DOI:
10.1039/C8RA08603F
(Paper)
RSC Adv., 2019,
9, 1895-1902
Electropolymerization of thienyl tethered comonomers and application towards the electrocatalytic reduction of nitrobenzene†
Received
17th October 2018
, Accepted 5th January 2019
First published on 15th January 2019
Abstract
The synthesis of different π-spacered thiophene comonomers via Suzuki cross-coupling in good synthetic yields was accomplished. Potentiodynamic electropolymerization of these precursors on ITO electrode by constant potential electrolysis results in the deposition of thin films of polymers between 0.05 and 0.2 μM. Interestingly, the as synthesized π-conjugated polymers exhibit electrochromic behaviour upon electrochemical oxidation. On the application side, the synthesized electropolymers showed catalytic activity better than glassy carbon towards electrochemical reduction of nitrobenzene.
Introduction
Considerable interest lies in conjugated polymers due to their unique properties such as planarity, semi-conductivity, optical band gap and so on.1 These properties in turn influence their potential applications in organic field effect transistors, actuators, organic light-emitting diodes, organic photovoltaics, etc.2 Molecular engineering allows precise tuning of the band gap by proper introduction of functionalities at the side chains.3 In addition, doping of external impurities with π-conjugated polymers is shown to enhance the electronic conductivity.4 Chemical method is quite often used to prepare polymer on large scale, but to study the electronic conductivity and/or electrochromic behaviour, deposition of polymer into a conducting matrix becomes necessary.5 In this regard, electrochemical polymerisation technique offer advantages such as synthesis under mild conditions, deposition of polymer directly onto the conducting substrate (electrode) and consequent electrochemical characterization of the deposited polymer.6 Electrochemical polymerization is frequently used for the synthesis of conducting polymers which show electrochromic behaviour. Thiophene and their derivatives were extensively investigated as monomers for electropolymerization over conducting substrates.7 Because of the electron rich C2-position, thiophene under electrochemical conditions undergoes a facile one electron oxidation to give highly reactive thiophene radical which is highly prone to polymerisation. Generally, electropolymerization is highly influenced by the solvent media and supporting electrolyte.8 Polymers electrosynthesized in different media or supporting electrolyte are shown to have different morphology.9 In some cases, supporting electrolytes are used as doping agents as well. Researchers have used this strategy to prepare thiophene polymers, copolymers and doped polymers to fine-tune their electronic properties.10 In this context, synthesis of thienyl based hybrid comonomers which are prone to undergo oxidative polymerization becomes essential as it would offer new electrochromic material with varying degree of physicochemical characteristics.11 As part of our research interest on small and macromolecular functional organic materials,12 we herein disclose the synthesis of thienyl end-capped hybrid monomers and their electropolymerization. Results pertaining to photophysical/electrochemical properties of the as prepared polymers, their surface morphology and application in catalytic reduction of nitroarenes are presented in this paper.
Results and discussion
Monomers synthesis and electropolymerization
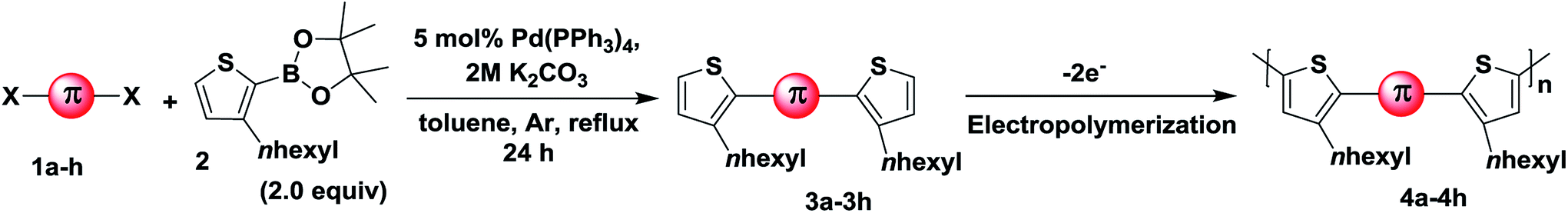
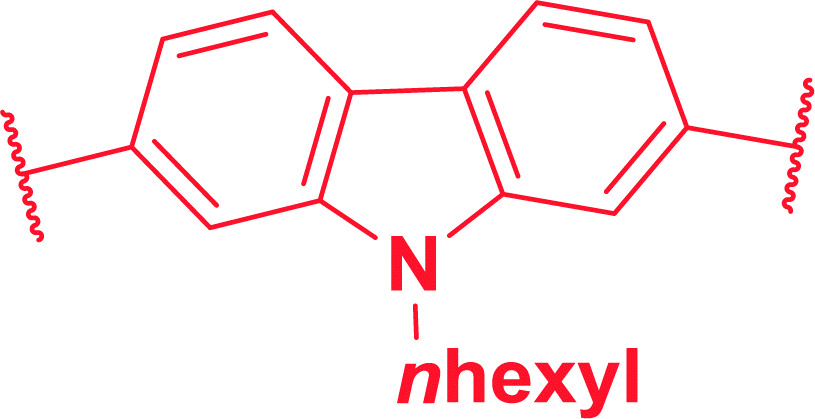
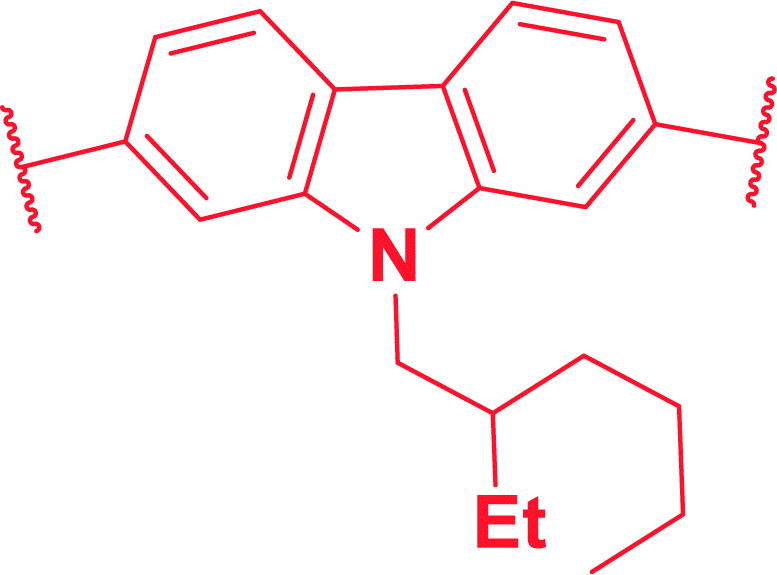
We initiated the synthesis of monomers by subjecting several di-halo aryls/heteroaryls (1a–f) with 3-hexylthiophene-2-boronic acid pinacol ester (2) under typical Suzuki–Miyaura conditions (Scheme 1). As provided in Table 1, all substrates essentially undergo cross-coupling at same conditions to afford good to excellent yields of 3-hexylthiophene end-capped monomers (3a–h). Keeping in mind that π-conjugated polymers possessing carbazole functionalized at the N-position exhibits unique electrochromic properties,13 we initially prepared three carbazole based monomers (3a, 3b and 3f). This was followed by the synthesis of monomers with aromatic spacers such as azulenyl, phenyl and biphenyl (3c–e). Finally, precursors having fused thiophene systems were (3g and 3h) prepared under identical reaction conditions. The presence of two hexyl chains in all monomers enhanced their solubility even in non-polar solvents. All synthesized monomers were purified by flash column chromatography in good to excellent yields and characterized by FTIR, 1H-NMR, 13C-NMR spectroscopy.
