
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh performance of PtCu@TiO2 nanocatalysts toward methanol oxidation reaction: from synthesis to molecular picture insight
Nina Dimitrovaa,
Marwa Dhifallahb,
Tzonka Mineva b,
Tzvetanka Boiadjieva-Scherzerc,
Hazar Guesmi*b and
Jenia Georgieva*a
b,
Tzvetanka Boiadjieva-Scherzerc,
Hazar Guesmi*b and
Jenia Georgieva*a
aRostislaw Kaischew Institute of Physical Chemistry, Bulgarian Academy of Sciences, Sofia 1113, Bulgaria. E-mail: jenia.georgieva@ipc.bas.bg
bInstitut Charles Gerhardt Montpellier, CNRS/ENSCM/UM, 240, Avenue du Professeur Emile Jeanbrau, 34090 Montpellier, France. E-mail: hazar.guesmi@enscm.fr
cCEST Kompetenzzentrum für Elektrochemische Oberflächentechnologie GmbH, Wiener Neustadt, Austria
First published on 21st January 2019
Abstract
The electrocatalytic production of hydrogen from methanol dehydrogenation successfully uses platinum catalysts. However, they are expensive and Pt has the tendency to be poisoned from the intermediate compounds, formed during the methanol oxidation reaction (MOR). For these two reasons, there has been active research for alternative bi- and tri-component Pt-based catalysts. Herein, PtCu nanoparticles deposited on titania were studied and proposed to be efficient MOR catalysts. The catalyst was prepared by photo-deposition of Cu on a high-surface-area TiO2 powder support, followed by a partial galvanic displacement of the Cu deposit by platinum. The morphology and structure of the catalyst were characterized by physicochemical methods. The PtCu@TiO2 electro-catalyst has higher intrinsic catalytic activity and comparable mass specific activity for MOR in comparison with a commercial Pt/C catalyst. The experimental analyses were complemented by density functional theory-based computations. The theoretical results revealed that the most energetically favorable Pt and Cu arrangement in the supported PtCu nanoparticles was core (Cu)–shell (Pt) and/or phase-separated. The inter-atomic interactions responsible for the bimetallic cluster stabilization on titania were highlighted from the computed electronic charge distribution.
1 Introduction
Methanol is widely used as a feedstock to produce many industrial chemicals. Methanol has also been proposed as a source of hydrogen due to its relatively high hydrogen content, making it attractive for a future hydrogen economy.1 Hydrogen can be produced from methanol through a catalytic dehydrogenation process, in particular, a photo-electrocatalytic process over titania-supported metal catalysts.2 It is known that Pt is the most widely used electro-catalyst in methanol oxidation reaction (MOR). However, the high cost of Pt and the tendency of Pt-based catalysts to be poisoned from MOR intermediates are the two main reasons that motivated the development of other two- and three-component catalysts.3,4Among the two-component Pt-based catalysts, the bimetallic PtCu systems have attracted an increasing attention. In addition to the formidable cost reductions that could be made, PtCu nanocatalysts have shown higher activity compared to the monometallic counterparts in a variety of energy-related catalytic processes, such as oxidation of CO5,6 and methanol,7,8 water–gas shift,9,10 and reduction of NO,11 O2,12,13 and CO2.14,15 These improved catalytic performances are often related to the structure of PtCu alloys, in terms of the electronic, geometric, or synergistic effects. For instance, PtCu (111) surface with Pt-skin and Cu-rich subsurface or PtCu nanoclusters with Cu-core/Pt-shell have been found to exhibit superior activities toward oxygen reduction reaction (ORR)12,13 and a significantly greater selectivity for N2 formation during NO reduction11 than that of pure Pt catalysts.
Our previous experimental results showed that PtCu and PtNi nanoparticles, supported on carbon black (Vulcan XC-72R) demonstrated comparable or improved catalytic activity for MOR than the commercial Pt/C electro-catalyst.16,17 These binary systems were prepared using galvanic displacement of electrolessly deposited Cu or Ni on carbon by Pt (transmetalation), due to the difference in the standard electrode potentials (E0).18,19 It was found that PtCu films are better electro-catalysts for methanol oxidation than PtNi.19 It is known that carbon is oxidized to CO2 at potentials above 0.97 V vs. reversible hydrogen electrode (RHE), resulting in the loss of a part of the noble metal and agglomeration. As a consequence, the electrocatalytic activity decreases. Lately, oxide-based supports, and in particular TiO2, have been tested as carriers for catalysts in fuel cells operating at low temperatures. In addition, it was found that Cu can be deposited on TiO2 by a simpler and low-temperature process of photodeposition.20
Herein, we have used a new and efficient catalyst that was obtained by a recently reported alternative two-step method: a photodeposition of Cu on a TiO2 powder support (containing mainly anatase), followed by a partial galvanic displacement of the Cu deposit by Pt, so that a practical catalyst is prepared.21 Physicochemical characterization of the obtained PtCu@TiO2 material was performed. Its electrochemical behavior towards methanol oxidation was also studied to evaluate its activity as a catalyst in fuel cell technology. In order to gain a better insight into the structure of the PtCu@TiO2 catalyst, a theoretical study at the molecular level was mandatory. For this purpose, the energetic stabilities of small free and supported PtCu nanoparticles were analyzed. Regular truncated octahedron clusters (rto) of Pt32Cu6 (Pt with less Cu content) representing different alloy types (core–shell, regular alloy, phase-separated) were considered.
