
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLarge-scale preparation of size-controlled Fe3O4@SiO2 particles for electrophoretic display with non-iridescent structural colors†
Wei Wangab,
Ang Zhengab,
Yifan Jianga,
Dongsheng Lana,
Fenghua Lua,
Lelin Zheng a,
Lin Zhuang*a and
Ruijiang Hong*a
a,
Lin Zhuang*a and
Ruijiang Hong*a
aInstitute for Solar Energy Systems, Guangdong Provincial Key Laboratory of Photovoltaics Technologies, School of Physics, Sun Yat-Sen University, Guangzhou 510275, People's Republic of China. E-mail: stszhl@mail.sysu.edu.cn; hongruij@mail.sysu.edu.cn
bSchool of Materials Science and Engineering, Sun Yat-Sen University, Guangzhou 510275, People's Republic of China
First published on 2nd January 2019
Abstract
Monodisperse colloidal particles have promising applications in electrophoretic displays with vivid colors, reversibility and low switching times. In this study, a facile, effective and large-scale strategy for preparing size-controlled Fe3O4@SiO2 particles is reported. Multiple Fe3O4 particles were synthesized by a modified solvothermal method using sodium citrate as a surface modifier with a binary solvent, and were then coated with a SiO2 layer to obtain a highly negatively charged surface via a modified Stöber method. Owing to the easily controlled sizes and sufficient surface charges, Fe3O4@SiO2 particles can be assembled into colloidal amorphous arrays with the balance of electrostatic repulsion and electrophoretic forces. The reflections cover wavelengths ranging from 802 to 453 nm, and were optimized by investigating the dependence of the particles on variables such as particle size, particle volume fraction, and electric field intensity. The large-scale preparation of electrically responsive Fe3O4@SiO2 particles facilitates an electrophoretic display with broad-range colors, showing the practical potential in industrial application.
Introduction
Recently, there have been numerous reports about full-color electrophoretic displays (EPDs) built with structural color materials.1–6 Commercial EPDs that consist of structural color materials face many challenges in realizing full-color display, including low yield, poorly controlled synthesis, and poor color tunability. The common EPD materials use organic particles with easy preparation, good monodispersity, and low refractive index, such as PS7 and PMMA.8 These spherical particles are limited by their low color visibility and iridescent color. Inorganic composite particles with high refractive indexes, such as Se@Ag2Se,9 ZnS,10 and TiO2,11 were employed to obtain bright structural colors. However, they are difficult to apply in EPDs due to high density, low surface charge, and low yield.Fe3O4, with a high refractive index (2.42), produces high-index contrast. Also, highly charged Fe3O4 particles with controlled sizes of 70–200 nm are suitable building blocks for structural color materials. Based on the factors mentioned above, Lee et al. developed silica-coated Fe3O4 particles suspended in propylene carbonate (PC) for electrically controlled particle arrays with only short-range order.12 However, due to the small-scale production (50 mg) via the hot-injection polyol method,13 the yield of Fe3O4 particles failed to meet the requirements for EPDs. Several synthesis routes have been developed to solve this problem.14–16 Co-precipitation and microemulsion methods can produce large numbers of Fe3O4 particles; however, their particles sizes are usually less than 20 nanometers, which are not suitable for EPD. Guan et al. synthesized Fe3O4 particles via a facile solvothermal method by adding poly(vinyl pyrrolidone, PVP), which resulted in approximately spherical clusters with only one diameter of around 150 nm.17 Although solvothermal methods afford a slight increase in the production of particles, the size of particles cannot be precisely controlled within a broad range. Also, these Fe3O4 particles are usually modified by varieties of polyelectrolytes (such as PAA, PVP, and PSSMA),18–20 due to which it is difficult to produce large numbers of products at a time, and are not cost-effective, stable, and biodegradable. For Fe3O4 particles, mass production and good size control still remain challenges for researchers.
Sodium citrate (Na3Cit) is employed as an ideal surface modifier in the modified solvothermal method to solve these problems. The low-cost Na3Cit is a stable antioxidant and does not harm humans and the environment, and is widely used in the chemical industry.21–23 These factors suggest that Na3Cit is of benefit to the large-scale preparation of Fe3O4. However, there are few reports about the synthesis of Fe3O4 for exhibiting structural color responses to external fields. In addition, SiO2 coatings can significantly improve the uniformity of particle morphology and size. To accommodate the large-scale preparation of core–shell particles, the Stöber method needs to be improved to avoid problems such as low surface negative charge, long-time reaction and aging processes.
Herein, we demonstrate a facile solvothermal method with a binary solvent to achieve the large-scale preparation of Fe3O4 particles modified by citrate. Diethylene glycol (DEG), with a high boiling point of 240 °C, was chosen as one binary solvent to raise the reaction temperature for better crystallinity in the massive preparation of Fe3O4. FeCl2·4H2O was added to shorten the reaction time. Besides, Fe3O4 particles with a size range of 70–170 nm can be effectively tuned by varying the mass ratio of Na3Cit/NaOAc in the reaction. This modified solvothermal method using Na3Cit with a binary solvent can achieve an eleven-fold improvement of the production, which means a great deal of Fe3O4 particles (3.02 g) with narrow size distribution can be obtained at one time. The Stöber method was also adapted to uniformly coat the SiO2 shell in a short time for the gram-scale preparation of core–shell particles. In the study, the particle sizes and particle volume fraction under the low voltage range are tuned to obtain vivid structural colors of the electrically responsive Fe3O4@SiO2 particles. In particular, when the concentration exceeds the critical volume fraction (7.8%), Fe3O4@SiO2 colloids show structural colors in different particle volume fractions even without the electric field, which provides another option for color display devices.
Experimental
Chemicals
