
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAggregation-induced chiroptical generation and photoinduced switching of achiral azobenzene-alt-fluorene copolymer endowed with left- and right-handed helical polysilanes†
Hailing Chena,
Lu Yina,
Meng Liua,
Laibing Wangb,
Michiya Fujiki *b,
Wei Zhang*a and
Xiulin Zhuac
*b,
Wei Zhang*a and
Xiulin Zhuac
aSuzhou Key Laboratory of Macromolecular Design and Precision Synthesis, Jiangsu Key Laboratory of Advanced Functional Polymer Design and Application, State and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou Industrial Park, Suzhou 215123, China. E-mail: weizhang@suda.edu.cn
bDivision of Materials Science, Nara Institute of Science and Technology, 8946-5, Takayama, Ikoma, Nara 630-0192, Japan. E-mail: fujikim@ms.naist.jp
cGlobal Institute of Software Technology, No. 5 Qingshan Road, Suzhou National Hi-Tech District, Suzhou 215163, China
First published on 8th February 2019
Abstract
The left and right helicities of azobenzene (Azo)-containing main-chain polymer (PF8Azo) were successfully controlled with an enantiomeric pair of rigid rod-like helical polysilanes carrying (S)- and (R)-2-methylbutyl groups (PSi-S and PSi-R, respectively) as their hetero-aggregates in a mixture of chloroform and methanol solvents and in the solid state. Optimizing the good and poor cosolvents and their volume fractions showed that the molar ratio of PF8Azo to PSi-S/-R and the molecular weight of PF8Azo were crucial to boost the CD amplitudes of PF8Azo/PSi-S and PF8Azo/PSi-R hetero-aggregates. The photoresponsive trans–cis transformation caused noticeable changes in the sign and magnitude of the chiroptical behavior due to the hetero-aggregates. Moreover, the optically active PF8Azo homo-aggregates were produced by complete photoscissoring reactions at 313 nm, which could be assigned to the Siσ–Siσ* transitions of PSi-S and PSi-R.
1 Introduction
Chirality plays key roles in living organisms, medicines, toxins, aromas and flavors (bitter and sweet), immunity, chiroptical functional materials and devices, and the origin of life.1–6 Generally, several chemical and physical origin biases are applied to efficiently generate and control the chirality of desired materials when achiral starting materials are employed. However, chiral chemical sources are usually well-defined molecules that specifically work toward achiral substances operated at controlled reaction conditions, while chiral physical sources are non-specific and often inefficient.To obtain more versatile and efficient chiral biases, scientists have investigated several chiral stimuli. For example, two types of chiral lights (circularly polarized light due to angular momentum and vortex light due to orbital angular momentum),7–15 chiral nematic liquid crystals and chiral terpenes,16–30 chiral molecules with functional groups,31,32 hydrodynamic swirling flow,33,34 and biological and artificial helical polymers35 induce supramolecular chirality when achiral and optically inactive molecules and polymers are employed. These driving forces are non-covalent intermolecular weak forces including π–π, C–H/π, C–H/O, C–H/F, cation/π, dipole–dipole, and van der Waals (London dispersion) interactions.
Among these strategies, chiral biological and synthetic molecules have been used as helix-inducible scaffolds and templates to construct supramolecular chiral and/or helical architectures from achiral or optically inactive macromolecular sources.
For example, the helical architecture of poly(1-phenylacetylene)-bearing functional groups was inducible in the presence of several chiral amines as scaffolds. This architecture displayed memory effect after removal of the chiral molecules, followed by replacement of achiral amines and amino alcohols.36,37 Notably, the memory effect is greatly affected by the number of methylene spacers in the achiral molecules. Also, supramolecular chirality of achiral porphyrin derivatives carrying long alkyl chains can be induced during the formation of a co-gel with a glutamate derivative acting as an efficient gelator in DMSO.38 Liu et al. and Ihara et al. demonstrated the transfer of molecular chirality to supramolecular gels using a series of chiral amphiphiles derived from L- and D-glutamide.39–42 Recently, one of the authors (WZ) reported for the first time that two achiral π-conjugated polymers (PSi8 and PCz8) could result in the corresponding helical co-gels with the help of L- and D-glutamide gelators through multiple weak interactions between the long n-alkyl chains, and the helicity of PSi8 and PCz8 was also maintained after the removal of the gelators despite the loss of weak interactions.43
Furthermore, naturally occurring and artificial chiral/helical compounds (e.g., DNA, polysaccharides, nucleic acids, synthetic polymers, oligopeptides, and carbon nanotubes (CNTs)) can serve as chirality-inducible templates and scaffolds.44–54 Recently, researchers found that a DNA-based origami supramolecular polymer was an efficient template to dynamically control the superstructure of gold nanorods, as shown by the changes in the chiroptical properties.55
Among the naturally occurring chiral/helical compounds, cellulose is the most abundant polysaccharide on earth. Recently, soluble cellulose triacetate (CTA) and cellulose acetate butyrate (CABu) were reported to be efficient scaffolds capable of transferring their chirality and/or helicity to achiral/non-helical semi-flexible non-charged oligo- and poly(dialkyfluorene)s without chiral catalysts.52–54 This strategy is of particular significance because of the established linkage between chiral biopolymers and artificial achiral/racemic materials.
Recently, one of the authors (MF) reported that non-charged poly(n-hexyl-(S)-2-methylbutylsiane) (PSi-S) and poly(n-hexyl-(R)-2-methylbutylsiane) (PSi-R) function as photoscissorable helical platforms to generate circularly polarized luminescent (CPL)-active and circular dichroism (CD)-active poly(dioctylfluorene) (PF8),56 poly(dioctyfluorene-alt-bis(thiophenyl)benzothiazole) (PF8DBT)57 and poly(dioctyfluorene-alt-bithiophene) (PF8T2)58 dispersed in a mixture of achiral good and poor solvents. Because of their photoscissorable nature, the CPL and CD activities of the resulting π-conjugated polymers were maintained even after the removal of helical PSis.
To further verify the scaffolding capability of PSi-R and PSi-S, we chose non-charged, non-helical poly((9,9-dioctylfluorenyl-2,7-diyl)-alt-4,4′-azobenzene) (PF8Azo, Fig. 1) as a photoinduced trans–cis isomerizable polymer. Azobenzene (Azo) derivatives are commonly used as photoresponsive building blocks, and their reversible photoinduced trans–cis isomerization causes significant changes in their physical and chemical properties.25–30,59 The combination of the chiroptical properties and photoisomerisability efficiently provides reversible chiroptical switching behavior. Herein, we reported unique photoinduced change in CD-active PF8Azo hetero-aggregates endowed with PSi-S and PSi-R under optimized conditions. The CD amplitudes of the PF8Azo aggregates were maintained even after the photoscissoring of PSi-S and PSi-R at 313 nm at the Si-Si main chain.