The most stable surface of anatase single crystals is the (101) plane.22,23 However, recently, it was shown24 that depending on the preparation and particle size, the (101) surface is less abundant on industrially produced nanostructured anatase particles. On these nanoparticles, other low-index surfaces, such as (100) and, to a lesser extent, (001) and (111), have been observed by high-resolution TEM. In the present study, we therefore focussed on the TiO2 (100) surface, which is usually regarded as a minority plane but has been shown to be active for several electrocatalytic reactions.25
2 Experimental and theoretical details
2.1 Preparation and characterization of the PtCu@TiO2 catalyst
The preparation of the catalyst was done according to the literature procedure.21 In brief, the photodeposition of Cu on TiO2 support was carried out as follows: 5 × 10−3 M CuSO4·5H2O (Merck, ACS reagent) was dissolved in 300 ml water (10 min stirring). Then, 0.2 g TiO2 powder (Degussa P25) was added to the solution of Cu precursor in the presence of formic acid (85%, ACS reagent). The photoreduction of Cu(II) was conducted in a reactor system, equipped with a quartz tube in which a lamp (9W/78 UV-A) was placed. The UV light irradiation was continued for 3 h under magnetic stirring and purging with nitrogen. Formate ions adsorbed on TiO2 reacted with positive holes, created in TiO2 under UV irradiation, and prevented the reoxidation of the reduced Cu(II). The prepared Cu/TiO2 precursor was filtered and washed with purified water.The PtCu@TiO2 catalyst was obtained by a galvanic replacement approach, which is based on the difference in E0 between Cu and Pt ions in the solution ([PtCl6]2−/Pt – +0.744 V vs. NHE and Cu2+/Cu – +0.340 V vs. NHE). The Cu/TiO2 precursor was put in a 25 ml solution, containing 5 × 10−3 M K2PtCl6 salt (Sigma-Aldrich, ACS reagent, >37.5% as Pt) and 0.1 M HCl (Aldrich). The experiment was performed under ultrasonic stirring and N2 deaeration for 45 min. Then, the obtained PtCu@TiO2 catalyst was filtered, washed, and dried.
The composition of the catalyst was analyzed by energy-dispersive spectrometry (EDS) using a scanning electron microscope JSM 6390 equipped with a system INCA Oxford Energy 350.
The morphology of the obtained catalytic material was evaluated with high resolution scanning transmission electron microscope (HR STEM) JEM 2100 with CCD camera GATAN Orius 832 SC1000 and GATAN Microscopy Suit Software.
X-ray photoelectron spectroscopy (XPS) was used to investigate the chemical state of the surface layers of the PtCu@TiO2 catalyst. A PHI 5600 (Physical Electronics) spectrometer was used, equipped with PHI Multipak 9.3 software. X-ray diffraction (XRD) characterization of the crystal structure was made by Empyrean diffractometer. Cu anode at 30 kV, 40 mA and Cu K-alpha radiation 1.5406 Å were used.
The electrocatalytic activity of the catalyst toward methanol oxidation was tested and compared with that of a commercial 10 wt% Pt/C catalyst, ETEK. The cyclic voltammetry (CV) experiments were performed in a three-electrode cell, using an Autolab 30 (EcoChemie) system. A Pt foil and a SCE were used as the auxiliary and the reference electrode, respectively. The CV experiments were conducted in a deaerated 0.1 M HClO4 solution at 50 mV s−1 and 0.1 M HClO4 + 0.5 M CH3OH solution at 5 mV s−1 potential sweep rate. Electrochemically active surface area (EASA) of Pt was defined from the charge amount of the hydrogen adsorption/desorption region (the charge associated with the formation or stripping of a hydrogen monolayer is 210 μC cm−2).26
2.2 Theoretical details
Spin-polarized density functional theory calculations, within periodic boundary conditions, were performed using the plane wave approach, as implemented in the Vienna ab initio simulation package (VASP) code.27,28 The generalized gradient approximation functional of the exchange–correlation energy was calculated within the Perdew, Burke, and Ernzerhof formulation of the generalized-gradient approximation (GGA-PBE).29,30 To correct for the well-known band-gap problem of GGA type of functionals, the Hubbard U parameter was added by using the Dudarev's approach (PBE+U)31 with an optimal value for the Coulomb interaction corrections of Ti electrons Ueff = 3.3 eV. The calculations included projector augmented-wave (PAW) pseudopotentials,32,33 where oxygen 2s and 2p, titanium 3p, 4s, and 3d, copper 3p and 3d and platinum 6s and 5d electrons were explicitly treated. The cut off energy was fixed to 400 eV.To model nanoparticles, a cubic cell with sides of 2 nm was chosen to provide enough spacing between metal clusters in the neighboring repeated cells. A cluster model of 38 atoms with cubo-octahedral shape was cut from bulk fcc metal to present (111) and (100) facets. The 38-atom cluster was selected because size 38 is a “magic” number for the regular truncated octahedron (rto) structure, which possesses high symmetry, and this simplifies the analysis of ligand adsorption.34 Moreover, as hexagonal local arrangements is a common occurrence on the surface of nanoparticles,35 a 38-atom cluster is often taken as a model of the crystalline-like structure and thus also as a representative of larger clusters.34,36–38 During geometry optimization, all atoms were allowed to relax. The gamma point was used in the Brillouin-zone integration. For bimetallic (BiM) Pt32–Cu6 nanoparticles three different nanoalloy clusters representing core–shell, phase-separated and regular alloy structures were considered.
Concerning TiO2 support, the stoichiometric surface was chosen because according to a recent scanning tunneling microscopy (STM) study39 almost no point defects on (100) surfaces were found. The (100) surface is flat and a channel is formed in the [0 1 0] direction. The outermost surface layer consists of O atoms with two different coordinations (a 3-fold-coordinated O (3f-O) located on the terrace and a 2-fold-coordinated Ti (2f-O) located in the channel) and Ti atoms (5-fold coordinated). The second layer exposes fully coordinated Ti and O atoms. To represent the anatase surface, a 5 × 2 supercell slab, two Ti–O double-layers thick, containing 240 atoms was modeled. Slabs up to 7 double layers of TiO2 were constructed, relaxed, and compared to the slab with two double layers, which was found to give similar electronic and geometric characteristics for both nanoparticles on TiO2 and bare TiO2. Therefore, a two double-layer slab was chosen to limit computational costs. A vacuum of 18 Å between the top of the nanoparticle and the backside of the periodic image was ensured. The associated k-point grids were defined from the bulk rule and the slab was modeled with the Monkhorst–Pack40 k-point mesh of (3 × 3 × 1). The active side of the slab was allowed to relax, while the atoms of the bottom two layers for all supercells were frozen in their bulk positions to provide a bulk-like boundary condition for the slab.
