
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 4-(1H-isochromen-1-yl)isoquinolines through the silver-catalysed homodimerization of ortho-alkynylarylaldehydes and subsequent condensation of the 1,5-dicarbonyl motif with NH3†
Minghui Guoa,
Xin Meng*a,
Yang Zhaoa,
Yuexia Donga,
Xuejun Suna,
Laijin Tian*ab and
Ziping Cao *ab
*ab
aSchool of Chemistry and Chemical Engineering, Qufu Normal University, Qufu 273165, P. R. China. E-mail: caozp_qfnu@163.com; mengxin1108@163.com; laijintian@163.com
bShandong Key Laboratory of Life-Organic Analysis, Key Laboratory of Pharmaceutical Intermediates and Analysis of Natural Medicine, Qufu 273165, P. R. China
First published on 21st January 2019
Abstract
4-(1H-Isochromen-1-yl)isoquinoline derivatives were synthesized in high yields via the AgBF4-catalyzed self-reaction of ortho-alkynylarylaldehydes to give isochromene intermediates, followed by the dehydration of the 1,5-dicarbonyl motif with NH3. Compared with electron-rich aromatic substituents, this strategy can provide the desired isochromene products with an electron-deficient isoquinoline unit. The reactions feature simple experimental operations, mild reaction conditions and high product yields.
Introduction
The 1H-isochromene skeleton is the core structure of many biologically active molecules possessing important pharmaceutical activities, such as the those involved in the inflammatory activation of microglia,1 antimicrobial activity against the Gram-positive bacteria Staphylococcus aureus and Streptococcus pneumoniae,2 selective receptor antagonists of neurokinin-1 (NK1),3 and antagonists of Mycobacterium bovis BCG.4 Isoquinoline derivatives are also a class of important heterocycles having acetylcholinesterase inhibitory activities,5 murine tumor cell cytotoxicity,6 antiplasmodial activities,7 and so on. Thus, compounds with a combination of these two structural motifs could possess important potential bioactivity properties. Therefore, the synthesis of these kinds of product is of great significance.Ortho-alkynylarylaldehydes are a class of important organic intermediates that can be converted into products possessing polycyclic skeletons.8,9 In particular, 1H-isochromenes can be readily constructed through metal- or non-metal-catalyzed reactions.10–17 Pioneering work by Yamamoto and co-workers revealed a Pd(OAc)2-catalyzed reaction of enynals with alcohols to afford alkoxyl-substituted 1H-isochromenes.10i From that time on, many methods were developed to access 1H-isochromene derivatives starting from ortho-alkynylarylaldehydes, with a series of nucleophilic reagents.8 Recently, electron-rich (hetero)arenes, such as alkoxylbenzenes and indoles, have been utilized as nucleophiles leading to the synthesis of functionalized 1H-isochromene compounds with high efficiency (Scheme 1(a)).17 However, electron-deficient heteroarene-substituted 1H-isochromenes could not be synthesized via direct methods due to weak nucleophilic abilities. In this context, and following our ongoing interest in the reactions of enynal substrates,17a,18,21a,c we envisaged that these heteroarene-substituted 1H-isochromenes could be obtained via indirect ways involving the late-stage synthesis of the cyclic skeletons. Herein, we provide an efficient strategy to achieve the synthesis of isoquinoline-substituted 1H-isochromenes through a two-step route, which contains the AgBF4-catalyzed self-reaction of ortho-alkynylarylaldehydes to give intermediate isochromenes with a 1,5-dicarbonyl motif followed by dehydration with NH3 to form the isoquinoline skeleton (Scheme 1(b)). It should be mentioned that the homodimerization of ortho-alkynylarylaldehydes was discovered by Porco Jr et al. in 2007, while only one example was provided for forming the isochromene efficiently.13c The present study exhibited better substrate compatibility and the efficient synthesis of isoquinoline-substituted 1H-isochromenes. Furthermore, the reactions feature mild reaction conditions, simple operation procedures and high efficiency.