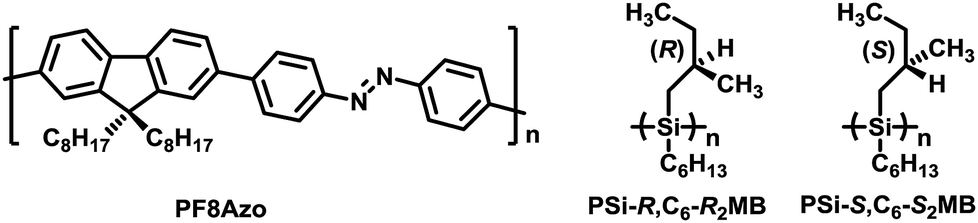 | ||
Fig. 1 Chemical structures of PF8Azo (Mn = 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 Da, Mw/Mn = 1.61, DPn = 42), PSi-R (Mn = 23900 Da, Mw/Mn = 1.64, DPn = 128), PSi-S (Mn = 20400 Da, Mw/Mn = 1.31, DPn = 110). 900 Da, Mw/Mn = 1.61, DPn = 42), PSi-R (Mn = 23900 Da, Mw/Mn = 1.64, DPn = 128), PSi-S (Mn = 20400 Da, Mw/Mn = 1.31, DPn = 110). | ||
2 Experimental section
Materials
Characterisation
980]/absorbance at the CD extremum of the aggregates. The gCD value refers to the degree of circular polarization in the ground state of the aggregates.Instrumentation
The CD and UV-vis spectra of the solution were obtained simultaneously on a JASCO (Hachioji-Tokyo, Japan) J-815 spectropolarimeter with a Peltier-controlled apparatus to control the sample temperature. A quartz cuvette (SQ-grade) with a 10 mm path length was used. The parameters for obtaining the CD/UV-vis spectra are as follows: scanning rate = 100 nm min−1, bandwidth = 2 nm, response time = 2 seconds, single accumulation and 1 nm interval sampling. The number-average molecular weight (Mn) and polydispersity (Đ = Mw/Mn) of the polymers were characterized by a TOSOH HLC-8320 gel permeation chromatograph (GPC) (Tokyo, Japan) equipped with a refractive index and UV detectors using two TSKgel Super Multipore HZ-N columns (4.6 × 150 mm, 3 μm particle size) enabling molecular weight analysis ranging from 7 × 102 to 2 × 105 g mol−1. THF was used as an eluent at a flow rate of 0.35 mL min−1 at 40 °C. These samples were calibrated with polystyrene standards (TOSOH). The recycling preparative HPLC Mode LC-9260NEXT equipped with a manual injector, UV-vis 4ch (200–800 nm) and RI-700NEXT detectors, using JAIGEL-2HH and JAIGEL-2.5HH (PS/DVB packing material) columns, was used to obtain a broad range of molecular weights of PF8Azo. The flow rate of THF is 6 mL min−1 at room temperature. Dynamic light scattering (DLS) measurements were performed with a Zetasizer Nano ZS instrument (Malvern, Worcestershire, UK) at room temperature. An ultra-high-pressure 500 W Hg lamp (Optiplex BA-H501 and USH-500SC2, Ushio [Tokyo, Japan]) with three narrow band-pass filters at 313 nm, 405 nm and 546 nm was irradiated to destruct PSi-S/-R and to photoisomerize the hetero-aggregate as a suspension in a cuvette at room temperature. The irradiation intensity was characterised as 128 μW cm−2 at 313 nm, 159 μW cm−2 at 405 nm and 589 μW cm−2 at 546 nm using a broadband photodetector Ophir Optronics with Nova with photodiode head PD300-UV (Tel-Aviv, Israel).3 Results and discussion
Emerging CD-active PF8Azo induced by chirality/helicity of PSi-S and PSi-R
Fig. 2 shows the distinct bisignate CD bands at 305 and 322 nm that are characteristic of an exciton couplet of an Siσ–Siσ* transition arising from the helically assorted aggregates of PSi-S and PSi-R. However, it should be noted that additional negative/positive CD bands at the corresponding broader UV-vis spectra in the range of 350–550 nm originated from the π–π* transition of the PF8Azo aggregate itself. Meanwhile, the signs of these CD bands were directly related to the chirality/helicity of employed PSi. The gCD values at 475 nm increased to +9.1 × 10−3 for PF8Azo/PSi-R and −10.5 × 10−3 for the PF8Azo/PSi-S hetero-aggregate. These spectral characteristics of the PSi-PF8Azo hetero-aggregates were nearly consistent with those of chiral light-induced polymer homo-aggregates.9,10 For comparison, the CD and UV-vis spectra of PF8Azo, PSi-S and PSi-R homo-aggregates were obtained after dispersion in CHCl3–MeOH (2/1, (v/v)) cosolvent at room temperature; the spectra are shown in the ESI (Fig. S1†). Evidently, PF8Azo homo-aggregates did not reveal a helix preference, while the PSi-S and PSi-R homo-aggregates exhibited significant bisignate CD band characteristics due to the exciton couplet in the near-UV region.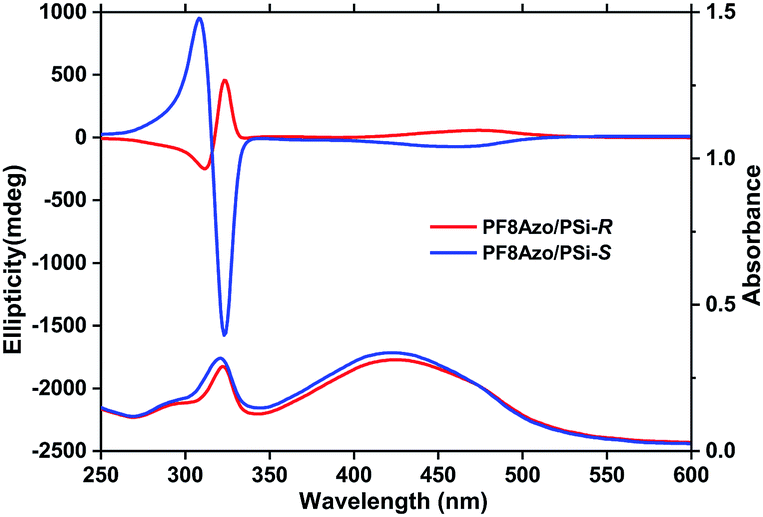 | ||
| Fig. 2 The CD and UV-vis spectra of PF8Azo/PSi-S (blue line) or PF8Azo/PSi-R (red line) hetero-aggregates. The hetero-aggregates were present at 1-to-1 molar ratio as repeating units in CHCl3–MeOH (2.0/1.0, (v/v)) cosolvent. The final concentrations of [PSi-S/-R]0 and [PF8Azo]0 were 1.0 × 10−5 M and 1.0 × 10−5 M, respectively. The single sign of the broad CD bands in the visible region was assigned to the π–π* transitions of the PF8Azo main chain induced by the chirality/helicity of the rigid rod-like helical PSi-S and PSi-R. | ||
These comparative measurements indicated that the helicity/chirality of PSi-S/-R can efficiently transfer to non-charged PF8Azo during hetero-aggregation. Clearly, the chirality/helicity of the hetero-aggregate could be maintained and increased by embedding in silicone grease (Fig. S2, ESI†), as indicated by a comparison of the corresponding gCD values. The gCD values at λext (320 nm) of PF8Azo/PSi-S and PF8Azo/PSi-R hetero-aggregates were −27.6 × 10−3 and +4.6 × 10−3 in the cosolvent, respectively, and −26.4 × 10−3 and +7.0 × 10−3 in the grease, respectively. However, the vinylpolymer containing azo-side-chain (PMMAzo) did not produce the corresponding CD-active PMMAzo during hetero-aggregation with PSi-S in any mixtures of CHCl3 and MeOH cosolvents (Fig. S3, ESI†). A plausible explanation is that floppy PMMAzo could not wrap efficiently around the semi-flexible PSi-S helical main chains due to inefficient intermolecular interactions. Also, PF8Azo that is a semiflexible π-conjugated polymer efficiently interacted with semi-flexible PSi-S. The main rigidity of two polymers was assumed to be crucial.