Geometry optimizations were performed using a convergence criterion for the electronic self-consistent cycle fixed to 10−6 eV per supercell. Atomic positions and lattice parameters were both relaxed using the conjugate gradient algorithm until all the forces were below 0.01 eV Å−1. Bader charge41 analysis was conducted using software from the Henkelman group.42
To evaluate the stability of free Pt–Cu nanoparticles the excess energy per atom (Eexc) was calculated.43,44 This energetic parameter is defined as follows:
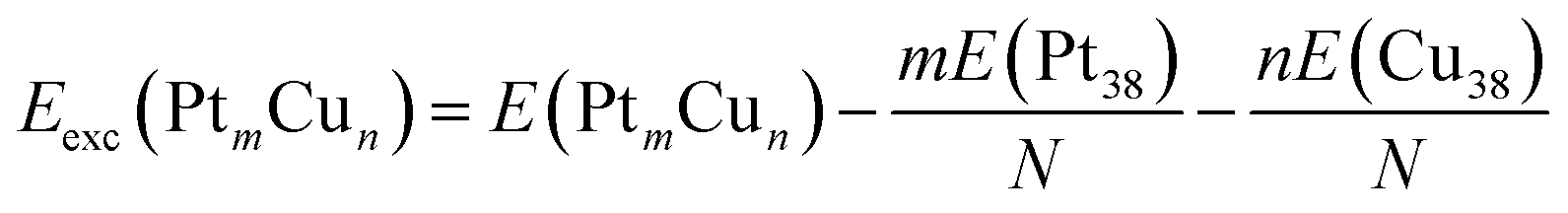 | (1) |
In order to evaluate the effect of the interaction of the nanoparticles with the oxide support, the energy gained by the system after the deposition of the gas-phase nanoparticle on the TiO2 oxide support was expressed as 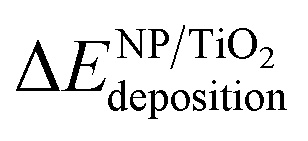 , and it was obtained as follows,
, and it was obtained as follows,
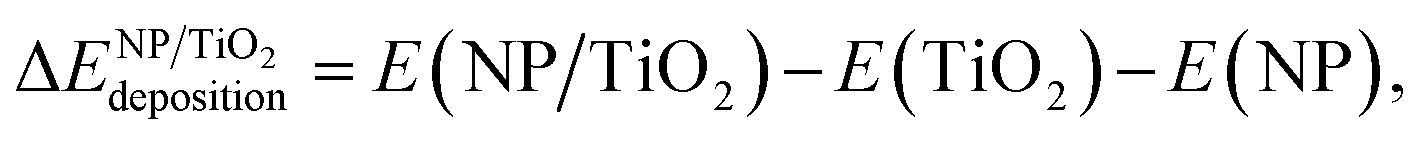 | (2) |
3 Results and discussions
3.1 Physicochemical characterization
The composition of the PtCu@TiO2 powder, obtained by EDS elemental analysis was: Ti 44.7 wt%, O 41.8 wt%, Cu 0.8 wt%, and Pt 12.7 wt%.Fig. 1a and b displays the TEM images of the Cu/TiO2 and the PtCu@TiO2 materials. The photodeposited Cu nanoparticles in the Cu/TiO2 precursor were in an oxidized state as Cu2O. After Pt deposition, the majority of Pt or PtCu crystallites, seen in Fig. 1b, uniformly dispersed on the TiO2 support, forming aggregates of different size.
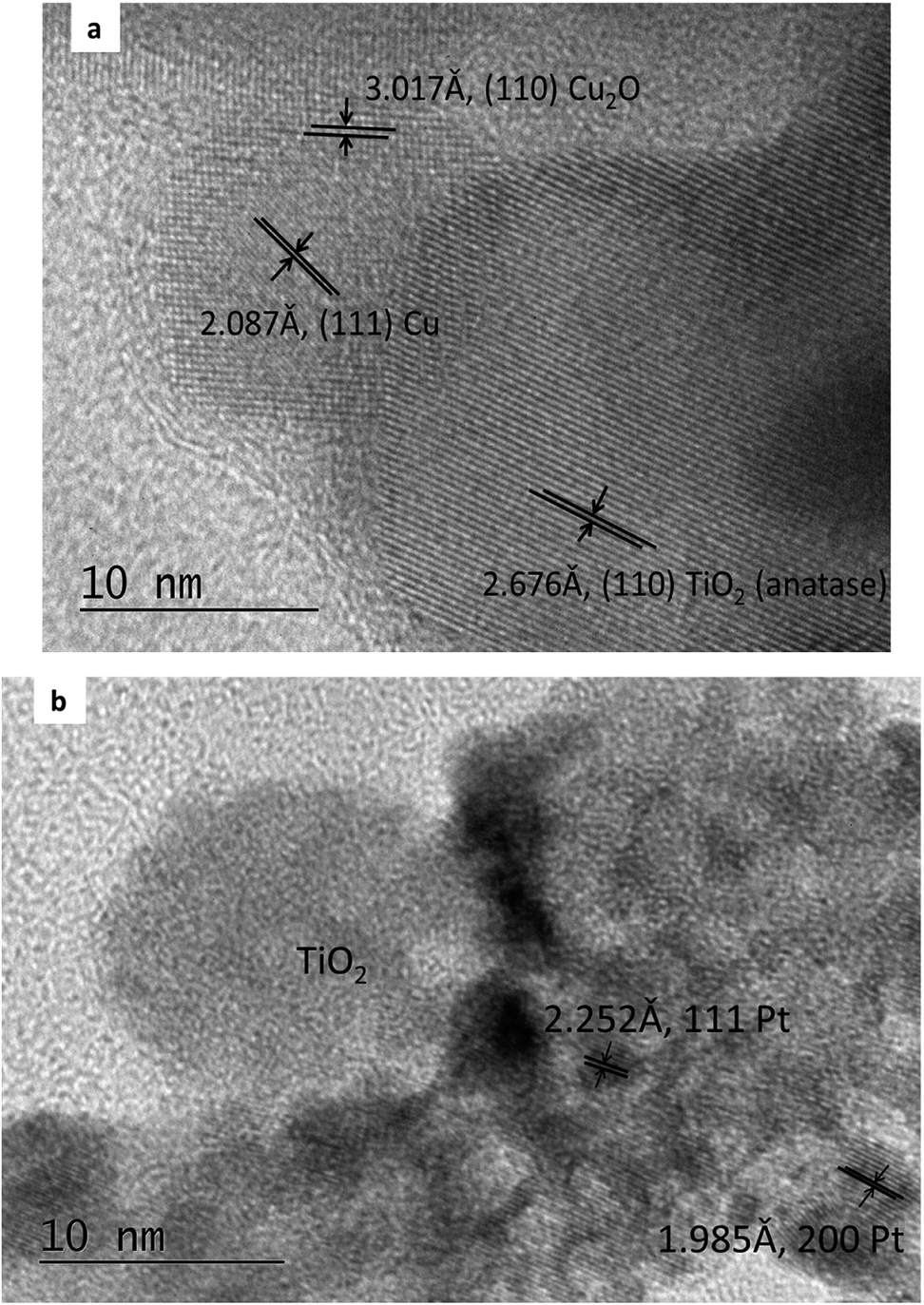 | ||
| Fig. 1 TEM images of Cu/TiO2 precursor (a) and PtCu@TiO2 catalyst (b). | ||
Fig. 2a shows the XRD pattern of the Cu/TiO2 precursor. Anatase and rutile phases (typical for Degussa P-25) can be seen. Peaks of Cu and small peaks of Cu2O are also observed.