Diethylene glycol (DEG), ethylene glycol (EG), ferric chloride (FeCl3·6H2O), sodium acetate (NaOAc), sodium citrate (Na3Cit), and ethanol were purchased from Aladdin Reagent. Ferrous chloride (FeCl2·4H2O) and ammonia solution (NH3·H2O, 28 wt%) were acquired from Shanghai Macklin Biochemical Co. Tetraethyl orthosilicate (TEOS) and propylene carbonate (PC) were purchased from Sinopharm Chemical Reagent.Characterization
A Lakeshore 7404 vibrating sample magnetometer (VSM) was used to measure the magnetic properties (M–H curves) at room temperature. The variation in the electric field was controlled by a DC electrometer (GW/INSTEK GPC-3060D) and a Rigol DG4000 Series function/arbitrary waveform generator. The morphology and size of the particles were investigated using the Tecnai G2 Spirit and the JEM-2010HR transmission electron microscopes (TEMs). X-ray diffraction (XRD) patterns were measured on an Empyrean diffractometer platform with CuKα radiation. The infrared spectra were obtained in the range of 400–7500 cm−1 with a Nicolet 6700-Continuum Fourier transform infrared (FT-IR) spectrometer using KBr wafers. The optical properties of colloidal amorphous arrays formed by particles in the external field were measured with an angular resolution spectrometer (Shanghai Ideaoptics) and a QE65000 optic fiber spectrometer (Ocean Optic, Inc.) coupled to a six-around-one fiber bundle reflection/backscattering probe.Synthesis of Fe3O4 particles
Fe3O4 particles were synthesized by the solvothermal method. Typically, 0.82 g of FeCl3·6H2O and 0.318 g of FeCl2·4H2O were dissolved in 20 mL of DEG. Next, 0.32 g of Na3Cit, 1.5 g of sodium acetate anhydrous, in turn, were added to 10 mL of the EG, then heated and stirred until they were dissolved. The two obtained solutions were transferred to a Teflon-lined stainless-steel autoclave, then sealed and left at 220 °C for 6 h. The products were thoroughly washed with ethanol and de-ionized (DI) water, then dispersed in DI water for the preparation of Fe3O4@SiO2 colloids.Preparation of Fe3O4@SiO2 particles
Fe3O4@SiO2 particles were synthesized using a modified Stöber method. A mixture of ethanol (160 mL) and ammonia solution (16 mL) was added to DI water (24 mL) and mixed before the addition of Fe3O4 particles (260 mg). The mixture was then transferred to a flask and subjected to vigorous mechanical stirring at 50 °C for 10 min to form a well-distributed system, and then a total of 400 μL of TEOS was added to the mixture at a rate of 200 μL every 30 min. After the reaction, Fe3O4@SiO2 particles were separated by a magnet. The as-obtained product was washed with DI water and ethanol. Fe3O4@SiO2 particles were suspended in a mixture of PC and ethanol by ultrasonication. The mixture was thoroughly dried at 100 °C in a vacuum oven, and the ethanol evaporated after 1 h. After the mixture was cooled to room temperature, a concentrated colloid was obtained.Preparation of devices
The EPD devices consisted of two pieces of indium tin oxides (ITO) glass, separated by a 50 μm-thick polyimide tape. A series of Fe3O4@SiO2 colloids were injected between the ITO electrodes.Results and discussion
As shown in Fig. 1a–d, the average diameter of Fe3O4 particles is influenced by the mass ratio of Na3Cit/NaOAc. These figures show that the average size of Fe3O4 particles increases from 70 to 170 nm when the mass ratio changes from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 to 1:7. In the high-resolution transmission electron microscopy (HRTEM) images, a single Fe3O4 particle consists of multiple nanocrystals with a diameter of 8 to 11 nm (Fig. 1e). The interplanar distances along a specific direction were measured to be 0.48 nm and 0.24 nm, which correspond well to the lattice spacing of the (111) and (311) planes of the cubic spinel structure (Fig. 1e, inset). The sharp diffraction rings are displayed in the selected area electron diffraction (SAED) pattern, suggesting a polycrystalline structure in the obtained products (Fig. 1f). The effects of Na3Cit and NaOAc on the size and morphology of Fe3O4 are attributed to both the electrostatic repulsion and the hydrolysis rate of the solvothermal method (Fig. 2). Na3Cit was selected due to the coordination of partial citrate groups with Fe cations on the Fe3O4 surface. The rest of the uncoordinated citrate groups are exposed in the solvent, leading to electrostatic repulsion between adjacent nanocrystals. Because of the high surface energy of Fe3O4 nanocrystals, the surface tension forces drive nanocrystals to aggregate to decrease the surface energy. When the concentration of Na3Cit is small in the mass ratio of Na3Cit and NaOAc, a few of the citrate molecules are modified on the surface of the nanocrystals. As a result, the electrostatic repulsion between adjacent nanocrystals decreases in the mixed solvent. Fe3O4 nanocrystals naturally form larger cluster particles in the mixed solvent because of their strong surface tension. As the concentration of Na3Cit increases, more citrate groups are anchored on the Fe3O4 nanocrystal surface, leading to stronger electrostatic repulsion between adjacent nanocrystals.24,25 In addition, the hydrolysis rate of Fe3+ can be decelerated by decreasing NaOAc to produce smaller Fe3O4 nanocrystals. Therefore, small Fe3O4 nanocrystals form smaller cluster particles in a solvothermal solvent by decreasing the levels of NaOAc. Fe3O4 particles depend not only on the interaction of strong surface tension and electrostatic repulsion but also on the hydrolysis rate. By controlling the mass ratio of Na3Cit and NaOAc in the solvothermal method, the size of Fe3O4 can be varied over a wide range.
:4 to 1:7. In the high-resolution transmission electron microscopy (HRTEM) images, a single Fe3O4 particle consists of multiple nanocrystals with a diameter of 8 to 11 nm (Fig. 1e). The interplanar distances along a specific direction were measured to be 0.48 nm and 0.24 nm, which correspond well to the lattice spacing of the (111) and (311) planes of the cubic spinel structure (Fig. 1e, inset). The sharp diffraction rings are displayed in the selected area electron diffraction (SAED) pattern, suggesting a polycrystalline structure in the obtained products (Fig. 1f). The effects of Na3Cit and NaOAc on the size and morphology of Fe3O4 are attributed to both the electrostatic repulsion and the hydrolysis rate of the solvothermal method (Fig. 2). Na3Cit was selected due to the coordination of partial citrate groups with Fe cations on the Fe3O4 surface. The rest of the uncoordinated citrate groups are exposed in the solvent, leading to electrostatic repulsion between adjacent nanocrystals. Because of the high surface energy