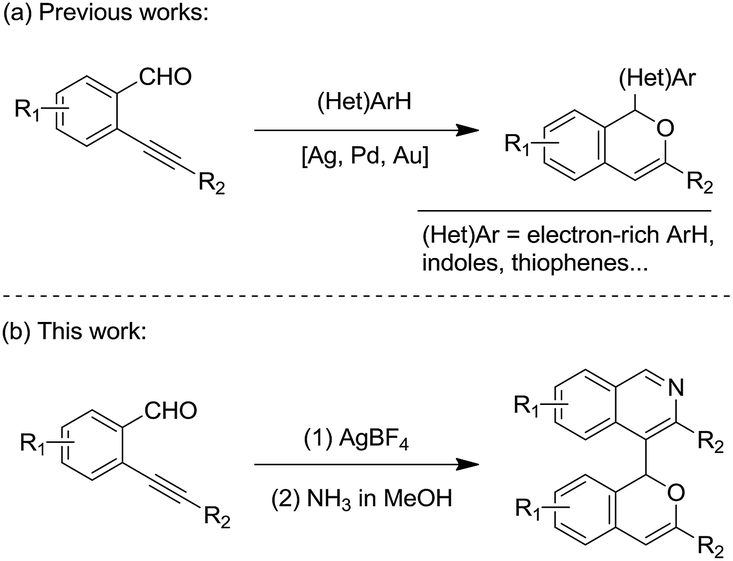 | ||
| Scheme 1 Access to (hetero)aromatic substituted 1H-isochromene derivatives. | ||
Results and discussion
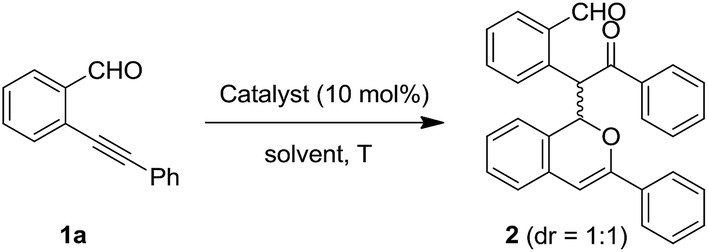
We started from a dimerized product 2, which has a 1H-isochromene ring and a 1,5-dicarbonyl motif. Compound 2 can be obtained in 80% yield as a mixture of two diastereoisomers (dr = 1/1 (ref. 19)) through the self-reaction of the ortho-alkynylbenzaldehyde 1a under conditions involving the presence of silver salt (AgBF4; 10 mol%) with 1,2-dichloroethane (DCE) as the solvent (Table 1, entry 1). Notably, the two isomers of 2 can be isolated carefully via column chromatography. In order to further improve the yield of 2, a series of metal salts were screened (Table 1, entries 2–8). The results showed that AgBF4 was the optimal catalyst. Other silver salts such as AgSbF6 and AgNO3 couldn't afford product 2 at all, whereas the use of an AgOTf catalyst could lead to the formation of 2 in 66% yield (Table 1, entries 2–4). Copper(I) and zinc(II) catalysts showed no catalytic reactivity under the presented conditions (Table 1, entries 5–7). Notably, Pd(OAc)2 could promote the formation of product 2, albeit with a slightly lower yield (Table 1, entry 8). Subsequently, several solvents were investigated, revealing that tetrahydrofuran (THF) was suitable for the present transformation and the yield of 2 could be improved to 89% (Table 1, entries 9–12). Furthermore, the temperature effects indicated that the homodimerization product 2 could be readily obtained in 91–93% yield when the reaction was carried out at room temperature or under heating (Table 1, entries 14–16), while lowering the temperature to 0 °C led to a slightly lower yield (Table 1, entry 13). In order to consider the water effect, control experiments were carried out. The use of completely dry THF led to a low yield of 2 under an N2 atmosphere (Table 1, entry 17). The addition of water (such as 1.0 equiv.) into dry THF did not obviously influence the reaction (Table 1, entry 18). In addition, the reaction couldn't proceed at 100 °C in the absence of AgBF4 (Table 1, entry 19). So, the final conditions were established as those shown by entry 14 in Table 1.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solv. | Temp. (°C) | Yieldb (%) |
| a All reactions were carried out with 1a (0.4 mmol) and catalyst (0.04 mmol, 0.1 equiv.) in the indicated solvent (1.6 mL) under air for about 7 and 8 hours.b The yield of isolated products.c Under an N2 atmosphere.d 1 equiv. of H2O was added. | ||||
| 1 | AgBF4 | DCE | 15 | 80 |
| 2 | AgSbF6 | DCE | 15 | 0 |
| 3 | AgNO3 | DCE | 15 | 0 |