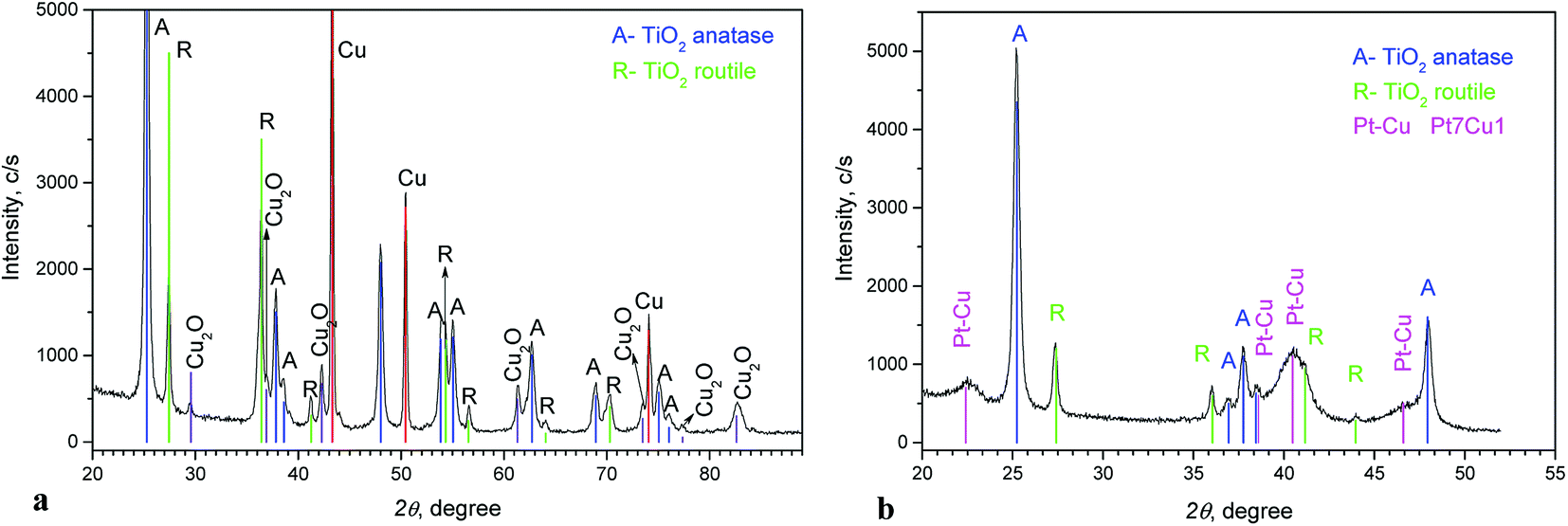 | ||
| Fig. 2 XRD diagrams of Cu/TiO2 precursor (a) and PtCu@TiO2 catalyst (b). | ||
The diffraction diagram of PtCu@TiO2, presented in Fig. 2b, displays that the PtCu@TiO2 catalyst contained some amount of alloyed Pt and Cu. No specific peaks of Pt and Cu were observed, which can be due to the small quantity of Cu remaining in the catalyst and the distorted structure of the small nanoparticles.
XPS gave information about the chemical state of the superficial coats of the PtCu@TiO2 catalyst. The Ti 2p region of the XPS spectrum (Fig. 3a) indicated that Ti(IV) was only present in the catalyst. Fig. 3b presents the Cu 2p photoelectron spectrum of the PtCu@TiO2. The main and the satellite peaks of Cu 2p3/2 and Cu 2p1/2 at the binding energies of 951.8 eV and 932.1 eV can be ascribed to Cu2O on the surface of copper, in accordance with TEM analysis. The Pt 4f part of the XPS spectrum of the PtCu@TiO2 catalyst (Fig. 3c) had peaks of 4f7/2 at 70.17 eV and 4f5/2 at 73.54 eV. It was found that Pt in the nanocatalyst was mainly in the zero valent metallic state, although it was oxidized partially on the surface.