 |
| | Scheme 1 Suzuki coupling of dihalo-π-spacers 1a–h with 3-hexylthiophene boronic acid 2. | |
Table 1 Synthesis and chemical yield of monomers 3a–f
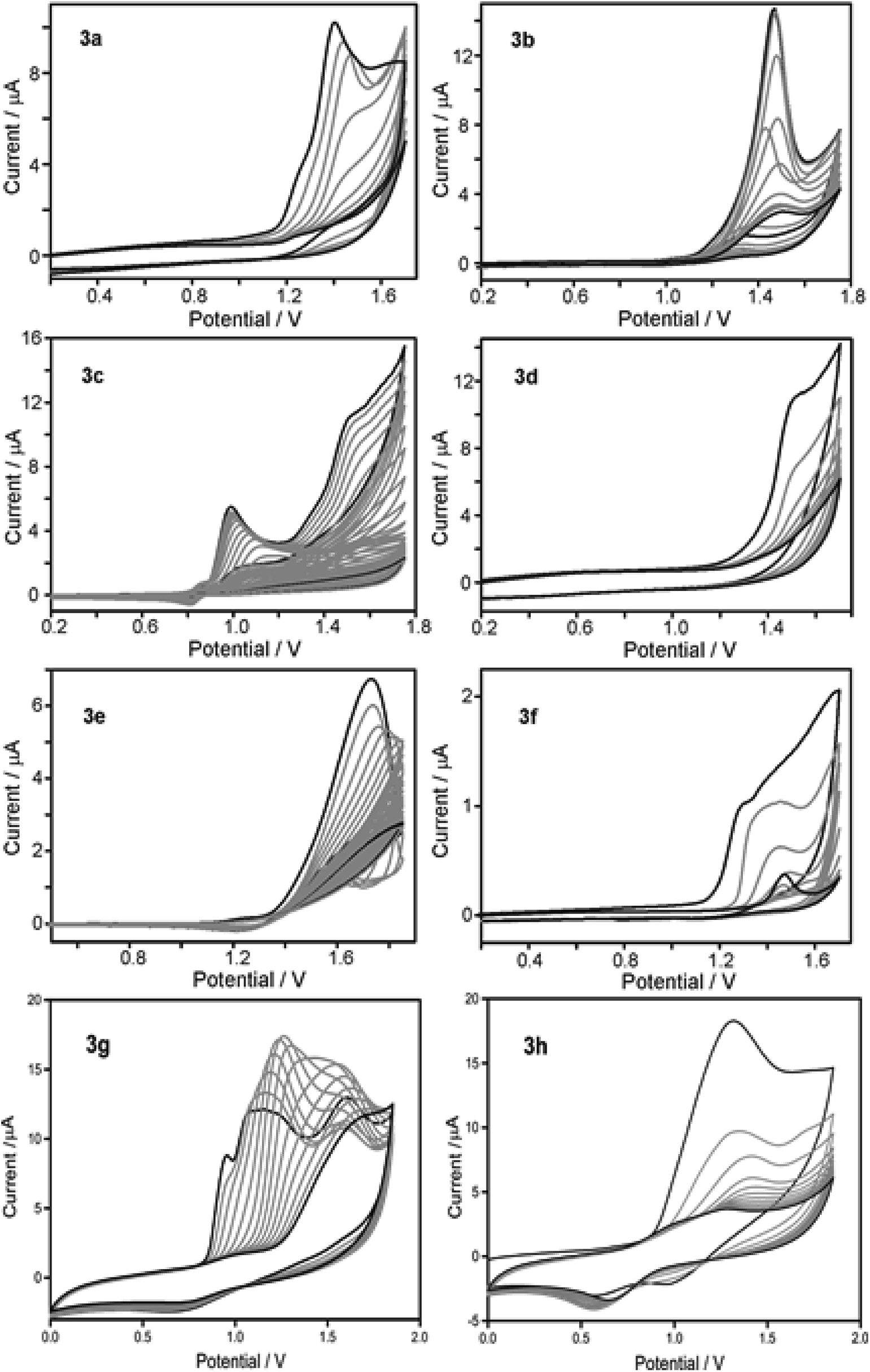
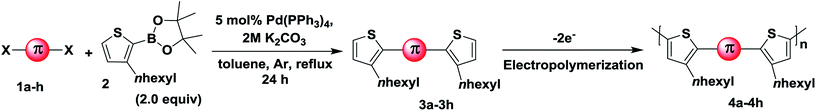
Having obtained the monomers in satisfactory yields, we next intended the electrochemical polymerisation of the prepared monomers. It is to be remembered that electrochemical strategy allows for the preparation of uniform thin films of thiophene/substituted-thiophene polymers on conducting substrates (as working electrode in monomer/electrolyte mixtures), the use of any catalysts can be circumvented. To this end, substituted thiophene polymers were synthesized by employing electrochemical oxidation method as shown in Scheme 1. The π spacer includes carbazole having alkyl chains such as n-hexyl (4a), 2-ethylhexyl (4b) n-octyl (4f) in addition to azulene (4c), phenyl (4d), biphenyl (4e), thienothiophene (4g) and dithienothiophene (4h) units. Since the monomer contains aromatic/heteroaromatic units, upon polymerisation it is expected to give rigid linear polymer which in turn would render poor solubility. To overcome this, we have pre-introduced hexyl units on C3-position of thiophene adjacent to the π-spacer. Despite these attempts, we obtained thin films of polymer on ITO with poor solubility. Initially, the monomers were characterized by cyclic voltammetry (Fig. S1†). Based on the oxidation potential obtained, multiscan CV was recorded on glassy carbon electrode (Fig. 1). In addition, constant potential electrolysis was carried out on ITO electrode at a fixed potential which is usually +200 mV higher than that of their corresponding oxidation potential. After obtaining uniform polymer films, the ITO electrode was rinsed with acetonitrile to remove any unreacted monomer impurities. The electrochemical characterisation of the polymer thin films was then carried out in nBu4PF6 (0.1 M) in dichloromethane, which revealed the possible electrochromic features of these samples (Fig. S2†). All the polymer samples showed electrochromic behaviour when electrochemically oxidized. The colour intensity of the polymers varied which is a consequence of differences in their π-substituent. When the polymer films are electrochemically reduced, the colour once again disappeared revealing their electrochromic nature. Moreover, a significant increase in the C–C stretching and decrease in the C–H stretching bands compared to the respective monomers clearly suggests the formation of polymers (Fig. S3†).