| 4 | AgOTf | DCE | 15 | 66 |
| 5 | CuI | DCE | 15 | 0 |
| 6 | ZnCl2 | DCE | 15 | 0 |
| 7 | ZnBr2 | DCE | 15 | 0 |
| 8 | Pd(OAc)2 | DCE | 15 | 71 |
| 9 | AgBF4 | DCM | 15 | 73 |
| 10 | AgBF4 | THF | 15 | 89 |
| 11 | AgBF4 | CH3CN | 15 | 76 |
| 12 | AgBF4 | 1,4-Dioxane | 15 | 84 |
| 13 | AgBF4 | THF | 0 | 85 |
| 14 | AgBF4 | THF | 25 | 93 |
| 15 | AgBF4 | THF | 45 | 91 |
| 16 | AgBF4 | THF | 60 | 91 |
| 17c | AgBF4 | Dry THF | 25 | 68 |
| 18c,d | AgBF4 | Dry THF | 25 | 92 |
| 19 | — | THF | 100 | 0 |
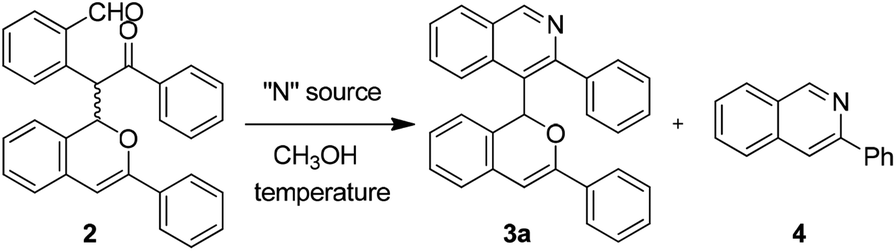
To achieve the cyclization of the 1,5-dicarbonyl motif of compound 2 to an isoquinoline skeleton, different nitrogen sources and reaction conditions were investigated, and these are shown in Table 2. It could be found that the use of NH3 solution in MeOH proved to be suitable, affording the desired product 3a in 62% yield with the decomposed product 4 present in 20% yield (Table 2, entry 3). Other nitrogen sources such as NH4OAc and NH3·H2O could lead to the formation of 3a but with more side-product 4 (Table 2, entries 1 and 2). Temperature studies showed that the cyclized product 3a could be obtained in 87% yield with a lower amount of 4 under room temperature conditions (Table 2, entry 5). In addition, lowering the loading of NH3 solution in MeOH could also add to the yield of 3a to 93%, with trace amounts of side-product 4 (Table 2, entry 7).
|
||||
|---|---|---|---|---|
| Entry | “N” source (equiv.) | Temp. (°C) | Yieldb (%) | |
| 3a | 4 | |||
| a All reactions were carried out with 2 (0.2 mmol) and the nitrogen source (0.2–0.6 mmol, 1.0–3.0 equiv.) in methanol (1.6 mL) at the indicated temperature for about 12 hours.b The yield of isolated products based on 2. | ||||
| 1 | NH4OAc (3) | 50 | 32 | 73 |
| 2 | NH3·H2O (3) | 50 | 47 | 50 |
| 3 | NH3 in CH3OH (3) | 50 | 62 | 20 |
| 4 | NH3 in CH3OH (3) | 40 | 75 | 14 |
| 5 | NH3 in CH3OH (3) | 25 | 87 | 7 |
| 6 | NH3 in CH3OH (3) | 0 | 82 | 7 |
| 7 | NH3 in CH3OH (1.5) | 25 | 93 | — |
| 8 | NH3 in CH3OH (1.0) | 25 | 75 | 8 |
The one-pot synthesis of the isoquinoline 3a with an isochromene unit was also subsequently examined. The AgBF4-catalyzed self-reaction was accomplished and the NH3 source was then injected in a straightforward manner, affording the final product 3a in only 31% yield. To our delight, after the filtration of the metal salt and the evaporation of solvent following the first reaction, the residue was dissolved in methanol and the NH3 source was then added to provide the isoquinoline 3a with higher efficiency (Scheme 2). The low yield for the direct one-pot reaction may be owing to the use of an improper solvent for the second step.
