnol., 2014, 98, 2545–2554 CrossRef CAS PubMed.
- N. E. Ivessa, C. Kitzmüller and M. de Virgilio, Protoplasma, 1999, 207, 16–23 CrossRef CAS.
- M. Wu, Q. Shen, Y. Yang, S. Zhang, W. Qu, J. Chen, H. Sun and S. Chen, J. Ind. Microbiol. Biotechnol., 2013, 40, 589–599 CrossRef CAS PubMed.
- Y. X. Xu, L. Liu, C. E. Caffaro and C. B. Hirschberg, J. Biol. Chem., 2010, 285, 24600–24608 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08121b |
| This journal is © The Royal Society of Chemistry 2019 |