 |
| | Fig. 1 Potentiodynamic electropolymerisation of 3a–h against Ag/AgCl on GC electrode using nBu4NPF6 (0.1 M) in CH2Cl2. | |
Photophysical properties
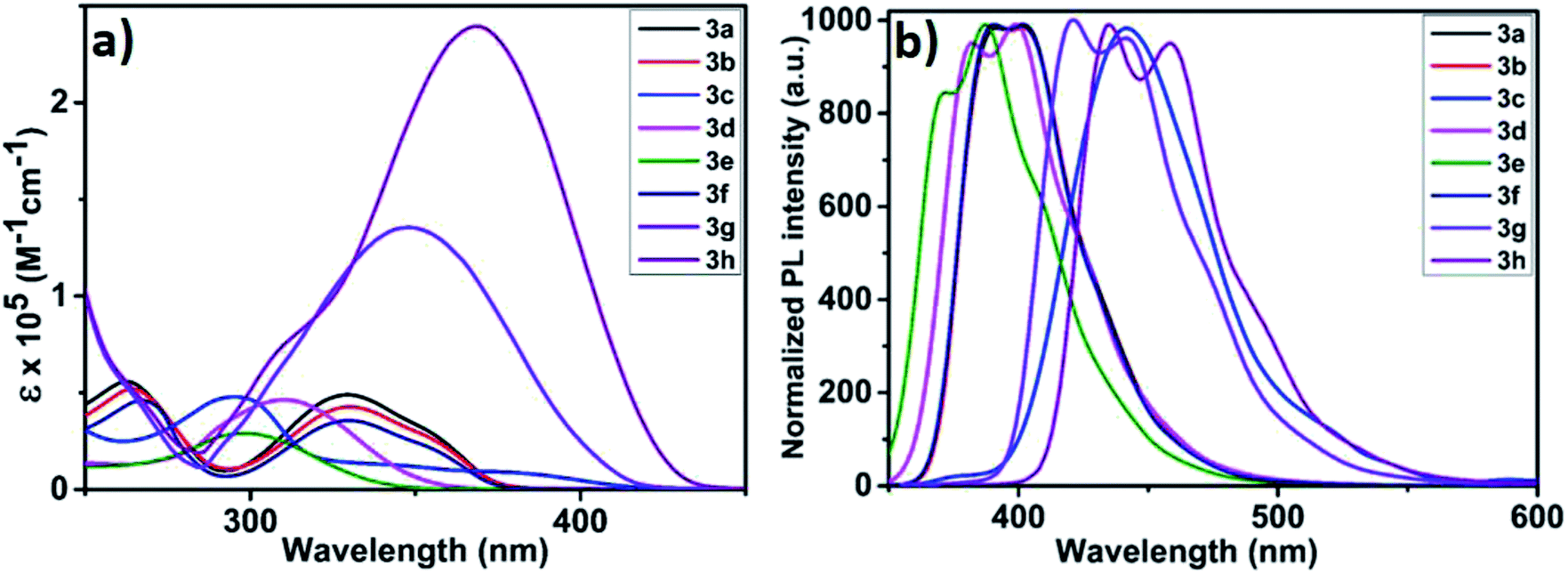
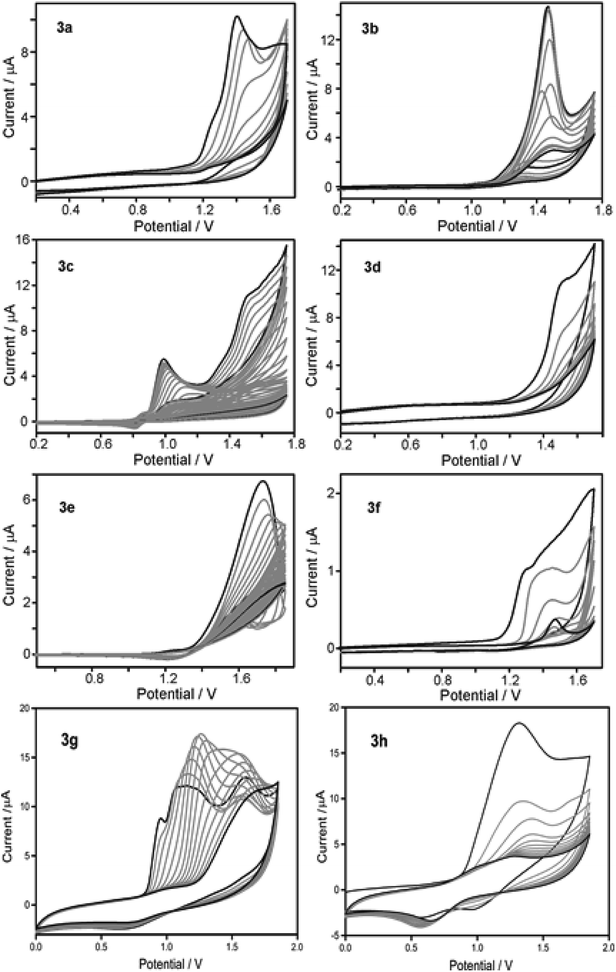
As depicted in Fig. 2, electronic absorption and emission spectra of all monomers (3a–h) were investigated as tetrahydrofuran solution (5 × 10−5 M) and the obtained optical data was disclosed in Table 2. At the outset, UV-vis absorption spectra of all monomers displayed π–π* transition between 295 and 329 nm with molar absorptivity (ε) ranging between 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 49200 M−1 cm−1 (Fig. 2a). Structurally similar carbazole monomers (3a, 3b and 3f) showed an identical peak maxima and optical absorption edge (λmax/λedge = 329/374 nm). The azulene containing monomer 3c showed a typical stretched wavelength (λedge = 417 nm) characteristic of thienyl-adjoined azulene system.14 For benzenoid systems, it was observed that those containing phenyl ring exhibited higher ε (46500 M−1 cm−1) and stretched wavelength (λmax/λedge = 309/352 nm). In comparison, the biphenyl counterpart 3e showed significant blue shift (λmax/λedge = 298/338 nm) with reduced ε (29000 M−1 cm−1). This distinctive behaviour can be attributed to the reduced conjugation effect offered by the different dihedral angle along the thiophene-phenyl and phenyl–phenyl plane.15 As expected, those monomers possessing fused thiophene units (3g and 3h) displayed fairly high ε values (136000 and 239900 M−1 cm−1) and reduced band gap (3.06 and 2.93 eV), which could be reasoned to their better planarity and extensive π-delocalization. To assess the solid state aggregation properties, thin film UV-vis spectra were recorded by drop casting THF solution of monomers on quartz plate (Fig. S4†). All monomers revealed a slightly reduced optical band gap (Eoptg = 2.35 to 3.55 eV) compared to their solution state spectra (Eoptg = 2.93 to 3.66 eV), which might be due to the likely J-aggregation in solid state. In comparison, the UV-vis spectra of polymers 4a–h (Fig. S5†) recorded in THF:CH2Cl2 (1:1) exhibit an extended wavelength by showing band gap (Eoptg = 2.13 to 2.86 eV) much lesser than the respective monomers (Eoptg = 2.97 to 3.66 eV). This reduced band gap clearly indicates the possibility of increased π-conjugation as in the case of semiconducting polymers by extending the optical absorption edge. On the other hand, photoluminescence spectra of monomers as THF solution (5 × 10−5 M) showed emission maxima (λem) between 387 and 458 nm with significant Stokes shift ranging from 5300 to 11300 cm−1 with the highest being for 3c (Fig. 2b). It is pertinent to mention that 3c showed a very weak S1–S0 fluorescence, hence its peak was normalized manifold.16 Likewise, the emission spectra of polymers suspended in a 1:1 mixture of THF:CH2Cl2 were recorded (Fig. S6†). The polymers displayed batho/hypsochromic shift in their emission spectra with λem varying from 387 to 546 nm. This can be reasoned to the varying degree of π–π interactions of the polymer backbone resulting in low/high energy emissions. For instance, polymers 4a, 4c, 4g and 4h exhibited Stokes shift of 550, 2000, 5200 and 5000 cm−1 respectively (Table S1†). Whereas, all other polymers showed substantial anti-Stokes shift (4b = 2900; 4d = 3500; 4e = 4100 and 4f = 3200 cm−1). This irregular energy dissipation of these polymers can be attributed to the different relaxation delay to the lowest vibrational level of the first excited state (S1).