of Fe3O4 nanocrystals, the surface tension forces drive nanocrystals to aggregate to decrease the surface energy. When the concentration of Na3Cit is small in the mass ratio of Na3Cit and NaOAc, a few of the citrate molecules are modified on the surface of the nanocrystals. As a result, the electrostatic repulsion between adjacent nanocrystals decreases in the mixed solvent. Fe3O4 nanocrystals naturally form larger cluster particles in the mixed solvent because of their strong surface tension. As the concentration of Na3Cit increases, more citrate groups are anchored on the Fe3O4 nanocrystal surface, leading to stronger electrostatic repulsion between adjacent nanocrystals.24,25 In addition, the hydrolysis rate of Fe3+ can be decelerated by decreasing NaOAc to produce smaller Fe3O4 nanocrystals. Therefore, small Fe3O4 nanocrystals form smaller cluster particles in a solvothermal solvent by decreasing the levels of NaOAc. Fe3O4 particles depend not only on the interaction of strong surface tension and electrostatic repulsion but also on the hydrolysis rate. By controlling the mass ratio of Na3Cit and NaOAc in the solvothermal method, the size of Fe3O4 can be varied over a wide range.
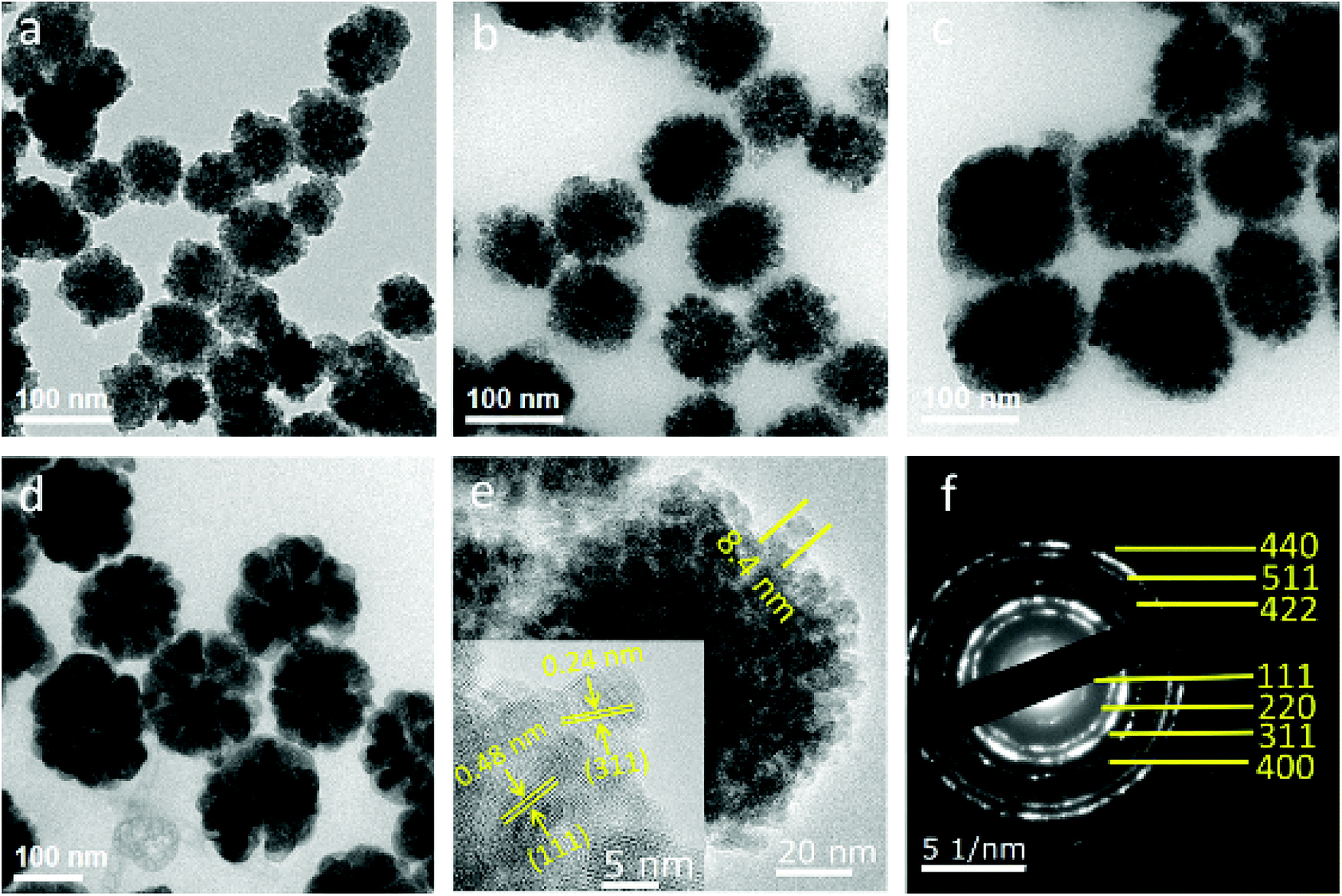 | ||
| Fig. 1 (a–d) TEM images of Fe3O4 particles with different mass ratios of Na3Cit/NaOAc (1:4, 1:5, 1:6, and 1:7). Average diameters of Fe3O4 particles are 73 nm for (a), 89 nm for (b), 121 nm for (c), and 165 nm for (d). (e) HRTEM image of the obtained Fe3O4 particles. (f) Corresponding SAED pattern of a single Fe3O4 particle. | ||
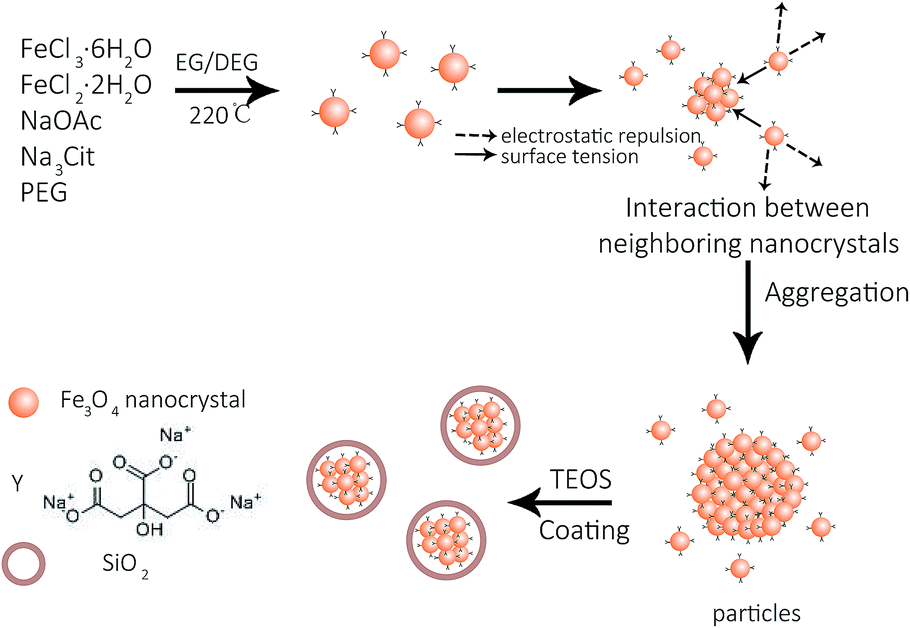 | ||
| Fig. 2 Formation of Fe3O4 and Fe3O4@SiO2. | ||
To study the effect of the magnetic properties of the Fe3O4 core in the stability of the colloids, their mass magnetizations were measured by applying a magnetic field from −13000 to 13000 Oe at 300 K. As shown in the illustration of magnetization versus magnetic field (Fig. 3a), the saturation magnetization (Ms) values of 73, 89, 121, and 165 nm Fe3O4 particles are 59.6, 66.2, 70.4, and 79 emu g−1, respectively. Since the sizes of primary nanocrystals can be smaller than the single domain size (about 20 nm), all nanocrystals with negligible coercive force and remanent magnetization exhibited superparamagnetism at room temperature. Furthermore, nanocrystals aggregate into spherical particles, maintaining the superparamagnetism with high saturation magnetization. Compared with polyelectrolyte-grafted Fe3O4, our samples possessed a higher quality of magnetism, which was due to the citrate group being chosen as a stabilizer and DEG with a high boiling point as a solvent. To further investigate the potential of the solvothermal reaction in mass production, the yield and particle size of the samples were studied with varying solution volumes, while other chemicals were kept at constant concentrations. It was observed that the particle yield increased linearly when the volume of reaction solvent increased from 50 to 600 mL (Fig. 3b). In addition, the size and size distribution of nanoparticles changed only slightly as the volume of the mixture solution increased from 50 to 600 mL. In this research, the reaction solutions were limited to 600 mL due to the experimental conditions, but it was shown that the modified solvothermal method could produce 8.7 mg of particles per milliliter of the reaction solution. Although the hot-inject method used the same ratio, the reactor and the additional NaOH amount both limited the production to 50 mg.13 FeCl2·4H2O was added to the reaction solution, shortening the reaction time to 6 h. The reaction required less time than the previous solvothermal method, which usually requires more than 10 h due to the reduction of Fe3+.26,27 The standard deviation of the particle size distributions showed a slight increase with the increase in reaction solution volume. The data clearly indicate that the novel solvothermal method provides a robust and scalable route to obtaining Fe3O4 particles from milligram to gram, which has sound potential for high-yield production for industrial use.
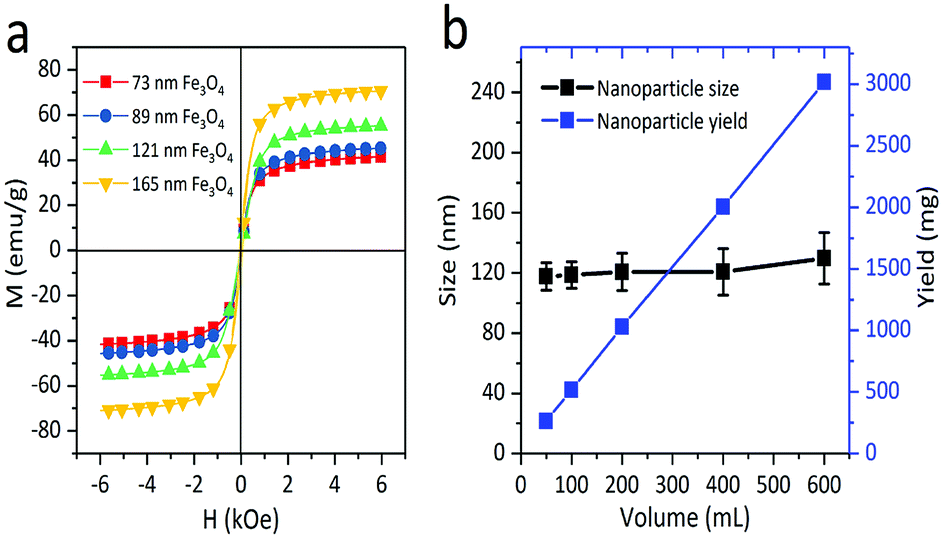 | ||
| Fig. 3 (a) Magnetization curves of 73 nm, 89 nm, 121 nm and 165 nm Fe3O4 particles at room temperature. (b) Yield and size of Fe3O4 nanoparticles obtained by a modified solvothermal method with varying volumes of the EG and DEG mixtures. The concentrations of other chemicals were constant while varying the total volume of the mixture solution. | ||
The SiO2 shell layer can improve the uniformity in the morphology and adjust the core–shell particle size at the same time (Fig. 4a–c). The growth of the shell layer can be controlled by adding TEOS. The TEM image in Fig. 4a suggests a 12 nm shell layer on the Fe3O4 particle, while Fig. 4b and c indicate a SiO2 coating of 25 and 48 nm. Therefore, the particle size can be adjusted by the core size or shell thickness. Fig. 4d indicates the dependence of the thickness of silica shell on the amount of TEOS added. The thickness of the silica shell first increases with the addition of TEOS, and then remains almost constant when the level of TEOS added exceeds 800 μL. Excessive amounts of TEOS do not promote an increase in the SiO2 growth rate. The powder XRD pattern of the Fe3O4@SiO2 sample is shown in Fig. 4e, which also confirms the presence of amorphous silica and the crystallinity of the Fe3O4 particle. Based on the Debye–Scherrer equation, the mean size of the nanocrystals (10–14 nm) obtained by calculating the strongest diffraction peak of (311) implies that the Fe3O4 particle has a double-stage hierarchical structure. The diffraction peaks located at 15–80° can be indexed to the corresponding (111), (220), (311), (400), (422), (511), and (440) planes of the cubic spinel structure of Fe3O4 (JCPDS no. 65-3107).26 In our modified solvothermal method, Fe2+ ions added to the precursor shortened the reaction time and improved the crystallization of nanocrystals. The red line in Fig. 4f shows the typical FT-IR spectrum of Fe3O4 particles, suggesting that the citrate group is successfully modified in the Fe3O4 nanocrystals. The absorption peaks at 1539 cm−1 and 1413 cm−1 correspond to both the symmetric vibration and anti-symmetric vibration of carboxyl groups, indicating that a large number of Na3Cit carboxylate groups are coordinated to the iron cations. As shown in the blue line of Fig. 4f, the absorption peaks at 567 cm−1 and 1071 cm−1 correspond to the Fe–O bending vibration and the Si–O–Si asymmetric stretching vibration.