To further optimize the hetero-aggregation conditions, we determined the best volume fractions of several good and poor solvents associated with the best molar ratio of PF8Azo to PSi-S/-R.
First, we systematically surveyed CHCl3 and THF as good solvents, while alcoholic solvents (ethanol, MeOH and isopropanol) and n-hexane were chosen as poor solvents. A series of CD/UV-vis spectral data sets (Fig. S4, ESI†) showed that the spectral amplitudes of the PF8Azo/PSi-S hetero-aggregate are very weak or nearly CD-silent except for the CHCl3–MeOH cosolvent. Alcoholic solvents were preferred over n-hexane as poor solvents to efficiently produce the CD-active hetero-aggregates.
Next, we optimized the molar ratios of the repeating units between PF8Azo and PSi-S/-R in their hetero-aggregates to reach the greatest CD amplitudes in the best volume fraction of the CHCl3 and MeOH (2:1) cosolvents. The gCD values as a function of the volume fractions of CHCl3 and MeOH cosolvents are shown in Fig. 3. Also, the gCD values as a function of hetero-aggregates, known as the Job's plot,60 are given in Fig. 4. The data were taken from the scattering-free CD spectra in Fig. S5 and S6 in the ESI.†
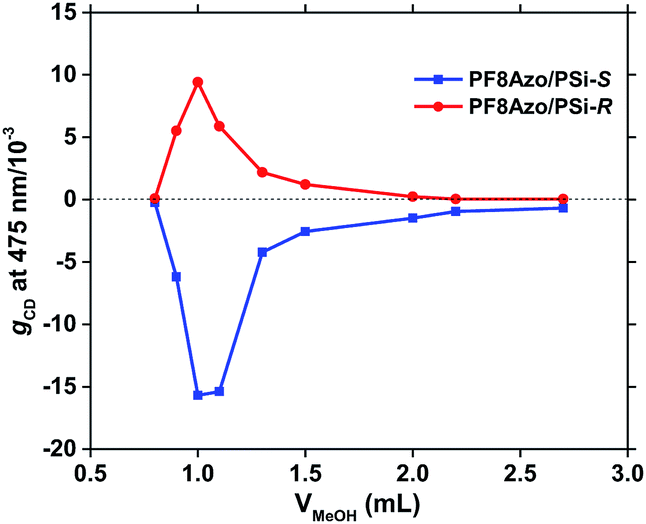 | ||
| Fig. 3 The gCD values at λext of 475 nm of PF8Azo/PSi-S (blue) and PF8Azo/PSi-R (red) hetero-aggregates as a function of the volume fraction of MeOH (poor solvent) in CHCl3 (good solvent) cosolvents (total volume: 3 mL). The molar ratio was 1:1. [PSi-S/-R]0 = [PF8Azo]0 = 1.0 × 10−5 M in the cuvette. | ||
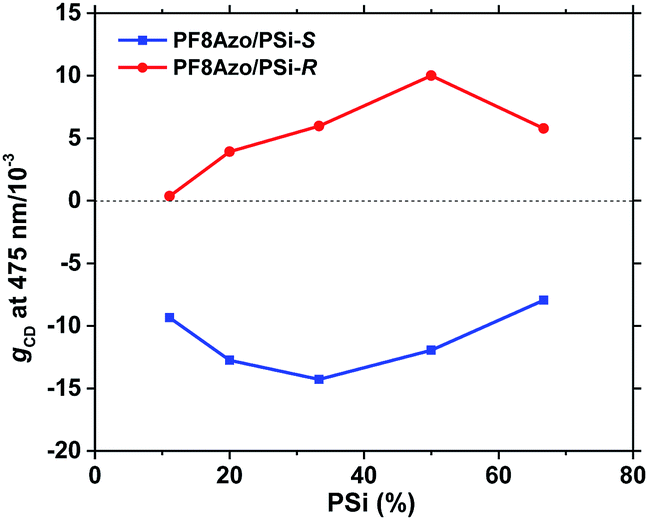 | ||
| Fig. 4 The gCD value at 475 nm vs. the molar ratio of PSi-S/-R in the PF8Azo/PSi-S (blue) and PF8Azo/PSi-R (red) hetero-aggregates. [PF8Azo]0 = 1.0 × 10−5 M. The volume fraction of CHCl3 and MeOH was 2:1 (total volume: 3 mL) in all experiments. | ||
From Fig. 3, we observe that the gCD value of the hetero-aggregates is greatly enhanced at a very specific volume fraction of CHCl3 (2.0 mL) and MeOH (1.0 mL) when the molar ratio of PSi-S/-R and PF8Azo is fixed at 1:1. This anomaly is ascribed to an optofluidic effect61 due to photon confinement in a polymeric optical cavity reported in several polymer aggregate systems. Alternatively, the Job's plot in Fig. 4 indicates that the PF8Azo/PSi-S/-R hetero-aggregates do not obey a sergeant-and-soldier scenario.62 The gCD values of the hetero-aggregates increased at very specific molar ratios: 1:1 for PSi-S and PF8Azo and 1:2 for PSi-R and PF8Azo. The difference in this ratio could be attributed to subtle differences in their Mn and Đ or other unresolved reasons.