000 to 49200 M−1 cm−1 (Fig. 2a). Structurally similar carbazole monomers (3a, 3b and 3f) showed an identical peak maxima and optical absorption edge (λmax/λedge = 329/374 nm). The azulene containing monomer 3c showed a typical stretched wavelength (λedge = 417 nm) characteristic of thienyl-adjoined azulene system.14 For benzenoid systems, it was observed that those containing phenyl ring exhibited higher ε (46500 M−1 cm−1) and stretched wavelength (λmax/λedge = 309/352 nm). In comparison, the biphenyl counterpart 3e showed significant blue shift (λmax/λedge = 298/338 nm) with reduced ε (29000 M−1 cm−1). This distinctive behaviour can be attributed to the reduced conjugation effect offered by the different dihedral angle along the thiophene-phenyl and phenyl–phenyl plane.15 As expected, those monomers possessing fused thiophene units (3g and 3h) displayed fairly high ε values (136000 and 239900 M−1 cm−1) and reduced band gap (3.06 and 2.93 eV), which could be reasoned to their better planarity and extensive π-delocalization. To assess the solid state aggregation properties, thin film UV-vis spectra were recorded by drop casting THF solution of monomers on quartz plate (Fig. S4†). All monomers revealed a slightly reduced optical band gap (Eoptg = 2.35 to 3.55 eV) compared to their solution state spectra (Eoptg = 2.93 to 3.66 eV), which might be due to the likely J-aggregation in solid state. In comparison, the UV-vis spectra of polymers 4a–h (Fig. S5†) recorded in THF:CH2Cl2 (1:1) exhibit an extended wavelength by showing band gap (Eoptg = 2.13 to 2.86 eV) much lesser than the respective monomers (Eoptg = 2.97 to 3.66 eV). This reduced band gap clearly indicates the possibility of increased π-conjugation as in the case of semiconducting polymers by extending the optical absorption edge. On the other hand, photoluminescence spectra of monomers as THF solution (5 × 10−5 M) showed emission maxima (λem) between 387 and 458 nm with significant Stokes shift ranging from 5300 to 11300 cm−1 with the highest being for 3c (Fig. 2b). It is pertinent to mention that 3c showed a very weak S1–S0 fluorescence, hence its peak was normalized manifold.16 Likewise, the emission spectra of polymers suspended in a 1:1 mixture of THF:CH2Cl2 were recorded (Fig. S6†). The polymers displayed batho/hypsochromic shift in their emission spectra with λem varying from 387 to 546 nm. This can be reasoned to the varying degree of π–π interactions of the polymer backbone resulting in low/high energy emissions. For instance, polymers 4a, 4c, 4g and 4h exhibited Stokes shift of 550, 2000, 5200 and 5000 cm−1 respectively (Table S1†). Whereas, all other polymers showed substantial anti-Stokes shift (4b = 2900; 4d = 3500; 4e = 4100 and 4f = 3200 cm−1). This irregular energy dissipation of these polymers can be attributed to the different relaxation delay to the lowest vibrational level of the first excited state (S1).