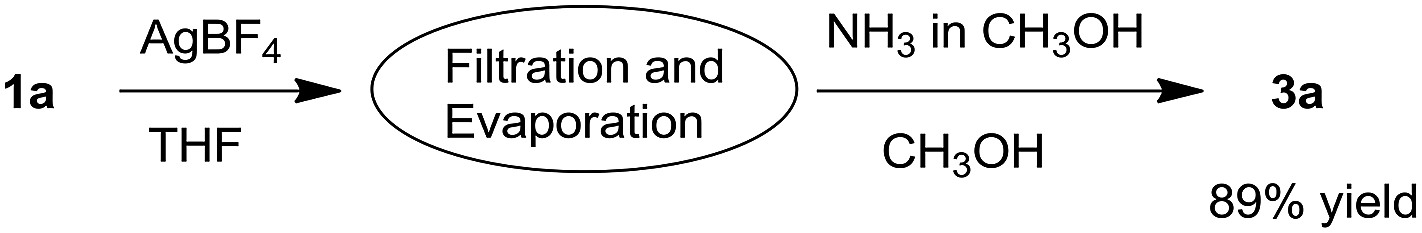 | ||
| Scheme 2 The convenient synthesis of the 4-(1H-isochromen-1-yl)isoquinoline 3a. | ||
With the optimal process for the synthesis of 3a in hand, the scope of ortho-alkynylarylaldehyde substrates was next investigated (Table 3). It could be found that a variety of ortho-alkynylbenzaldehyde derivatives could be applied to the present reaction, leading to the synthesis of the 4-(1H-isochromen-1-yl)isoquinolines 3a–p in 31–91% yields. The substrates 1b–c, having an electron-rich group at the para-position of the alkynylaryl part, were suitable for this transformation, giving the corresponding products 3b–c with good efficiency. The halo-substituted (F, Cl and Br) phenylethynylbenzaldehydes 1d–f could be also converted to the desired isoquinolines 3d–f efficiently. The substrate 3g, with an electron-deficient cyano group, could afford the desired isoquinoline 3g, albeit with a low yield. The meta-substituted-phenylethynylbenzaldehydes 1h–j were also good substrates, leading to the formation of the 4-(1H-isochromen-1-yl)isoquinolines 3h–j in 70–86% yields. In addition, the alkynylarylaldehydes 1k–p, having groups with different electronic natures in the arylaldehyde part, could generate efficiently the cyclized products 3k–p under the current conditions. Alkyl-substituted-alkynylbenzaldehydes such as 1q were also examined. However, the synthesis of the desired polycycle 3q was not observed.
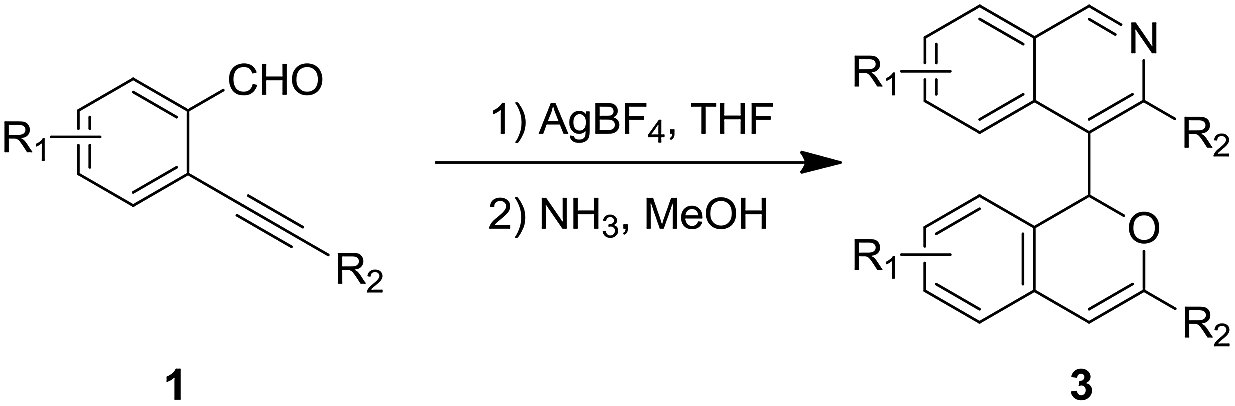

A possible mechanism for the synthesis of the isoquinoline 3a is proposed in Scheme 3.13c The coordination of the alkyne motif of 1a to a silver(I) cation could give complex A, which then undergoes 6-endo-dig cycloisomerization to form the vinyl metal benzopyrylium B. The exo[3+2]-cycloaddition of B could afford the polycyclic intermediate D via C.20,21 The isomerization of the double bonds of D with the departure of Ag(I)+ (or H+) leads to the formation of intermediate E, followed by fragmentation to yield the oxonium species F.22 The addition of water to the oxonium F and its protodemetalation could form the hemiketal G, which could be converted to the ketoaldehyde product 2 via aromatization. The condensation of the 1,5-dicarbonyl motif of 2 with NH3 could afford the final isoquinoline product 3a.
 | ||
| Scheme 3 The proposed mechanism for the formation of 3a. | ||
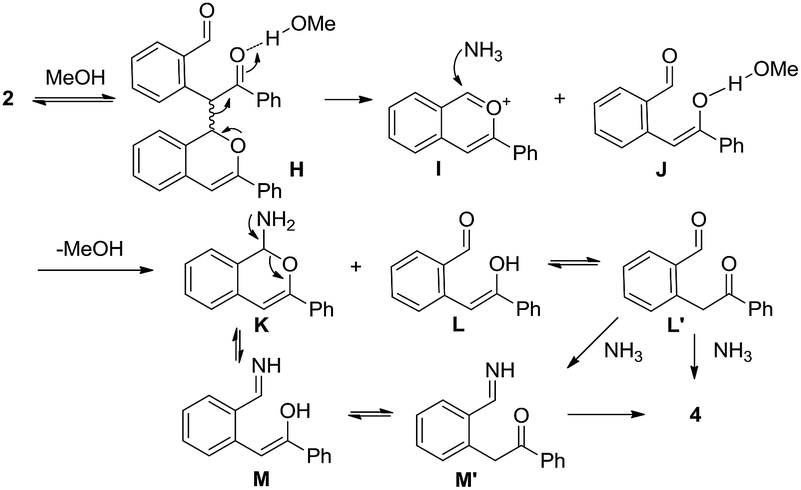
Scheme 4 outlines a proposed pathway for the formation of the isoquinoline 4. The homodimer 2 could give species H through the solvation of methanol to activate the ketocarbonyl, followed by fragmentation (retro-aldol type reaction) to yield the benzopyrylium I and species J.23 The addition of NH3 to I and the desolvation of J could afford the intermediates K and L, respectively. The fragmentation of the N,O-acetal K could give M (or the tautomer M′), leading to the formation of 4 through dehydration. Furthermore, the condensation of L (or the tautomer L′) with NH3 via the intermediate M′ affords the final isoquinoline 4.
 | ||
| Scheme 4 The proposed mechanism for the formation of the side-product 4. | ||
Experimental
Representative procedure for the synthesis of 4-(1H-isochromen-1-yl)isoquinolines
To a stirred solution of the ortho-alkynylbenzaldehyde 1a (82.5 mg, 0.4 mmol, 1.0 equiv.) in THF (1.6 mL), AgBF4 (7.8 mg, 0.04 mmol, 0.1 equiv.) was added at room temperature. The reaction proceeded for about 8 hours. The mixture was filtered using a short silica gel column and volatiles were then evaporated off. The resulting crude 1,5-dicarbonyl 2 was dissolved in methanol (1.6 mL), followed by the injection of a solution of NH3 in methanol (0.15 mL, 2.0 mol L−1, 1.5 equiv.). The mixture was then stirred at room temperature for about 12 hours. After the evaporation of the solvent, the residue was then subjected to column chromatography to afford the desired isoquinoline 3a.Conclusions
In summary, we have developed a new method to prepare 4-(1H-isochromen-1-yl)isoquinoline derivatives in high yields. This method contains two reactions involving the silver-catalyzed self-reaction of ortho-alkynylarylaldehydes and the subsequent condensation of the 1,5-dicarbonyl motif with NH3, without the chromatographic purification of the intermediates. Notably, this method could provide the desired isochromene products with an electron-deficient isoquinoline unit, which can't be obtained through the direct reaction of isoquinolines with ortho-alkynylarylaldehydes. Further work will focus on potential bioactivity studies of these heterocycles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the NSFC (21402106), the Shandong Natural Science Foundation (ZR2018JL013 and ZR2014BQ024), and the Research Start-up Foundation of Qufu Normal University (bsqd20130115).Notes and references
- A. R. Togna, V. Latina, G. Trefiletti, M. Guiso, S. Moschini and G. I. Togna, Brain Res. Bull., 2013, 95, 33 CrossRef CAS PubMed.
- Y. Li, S. Niu, B. Sun, S. Liu, X. Liu and Y. Che, Org. Lett., 2010, 12, 3144 CrossRef CAS PubMed.
- Y. Shishido, H. Wakabayashi, H. Koike, N. Ueno, S. Nukui, T. Yamagishi, Y. Murata, F. Naganeo, M. Mitzutani, K. Shimada, Y. Fujiwara, A. Sakakibara, O. Suga, R. Kusano, S. Ueda, Y. Kanai, M. Tsuchiya and K. Satake, Biomed. Chem., 2008, 16, 7193 CAS.
- C. W. Brown, S. Liu, J. Klucik, K. D. Berlin, P. J. Brennan, D. Kaur and D. M. Benbrook, J. Med. Chem., 2004, 47, 1008 CrossRef CAS PubMed.
- J. Zhang, Q.-Y. Zhang, P.-F. Tu, F.-C. Xu and H. Liang, J. Nat. Prod., 2018, 81, 364 CrossRef CAS PubMed.
- D. J. Milanowski, K. R. Gustafson, J. A. Kelley and J. B. McMahon, J. Nat. Prod., 2004, 67, 70 CrossRef CAS PubMed.
- C. W. Wright, S. J. Marshall, P. F. Russell, M. M. Anderson, D. Phillipson, G. C. Kirby, D. C. Warhurst and P. L. Schiff Jr., J. Nat. Prod., 2000, 63, 1638 CrossRef CAS.
- For leading reviews on the reactions of enynal substrates, see: (a) L. Chen, K. Chen and S. Zhu, Chem, 2018, 4, 1208 CrossRef CAS; (b) J. Ma, L. Zhang and S. Zhu, Curr. Org. Chem., 2016, 20, 102 CrossRef CAS; (c) A. L. S. Kumari, A. S. Reddy and K. C. K. Swamy, Org. Biomol. Chem., 2016, 14, 6651 RSC; (d) H. Wang, Y. Kuang and J. Wu, Asian J. Org. Chem., 2012, 1, 302 CrossRef CAS; (e) N. T. Patil and Y. Yamamoto, ARKIVOC, 2007, 5, 6 Search PubMed; (f) N. Asao, Synlett, 2006, 1645 CrossRef CAS.
- For recent examples, see: (a) H. Luo, R. Liang, L. Chen, H. Jiang and S. Zhu, Org. Chem. Front., 2018, 5, 1160 RSC; (b) M. Michalska, K. Grudzień, P. Małecki and K. Grela, Org. Lett., 2018, 20, 954 CrossRef CAS PubMed; (c) C. R. Reddy and K. Mallesh, Org. Lett., 2018, 20, 150 CrossRef PubMed; (d) X. Zhang, C. Lyu, P. Li, W. Yong, J. Li and X. Zhu, Chin. J. Org. Chem., 2018, 38, 208 CrossRef; (e) S. Qiu, L. Chen, H. Jiang and S. Zhu, Org. Lett., 2017, 19, 4540 CrossRef CAS PubMed; (f) W.-J. Hao, Y. Du, D. Wang, B. Jiang, Q. Gao, S.-J. Tu and G. Li, Org. Lett., 2016, 18, 1884 CrossRef CAS PubMed; (g) T. Arto, P. Fernández, F. J. Fañanás and F. Rodríguez, Chem. Commun., 2016, 52, 13405 RSC.
- For examples with oxygen nucleophiles: (a) S. Handa and L. M. Slaughter, Angew. Chem., Int. Ed., 2012, 51, 2912 CrossRef CAS PubMed; (b) L.-P. Liu and G. B. Hammond, Org. Lett., 2010, 12, 4640 CrossRef CAS PubMed; (c) A. Kotera, J. I. Uenishi and M. Uemura, Tetrahedron Lett., 2010, 51, 1166 CrossRef CAS; (d) T. Godet, C. Vaxelaire, C. Michel, A. Milet and P. Belmont, Chem.–Eur. J., 2007, 13, 5632 CrossRef CAS PubMed; (e) N. T. Patil and Y. Yamamoto, J. Org. Chem., 2004, 69, 5139 CrossRef CAS PubMed; (f) D. Yue, N. D. Cá and R. C. Larock, Org. Lett., 2004, 6, 1581 CrossRef CAS PubMed; (g) S. Mondal, T. Nogami, N. Asao and Y. Yamamoto, J. Org. Chem., 2003, 68, 9496 CrossRef CAS PubMed; (h) J. Barluenga, H. V. Villa, A. Ballesteros and J. M. González, J. Am. Chem. Soc., 2003, 125, 9028 CrossRef CAS PubMed; (i) N. Asao, T. Nogami, K. Takahashi and Y. Yamamoto, J. Am. Chem. Soc., 2002, 124, 764 CrossRef CAS PubMed.
- For examples with nitrogen nucleophiles: (a) A. K. Verma, D. Choudhary, R. K. Saunthwal, V. Rustagi, M. Patel and R. K. Tiwari, J. Org. Chem., 2013, 78, 6657 CrossRef CAS PubMed; (b) G. Dyker, D. Hildebrandt, J. Liu and K. Merz, Angew. Chem., Int. Ed., 2003, 42, 4399 CrossRef CAS PubMed.
- For examples with phosphite nucleophiles: X. Yu, Q. Ding, W. Wang and J. Wu, Tetrahedron Lett., 2008, 49, 4390 CrossRef CAS.
- For examples with activated methylene nucleophiles: (a) D. Malhotra, L.-P. Liu, M. S. Mashuta and G. B. Hammond, Chem.–Eur. J., 2013, 19, 4043 CrossRef CAS PubMed; (b) S. Obika, H. Kono, Y. Yasui, R. Yanada and Y. Takemoto, J. Org. Chem., 2007, 72, 4462 CrossRef CAS PubMed; (c) A. B. Beeler, S. Su, C. A. Singleton and J. A. Porco Jr., J. Am. Chem. Soc., 2007, 129, 1413 CrossRef CAS PubMed.
- For examples with terminal alkyne nucleophiles: X. Yao and C.-J. Li, Org. Lett., 2006, 8, 1953 CrossRef CAS PubMed.
- For examples with allylsilane nucleophiles: (a) S. Bhunia, K.-C. Wang and R.-S. Liu, Angew. Chem., Int. Ed., 2008, 47, 5063 CrossRef CAS PubMed; (b) N. Asao, C. S. Chan, K. Takahashi and Y. Yamamoto, Tetrahedron, 2005, 61, 11322 CrossRef CAS.
- For examples with hydride nucleophiles: (a) E. Tomás-Mendivil, J. Starck, J.-C. Ortuno and V. Michelet, Org. Lett., 2015, 17, 6126 CrossRef PubMed; (b) M. Terada, F. Li and Y. Toda, Angew. Chem., Int. Ed., 2014, 53, 235 CrossRef CAS PubMed; (c) K. Saito, Y. Kajiwara and T. Akiyama, Angew. Chem., Int. Ed., 2013, 52, 13284 CrossRef CAS PubMed.
- For examples with (hetero)arene nucleophiles: (a) H. Zhu, X. Meng, Z. Cao, G. Chen, X. Sun and J. You, Synth. Commun., 2017, 47, 463 CrossRef CAS; (b) G. Mariaule, G. Newsome, P. Y. Toullec, P. Belmont and V. Michelet, Org. Lett., 2014, 16, 4570 CrossRef CAS PubMed; (c) H. Wang, X. Han and X. Lu, Chin. J. Chem., 2011, 29, 2611 CrossRef CAS; (d) B. Ouyang, J. Yuan, Q. Yang, Q. Ding, Y. Peng and J. Wu, Heterocycles, 2011, 82, 1239 CAS; (e) R.-Y. Tang and J.-H. Li, Chem.–Eur. J., 2010, 16, 4733 CrossRef CAS PubMed; (f) J. Barluenga, V.-V. Henar, I. Merino, A. Ballesteros and J. M. González, Chem.–Eur. J., 2006, 12, 5790 CrossRef CAS PubMed; (g) D. Yue, N. D. Cá and R. C. Larock, J. Org. Chem., 2006, 71, 3381 CrossRef CAS PubMed.
- For selected examples: (a) X. Meng, H. Zhu, G. Chen, L. Tian, X. Sun, Z. Cao and J. You, Asian J. Org. Chem., 2017, 6, 921 CrossRef CAS; (b) R. Zhang, H. Zhu, X. Meng, Z. Cao, G. Chen, L. Tian, X. Sun and J. You, Eur. J. Org. Chem., 2017, 2615 CrossRef CAS; (c) H. Zhu, Z. Cao, X. Meng, L. Tian, G. Chen, X. Sun and J. You, Tetrahedron, 2017, 73, 3310 CrossRef CAS; (d) Z. Cao, H. Zhu, X. Meng, J. Guan, Q. Zhang, L. Tian, X. Sun, G. Chen and J. You, Chem.–Eur. J., 2016, 22, 16979 CrossRef CAS PubMed; (e) Z. Cao, R. Zhang, X. Meng, H. Li, J. Li, H. Zhu, G. Chen, X. Sun and J. You, RSC Adv., 2016, 6, 74582 RSC; (f) Z. Cao, H. Zhang, X. Zhang, L. Zhang, X. Meng, G. Chen, X.-E. Zhao, X. Sun and J. You, RSC Adv., 2015, 5, 103155 RSC.
- The dr value of 2 was determined by 1H NMR analysis of crude reaction mixtures..
- The intermediate B could be very prone to protodemetalation, also see (a) J. T. R. Liddon, M. J. James, A. K. Clarke, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Chem.–Eur. J., 2016, 22, 8777 CrossRef CAS PubMed; (b) J. T. R. Liddon, J. A. Rossi-Ashton, A. K. Clarke, J. M. Lynam, R. J. K. Taylor and W. P. Unsworth, Synthesis, 2018, 50, 4829 CrossRef CAS.
- For the case of carbenoid type intermediates, see ref. 13c, also, (a) Z. Cao, H. Zhu, X. Meng, L. Tian, X. Sun, G. Chen and J. You, Chem.–Eur. J., 2016, 22, 9125 CrossRef CAS PubMed; (b) R. Liang, T. Ma and S. Zhu, Org. Lett., 2014, 16, 4412 CrossRef CAS PubMed; (c) Z. Cao, H. Zhu, X. Meng, L. Tian, G. Chen, X. Sun and J. You, J. Org. Chem., 2016, 81, 12401 CrossRef CAS PubMed.
- Another possible mechanism involving the direct self-addition of intermediate B (or protodemetalated type) to give F could not be ignored..
- For leading examples on acid or solvent-promoted retro-Aldol reaction, see: (a) K. L. Ivanov, I. M. Vatsouro, S. I. Bezzubov, M. Y. Melnikov and E. M. Budynina, Org. Chem. Front., 2018, 5, 1655 RSC; (b) M. Sasaki, K. Goto, K. Tajima, T. Adschiri and K. Arai, Green Chem., 2002, 4, 285 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analysis data for new compounds. See DOI: 10.1039/c8ra09269a |
| This journal is © The Royal Society of Chemistry 2019 |