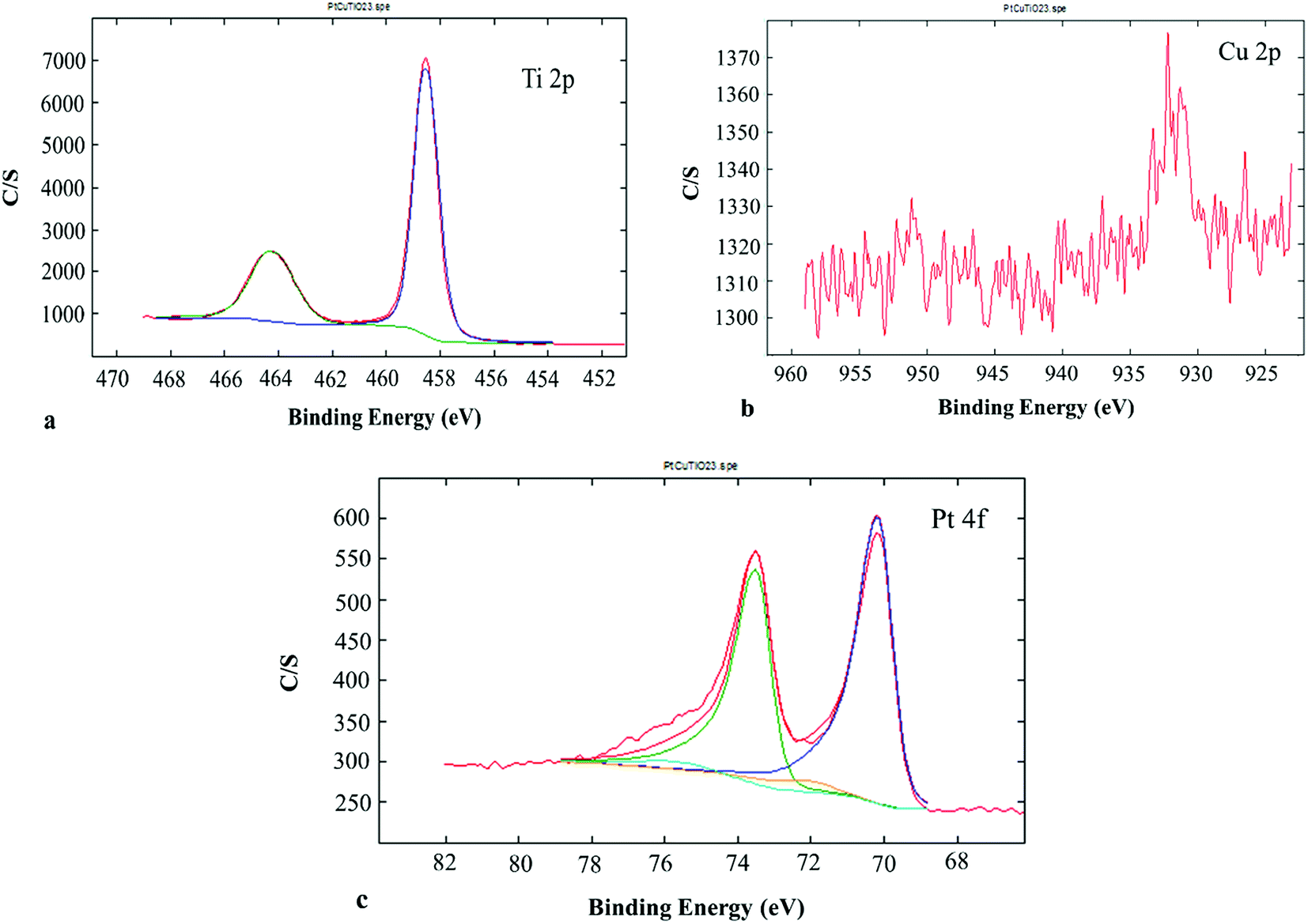 | ||
| Fig. 3 XPS spectra in the Ti 2p (a), Cu 2p (b) and Pt 4f (c) binding energy range. | ||
3.2 Electrocatalytic activity
The catalytic activity of the PtCu@TiO2 powder catalyst for the MOR was evaluated against a Pt/C catalyst. Before performing the tests, the PtCu@TiO2-coated graphite electrode was scanned many times in 0.1 M HClO4 between the evolution potentials of hydrogen and oxygen. During repetitive scanning, any Cu or Cu2O particles, not protected by Pt, were anodically dissolved. Finally, the Pt or Pt-covered Cu particles existing on the surface were the only electrically active species.The experiments for MOR were carried out in deaerated 0.1 M HClO4 + 0.5 M MeOH solutions. Fig. 4a shows the CV curves, obtained at 5 mV s−1 scan rate. The methanol oxidation currents were normalized by the Pt EASA (je), which gives the intrinsic activity of the catalysts. It was seen that the PtCu@TiO2 electrode had higher intrinsic catalytic activity for MOR than Pt/C catalyst. The enhanced activity can be explained as a result of interactions between Pt and Cu, Pt particles and the TiO2 support and the synergy effect of the latter on CO oxidative removal.
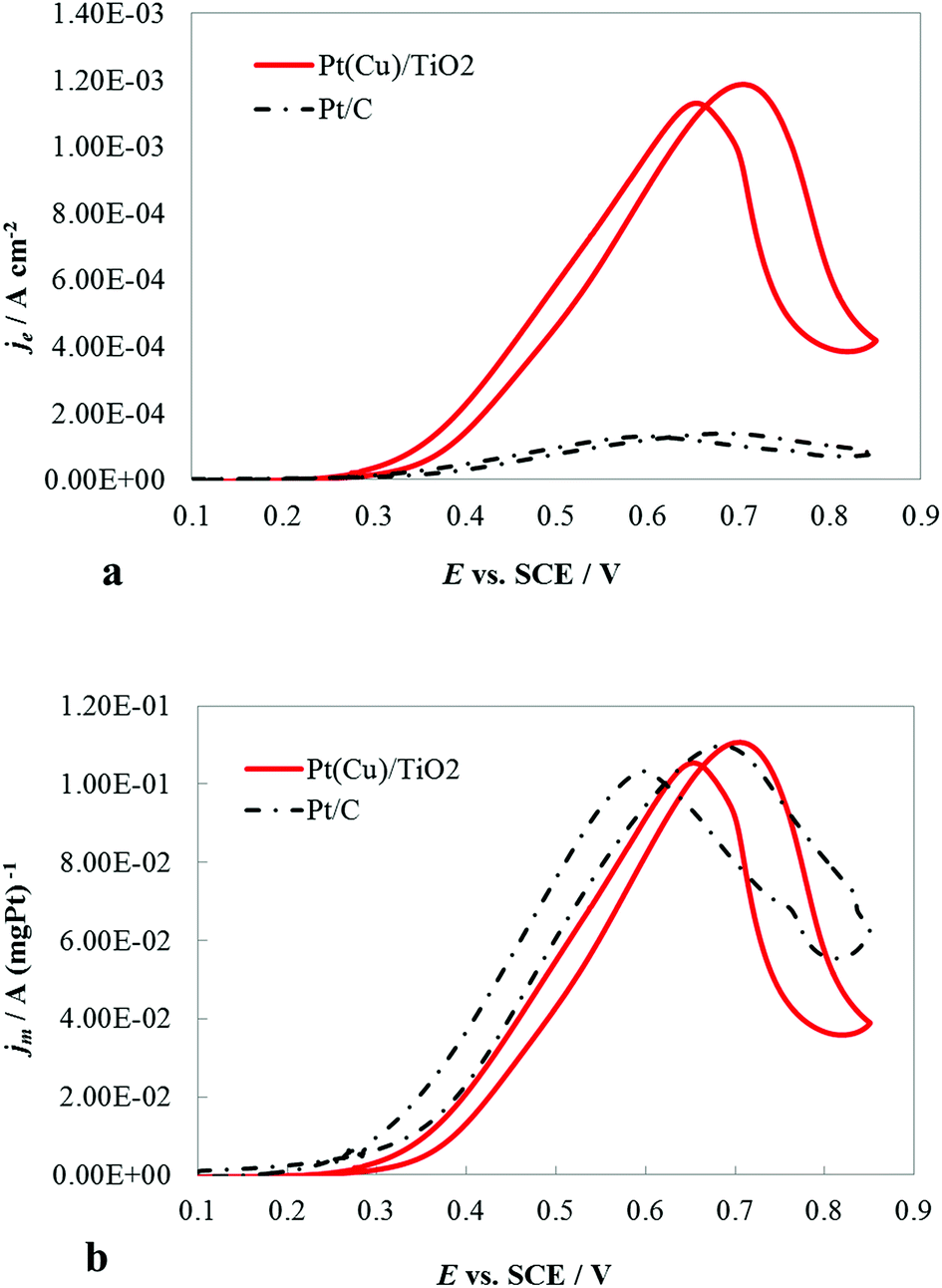 | ||
| Fig. 4 CV curves at 5 mV s−1 of PtCu@TiO2- and Pt/C-coated graphite electrodes in a deaerated 0.1 M HClO4 + 0.5 M MeOH solution: (a) the currents were normalized by the Pt electroactive surface area (je); (b) the currents were normalized by the mass of Pt in the catalyst (jm). | ||
The CV curves (Fig. 4b) demonstrated the specific mass activity of the PtCu@TiO2 and Pt/C catalysts. For this, the currents were normalized per mass of Pt (jm). The mass activity of PtCu@TiO2 was similar to that of the commercial Pt/C catalyst. The obtained result can be attributed to the more effective dispersion and better utilization of Pt, deposited by the photodeposition – galvanic replacement method and alloy formation between Pt and Cu.