 |
| | Fig. 2 (a) UV-visible absorption spectra of 3a–h in THF solution (5 × 10−5 M); (b) normalized emission spectra of 3a–h in THF solution. | |
Table 2 Optical properties of monomers 3a–f
| Monomer |
λmaxa/λedge (nm) |
ε (M−1 cm−1) |
Eoptgb (eV) |
λmaxc/λedge (nm) |
Eoptgd (eV) |
λeme (nm) |
Stokesf shift (cm−1) |
Polymer |
λmax/λedge (nm) |
Eoptgb (eV) |
| Absorption in solution. Band gap in solution was determined from λedge using, Eoptg (eV) 1240/λedge. Absorption in thin films. Optical band gap was determined from λedge in thin solid films using, Eoptg (eV) = 1240/λedge. Emission in solution. Stokes shift was calculated from the difference between λmax and λem. |
| 3a |
329/374 |
49200 |
3.31 |
335/384 |
3.22 |
403 |
5600 |
4a |
461/579 |
2.14 |
| 3b |
329/374 |
42800 |
3.31 |
337/384 |
3.22 |
402 |
5500 |
4b |
459/572 |
2.16 |
| 3c |
295/417 |
48000 |
2.97 |
302/425 |
2.91 |
442 |
11300 |
4c |
373/433 |
2.86 |
| 3d |
309/352 |
46500 |
3.52 |
309/389 |
3.18 |
398 |
7200 |
4d |
462/577 |
2.14 |
| 3e |
298/338 |
29000 |
3.66 |
302/349 |
3.55 |
387 |
7700 |
4e |
460/581 |
2.13 |
| 3f |
329/374 |
35700 |
3.31 |
341/394 |
3.14 |
401 |
5500 |
4f |
462/574 |
2.16 |
| 3g |
348/405 |
136000 |
3.06 |
360/433 |
2.86 |
441 |
6060 |
4g |
412/510 |
2.43 |
| 3h |
368/422 |
239900 |
2.93 |
415/527 |
2.35 |
458 |
5300 |
4h |
429/534 |
2.32 |
Surface morphology and polymers thickness
The as synthesized electropolymers 4a–h were subjected to SEM analysis and all samples revealed different morphology and porosity (Fig. S7†). As evident from the SEM images, most of these polymer samples revealed porous network structure which could result from the subsequent in and out movement of electrolytes during the redox process. For instance, SEM of polymer 4a reveals that the polymer exhibits porous network structure with cauliflower like morphology. Polymer 4b reveals the micro-porosity and exhibits an accumulation state of clusters of random granules. Polymer 4c was obtained as a well-adhered polymer film whose surface was smooth and homogeneous. However, polymer 4d appeared to be irregularly shirked smooth surface with multiple pores on it. Polymer 4e exhibited a homogeneous and porous structure. At low resolution, it is evident that there is an accumulation of small globules with dense holes among the clusters. However, at higher resolution, plenty of tiny holes were observed on globular surfaces indicating the microporous nature of the sample. Polymer 4f revealed islands of globular lumps with irregularities whose sizes were in the range of 200–400 nm. Finally, polymers 4g and 4h showed well defined porous network and uneven microporous layer respectively. In continuation, surface morphology of the synthesized polymers was probed by atomic force microscopy (AFM) by measuring the thickness of deposited polymer on ITO electrode. The corresponding three dimensional topographic images and height profile analysis were depicted in Fig. S8.† Analysis of the images revealed that polymers derived from linear alkyl chain carbazole units showed maximum thickness (4a = ∼0.2 μm; 4f = ∼0.1 μm) compared to the branched chain analogue (4b = ∼0.05 μm). This could possibly due to the enhanced solubility of 4b in THF leading to weak deposition. In a similar vein, polymer with biphenyl backbone (4e = ∼0.2 μm) deposited well than the phenyl counterpart (4d = ∼0.1 μm). However, azulene polymer 4c showed uneven deposition with a thickness of ∼0.1 μm. In the case of thienyl polymers 4g and 4h, the thickness ranges from 0.15 to 0.22 μM. The thickness variation in the electrodeposited polymers may be because of the dissimilarities in the monomer concentration and solubility.
Electrochemical reduction of nitrobenzene
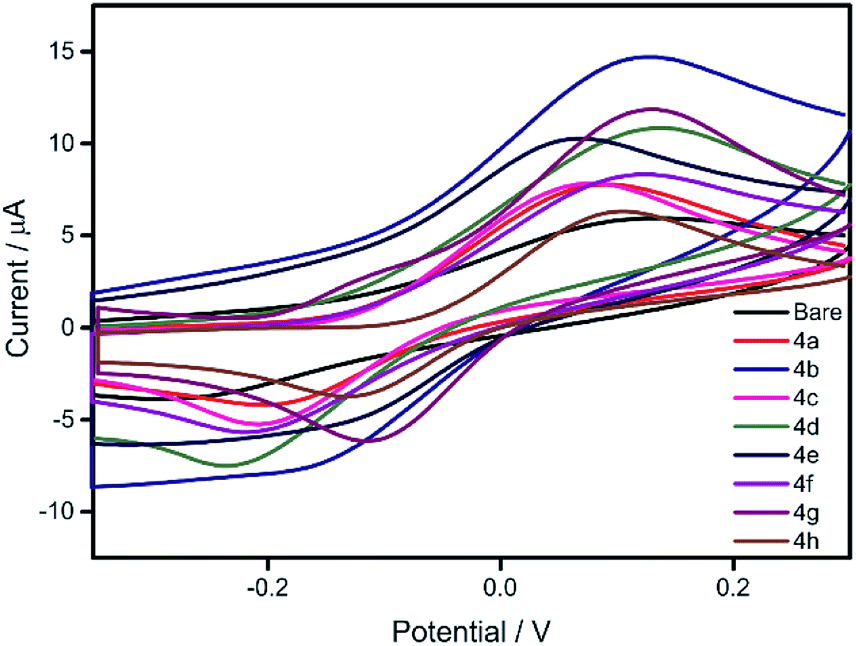
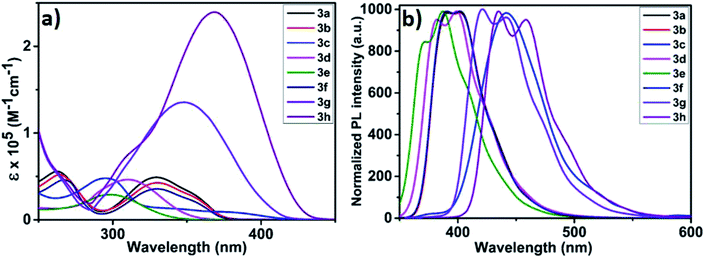
Reduction of nitrobenzene and its derivatives has industrial importance as it involves hazardous waste treatment.12e The reduction of nitrobenzene yields aniline derivatives which are mainly used as synthetic precursors for pharmaceutical products. The catalytic activities of the electro-polymers 4a–h were explored for the electrochemical reduction of nitrobenzene. These polymers are expected to show semi-conducting behaviour; therefore their conductivity is enhanced by doping with molecular iodine. Electrodes for nitrobenzene reduction were prepared by performing the electropolymerisation of monomers 3a–h in the presence of iodine (multiscan CV, 50 mV s−1, 10 cycles on GC electrode). The resulting polymer modified electrodes were then washed with distilled water and tested for catalytic activity towards the reduction of nitrobenzene in phosphate buffer solution (PBS). The results are compared with that obtained for bare GC electrode as shown in Fig. 3. It is worth mentioning that the polymer modified GC electrodes showed better onset potential and current intensity compared to bare GC, thus highlights their superior catalytic activity than GC electrodes. Among the tested electrodes, the one with polymer 4b exhibited excellent catalytic activity followed by 4d, 4g, 4e, 4f, 4c, 4a and 4h.
 |
| | Fig. 3 Reduction of nitrobenzene on GC and polymer modified GC electrodes in PBS. | |
In order to investigate the effect of scan rate on peak currents, CV was recorded at various scan rates using polymer (4g and 4h) modified GC electrode. The scan rate dependence of both cathodic reduction and anodic oxidation are shown in Fig. S9.† A linear variation was observed for potential scan rates from 20 to 250 mV s−1 signifying that the electroactive species is involved in the reaction.17 With an increase in scan rate, oxidation is shifted towards positive potential and the reduction towards negative potential which is indicative of the kinetic limitations of the reduction process. With an increase in scan rate, all peak currents varied linearly which signifies a typical diffusion-controlled electrochemical process. Further investigations on improving the conductivity and electrochemical performance by fine tuning with other dopants are actively underway.
Experimental section
Materials, methods and measurements
All commercially available reagents were used without further purification unless otherwise stated. Solutions in organic solvents were dried with anhydrous sodium sulphate. Solvents were evaporated under reduced pressure. Column chromatography was performed using thick-walled glass columns along with a mixture of petroleum ether and ethyl acetate on silica gel (100–200 mesh, SRL, India). The relative proportions of solvents in chromatography solvent mixtures refer to the volume to volume ratio. Analytical TLC was performed on precoated plastic sheets of silica gel G/UV-254 of 0.2 mm thickness (Macherey-Nagel, Germany) using analytical grade solvents and visualized with iodine spray (10% w/w I2 in silica gel), UV light (λ = 254 and 365 nm) and alkaline KMnO4 solution. Melting points were recorded on micro controller based melting point apparatus using open capillaries. Infrared (IR) spectra for all solid compounds were recorded on a Bruker Optik GmbH, TENSOR 27 FT-IR spectrophotometer as KBr pellets. 1H and 13C NMR spectra were obtained in benzene-d6 and CDCl3 on a BRUKER spectrometer at 400, 500 and 100, 125 MHz respectively. Proton chemical shifts (δ) are relative to tetramethylsilane (TMS, δ = 0.00) as internal standard and expressed in parts per million. The number of protons (n) for a given resonance was indicated as nH. Spin multiplicities are given as s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). Coupling constants (J) are given in hertz. Dihalo precursors are prepared following standard procedures18 and procured from Sigma-Aldrich. UV-visible spectra were measured as chloroform solution and thin film on SHIMADZU UV-3600 spectrophotometer. Cyclic voltammetry (CV) experiments were performed with Autolab potentiostat-galvanostat (Model PGSTAT-30). All CV measurements were carried out at room temperature with a three-electrode configuration employing glassy carbon as working electrode, silver/silver nitrate as reference electrode, and platinum as counter electrode. Degassed acetonitrile or dichloromethane was used as medium with tetrabutylammonium hexafluorophosphate (0.1 M) as the supporting electrolyte at a scan rate of 50 mV s−1. Atomic force microscopic images were captured using 5500 series, Agilent Technologies.
General experimental procedure for the synthesis of 3a–h
1.0 eq. of dihalo compound (1a–h) was dissolved in 25 mL of toluene. To this mixture, 2.2 eq. of 3-hexylthiopheneboronic acid pinacol ester (2) and 20 mL of 2 M K2CO3 solution were added one another in the interval of 10 min under argon medium. Then 5 mol% of (Ph3P)4Pd was added 1 hour deferred from the prerequisite addition and then refluxed for 24 h. The crude product was extracted with chloroform and water. The organic fraction was concentrated under reduced pressure and then chromatographed on silica column (100–200 mesh) using 5% EA/PE as an eluent. Pure monomer (3a–h) was obtained.
9-Hexyl-2,7-bis(3-hexylthiophen-2-yl)-9H-carbazole (3a). (2.41 g, 95%), bright yellow solid; mp 48–50 °C; Rf = 0.17 (PE); IR (KBr): ν = 2923, 2855, 1599, 1457, 1327, 1176, 854, 804, 724, 661 cm−1; 1H NMR (400 MHz, C6D6): δ = 0.77–0.90 (m, 9H), 0.99–1.11 (m, 6H), 1.16–1.31 (m, 12H), 1.49–1.56 (m, 2H), 1.60–1.68 (m, 4H), 2.80 (t, J = 8.0 Hz, 4H), 3.80 (t, J = 8.0 Hz, 2H), 6.92 (d, J = 4.0 Hz, 2H), 7.01 (d, J = 4.0 Hz, 2H), 7.48 (d, 1H), 7.50 (d, 1H), 7.51 (s, 2H), 7.95–7.97 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, C6D6): δ = 13.8, 13.9, 22.4, 22.6, 26.9, 28.8, 28.9, 31.2, 31.4, 31.7, 42.6, 109.7, 120.5, 121.1, 122.3, 123.6, 128.3, 129.4, 132.6, 138.5, 139.2, 141.2; HRMS m/z calculated for C38H49NS2 MW: 583.3306, found: 583.3304.