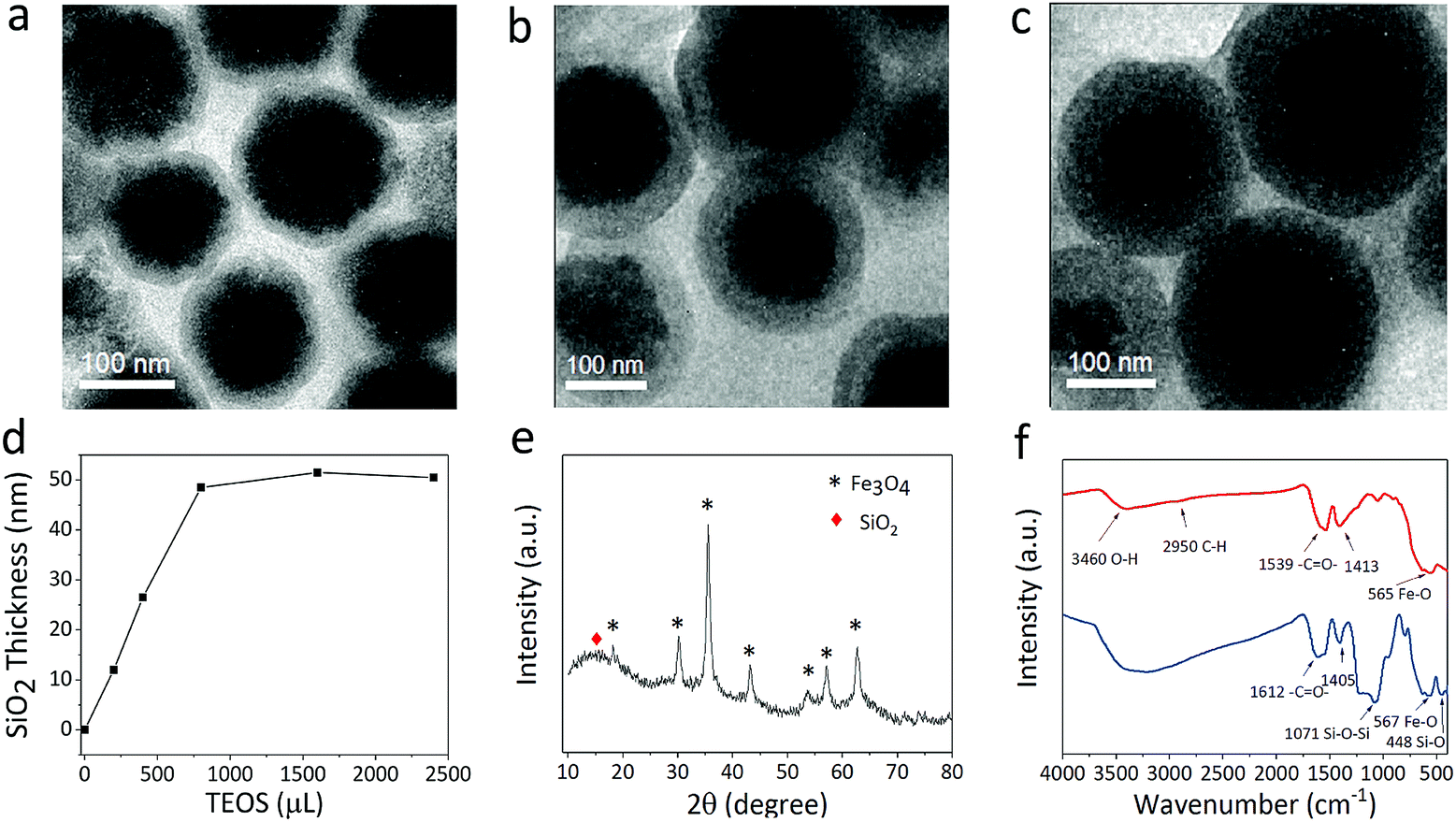 | ||
| Fig. 4 (a–c) TEM images of Fe3O4@SiO2 core–shell structures with increased SiO2 thickness formed by adding increasing amounts of TEOS during the synthesis. (d) Shell thickness of Fe3O4@SiO2 obtained with different amounts of TEOS. (e) XRD patterns of the Fe3O4@SiO2 particles. (f) FT-IR spectra of Fe3O4 particles and Fe3O4@SiO2 particles. | ||
In the modified Stöber method, the temperature during the reaction process plays an important role in the SiO2 coating. The optimal temperature is 50 °C, which ensures that the silicon shell grows rapidly and maintains uniform morphology (Fig. S1a–c†). When the reaction temperature is too low, SiO2 cannot form within half an hour. When the reaction temperature is high enough, the magnetic particles agglomerate easily, so that a uniform core–shell sphere cannot be obtained. Other modified Stöber methods require at least 3 h of aging or show little improvement in surface charge without further surface modification.28,29 A suitable coating temperature leads to an enhanced negatively charged surface on the particles in a short time. The zeta potential of the Fe3O4 and Fe3O4@SiO2 was measured to be about −38.0 mV and −55.9 mV (Fig. S1d†), which demonstrates that the SiO2 layer can significantly enhance the surface charge of the Fe3O4@SiO2 particles. The Fe3O4@SiO2 core–shell particles show excellent monodispersity and negatively charged surfaces. The silica shell layer allows particles to be dispersed in a variety of polar solvents, including water, ethanol and PC. Due to their high surface charge, Fe3O4@SiO2 particles of controllable size can effectively be assembled by adjusting the electric field.
The EPD device containing the Fe3O4@SiO2 colloid response to the electric field was then tested for the production of structural colors. As shown in the schematic diagram of Fig. 5a, the Fe3O4@SiO2 colloid in the display cell is a Brownian fluid in which every particle moves randomly. After applying an electric field, the particles are driven by electrophoretic force toward the positive electrode of the ITO. A balance of electrophoretic movement and electrostatic repulsion assembles particles into periodic nanostructures. Due to Bragg's law,30 a range of the electromagnetic spectrum is forbidden or allowed for propagation, which may lead to specific reflections or transmission of visible light. Dynamically adjusting the spacing of periodic nanostructures can change these propagation properties, leading to the modulation necessary to produce structural colors. For example, an increase in electric field strength enables a blue-shift in the reflection spectra, resulting in structural colors changing from a coffee color to black. Corresponding photographs taken of display cells from 0 V to 3 V are shown in Fig. 5b. Because Fe3O4@SiO2 particles with a negative charge are driven by the applied voltage, color variations are evident at the positive ITO electrode. As the applied voltage increases, the structural colors gradually change. When the voltage is above the critical value, the aggregation of particles is displayed as black. Because the applied voltage is reversible, the reflection peak is reproduced well at a fixed voltage. The reversibility of the EPD was proved by reflection spectra with a 0.5 Hz square wave signal, as shown in Fig. S2.† Reflection spectra of EPD based on Fe3O4@SiO2 particles were continuously recorded as the voltage was reversibly turned on and off for 30 cycles. There were no significant changes in the reflectance or structural colors of EPD. As shown in Fig. S3,† the EPD has a fast response to the external electric field. The response time measurement showed that the on-switching (2 V) and off-switching (0 V) were 472 ms and 251 ms, respectively. In every on–off process, particles achieving balance in electrostatic repulsion and electrophoretic forces need to spend a little time (about 30 to 100 ms), since the reflectance shows irregular fluctuation within a short time.