3.3 DFT geometric and energetic properties
Nanoclusters of regular orthorhombic tetrahedral shape were considered to model Pt38, Cu38 and three types of alloyed Pt38−x–Cux (x = 0 and 6) nanoparticles: (core–shell (CS(Pt32Cu6))), phase-separated (PhSep(Pt32Cu6)) and regular alloy (RA(Pt32Cu6)) structures. The optimized structures of these clusters, obtained with the DFT+U method are reported in Fig. 5. For each type of deposited nanoclusters, two different deposition orientations on the surface were considered: (1) through the fcc face and (2) through the square face. All the PtCu@TiO2 models were relaxed to determine the energetically most stable configuration. In Table 1 the calculated excess energies (Eexc) of gas-phase nanoalloy clusters and their optimized geometric characteristics in comparison with the monometallic counterparts are depicted. From this analysis it was clear that the computed Eexc were negative, which indicated that mixing was favorable at the considered composition. This result was expected because in the bulk PtCu alloy the mixing was favorable. Moreover, the core–shell (CS(Pt32Cu6)) structure with Cu in the core and Pt in the shell was predicted to be the most stable nanoalloy structure, followed by the regular alloy (RA(Pt32Cu6)) structure, where the six cooper atoms occupied the centers of the hexagonal facets. In the phase-separated structure (PhSep(Pt32Cu6)) copper atoms occupied the surface of the cluster hexagonal facet (see Fig. 5). It, therefore, followed that the energetically preferred organization between the two metal components in the small PtCu nanoparticles was the core–shell Cu–Pt with the highest possible coordination of the Cu-atoms. Geometric analysis showed a slight volume contraction (about 1.5%) of nanoalloy clusters with respect to pure Pt. | ||
| Fig. 5 DFT-computed nanoalloy cluster models, from the left to the right: monometallic Pt38, core–shell (CS(Pt32Cu6)), phase-separated (Ph_Sep(Pt32Cu6)) and regular alloy (RA(Pt32Cu6)) clusters, and monometallic Cu38. | ||
| Nanoparticle | Eexc (eV) | dM–M (Å) | V (Å3) |
|---|---|---|---|
| Pt38 | 0 | 2.68 | 2133.40 |
| Cu38 | 0 | 2.48 | 1694.91 |
| Ph_Sep(Cu6Pt32) | −0.017 | 2.66 | 2102.86 |
| RA(Cu6Pt32) | −0.036 | 2.66 | 2101.24 |
| CS(Cu6Pt32) | −0.038 | 2.66 | 2102.86 |
The calculated deposition energies 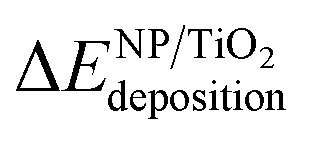 of the optimized Pt38, Cu38, and Pt38−xCux clusters, supported on TiO2 anatase surface, are reported in Fig. 6. For the monometallic Pt38 cluster, the deposition energy was found to be positive (0.68 eV), while it was negative for copper (−2.7 eV). For alloy clusters, DFT calculations predicted both (CS(Pt32Cu6)) and (PhSep(Pt32Cu6)) to be the most stable structures on the support, with a slight energy preference (<0.1 eV) for the core–shell structure. In contrast with its high stability in gas phase, the regular alloy structure was revealed to be the least stable when supported on titania. Regarding, the two competitive CS(Pt32Cu6) and PhSep(Pt32Cu6) structures, copper was predicted to be either in the core or in the interface between the Pt and the support. This result strongly supported our experimental observation showing that for PtCu clusters, Pt mainly covered Cu.
of the optimized Pt38, Cu38, and Pt38−xCux clusters, supported on TiO2 anatase surface, are reported in Fig. 6. For the monometallic Pt38 cluster, the deposition energy was found to be positive (0.68 eV), while it was negative for copper (−2.7 eV). For alloy clusters, DFT calculations predicted both (CS(Pt32Cu6)) and (PhSep(Pt32Cu6)) to be the most stable structures on the support, with a slight energy preference (<0.1 eV) for the core–shell structure. In contrast with its high stability in gas phase, the regular alloy structure was revealed to be the least stable when supported on titania. Regarding, the two competitive CS(Pt32Cu6) and PhSep(Pt32Cu6) structures, copper was predicted to be either in the core or in the interface between the Pt and the support. This result strongly supported our experimental observation showing that for PtCu clusters, Pt mainly covered Cu.