Meanwhile, we characterized the hydrodynamic particle sizes of the PSi-S and PF8Azo homo-aggregates at different volume fractions of CHCl3–MeOH cosolvents by DLS methods (Tables S1 and S2, ESI†) to monitor the change in the sizes of the homo-aggregates. The particle size of the homo-aggregates gradually decreased as the fraction of CHCl3 increased. A mixture of PSi-S and PF8Azo (1:1) and a mixture of PSi-R and PF8Azo (1:1) produced similar sizes (≈700 nm) of hetero-aggregates (Fig. 5). From these results, we concluded that the helicity of the non-helical and non-charged PF8Azo is successfully induced by the helicity/chirality of non-charged PSi-S/-R during hetero-aggregation in CHCl3–MeOH cosolvents.
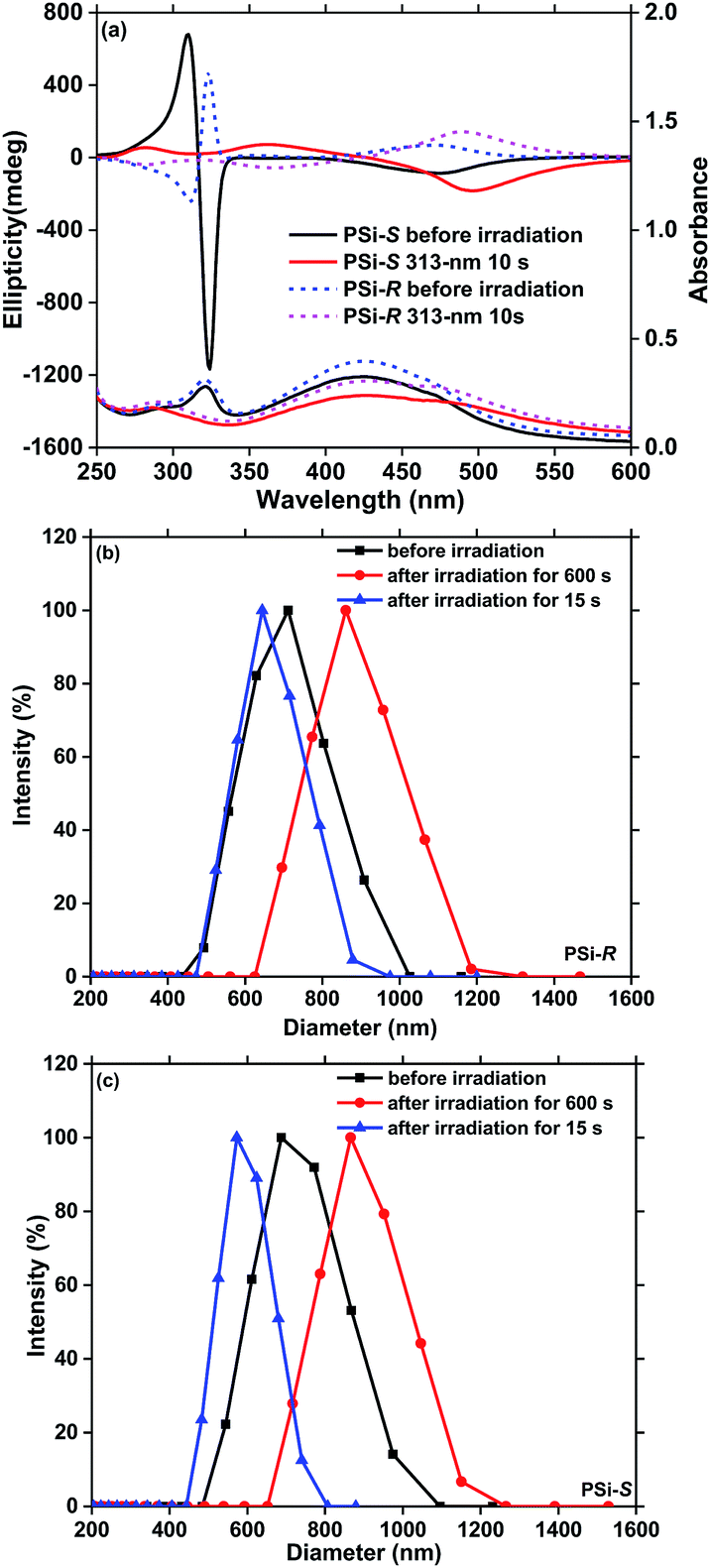 | ||
| Fig. 5 (a) Changes in the CD and UV-vis spectra of the PF8Azo/PSi-R and PF8Azo/PSi-S hetero-aggregates before and after the irradiation at 313 nm. Comparisons of hydrodynamic sizes of hetero-aggregates for (b) PF8Azo/PSi-R (c) PF8Azo/PSi-S before (black line) and after the 313 nm irradiation for 15 s (blue line) and 600 s (red line) obtained via DLS. The hetero-aggregates were produced with a volume ratio (CHCl3–MeOH) of 2:1 and a molar ratio of 1:1 ([PSi-S/-R]0 = [PF8Azo]0 = 1.0 × 10−5 M). | ||
Si–Si bond selective photoscissoring reaction of PSi in the PF8Azo/PSi hetero-aggregates
Previous studies have demonstrated the helix scaffolding capability of PSi-S and PSi-R in the presence of non-charged achiral π-conjugated polymers by rapid photoscissoring of several polysilanes upon near-UV irradiation at the Siσ–Siσ* transition.56–58,63–65 The result prompted us to further explore the photoscissoring characteristics of rigid helical PSi-S and PSi-R. The hetero-aggregates of PF8Azo/PSi-S and PF8Azo/PSi-R exhibited originally intense bisignate (+/−) or (−/+) CD characteristics at ≈320 nm due to the sharper Siσ–Siσ* bands that are parallel to the main chain axis of the Si–Si bond, as seen in Fig. 5(a). Upon irradiation at 313 nm for 10 s, the bisignate CD bands and the corresponding Siσ–Siσ* bands significantly weakened, followed by a distinct red shift (≈10–20 nm) at the first Cotton band (≈460 nm) of the PF8Azo aggregate (Fig. 5(a)). These results indicated that PSi-S and PSi-R decomposed rapidly upon irradiation at 313 nm within 10 s. Enhanced CD signal amplitudes associated with the noticeable red-shift of the PF8Azo/PSi-S and PF8Azo/PSi-R aggregates suggested a significant structural transition from ill-ordered to well-ordered π–π stacks of PF8Azo by photochemically removing the polysilanes. The origin of the red shift of PF8Azo indicated the production of J-aggregation. Although the near-UV irradiation allowed efficient decomposition of the polysilanes, the chirality of PF8Azo was maintained. The bisignate CD spectral profiles of the PF8Azo aggregates were nearly mirror symmetric (Fig. 5(a)), while the corresponding π–π* CD bands were monosignate spectral profiles for the hetero-aggregates before PSi-S and PSi-R were photochemically removed (Fig. 2).Next, we optimized the best irradiation time of the near-UV light source to fully decompose PSi-S and PSi-R and to avoid undesirable trans-to-cis transformation of PF8Azo. Sequential alterations in the CD spectral data associated with the aggregate sizes of the PF8Azo/PSi-S and PF8Azo/PSi-R hetero-aggregates as a function of irradiation time (5 s, 10 s, 15 s and 600 s) of the 313 nm light source are given in Fig. 5(a), S7, S8 and Table S3.† Importantly, PSi-S and PSi-R decomposed within 5 s. The polysilanes substantially decomposed after 15 s. A prolonged UV-irradiation (600 s) led to definitive decomposition of these polysilanes based on the complete disappearance of the Siσ–Siσ* bands, while the magnitude of the CD-active π–π* bands due to PF8Azo progressively increased and red shifted. These CD/UV-vis spectral alterations at the Siσ–Siσ* and π–π* bands were almost identical to previous results for the non-azobenzene π-conjugated polymers with PSi-S and PSi-R.56–58 However, uncharacterized CD-silent species remaining in the solution may afford CD-active PF8Azo.
To characterize the hydrodynamic sizes of the aggregates, we analysed the sizes of the F8Azo/PSi-R (Fig. 5(b)) and F8Azo/PSi-S (Fig. 5(c)) hetero-aggregates for three irradiation times (0 s, 15 s, and 600 s) of the 313 nm light source by means of the DLS method. Clearly, the original sizes of these hetero-aggregates greatly changed at 15 s and 600 s.
The initial size of the PF8Azo/PSi-R hetero-aggregate was ≈650 nm, followed by slight reduction in the size (≈620 nm) after 15 s of UV irradiation. Eventually, the aggregate size increased slightly to ≈810 nm after 600 s of UV irradiation. Similarly, the initial size (≈690 nm) of F8Azo/PSi-S hetero-aggregate reduced to ≈580 nm, followed by increase to ≈830 nm after 600 s of 313 nm irradiation. The decreases in the aggregate size were due to the photoscissoring reaction at 313 nm, rapidly breaking the Si–Si bond, followed by dissolution of the resulting fragments with low molecular weights from the aggregates to the surrounding cosolvents. This scenario is similar to a rinsing process with proper solvents when positive-type photoresist materials are employed.
Although these fragments may contribute to smaller aggregates, the increase in the aggregate size is due to the so-called Ostwald ripening,66 which increases the initial size of the aggregate after the removal of polysilane with the help of the prolonged UV light irradiation and increasing time. By dissolving the smaller aggregates into the solution, the larger aggregates are easy to grow, resulting in apparent increase in the aggregate size.
The Ostwald ripening scenario indicates that (i) smaller particles are easily dissolved into solution but larger particles are not; (ii) smaller particles can deposit onto the surface of larger particles; and (iii) larger particles become much larger, while smaller particles disappear. Actually, in the cases of the homo-aggregates, the hydrodynamic aggregate sizes increased with increasing time (Table S4, ESI†).
Dual wavelength-controlled photoisomerization of PF8Azo in the PF8Azo/PSi hetero-aggregate – all photon-mode chiroptical switching and memory
The advantages of azobenzene-containing polymers, oligomers, and molecules are reversibility and smart functions in photophysical and photochemical properties owing to the trans–cis isomerization of the azobenzene framework in response to external photochemical and thermal biases. Previously, we demonstrated thermo-and photoisomerization of the main-chain and side-chain azo-containing polymers in cosolvents. PF8Azo dissolved in CHCl3 showed clear trans–cis–trans isomerization capability by applying two different wavelengths of 405 nm and 546 nm (Fig. S9, ESI†). These results prompted us to further investigate the chiroptical switching characteristics of CD-active PF8Azo aggregates as the main-chain type azo polymer was endowed with chirality/helicity of PSi-S and PSi-R.Fig. 6 shows the changes in the gCD values at 475 nm of trans-PF8Azo/PSi-R (red line) and trans-PF8Azo/PSi-S (blue line) hetero-aggregates in CHCl3–MeOH cosolvent by applying two different wavelengths of 405 nm and 546 nm. Importantly, the 405 nm and 546 nm wavelengths were insensitive to PSi-S and PSi-R due to the lack of the corresponding transitions in the visible region. For comparison, to obtain the corresponding cis-PF8Azo/PSi-R and cis-PF8Azo/PSi-S hetero-aggregates, cis-PF8Azo in homogeneous CHCl3 solution was produced by photoirradiation at 405 nm light, followed by hetero-aggregation with PSi-S and PSi-R. These changes in cis- and trans-PF8Azo can be recognized by the noticeable changes in the gCD values along with the corresponding CD/UV-vis spectral profiles (Fig. S10, ESI†). These cis-PF8Azo/PSi aggregates were initially CD-silent states at the π–π* transition in the visible region. The subsequent 546 nm irradiation for 5 min produced the corresponding trans-PF8Azo/PSi hetero-aggregates. Scheme S1† shows the proposed mechanism for this interesting phenomenon, which involves the cis–trans photoisomerization of the azobenzene group causing changes in the gCD value.68,69
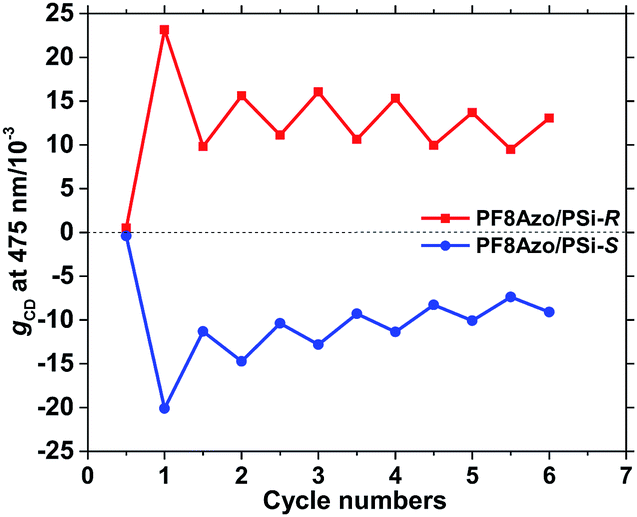 | ||
| Fig. 6 The gCD values at 475 nm of PF8Azo/PSi-R (red line) and PF8Azo/PSi-S (blue line) hetero-aggregates upon photoirradiation cycles at 405 nm and 546 nm in CHCl3–MeOH cosolvent (2.0/1.0, (v/v)). Irradiation time: 5 min. ([PSi-S or PSi-R]0 = [PF8Azo]0 = 1.0 × 10−5 M). A CHCl3 solution of PF8Azo was irradiated for 15 min at 405 nm, followed by aggregation with PSi-S and PSi-R in CHCl3–MeOH cosolvent to obtain the cis-PF8Azo/PSi aggregates. | ||
These results allowed us to further investigate the chiroptical switching capability of the CD-active PF8Azo ‘homo-aggregate’. The scaffold-induced chiral system can be manifested by the photoresponse results of PF8Azo. By alternating the photoirradiation of the hetero-aggregate between 405 nm and 546 nm, the trans-to-cis-to-trans isomerization was partly possible because the process is not fully reversible. The reason for this incomplete process is the Ostwald ripening effect during irradiation in the visible region. A modern concept of optofluidics67 indicates that external and internal lights cannot penetrate large-sized aggregates consisting of π-conjugated chromophores with high absorptivity according to the Lambert–Beer law. Even prolonged photoirradiation did not allow complete cis–trans isomerization.
As a comparison, we investigated the chiral side chain effect using two different helical polysilanes to realize the reversibility of photoisomerization. We tested semi-flexible helical polysilane, C6-S3MePe (Fig. S11(a), ESI†), in place of rigid helical PSi-S (C6-S2MB). The side-chain branching of C6-S3MePe is located at the γ-position from the Si–Si bond, while that of PSi-S (C6-S2MB) is located at the β-position. The subtle difference in the branching points definitively determined the main chain flexibility.63 First, we optimized the volume fraction of the CHCl3–MeOH cosolvents and the molar ratio of PF8Azo-to-C6-S3MePe to afford significantly boosted Cotton effect (Fig. S11(b) and (c), ESI†). The optimum conditions for PF8Azo/C6-S3MePe were the same as those of PF8Azo/PSi-S (C6-S2MB) for subsequent photoisomerization studies, but the absolute (+)-sign gCD values around ≈480 nm of PF8Azo greatly decreased.
Owing to C6-S3MePe, by alternatively irradiating at 405 nm and 546 nm, the main-chain chirality of PF8Azo around 470 nm of the cis-PF8Azo/C6-S3MePe hetero-aggregate was reversed compared to that of the trans-PF8Azo/PSi-S (C6-S2MB) hetero-aggregate. A similar cis–trans transformation occurred in response to the alternate photoirradiation at 405 nm and 546 nm of the hetero-aggregates (Fig. S11(d), ESI†).
Next, we investigated the concentration dependence of PF8Azo and the molar fraction of PSi-S for the PF8Azo/PSi-S hetero-aggregate (Fig. S12, ESI†). The photoisomerization capability was similar to the previous conditions mentioned before because a partial cis–trans transformation change occurred similarly. However, a PF8Azo sample with a lower molecular weight of 7300 Da (Mn,GPC) showed a suppressed CD signal associated with the corresponding UV-visible absorption due to PF8Azo that greatly decreased with the irradiation time; the bisignate CD bands due to the Siσ–Siσ* bands were nearly unchanged (Fig. S13, ESI†). The cis-PF8Azo aggregate did not undergo any red shift even after 546 nm irradiation. The cis–trans photoisomerization of PF8Azo was not significant even upon alternating the irradiation between two wavelengths. Possibly, PF8Azo having lower molecular weight has poor encapsulation capability toward rigid helical PSi-S.
Marked Mn effect of CD-active PF8Azo
To know how Mn of PF8Azo affects the CD amplitude of the resulting PF8Azo/PSi hetero-aggregates, indicating the scaffolding capability of PSi-S and PSi-R to PF8Azo, we investigated the relationship between Mn of PF8Azo and the resulting gCD magnitude of PF8Azo/PSi hetero-aggregates when the Mns values of PSi-S and PSi-R were fixed. Fractionated PF8Azo samples were obtained via cyclic preparative HPLC. The molar ratios of PF8Azo/PSi-S and PF8Azo/PSi-R were fixed at 1:1. The volume fractions of CHCl3 and MeOH were varied to efficiently enhance the Cotton effect of PF8Azo with different Mn values of the hetero-aggregates. As the value of Mn increased, the solubility of PF8Azo decreased. Thus, the volume fraction of CHCl3 gradually increased to efficiently boost the corresponding CD signal of the hetero-aggregate. Generally, polymers having lower Mn values show better solubility than those having higher Mn values. Nevertheless, from Table S1† and the CD/UV-vis spectra (Fig. S14, ESI†), we consider that the aggregations of PSi-S and PSi-R were incomplete when the volume fraction of MeOH was less than 23.3%. This condition prevented the efficient generation of PF8Azo hetero-aggregates.
Fig. 7 shows the gCD value at the first Cotton band of the PF8Azo/PSi-S and PF8Azo/PSi-R hetero-aggregates as a function of Mn of PF8Azo in CHCl3-MeOH cosolvent (2.0/1.0, (v/v)). Clearly, gCD value largely depends on the Mn value. Furthermore, when the value of Mn was greater than 30000 Da, the hetero-aggregate exhibited small chirality induction capability. PF8Azo with the best Mn value could efficiently interact with PSi-S and PSi-R at their fixed molecular weights. More detailed Mn and MW/Mn characteristics of PF8Azo are given in ESI (Table S5†). Thus, the PF8Azo/PSi-S and PF8Azo/PSi-R hetero-aggregates showed enhanced gCD values due to the π–π* transitions when Mn,GPC of PF8Azo ranged from 10000 Da to 20000 Da. The Da values of PF8Azo corresponding to the DPn values were 1/4-to-1/5 times that of PSi-S/-R with fixed Mn.
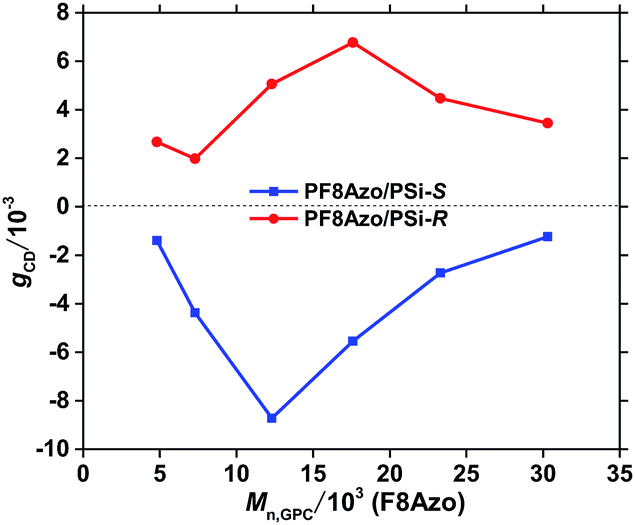 | ||
| Fig. 7 The gCD values at the first Cotton band of PF8Azo/PSi-S and PF8Azo/PSi -R hetero-aggregates as a function of Mn of PF8Azo in CHCl3–MeOH cosolvent (2.0/1.0, (v/v)). [PSi-S or PSi-R]0 = [PF8Azo]0 = 1.0 × 10−5 M. | ||
4 Conclusions
We efficiently induced CD activity at the π–π* transitions of PF8Azo when achiral PF8Azo and rigid rod-like helical PSi-S and PSi-R were employed as starting materials to produce hetero-aggregates. To efficiently boost the CD signal amplitudes of PF8Azo/PSi-S and PF8Azo/PSi-R hetero-aggregates, optimizations of the volume fractions of CHCl3 and MeOH as good and poor solvents and the molar ratios of PF8Azo-to-PSi-S and PF8Azo-to-PSi-R were conducted. By optimizing several parameters, CD-active PF8Azo homo-aggregates were produced by photoscissoring at 313 nm due to the Siσ–Siσ* transition of the PSi-S and PSi-R main chains. The CD activity of PF8Azo remained unchanged after photoscissoring. The PF8Azo/PSi-S and PF8Azo/PSi-R hetero-aggregates underwent trans–cis photoisomerization by alternating irradiations at 405 nm and 546 nm.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful for the financial support from the National Science Foundation of China (21574089), the JSPS KAKEN-HI (16H04155), the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions and the Program of Innovative Research Team of Soochow University.Notes and references
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed
.
- M. A. Mateos-Timoneda, M. Crego-Calama and D. N. Reinhoudt, Chem. Soc. Rev., 2004, 33, 363–372 RSC
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed
- M. M. Green, C. Khatri and N. C. Peterson, J. Am. Chem. Soc., 1993, 115, 4941–4942 CrossRef CAS
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS PubMed
- E. Schwartz, S. Le Gac, J. J. L. M. Cornelissen, R. J. M. Nolte and A. E. Rowan, Chem. Soc. Rev., 2010, 39, 1576–1599 RSC
- Y. Wang, T. Sakamoto and T. Nakano, Chem. Commun., 2012, 48, 1871–1873 RSC
- L. Wang, L. Yin, W. Zhang, X. Zhu and M. Fujiki, J. Am. Chem. Soc., 2017, 139, 13218–13226 CrossRef CAS PubMed
- M. Fujiki, K. Yoshida, N. Suzuki, J. Zhang, W. Zhang and X. Zhu, RSC Adv., 2013, 3, 5213 RSC
- M. Fujiki, Y. Donguri, Y. Zhao, A. Nakao, N. Suzuki, K. Yoshida and W. Zhang, Polym. Chem., 2015, 6, 1627–1638 RSC
- G. Zou, H. Jiang, H. Kohn, T. Manaka and M. Iwamoto, Chem. Commun., 2009, 0, 5627–5629 RSC
- T. Kawasaki, M. Sato, S. Ishiguro, T. Saito, Y. Morishita, I. Sato, H. Nishino, Y. Inoue and K. Soai, J. Am. Chem. Soc., 2005, 127, 3274–3275 CrossRef CAS PubMed
- M. Watabe, G. Juman, K. Miyamoto and T. Omatsu, Sci. Rep., 2014, 4, 4281 CrossRef PubMed
- Y. Taira and M. Katoh, Astrophys. J., 2018, 860, 45 CrossRef
- L. Yin, T. Miao, X. Cheng, Y. Zhao, J. Li, W. Zhang and X. Zhu, J. Funct. Polym., 2018, 5, 387–401 Search PubMed
- K. Akagi, G. Piao, S. Kaneko, K. Sakamaki, H. Shirakawa and M. Kyotani, Science, 1998, 282, 1683–1686 CrossRef CAS PubMed
- H. Goto and K. Akagi, Angew. Chem., Int. Ed., 2005, 44, 4322–4328 CrossRef CAS PubMed
- K. Akagi, Chem. Rev., 2009, 109, 5354–5401 CrossRef CAS PubMed
- Y. Kawagoe, M. Fujiki and Y. Nakano, New J. Chem., 2010, 34, 637 RSC
- Y. Zhao, N. A. Abdul Rahim, Y. Xia, M. Fujiki, B. Song, Z. Zhang, W. Zhang and X. Zhu, Macromolecules, 2016, 49, 3214–3221 CrossRef CAS
- Y. Nakano, Y. Liu and M. Fujiki, Polym. Chem., 2010, 1, 460–469 RSC
- M. Fujiki, Y. Kawagoe, Y. Nakano and A. Nakao, Molecules, 2013, 18, 7035–7057 CrossRef CAS PubMed
- J. Liu, J. Zhang, S. Zhang, N. Suzuki, M. Fujiki, L. Wang, L. Li, W. Zhang, N. Zhou and X. Zhu, Polym. Chem., 2014, 5, 784–791 RSC
- L. Wang, N. Suzuki, J. Liu, T. Matsuda, N. A. A. Rahim, W. Zhang, M. Fujiki, Z. Zhang, N. Zhou and X. Zhu, Polym. Chem., 2014, 5, 5920–5927 RSC
- L. Yin, Y. Zhao, S. Jiang, L. Wang, Z. Zhang, J. Zhu, W. Zhang and X. Zhu, Polym. Chem., 2015, 6, 7045–7052 RSC
- W. Zhang, K. Yoshida, M. Fujiki and X. Zhu, Macromolecules, 2011, 44, 5105–5111 CrossRef CAS
- S. Jiang, Y. Zhao, L. Wang, L. Yin, Z. Zhang, J. Zhu, W. Zhang and X. Zhu, Polym. Chem., 2015, 6, 4230–4239 RSC
- L. Yin, Y. Zhao, M. Liu, N. Zhou, W. Zhang and X. Zhu, Polym. Chem., 2017, 8, 1906–1913 RSC
- L. Yin, M. Liu, Y. Zhao, S. Zhang, W. Zhang, Z. Zhang and X. Zhu, Polym. Chem., 2018, 9, 769–776 RSC
- T. Miao, L. Yin, X. Cheng, Y. Zhao, W. Hou, W. Zhang and X. Zhu, Polymers, 2018, 10, 612 CrossRef
- K. Maeda, M. Ishikawa and E. Yashima, J. Am. Chem. Soc., 2004, 126, 15161–15166 CrossRef CAS PubMed
- H. Nakashima, J. R. Koe, K. Torimitsu and M. Fujiki, J. Am. Chem. Soc., 2001, 123, 4847–4848 CrossRef CAS PubMed
- E. Ohta, H. Sato, S. Ando, A. Kosaka, T. Fukushima, D. Hashizume, M. Yamasaki, K. Hasegawa, A. Muraoka, H. Ushiyama, K. Yamashita and T. Aida, Nat. Chem., 2010, 3, 68 CrossRef PubMed
- K. Okano, M. Taguchi, M. Fujiki and T. Yamashita, Angew. Chem., Int. Ed., 2011, 50, 12474–12477 CrossRef CAS PubMed
- L. Zhang and M. Liu, J. Phys. Chem. B, 2009, 113, 14015–14020 CrossRef CAS PubMed
- E. Yashima, T. Matsushima and Y. Okamoto, J. Am. Chem. Soc., 1997, 119, 6345–6359 CrossRef CAS
- E. Yashima, K. Maeda and Y. Okamoto, Nature, 1999, 399, 449–451 CrossRef CAS
- Y. Li, T. Wang and M. Liu, Soft Matter, 2007, 3, 1312 RSC
- P. Duan, X. Zhu and M. Liu, Chem. Commun., 2011, 47, 5569–5571 RSC
- P. Duan, Y. Li, L. Li, J. Deng and M. Liu, J. Phys. Chem. B, 2011, 115, 3322–3329 CrossRef CAS PubMed
- Q. Jin, L. Zhang and M. Liu, Chem.–Eur. J., 2013, 19, 9234–9241 CrossRef CAS PubMed
- H. Jintoku, T. Sagawa, T. Sawada, M. Takafuji, H. Hachisako and H. Ihara, Tetrahedron Lett., 2008, 49, 3987–3990 CrossRef CAS
- D. Yang, Y. Zhao, K. Lv, X. Wang, W. Zhang, L. Zhang and M. Liu, Soft Matter, 2016, 12, 1170–1175 RSC
- S. Le Gac, E. Schwartz, M. Koepf, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem.–Eur. J., 2010, 16, 6176–6186 CrossRef CAS PubMed
- J. D. Le, Y. Pinto, N. C. Seeman, K. Musier-Forsyth, T. A. Taton and R. A. Kiehl, Nano Lett., 2004, 4, 2343–2347 CrossRef CAS
- Y. Guo, H. Oike and T. Aida, J. Am. Chem. Soc., 2004, 126, 716–717 CrossRef CAS PubMed
- G. Fukuhara, K. Iida, Y. Kawanami, H. Tanaka, T. Mori and Y. Inoue, J. Am. Chem. Soc., 2015, 137, 15007–15014 CrossRef CAS PubMed
- S. Haraguchi, M. Numata, C. Li, Y. Nakano, M. Fujiki and S. Shinkai, Chem. Lett., 2009, 38, 254–255 CrossRef CAS
- T. Shiraki, A. Dawn, Y. Tsuchiya and S. Shinkai, J. Am. Chem. Soc., 2010, 132, 13928–13935 CrossRef CAS PubMed
- S. Haraguchi, T. Hasegawa, M. Numata, M. Fujiki, K. Uezu, K. Sakurai and S. Shinkai, Org. Lett., 2005, 7, 5605–5608 CrossRef CAS PubMed
- M. Ikeda, T. Hasegawa, M. Numata, K. Sugikawa, K. Sakurai, M. Fujiki and S. Shinkai, J. Am. Chem. Soc., 2007, 129, 3979–3988 CrossRef CAS PubMed
- S. Guo, N. Suzuki and M. Fujiki, Macromolecules, 2017, 50, 1778–1789 CrossRef CAS
- S. Guo, H. Kamite, N. Suzuki, L. Wang, A. Ohkubo and M. Fujiki, Biomacromolecules, 2018, 19, 449–459 CrossRef CAS PubMed
- T. Shiraki, Y. Tsuchiya, T. Noguchi, S. Tamaru, N. Suzuki, M. Taguchi, M. Fujiki and S. Shinkai, Chem.–Asian J., 2014, 9, 218–222 CrossRef CAS PubMed
- X. Lan, T. Liu, Z. Wang, A. O. Govorov, H. Yan and Y. Liu, J. Am. Chem. Soc., 2018, 140, 11763–11770 CrossRef CAS PubMed
- N. A. A. Rahim and M. Fujiki, Polym. Chem., 2016, 7, 4618–4629 RSC
- M. Fujiki and S. Yoshimoto, Mater. Chem. Front., 2017, 1, 1773–1785 RSC
- S. T. Duong and M. Fujiki, Polym. Chem., 2017, 8, 4673–4679 RSC
- A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139–4175 CrossRef CAS PubMed
- A. Munoz and A. Virgili, Tetrahedron: Asymmetry, 2002, 13, 1529–1534 CrossRef CAS
- P. Domachuk, F. G. Omenetto, B. J. Eggleton and M. Cronin-Golomb, J. Opt. A: Pure Appl. Opt., 2007, 9, S129–S133 CrossRef CAS
- M. M. Green, M. P. Reidy, R. J. Johnson, G. Darling, D. J. Oleary and G. Willson, J. Am. Chem. Soc., 1989, 111, 6452–6454 CrossRef
- M. Fujiki, J. R. Koe, K. Terao, T. Sato, A. Teramoto and J. Watanabe, Polym. J., 2003, 35, 297–344 CrossRef CAS
- R. D. Miller and J. Michl, Chem. Rev., 1989, 89, 1359–1410 CrossRef CAS
- A. Saxena, K. Okoshi, M. Fujiki, M. Naito, G. Guo, T. Hagihara and M. Ishikawa, Macromolecules, 2004, 37, 367–370 CrossRef CAS
- P. W. Voorhees, J. Stat. Phys., 1985, 38, 231–252 CrossRef
- C. Monat, P. Domachuk and B. J. Eggleton, Nat. Photonics, 2007, 1, 106–114 CrossRef CAS
- A. Bobrovsky, V. Shibaev, A. Bubnov, V. Hamplová, M. Kašpar and M. Glogarová, Macromolecules, 2013, 46, 4276–4284 CrossRef CAS
- H. Sogawa, M. Shiotsuki and F. Sanda, Macromolecules, 2013, 46, 4378–4387 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: CD spectra and DLS etc. See DOI: 10.1039/c8ra09345h |
| This journal is © The Royal Society of Chemistry 2019 |