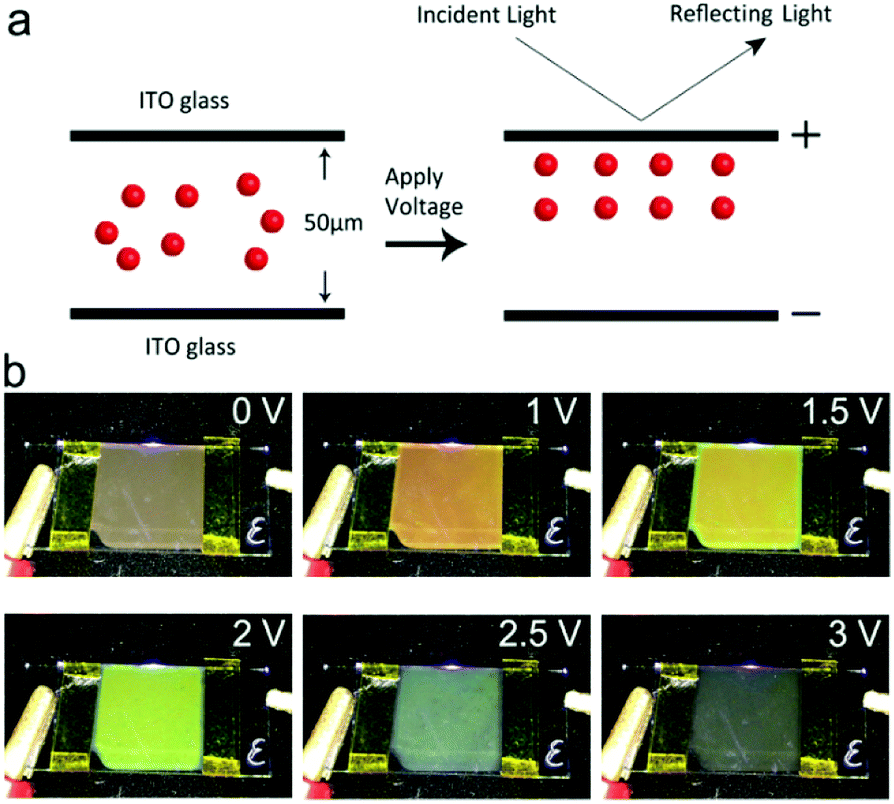 | ||
| Fig. 5 (a) Schematic illustration of the procedure for EPD in an electric field. (b) Photographs taken from 0 V to 3 V. A display cell of size 150 mm × 150 mm exhibits uniform color, which is dependent on the voltage. | ||
The particle size of electrophoretic particles is also critical to EPD. Fig. 6 exhibits the reflectance spectra of Fe3O4@SiO2 particles in three sizes under varied voltage. As shown in Fig. 6a, the reflection intensity of Fe3O4@SiO2 particles dispersed in PC first increases gradually and then decreases. The large particles are driven by the voltage to produce more stable particle array structures, resulting in an initial increase in reflection intensity. As the voltage increases, the electrostatic repulsion is insufficient to maintain the structure. More and more defects are found in the particle array structures, leading to a decrease in reflection intensity. Generally, the reflection peak of S1 (Fe3O4 165 nm, SiO2 12 nm) undergoes a blue shift and changes position from 802 to 571 nm, with the applied positive voltage increasing to 2.5 V. Fig. 6b shows the optical properties of the positive electrode of S2 (Fe3O4 121 nm, SiO2 12 nm). The reflectance spectral range of S2 shows a blue shift, and the reflection intensity follows a downward trend; when the applied positive voltage increases to 2.5 V, the reflection peak shifts from 679 to 514 nm. The decrease in the reflection intensity is due to particle disorder resulting from the increasing voltage. A higher specific surface area in small particles causes fast particle movement, then particle array structures are rapidly produced, which does not produce any rise in reflection intensity. Similarly, Fig. 6c shows that the reflection peaks of S3 (core 89 nm, shell 12 nm) undergo a blue shift from 646 to 453 nm. Because of the greater number of defects present in the particle array structures of S3, its reflection intensity is lower than that of S2. In particular, all particles have a high surface charge, which leads to particles being driven with low voltage, and this reduces the voltage demand by at least 1 V as compared with the previous Fe3O4@SiO2 report.12,31
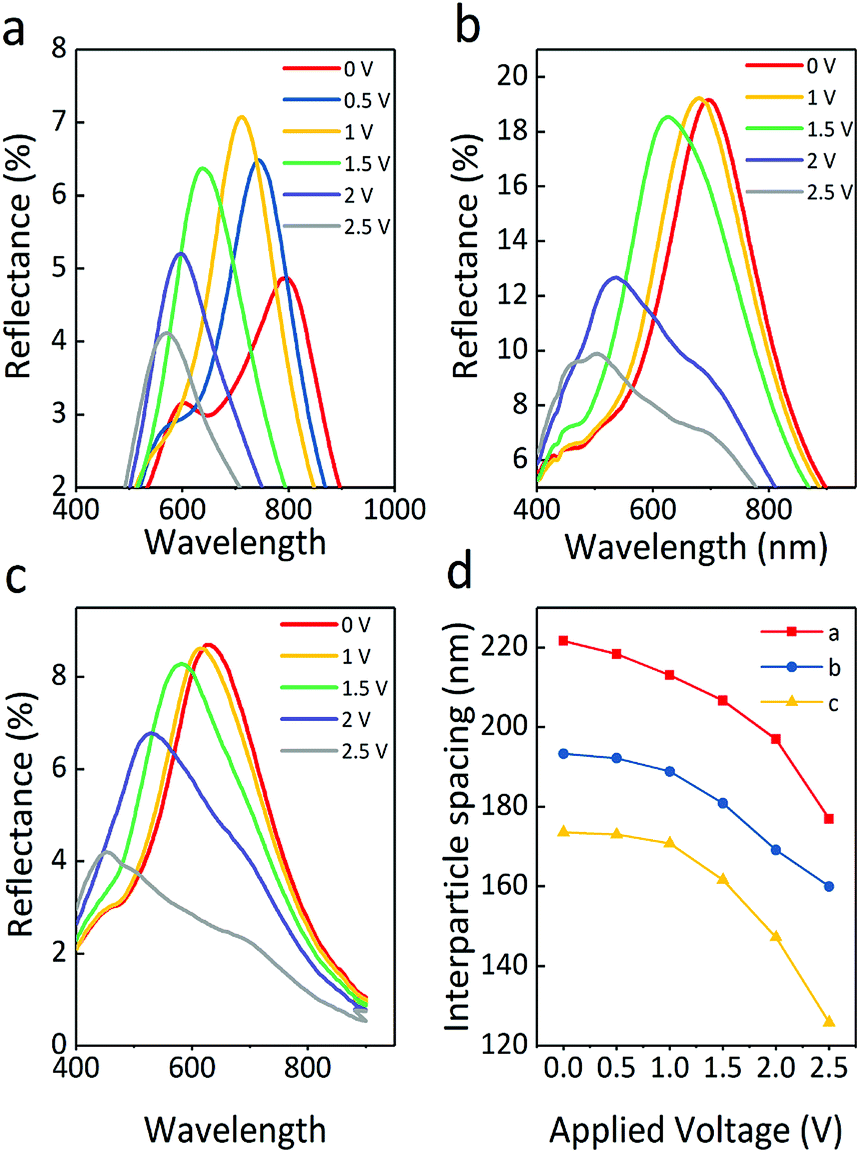 | ||
| Fig. 6 Reflection spectra with changing voltage for the Fe3O4@SiO2 colloid with particle sizes as follows: (a) S1, core 165 nm, shell 12 nm; (b) S2, core 121 nm, shell 12 nm; (c) S3, core 89 nm, shell 12 nm. (d) Plots of interparticle spacing obtained from Bragg's law, calculated from S1, S2, and S3 with voltage ranging from 0 to 2.5 V. | ||
It is worth noting that an increasing electric field causes a stronger electrostatic repulsion among the surfaces of the Fe3O4 particles, which is essential to form a balance between the electrostatic repulsion and the electrophoretic force. Without a magnetic field, dipole forces are absent because of the superparamagnetism of particles, which means the balance between the forces is mainly determined by the electrostatic repulsion and the electrophoretic force. The reflection intensity of structural color materials is positively related to the periodic number of ordered structures. The suitable particle size of S2 is beneficial to form more ordered structures as compared to the others. When the incident light propagates into the display cell, a large coherent periodic number of ordered structures interact with the incident light, leading to the highest reflectance. As the smallest particles, there is a larger space for S3 to move in the applied electric field, which results in the largest shift width of colors. However, the particles with the smallest size will reduce the reflection intensity due to partial disorder resulting from the inhomogeneous spacing between the periodic numbers of the ordered structures. On summarizing the optical properties of the three samples, it was found that the total reflection intensity of sample S2 was the strongest, and the blue-shift range of S3 was the widest.
Moreover, when the particle size decreased, the range of the reflection wavelengths showed a blue-shift. The average refractive index was calculated using Gladstone and Dale's law:32
| navg = npϕp + nm(1 − ϕp) | (1) |
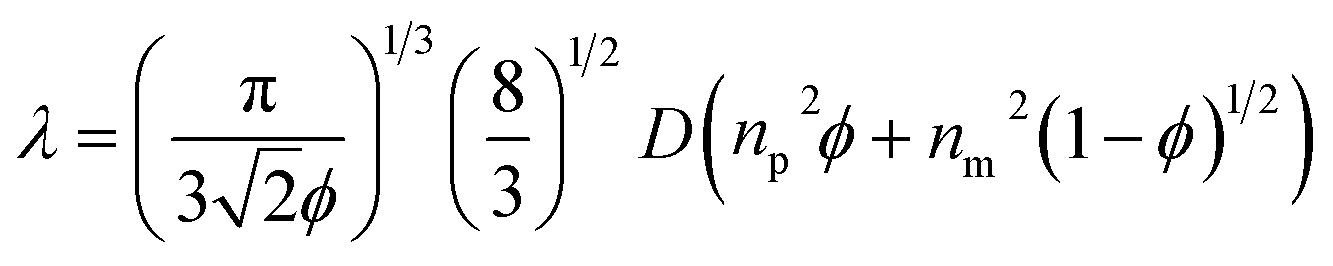 | (2) |
The particle volume fraction can remarkably influence the structural color, even when the voltage is not applied to the particles. The superparamagnetism of the Fe3O4 core prevents particles from attractive dipole–dipole aggregation in the absence of an electric field. No significant changes in color were observed for at least six months in the sealed glass bottle. The reflection peaks of the colloids shift from 727 to 468 nm with increasing particle volume fraction (Fig. 7a), which is another significant parameter that drives the assembly of the sample, since the various volume ratios of particles to PC can change the particle distance of the colloidal array structure. At the lower volume fraction, the Fe3O4@SiO2 colloid is a Brownian fluid that shows the native coffee color. Once the concentration exceeds the critical volume fraction (7.8%), the colloidal Fe3O4@SiO2 particle arrays are driven by electrostatic interactions. The highly negatively charged surface provides the Fe3O4@SiO2 particles with stronger electrostatic interaction, leading to the display of various colors that can be obtained by decreasing the particle volume fraction, including blue, green, yellow, red, and black (Fig. 7b). Because a sufficient surface charge can maintain the particle–particle spacing within a narrow range to reflect short wavelengths, our samples without applied voltage had a variety of basic colors, as reported previously.12 As the volume fraction decreases, increasing interparticle separation results in a red-shift of λ. The lower volume fraction results in broad particle spacing, which leads to a red shift in the reflection peak. The reflection bandwidth of the colloid without applied voltage is broader (FWHM = 150–200 nm) than in other highly ordered colloidal arrays33–36 (FWHM < 50 nm), thanks to the high contrast in refractive index of particles, producing a relatively broad reflection spectrum. Previous reports suggest that the refractive index contrast between Fe3O4@SiO2 particles and PC influences the bandwidth.37,38
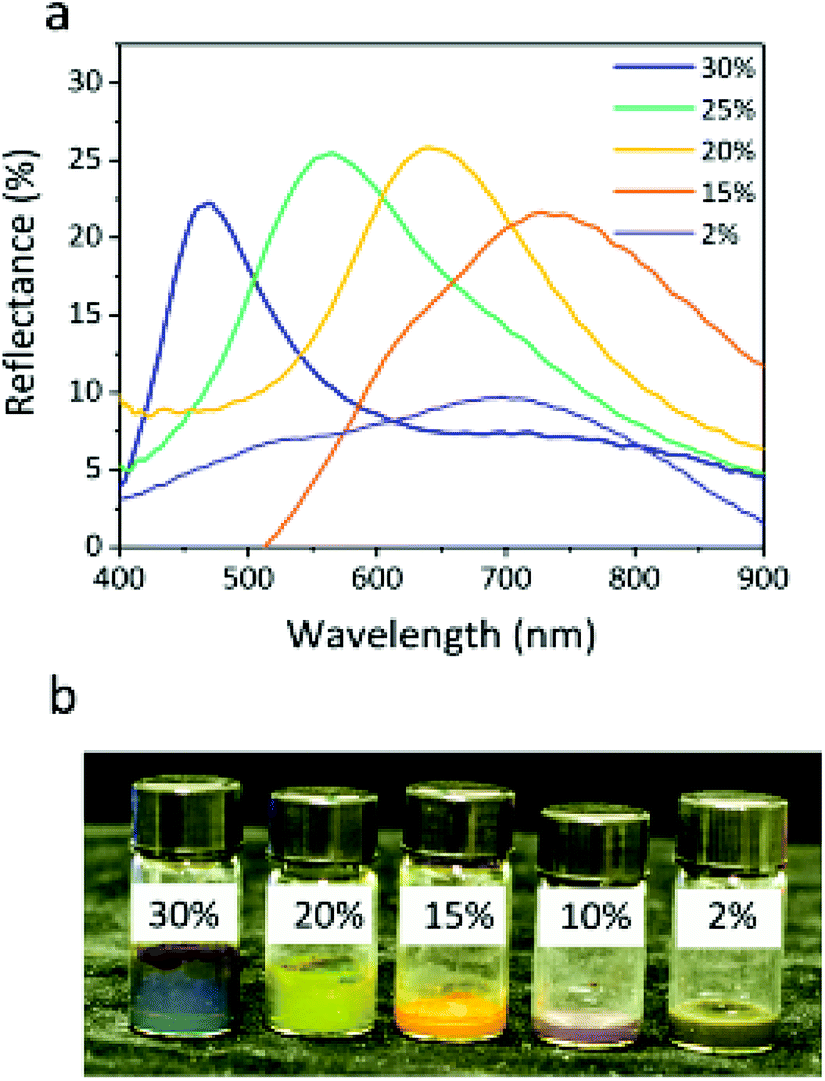 | ||
| Fig. 7 Reflection spectra (a) and photograph (b) of the Fe3O4@SiO2 particles (core 121 nm, shell 12 nm) without applied voltage at different particle volume fractions (30%, 20%, 15%, 10%, and 2%). | ||
The reflection spectra of four colloids with volume fractions of 10%, 15%, 20%, and 30% under various voltages with a particle size fixed at 145 nm (core 121 nm, shell 12 nm) are presented in Fig. 8a–d, respectively. All reflection spectra show a blue shift as the applied voltage increases from 0 to 3 V, but the range of the shift of the reflection spectra tends to be narrow. In Fig. 8a, the reflection has a broad shift range of approximately 230 nm from 680 to 450 nm. When the volume fraction is 10% and 15%, the reflection still covers visible wavelengths (Fig. 8b). However, Fig. 8c and d show a narrow shift range for reflection, suggesting that the sample fails to display a variety of colors. Due to the decreasing distance between particles, the blue shift range of reflection wavelength for these colloids gradually decreases from 100 nm to 50 nm. It is worth noting that the increase in volume fraction leads to an increase in reflection intensity at the same voltage. This is mainly attributed to the additional layers of colloidal amorphous arrays generated by a large number of particles. A volume fraction ranging from 10% to 15% is chosen as the optimal concentration for colloids. The colloids with volume fraction higher than 15% would not be chosen as building blocks because they produce a narrower reflection wavelength range. The above results suggest that the particle volume fraction has an impact on the structural color of colloids.
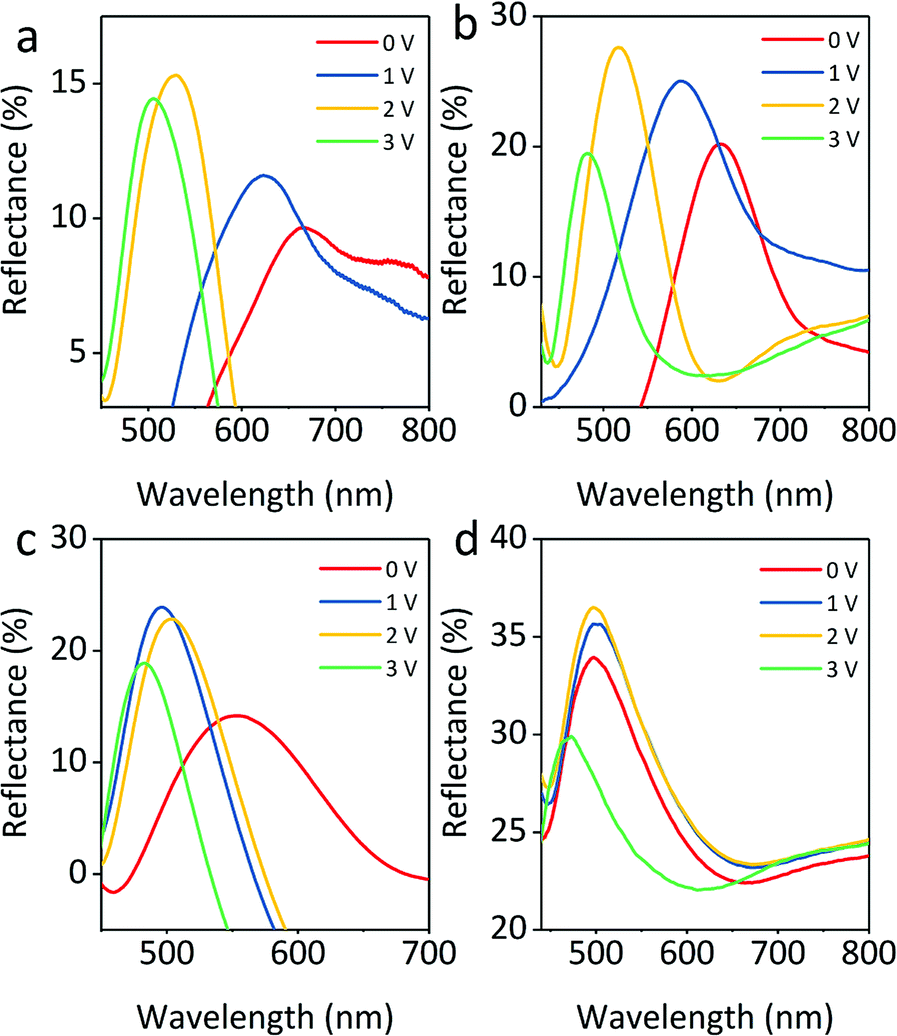 | ||
| Fig. 8 Blue-shift of reflection peaks for 145 nm particles with volume fractions of (a) 10%, (b) 15%, (c) 20%, and (d) 30% when the electric field increases from 0 to 4 V. | ||
As shown in the SEM images (Fig. 9a), the particle arrays show spatial periodicity in short-range order. Meanwhile, the average interparticle spacing narrows with a decrease in the particle size. The 2D Fourier power spectra were used to examine the SEM spatial arrangement. Prum et al.39 investigated the spatial variation in the refractive index of animal fur by the 2D FFT method. Takeoka et al.40 first evaluated artificial amorphous arrays by observing the SEM image of submicrometer-sized silica particles. The diffraction pattern provided by 2D Fourier power spectra shows that all three samples have circular diffraction rings (Fig. 9b), which suggests that the particle arrays are composed of identical nanostructures in every direction with respect to visible wavelengths and results in the scattering of coherent light. The periodicity of the spatial variation in the refractive index is equivalent in every direction in the plane within the colloids, which explains the optical properties of non-iridescent structural colors. These phenomena are significantly similar to those investigated for colloidal amorphous arrays in animal fur and artificial materials.41–44 Also, the optical properties of colloidal amorphous arrays have been proven in theoretical treatments. Jin reported that short-range order in 2D amorphous photonic structures led to almost unchanged spectral peaks using the multiple-scattering method.45 The peak of the angle-resolved specular reflection from the S1 sample appeared at nearly the same reflection peak when the detection angle was changed from 0° to 60° (Fig. 9c). Colloidal amorphous arrays are equivalently nanostructured in all directions so that the peaks of the angle-resolved specular reflection have no significant shifts. This demonstrates that the structural colors of the display cells with normal incidence of white light are nearly angle-independent, or non-iridescent, and samples S2 and S3 had similar results. The peak of S3 observed at 0° was sharper as compared to others, but the peak position was almost the same. Sample S3, with small particles and high volume fraction, had some defects in the colloidal amorphous arrays, and its reflection peak had a wider bandwidth. These results suggest that non-iridescence does not change with the size and volume fraction of the particles.
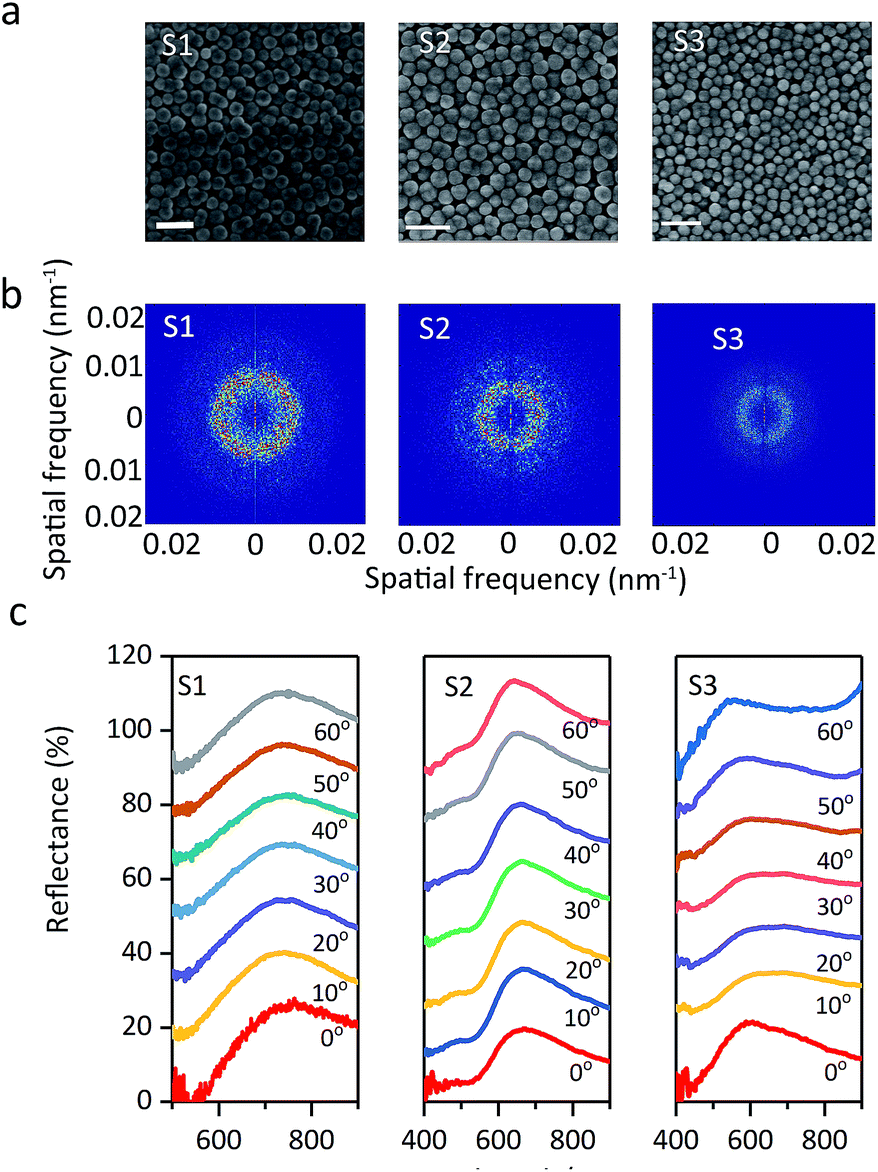 | ||
| Fig. 9 (a) SEM images, (b) Fourier power spectra, (c) angle-resolved specular reflection of S1, S2, and S3. The scale bar is 0.5 μm. | ||
Conclusions
We have demonstrated the gram-scale preparation of Fe3O4@SiO2 particles by selecting Na3Cit as the modifier with a binary solvent, and they were assembled into colloidal amorphous arrays with unique and attractive optical properties for EPD. As a result of their well-controlled size and adequate negative surface charges, Fe3O4@SiO2 particles promptly assembled into colloidal amorphous arrays and exhibited vivid structural colors at low voltages ranging from 0.5 V to 2.5 V, covering the whole visible spectrum from 802 to 453 nm. The particle volume fraction also has an influence on the display of the structural color of Fe3O4 colloids by regulating the interparticle distance with or without an electric field. Moreover, the colloidal amorphous arrays with short-range order help the full-color EPD to show non-iridescent colors. The effective and efficient synthesis of Fe3O4@SiO2 particles on the large-scale for full-color EPD will facilitate their ready integration into optical devices that modulate electromagnetic wave diffraction in industrial applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project was supported by the Natural Science Foundation of China (Grant No. 61775242 and 61376014), the Key Program of Natural Science Foundation of China (No. 61835013), SPRPCAS under grants no. XDB01020300, XDB21030300, the National Key R&D Program of China under grants no. 2016YFA0301500. L. Zhuang thanks Prof. Prum for providing the Fourier tool.References
- A. C. Arsenault, D. P. Puzzo, I. Manners and G. A. Ozin, Nat. Photonics, 2007, 1, 468 CrossRef CAS.
- H. Shim, C. Gyun Shin, C. Heo, S. Jeon, H. Jin, J. Woo Kim, Y. Jin, S. Lee, J. Lim, M. Gyu Han and J. Lee, Appl. Phys. Lett., 2014, 104, 51104 CrossRef.
- K. Chen, Q. Fu, S. Ye and J. Ge, Adv. Funct. Mater., 2017, 27, 1702825 CrossRef.
- A. A. Shah, M. Ganesan, J. Jocz and M. J. Solomon, ACS Nano, 2014, 8, 8095–8103 CrossRef CAS.
- L. Nucara, F. Greco and V. Mattoli, J. Mater. Chem. C, 2015, 3, 8449–8467 RSC.
- M. G. Han, C. J. Heo, G. S. Chang, H. S. Shim, J. W. Kim, Y. W. Jin and S. Y. Lee, J. Mater. Chem. C, 2013, 1, 5791–5798 RSC.
- J. J. Walish, Y. Kang, R. A. Mickiewicz and E. L. Thomas, Adv. Mater., 2009, 21, 3078–3081 CrossRef CAS.
- T. Kuno, Y. Matsumura, K. Nakabayashi and M. Atobe, Angew. Chem., 2016, 55, 2503–2506 CrossRef CAS.
- U. Jeong and Y. Xia, Angew. Chem., 2005, 44, 3099–3103 CrossRef CAS.
- F. Wang, X. Zhang, Y. Lin, L. Wang and J. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 5009–5016 CrossRef CAS PubMed.
- Y. Luo, J. Zhang, A. Sun, C. Chu, S. Zhou, J. Guo, T. Chen and G. Xu, J. Mater. Chem. C, 2014, 2(11), 1990–1994 RSC.
- I. Lee, D. Kim, J. Kal, H. Baek, D. Kwak, D. Go, E. Kim, C. Kang, J. Chung, Y. Jang, S. Ji, J. Joo and Y. Kang, Adv. Mater., 2010, 22, 4973–4977 CrossRef CAS.
- J. Ge, Y. Hu, M. Biasini, W. P. Beyermann and Y. Yin, Angew. Chem., 2007, 46, 4342–4345 CrossRef CAS.
- Y. Dong, B. Wen, Y. Chen, P. Cao and C. Zhang, RSC Adv., 2016, 6, 64434–64440 RSC.
- M. Chen, L. L. Shao, J. J. Li, W. J. Pei, M. K. Chen and X. H. Xie, RSC Adv., 2016, 6, 35228–35238 RSC.
- I. Rosapardo, M. Roigpons, A. A. Heredia, J. V. Usagre, A. Ribera, R. E. Galian and J. Pérezprieto, Nanoscale, 2017, 9, 10388–10396 RSC.
- W. Luo, H. Ma, F. Mou, M. Zhu, J. Yan and J. Guan, Adv. Mater., 2014, 26, 1058–1064 CrossRef CAS.
- J. Gao, X. Ran, C. Shi, H. Cheng, T. Cheng and Y. Su, Nanoscale, 2013, 5, 7026 RSC.
- H. Ma, K. Tang, W. Luo, L. Ma and Q. Cui, Nanoscale, 2017, 3105–3113 RSC.
- J. Liang, H. Ma, W. Luo and S. Wang, Mater. Chem. Phys., 2013, 139, 383–388 CrossRef CAS.
- F. Dong, W. Guo, J. Bae, S. Kim and C. Ha, Chem.–Eur. J., 2011, 17, 12802–12808 CrossRef CAS.
- A. Dikhtiarenko, S. A. Khainakov, J. R. García, J. Gimeno, I. de Pedro, J. R. Fernández and J. A. Blanco, J. Alloys Compd., 2012, 536, S437–S440 CrossRef CAS.
- H. Kochkar, M. Aouine, A. Ghorbel and G. Berhault, J. Phys. Chem. C, 2011, 115, 11364–11373 CrossRef CAS.
- C. Cheng, Y. Wen, X. Xu and H. Gu, J. Mater. Chem., 2009, 19, 8782–8788 RSC.
- F. Dong, W. Guo, J. Bae, S. Kim and C. Ha, Chem.–Eur. J., 2011, 17, 12802–12808 CrossRef CAS.
- H. Deng, X. Li, Q. Peng, X. Wang, J. Chen and Y. Li, Angew. Chem., 2005, 117, 2842–2845 CrossRef.
- H. Wang, Y. Sun, Q. Chen, Y. Yu and K. Cheng, Dalton Trans., 2010, 39, 9565 RSC.
- J. Wang, S. Zheng, Y. Shao, J. Liu, Z. Xu and D. Zhu, J. Colloid Interface Sci., 2010, 349, 293–299 CrossRef CAS PubMed.
- Y. Hu, L. He and Y. Yin, Small, 2012, 8, 3795–3799 CrossRef CAS PubMed.
- T. S. Shim, S. Kim, J. Y. Sim, J. Lim and S. Yang, Adv. Mater., 2010, 22, 4494–4498 CrossRef CAS.
- S. Lee, J. Y. Kim, S. Cheon, S. Kim, D. Kim and H. Ryu, RSC Adv., 2017, 7, 6988–6993 RSC.
- G. S. Zhang and Z. Q. Huang, Opt. Express, 2010, 18, 13361 CrossRef CAS PubMed.
- X. Xu, G. Friedman, K. D. Humfeld, S. A. Majetich and S. A. Asher, Chem. Mater., 2002, 14, 1249–1256 CrossRef CAS.
- K. Ueno, A. Inaba, Y. Sano, M. Kondoh and M. Watanabe, Chem. Commun., 2009, 24, 3603 RSC.
- W. Wang, B. Gu, L. Liang and W. Hamilton, J. Phys. Chem. B, 2003, 107, 3400–3404 CrossRef CAS.
- D. Go, D. Rommel, L. Chen, F. Shi, J. Sprakel and A. J. C. Kuehne, Langmuir, 2017, 33, 2011–2016 CrossRef CAS.
- L. He, V. Malik, M. Wang, Y. Hu, F. E. Anson and Y. Yin, Nanoscale, 2012, 4, 4438–4442 RSC.
- M. G. Han, C. G. Shin, S. Jeon, H. Shim, C. Heo, H. Jin, J. W. Kim and S. Lee, Adv. Mater., 2012, 24, 6438–6444 CrossRef CAS.
- R. O. Prum and R. H. Torres, Integr. Comp. Biol., 2003, 43, 591–602 CrossRef.
- Y. Takeoka, S. Yoshioka, A. Takano, S. Arai, K. Nueangnoraj, H. Nishihara, M. Teshima, Y. Ohtsuka and T. Seki, Angew. Chem., 2013, 52, 7261–7265 CrossRef CAS.
- J. Teyssier, S. V. Saenko, D. Van Der Marel and M. C. Milinkovitch, Nat. Commun., 2015, 6, 6368 CrossRef CAS.
- V. Welch, J. P. Vigneron, V. Lousse and A. Parker, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2006, 73, 41916 CrossRef.
- P. Simonis and J. P. Vigneron, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 11908 CrossRef PubMed.
- M. Iwata, M. Teshima, T. Seki, S. Yoshioka and Y. Takeoka, Adv. Mater., 2017, 29, 1605050 CrossRef PubMed.
- C. Jin, X. Meng, B. Cheng, Z. Li and D. Zhang, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 195107 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08352e |
| This journal is © The Royal Society of Chemistry 2019 |