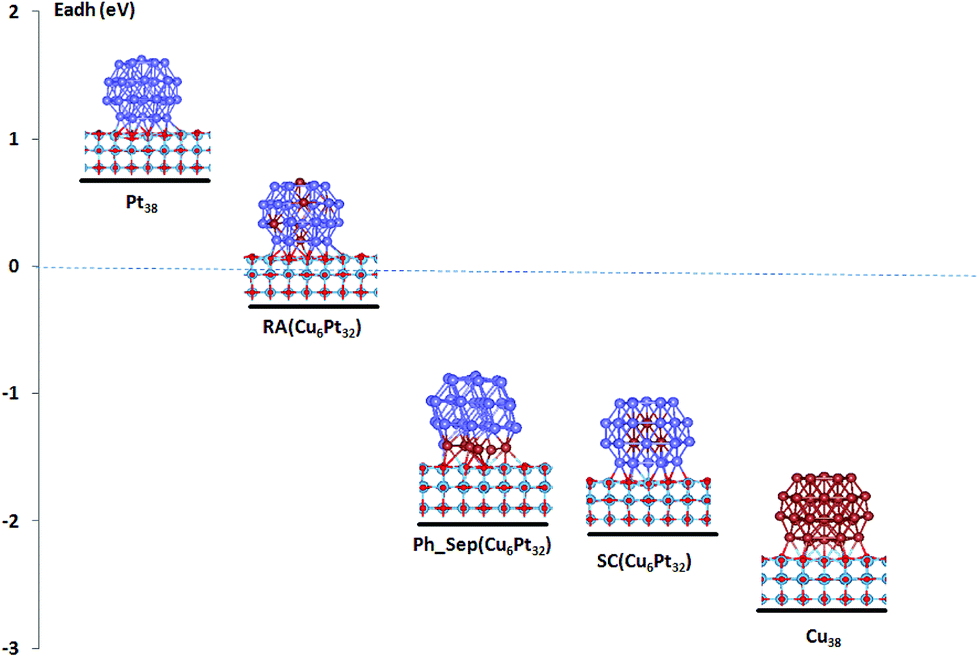 | ||
| Fig. 6 Computed adhesion energies of pure and bimetallic PtCu clusters on TiO2 anatase support. | ||
3.4 Electronic charge distribution
In order to understand the superior stability of the supported CS(Pt32Cu6) and PhSep(Pt32Cu6) structures with respect to the regular alloy one, we calculated and compared the Bader charges (q) in the three clusters. This analysis revealed that all the Cu atoms were positively charged, independent of the particle structure, with q ranging from +0.39 e− to +0.50 e−. The most positive Cu atoms were found in the PhSep(Pt32Cu6) particle with Cu bound to the anatase surface. The Pt atoms in all the supported structures were predominantly negatively charged with q in the interval of −0.01 e− to −0.30 e−. The most negative Pt atoms were those in the core–shell cluster neighboring the copper atoms and in the regular alloy structure, bound to the titania surface. Several Pt atoms in CS(Pt32Cu6) bound to TiO2 surface were; however, positively charged with the maximum charge q ∼ +0.15 e−. Different charge distribution was obtained in the supported RA(Pt32Cu6), where five of the six surface-bound Pt atoms were negatively charged with q ranging from −0.05 e− to −0.3 e−. Note that the only one positive Pt atom had q = +0.06 e−. In the same structure, the innermost Pt had positive charges (q in the interval of +0.1 e− to +0.2 e−).The Bader atomic charges can be rationalized in view of the Pauling electronegativity (χ) scale. Pt is a more electronegative atom with χ = 2.28 as compared to χ = 1.90 of Cu. This explains the largest charge transfer from the core Cu to the shell Pt atoms leading to the strongest Cu (in core)–Pt (in shell) interaction. In addition, the obtained larger charge transfer (∼0.1 e−) from the CS(Pt32Cu6) and PhSep(Pt32Cu6) nanoclusters to the support (positively charged Pt and Cu bound to titania) augments the energetic stability of these two structures on titania. The established majority of negative surface-bound Pt atoms in RA(Pt32Cu6)/TiO2 (vide supra) suggests that the RA(Pt32Cu6)/TiO2 cluster is destabilized because of the predominant charge transfer from the oxide to the first layer Pt atoms, which is not compensated by the amount of charge transferred from Cu to Pt inside the cluster. This conclusion is also corroborated from the computed positive adhesion energy of the pure Pt38 cluster on anatase surface (see Fig. 6), showing that the small Pt clusters cannot be stabilized on the TiO2 support. The alloying of Pt with Cu allows the platinum atoms to gain back some charge from copper, because of the stronger electronegativity of Pt. The amount of the charge transferred from Cu to Pt depends on the particular arrangements between the two metal components in the alloy cluster and determines the relative stability of the supported PtCu nanoparticles.
In conclusion and following the above DFT structural and energetic results, we suggest that the Pt and Cu arrangement in the experimental bimetallic nanoparticle catalyst is of core–shell and/or phase-separated types. A PtCu catalyst with a regular alloyed like structure can be ruled out because of its significantly low energy stability when deposited on titania.
4 Conclusions
PtCu nanoparticles were deposited on TiO2 powder support using a two-step method: Cu photodeposition – Pt galvanic replacement. The PtCu@TiO2 catalyst demonstrated higher intrinsic catalytic activity and similar mass specific activity for MOR, in comparison with a commercial Pt/C catalyst. The improved catalytic activity may be due to an effective dispersion and utilization of Pt, Pt–Cu and Pt–TiO2 interactions.The interatomic interactions of small clusters of Pt, Cu and PtCu supported on titanium oxide surface were studied theoretically using DFT quantum chemistry computations. The catalyst was modeled by core–shell CS(Pt32Cu6), phase-separated (PhSep(Pt32Cu6)) and regular alloy RA(Pt32Cu6) clusters, supported on (100) anatase TiO2 surface. The computed adhesive energies revealed the former two clusters as energetically favorable, which allows us to conclude that the core–shell and/or phase-separated like nanoparticles are formed in the here presented PtCu@TiO2 catalyst. An additional analysis of the computed charge distributions in the three BiM models on (100) TiO2, highlighted the effect of a particular inter-atomic arrangement in the alloy clusters on their energetic stabilization on titania surface and confirmed the experimental observations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was granted access to the HPC resources of [CCRT/CINES/IDRIS] under the allocation 2017 [x2017087369] made by GENCI (Grand Equipement National de Calcul Intensif). Financial support of CEST Kompetenzzentrum für elektrochemische Oberflächentechnologie GmbH, Austria in the frame of the COMET program is gratefully acknowledged.References
- P. J. Wild and M. J. F. M. Verhaak, Catal. Today, 2000, 60, 3–10 CrossRef.
- Y. Han, C. J. Liu and Q. Ge, J. Phys. Chem. C, 2009, 113, 20674–20682 CrossRef CAS.