9-(2-Ethylhexyl)-2,7-bis(3-hexylthiophen-2-yl)-9H-carbazole (3b). (1.31 g, 94%), bright yellow semi solid, Rf = 0.12 (PE); IR (KBr): ν = 2926, 2859, 2400, 2312, 1737, 1602, 1456, 1327, 1247, 807, 720, 657 cm−1; 1H NMR (400 MHz, CDCl3): δ = 0.74–0.78 (m, 10H), 0.84 (t, J = 7.2 Hz, 4H), 1.16–1.16 (m, 13H), 1.32 (t, J = 7.2 Hz, 3H), 1.54–1.61 (m, 5H), 2.03 (t, J = 6 Hz, 1H), 2.67 (t, J = 7.6 Hz, 5H), 4.11 (dd, J1 = 2.0 Hz, J2 = 7.2 Hz, 2H), 6.95 (d, J = 5.2 Hz, 3H), 7.20 (s, 1H), 7.23 (d, J = 1.6 Hz, 1H), 7.25 (d, J = 1.6 Hz, 1H), 7.36 (d, J = 0.8 Hz, 2H), 8.01 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 9.9, 12.9, 13.0, 21.6, 22.0, 23.44, 27.7, 27.9, 28.3, 28.6, 30.1, 30.6, 38.4, 46.6, 108.8, 119.1, 119.8, 120.7, 122.5, 128.5, 131.2, 137.4, 137.9, 140.4; HRMS m/z calculated for C40H53NS2 MW: 611.3619, found: 611.3611.
1,3-Bis(3-hexylthiophen-2-yl)azulene (3c). (123.2 mg, 92%), dark blue semi solid; Rf = 0.23 (PE); IR (KBR): ν = 3058, 2925, 2856, 1732, 1570, 1456, 1401, 1220, 1088, 946, 870, 729, 652, 574 cm−1; 1H NMR (500 MHz, CDCl3) δ = 0.68 (t, J = 10.0 Hz, 6H), 1.02–1.17 (m, 12H), 1.43–1.49 (m, 4H), 2.47 (t, J = 10.0 Hz, 4H), 7.00 (d, J = 5 Hz, 4H), 7.05 (t, J = 10.0 Hz, 2H), 7.25 (d, J = 5.1 Hz, 2H), 7.49 (d, J = 10.0 Hz, 2H), 7.84 (s, 1H), 8.29 (d, J = 10.0 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ = 14.0, 22.6, 29.0, 29.0, 30.9, 31.64, 122.0, 123.7, 124.4, 129.0, 132.0, 136.5, 138.5, 138.9, 140.33, 140.5; HRMS m/z calculated for C30H36S2 MW: 460.2258, found: 460.2257.
4,4′-Bis(3-hexylthiophen-2-yl)-1,1′-biphenyl (3d). (534 mg, 89%), yellow solid; mp: 51–53 °C; Rf = 0.17 (PE); IR (KBR): ν = 3051, 2923, 2855, 1911, 1729, 1668, 1543, 1479, 1384, 1135, 1084, 947, 826, 688, 644, 531, 484 cm−1; 1H NMR (500 MHz, CDCl3) δ = 0.78 (t, J = 5.0 Hz, 6H), 1.19–1.27 (m, 12H), 1.52–1.58 (m, 4H), 2.62 (t, J = 5.0 Hz, 4H), 6.91 (d, J = 5.0 Hz, 2H), 7.15 (d, J = 5.0 Hz, 2H), 7.44 (d, J = 5.0 Hz, 4H), 7.59 (d, J = 5.0 Hz, 4H); 13C NMR (125 MHz, CDCl3) δ = 14.1, 22.6, 28.8, 29.2, 31.0, 31.7, 123.8, 127.0, 129.7, 129.8, 134.0, 137.4, 138.9, 139.4; HRMS m/z calculated for C32H38S2 MW: 486.2415, found: 486.2410.
1,4-Bis(3-hexylthiophen-2-yl)benzene (3e). (1.21 g, 97%), yellow semi solid; Rf = 0.33 (PE); IR (KBR): ν = 3063, 2927, 2858, 1550, 1454, 1373, 1093, 964, 836, 718, 654 cm−1; 1H NMR (500 MHz, CDCl3) δ = 0.92 (t, J = 5.0 Hz, 6H), 1.30–1.41 (m, 12H), 1.65–1.71 (m, 4H), 2.75 (t, J = 5.0 Hz, 4H), 7.05 (d, J = 5.0 Hz, 2H), 7.29 (d, J = 5.0 Hz, 2H), 7.52 (s, 4H); 13C NMR (125 MHz, CDCl3) δ = 14.3, 22.8, 28.9, 29.4, 31.2, 31.8, 123.9, 129.5, 129.7, 133.99, 137.5, 138.9; HRMS m/z calculated for C26H34S2 MW: 410.2102, found: 410.2106.
2,7-Bis(3-hexylthiophen-2-yl)-9-octyl-9H-carbazole (3f). (2.73 g, 76%), pale yellow solid, mp 49–52 °C; Rf = 0.12 (PE); IR (KBR): ν = 3064, 2924, 2858, 1602, 1563, 1456, 1365, 1325, 1251, 1133, 1090, 847, 806, 720, 659 cm−1; 1H NMR (400 MHz, CDCl3): δ = 0.88 (t, J = 8.0 Hz, 9H), 1.27–1.46 (m, 22H), 1.65–1.73 (m, 4H), 1.89–1.96 (m, 2H), 2.78 (t, J = 8.0 Hz, 4H), 4.34 (t, J = 8.0 Hz, 2H), 7.06 (d, J = 4.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 4.0 Hz, 1H), 7.36 (d, J = 4.0 Hz, 1H), 7.48 (s, 2H), 8.12 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 14.1, 22.6, 22.6, 27.4, 28.9, 29.1, 29.2, 29.3, 29.4, 31.2, 31.7, 31.8, 43.2, 109.5, 120.2, 120.9, 121.8, 123.6, 129.6, 132.3, 138.6, 138.9, 141.0; HRMS m/z calculated for C40H53NS2 MW: 611.3619, found: 611.3613.
2,5-Bis(3-hexylthiophen-2-yl)thieno[3,2-b]thiophene (3g). (1.11 g, 82%), green semisolid, Rf = 0.64 (5% EA/PE), IR (KBR): ν = 3084, 2979, 2843, 2721, 2339, 1731, 1625, 1541, 1459, 1393, 1177, 1088, 918, 814, 692 cm−1; 1H NMR (500 MHz, CDCl3, δ in ppm): 0.81 (t, 6H, J = 5.0 Hz), 1.18–1.31 (m, 13H), 1.51–1.61 (m, 4H), 2.71 (t, 3H, J = 5.0 Hz), 6.89 (d, 2H, J = 5.0 Hz), 7.14 (d, 2H, J = 5.0 Hz), 716 (s, 2H); 13C NMR (125 MHz, CDCl3, δ in ppm): 14.0, 22.6, 29.2, 30.8, 31.7, 118.0, 124.3, 130.0, 130.7, 137.6, 139.1, 140.3; HRMS m/z calculated for C26H32S4 MW: 472.1387, found: 472.1384.
2,6-Bis(3-hexylthiophen-2-yl)dithieno[3,2-b:2′,3′-d]thiophene (3h). (0.63 g, 85%), yellow solid, mp 160–165 °C; Rf = 0.61 (5% EA/PE), IR (KBR): ν = 3092, 2934, 2869, 2382, 1750, 1661, 1583, 1471, 1423, 1373, 1245, 1181, 1093, 907, 811, 699, 607 cm−1; 1H NMR (500 MHz, CDCl3, δ in ppm): 0.9 (t, 6H, J = 5.0 Hz), 1.32–1.42 (m, 12H), 1.69 (p, 4H, J = 5.0 Hz), 2.83 (t, 4H, J = 10.0 Hz), 6.99 (d, 2H, J = 5.0 Hz), 7.24 (d, 2H, J = 5.0 Hz), 7.31 (s, 2H); 13C NMR (125 MHz, CDCl3, δ in ppm): 14.0, 22.6, 29.2, 29.2, 30.7, 31.6, 119.2, 124.4, 130.1, 130.5, 130.6, 136.9, 140.3, 140.9; HRMS m/z calculated for C28H32S5 MW: 528.1108, found: 528.1101.
Conclusions
Comonomers end-capped with thienyl units were successfully prepared via Suzuki–Miyaura coupling reaction in synthetically excellent yields. Potentiodynamic electropolymerisation of these monomers on ITO substrate afforded thin films of electrodeposited π-conjugated polymers between 0.05 and 0.2 μM. Microstructure of the polymers revealed different surface morphology and porosity. The electrochemically synthesized polymers were found to exhibit excellent catalytic activity in the reduction of nitrobenzene than bare GC electrode. Further studies to delineate the scope of our monomers and polymers for other applications are on the way.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support by the Department of Science & Technology (DST), India to carry out this work through INSPIRE faculty award (IFA-14/MS-27) and (IFA-13/CH-133) is greatly acknowledged.
Notes and references
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832 CrossRef CAS.
- A. Facchetti, Chem. Mater., 2011, 23, 733 CrossRef CAS.
-
(a) J. Roncali, Chem. Rev., 1997, 97, 173 CrossRef CAS;
(b) Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS PubMed.
-
(a) I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370 CrossRef CAS PubMed;
(b) B. Lüssem, C.-M. Keum, D. Kasemann, B. Naab, Z. Bao and K. Leo, Chem. Rev., 2016, 116, 13714 CrossRef PubMed.
- B. B. Berkes, A. S. Bandarenka and G. Inzelt, J. Phys. Chem. C, 2015, 119, 1996 CrossRef CAS.
-
(a) Q. Zhang, H. Dong and W. Hu, J. Mater. Chem. C, 2018, 6, 10672 RSC;
(b) Electropolymerization: Concepts, Materials and Applications, ed. S. Cosnier and A. Karyakin, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010 Search PubMed.
- Electropolymerization: Concepts, Materials and Applications, ed. S. Cosnier and A. Karyakin, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010 Search PubMed.
- C. A. Ferreira, S. Aeiyach, M. Delamer and P. C. Lacaze, J. Electroanal. Chem., 1990, 284, 351 CrossRef CAS.
- E. Poverenov, M. Li, A. Bitler and M. Bendikov, Chem. Mater., 2010, 22, 4019 CrossRef CAS.
- B. Lu, S. Zhen, S. Ming, J. Xu and G. Zhao, RSC Adv., 2015, 5, 70649 RSC.
- J. A. Carrillo, M. J. Ingleson and M. L. Turner, Macromolecules, 2015, 48, 979 CrossRef CAS.
-
(a) A. A. Raheem, S. Kamaraj, V. Sannasi and C. Praveen, Org. Chem. Front., 2018, 5, 777 RSC;
(b) S. Gopi, K. Giribabu and M. Kathiresan, ACS Omega, 2018, 3, 6251 CrossRef CAS;
(c) K. Madasamy, V. M. Shanmugam, D. Velayutham and M. Kathiresan, Sci. Rep., 2018, 8, 1354 CrossRef PubMed;
(d) M. Kathiresan, H.-J. Steinhoff and L. Walder, Macromol. Chem. Phys., 2017, 218, 1700142 CrossRef;
(e) S. Gopi and M. Kathiresan, Polymer, 2017, 109, 315 CrossRef CAS;
(f) K. Madasmy and M. Kathiresan, ChemistrySelect, 2016, 3, 354 CrossRef.
-
(a) S. Koyuncu, O. Usluer, M. Can, S. Demic, S. Icli and N. S. Sariciftci, J. Mater. Chem., 2011, 21, 2684 RSC;
(b) F. B. Koyuncu, S. Koyuncu and E. Ozdemir, Org. Electron., 2011, 12, 1701 CrossRef CAS;
(c) F. B. Koyuncu, S. Koyuncu and E. Ozdemir, Electrochim. Acta, 2010, 55, 4935 CrossRef CAS.
- E. Amir, M. Murai, R. J. Amir, J. S. Cowart Jr, M. L. Chabinyc and C. J. Hawker, Chem. Sci., 2014, 4, 4483 RSC.
-
(a) T.-J. Lin and S.-T. Lin, Phys. Chem. Chem. Phys., 2015, 17, 4127 RSC;
(b) H. Huang, L. Yang, A. Facchetti and T. J. Marks, Chem. Rev., 2017, 117, 10291 CrossRef CAS PubMed.
- E. Amir, R. J. Amir, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2011, 133, 10046 CrossRef CAS PubMed.
- W. Richard, D. Evrard and P. Gros, J. Electroanal. Chem. Interfacial Electrochem., 2012, 685, 109 CrossRef CAS.
-
(a) T.-H. Lee, K.-Y. Wu, T.-Y. Lin, J.-S. Wu, C.-L. Wang and C.-S. Hsu, Macromolecules, 2013, 46, 7687 CrossRef CAS;
(b) H. Wakabayashi, T. Kurihara, K. Shindo, M. Tsukada, P.-W. Yang, M. Yasunami and T. Nozoe, J. Chin. Chem. Soc., 1998, 45, 391 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR, 13C NMR spectra of monomers, FTIR, UV-vis, PL, SEM and AFM images of polymers. See DOI: 10.1039/c8ra08603f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bc and
Chandrasekar Praveen
*bc and
Chandrasekar Praveen