- L. Li and Y. Xing, J. Phys. Chem. C, 2007, 111, 2803–2808 CrossRef CAS.
- E. Antolini, J. R. C. Salgado and E. R. Gonzalez, Appl. Catal., B, 2006, 63, 137–149 CrossRef CAS.
- C. J. Zhang, R. J. Baxter, P. Hu, A. Alavi and M. H. Lee, J. Chem. Phys., 2001, 115, 5272–5277 CrossRef CAS.
- N. N. Hoover, B. J. Auten and B. D. Chandler, J. Phys. Chem. B, 2006, 110, 8606–8612 CrossRef CAS PubMed.
- D. Xu, Z. P. Liu, H. Z. Yang, Q. S. Liu, J. Zhang, J. Y. Fang, S. Z. Zou and K. Sun, Angew. Chem., Int. Ed., 2009, 48, 4217–4221 CrossRef CAS PubMed.
- B. Y. Xia, H. B. Wu, X. Wang and X. W. Lou, J. Am. Chem. Soc., 2012, 134, 13934–13937 CrossRef CAS PubMed.
- J. Knudsen, A. U. Nilekar, R. T. Vang, J. Schnadt, E. L. Kunkes, J. A. Dumesic, M. Mavrikakis and F. Besenbacher, J. Am. Chem. Soc., 2007, 129, 6485–6490 CrossRef CAS PubMed.
- J. Kugai, J. T. Miller, N. Guo and C. S. Song, J. Catal., 2011, 277, 46–53 CrossRef CAS.
- S. Zhou, B. Varughese, B. Eichhorn, G. Jackson and K. Mcllwrath, Angew. Chem., Int. Ed. Engl., 2005, 44, 4539–4543 CrossRef CAS PubMed.
- I. E. L. Stephens, A. S. Bondarenko, F. J. Perez-Alonso, F. CalleVallejo, L. Bech, T. P. Johansson, A. K. Jepsen, R. Frydendal, B. P. Knudsen and J. Rossmeisl, et al., J. Am. Chem. Soc., 2011, 133, 5485–5491 CrossRef CAS PubMed.
- I. E. L. Stephens, A. S. Bondarenko, U. Grønbjerg, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 6744–6762 RSC.
- I. L. Escalante-Garcia, J. S. Wainright and R. F. Savinell, ECS Trans., 2013, 50, 95–101 CrossRef.
- R. Reske, M. Duca, M. Oezaslan, K. J. P Schouten, M. T. M. Koper and P. Strasser, J. Phys. Chem. Lett., 2013, 4, 2410–2413 CrossRef CAS.
- I. Mintsouli, J. Georgieva, S. Armyanov, E. Valova, G. Avdeev, A. Hubin, O. Steenhaut, J. Dille, D. Tsiplakides, S. Balemenou and S. Sotiropoulos, Appl. Catal., B, 2013, 136–137, 160–167 CrossRef CAS.
- I. Mintsouli, J. Georgieva, E. Valova, S. Armyanov, A. Kakaroglou, A. Hubin, O. Steenhaut, J. Dill, A. Papaderakis, G. Kokkinidis and S. Sotiropoulos, J. Solid State Electrochem., 2013, 17, 435–443 CrossRef CAS.
- S. R. Brankovic, J. X. Wang and R. R. Adzic, Surf. Sci., 2001, 474, L173–L179 CrossRef CAS.
- S. Papadimitriou, S. Armyanov, E. Valova, A. Hubin, O. Steenhaut, E. Pavlidou, G. Kokkinidis and S. Sotiropoulos, J. Phys. Chem. C, 2010, 114, 5217–5223 CrossRef CAS.
- A. J. J. Lennox, P. Bartels, M.-M. Pohl, H. Junge and M. Beller, J. Catal., 2016, 340, 177–183 CrossRef CAS.
- N. Dimitrova, J. Georgieva, S. Sotiropoulos, Tz. Boiadjieva-Scherzer, E. Valova, S. Armyanov, O. Steenhaut, A. Hubin and D. Karashanova, J. Electroanal. Chem., 2018, 823, 624–632 CrossRef CAS.
- R. L. Penn and J. F. Banfield, Geochim. Cosmochim. Acta, 1999, 63, 549–1557 CrossRef.
- A. Vittadini, M. Casarin and A. Selloni, Theor. Chem. Acc., 2007, 117, 663–671 Search PubMed.
- A. Feldhoff, C. Mendive, T. Bredow and D. Bahnemann, ChemPhysChem, 2007, 8, 805–809 CrossRef CAS PubMed.
- X. Ma, Y. Dai, M. Guo and B. Huang, Langmuir, 2013, 29, 13647–13654 CrossRef CAS PubMed.
- V. S. Bagotzky and Y. B. Vassilyev, Electrochim. Acta, 1967, 12, 1323–1343 CrossRef.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B, 1998, 57, 1505–1509 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef CAS.
- P. S. West, R. L. Johnston, G. Barcaro and A. Fortunelli, J. Phys. Chem. C, 2010, 114, 19678–19686 CrossRef CAS.
- E. Aprá, F. Baletto, R. Ferrando and A. Fortunelli, Phys. Rev. Lett., 2004, 93, 065502–065506 CrossRef PubMed.
- L. O. Paz-Barbon, R. L. Johnston, G. Barcaro and A. Fortunelli, J. Chem. Phys., 2008, 128, 134517 CrossRef PubMed.
- R. Ismail and R. L. Johnston, Phys. Chem. Chem. Phys., 2010, 12, 8607–8619 RSC.
- F. Pittaway, L. O. Paz-Borbon, R. L. Johnston, H. Arslan, R. Ferrando, C. Mottet, G. Barcaro and A. Fortunelli, J. Phys. Chem. C, 2009, 113, 9141–9152 CrossRef CAS.
- N. Ruzycki, G. S. Herman, L. A Boatner and U. Diebold, Surf. Sci. Rep., 2003, 529, L239–L244 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford Science, Oxford, UK, 1990 Search PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- C. L. Cleveland and U. Landman, J. Chem. Phys., 1991, 94, 7376–7396 CrossRef CAS.
- A. Rapallo, G. Rossi, R. Ferrando, A. Fortunelli, B. C. Curley, L. D. Lloyd, G. M. Tarbuck and R. L. Johnston, J. Chem. Phys., 2005, 122, 194308 CrossRef PubMed.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |