
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of pH-responsive block glycopolymers with different structures for the delivery of doxorubicin†
Ibrahim Abdalla ac,
Jiaming Xua,
Danyue Wanga,
Han Tonga,
Bin Suna,
Bin Dingbc,
Xiaoze Jiang*a and
Meifang Zhua
ac,
Jiaming Xua,
Danyue Wanga,
Han Tonga,
Bin Suna,
Bin Dingbc,
Xiaoze Jiang*a and
Meifang Zhua
aState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China. E-mail: xiaozejiang@dhu.edu.cn
bKey Laboratory of Textile Science and Technology, Ministry of Education, College of Textiles, Donghua University, Shanghai 201620, China
cInnovation Center for Textile Science and Technology, Donghua University, Shanghai 200051, China
First published on 15th January 2019
Abstract
To understand the influence of the construction of pH-responsive glycopolymer carriers on loading and release behaviors of the drug, three types of block glycopolymers with similar compositions but different constructions, PEG-b-P(DEA-co-GAMA), PEG-b-PDEA-b-PGAMA and PEG-b-PGAMA-b-PDEA, were successfully synthesized via atom transfer radical polymerization (ATRP) method. The compositions and structures of the three glycopolymers were characterized using 1H NMR (nuclear magnetic resonance) and GPC (gel permeation chromatography), while the morphology and size of aggregates from pH-sensitive block glycopolymers were measured using TEM (transmission electron microscopy) and DLS (dynamic light scattering). The results indicated that the micelles prepared from PEG-b-PGAMA-b-PDEA had a more compact shell structure. The drug-loaded micelles were prepared using the diafiltration method at pH 10, and the loading content and loading efficiency were analyzed using a UV-visible spectrophotometer. DOX-loaded micelles formed by PEG-b-PGAMA-b-PDEA with the more compact shell construction showed the highest loading content and loading efficiency (12.0 wt% and 58.0%) compared with the other two micelles. Moreover, the DOX release tests of these micelles were carried out under two PBS conditions (pH 7.4 and pH 5.5), and the DOX release amount in a certain time was analyzed using a UV-visible spectrophotometer. The results showed that the more compact shell construction of the three layered micelle obstructed the diffusion of a proton into the PDEA core at pH 5.5 and delayed the drug from releasing under both conditions. Moreover the two-layered micelle with a PDEA and PGAMA mixed core showed a relatively high release amount owing to the porous core permitting unimpeded releasing at pH 7.4 and promoted the protonation of PDEA at pH 5.5. Insights gained from this study show that the structure of block copolymers, leading to different constructions of micelles, could adjust the drug loading and release behavior to certain extent, thus it may contribute to improving the design of desirable drug delivery systems.
1 Introduction
Driven by the need to reduce the side effects of chemotherapy and enhance the specific cytotoxicity of drugs against cancer cells, numerous drug delivery systems for controlled release of drugs were designed over the past several decades.1,2 Shell–core structured micelles, self-assembled by block copolymers, are a classic drug delivery system, which loads the drug and delivers it to the location of the lesion. The shell provides stability in aqueous conditions due to its hydrophilicity and biocompatibility, while the core stores the hydrophobic drugs such as doxorubicin via hydrophobic interactions.3,4An ideal drug-delivery nano-platform could overcome the dilemma as it maintains the drug in the core under normal physical conditions, while releasing the drug at tumor sites. Thus, stimuli-responsive drug carriers have been investigated aiming at the controlled release of anti-cancer drugs according to the difference between normal tissue and tumor sites.5–7 Among the most popular stimuli-responsive candidates for drug delivery systems, pH-responsiveness is one of the most significant methods owing to the distinction between acidic cancer cells (pH 6.5–5.0) and normal physical conditions (pH 7.4). Poly-(2-(diethylamino)ethyl methacrylate) (PDEA), as a typical pH-responsive cationic polymer with excellent biocompatibility, has attracted much attention for its responsive scope (pH 6–8) matching well with the gap between tumor and normal tissues.8 Moreover, in order to prolong the circulation time of carriers under physical conditions and enrich the accumulation at the tumor sites according to the enhanced permeation and retention (EPR) effect of tumor sites,9,10 the extraordinary biocompatibility and hydrophilicity of the shell component were required. Poly-(ethylene glycol) (PEG), as the most popular hydrophilic segment applied in drug delivery systems, has a mature clinical application and good therapeutic effect to certain extent.11,12 In recent years, in biological fields such as biological recognition and drug delivery, much attention has been paid to glycopolymer as its excellent hydrophilicity, ability to mimic biological functions of natural polymers containing carbohydrates and modification ability due to the polyhydroxy structure.13,14 Narain and Armes reported the facile preparation of a series of methacrylate-based glycopolymers without recourse to protecting group chemistry via ATRP,15 therefore, glycopolymers like poly(D-gluconamidoethyl methacrylate) (PGAMA) had already been widely applied in drug delivery systems16 as a hydrophilic segment to supply the biocompatibility or functional component to form complexes with other chemicals for drug delivery or biological recognition.
In the current stage, linear copolymer self-assembled micelles, such as ABC-block copolymers in which A and B were hydrophilic while C was hydrophobic, have three typical structures,17 with three layer “onion-like” morphologies,18,19 a two-layered structure with mixed shell of A and B20 and a two-layered structure with a mixed core of B and C.21,22 The diversity of polymers provides abundant functionalities and characteristics for choosing as one of the constituents in the polymeric carriers, cooperating with the ratio governing to realize the tunable drug loading and release.23 However, whether the structures of copolymers, such as random or block and the order of chain segments, which leads to different constructions of carriers play important roles in drug delivery needed further investigation.
Herein, the pH-responsive glycopolymers with three different structures corresponding attempt were synthesized to prepare three classical micelles in case of ABC-block copolymers, which contained PEG and PGAMA as hydrophilic segments and pH-sensitive hydrophobic PDEA with fixed proportion (Scheme 1). Furthermore, the copolymers mentioned above were used to encapsulate the hydrophobic anti-cancer drugs doxorubicin (DOX) as the model drug, and the release profiles of DOX in different pH (at pH 7.4 and 5.5) were studied to clarify the influence of structures for drug delivery behaviors.
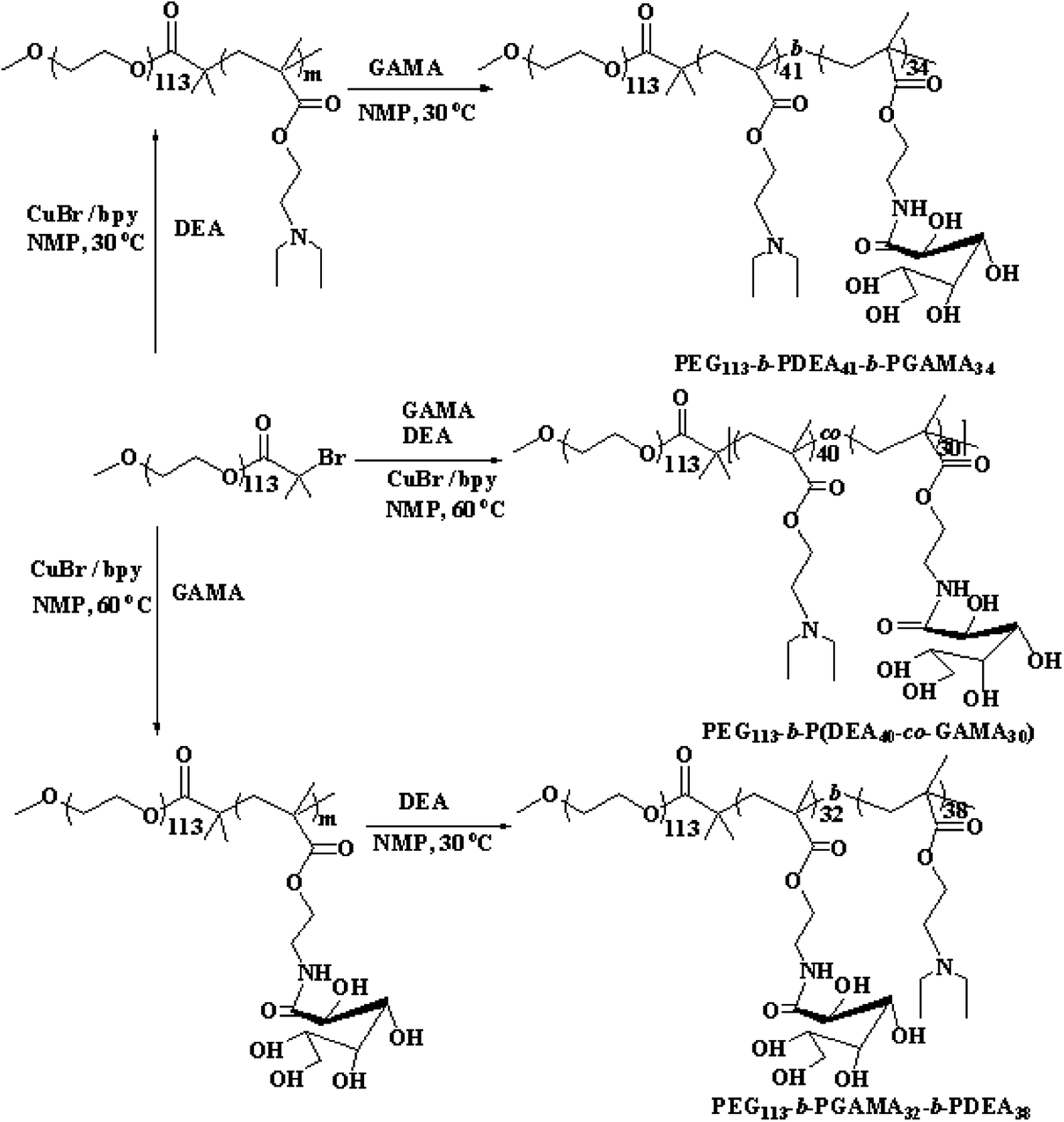 | ||
| Scheme 1 Synthetic routes for the preparation of PEG113-b-P(DEA40-co-GAMA30), PEG113-b-PGAMA32-b-PDEA38 and PEG113-b-PDEA41-b-PGAMA34 pH-responsive block glycopolymers. | ||
2 Experimental
2.1 Materials
Poly(ethyleneglycol) monomethyl ether (MeO-PEG-OH, Sigma-Aldrich) with a molecular (Mn) weight of 5 kDa was dried at 35 °C in vacuum overnight before using. 2-(Diethylamino) ethyl methacrylate (DEA, Sigma-Aldrich) was passed through a basic alumina column to remove the inhibitor. Dichloromethane, triethylamine (TEA), N-methyl pyrrolidone (NMP) and dimethyl sulfoxide (DMSO) (Sinopharm Chemical Reagent Co. Ltd.) were purified according to standard procedures. Doxorubicin hydrochloride (DOX·HCl, J&K Scientific) was deprotonated by excess triethylamine. CuBr, 2,2-bipyridine (bpy), D-gluconolactone, hydroquinone, 2-bromoisobutyryl bromide, 2-aminoethyl methacrylate hydrochloride were purchased from J&K Scientific without further purification before use. The macro-initiator PEG-Br24,25 and the monomer 2-gluconamidoethylmethacrylate (GAMA) were prepared according to reported procedures.26–282.2 Synthesis of PEG-b-P(DEA-co-GAMA) diblock glycopolymer
PEG-b-P(DEA-co-GAMA) was prepared by ATRP method with PEG-Br used as a macro-initiator and catalyzed by CuBr/bpy. PEG-Br (0.500 g, 0.097 mmol), bpy (0.030 g, 0.194 mmol), DEA (0.78 mL, 3.88 mmol), and GAMA (0.894 g, 2.91 mmol) were dissolved in NMP (2.0 mL), and the solution was degassed by purging with N2 gas for at least 30 minutes before adding the catalyst CuBr (0.013 mg, 0.097 mmol). Polymerization was proceeded at 60 °C for 24 hours under the protection of nitrogen, and then the polymerization mixture was freezed by liquid nitrogen to interrupt the reaction. After warming up to room temperature, the solution was dialyzed in deionized water for 2 days and was freeze-dried overnight to obtain the PEG-b-P(DEA-co-GAMA) diblock glycopolymer (1.71 g, yield 81%).2.3 Synthesis of PEG-b-PDEA-b-PGAMA and PEG-b-PGAMA-b-PDEA triblock glycopolymers
The pH-responsive triblock copolymers were prepared via successive ATRP technique using PEG-Br as the initiator. The procedure is as follows: PEG-Br (0.50 g, 0.097 mmol), bpy (0.030 g, 0.194 mmol), DEA monomer (0.78 mL, 3.88 mmol) were dissolved in NMP (1.0 mL), and the catalyst CuBr (0.013 mg, 0.097 mmol) was added to the solution after purging with N2 gas for 30 minutes. The reaction was performed at 60 °C for 24 hours (conversion rate achieved 90%) before adding degassed GAMA (0.894 g, 2.91 mmol) and NMP (1.0 mL) mixture. Liquid nitrogen was used to terminate the polymerization after exposing to air about 2 hours, and then the mixture was purified by dialysis method and was freeze-dried overnight to prepare the PEG-b-PDEA-b-PGAMA triblock glycopolymer (1.79 g, yield 85%).According to the above procedure, the first monomer changed to GAMA (0.894 g, 2.91 mmol), the first step of the reaction carried on at 30 °C for 4 hours to achieve 90% conversion rate and then added the second monomer DEA (0.78 mL, 3.88 mmol) and heated up to 60 °C stirring for 24 hours. The other conditions were consisted with the former procedure and yielded the PEG-b-PGAMA-b-PDEA triblock glycopolymer (1.75 g, yield 83%).
2.4 Polymers characterization
The block glycopolymers prepared were dissolved in D2O and were recorded on a Bruker 400 MHz spectrometer to get the 1H NMR spectra to characterize their compositions and structures. Molecular weight and molecular weight distribution of glycopolymers were assessed by gel permeation chromatography (GPC) in DMF containing 0.5 g L−1 LiBr at a flow rate of 1.0 mL min−1 using a series of three linear Styragel columns (HT2, HT4, and HT5) or two linear Styragel columns (HT4, and HT5), a Waters 1515 pump and Waters 2414 differential refractive index detector (set at 30 °C) and an oven temperature of 60 °C. Calibration was performed using a series of near-monodisperse PEG standards.2.5 Glycopolymer assemblies' characterization
The critical micelle concentration (CMC) of micelles self-assembled by glycopolymers prepared were characterized using a fluorescence spectrophotometer with Nile Red as probe and the excitation wavelength was 550 nm. The glycopolymers were first dissolved in water and were diluted to a series of concentrations, 0.5 mg mL−1 to 0.1 μg mL−1; polymer solutions (above pH 7.4) and 10 mL of the solutions added into Nile Red filmed sample vials stirring for 24 h in darkness, the final concentration of Nile Red was 1 × 10−6 M.The morphology of the micelles was measured by Dynamic light scattering (DLS, Nano Zetasizer) and transmission electron microscope (TEM, JEM-2100). Three types of glycopolymers were dissolved in acidic water as unimers and were adjusted to a series of different pH solutions from pH 3 to pH 12 with a 2 mg mL−1 concentration of glycopolymers. The sizes and zeta potentials of assemblies at different pH were detected by DLS and the morphology was characterized by TEM using the diluted pH 10 samples (below 2 mg mL−1).
2.6 Preparation of DOX-loaded micelles
The diafiltration method was used to prepare DOX-loaded glycopolymer micelles. Glycopolymers (15 mg) were dissolved in 8 mL DMSO and were mixed with DOX base solutions (3 mg mL−1, DMSO) treated with excess triethylamine (five mole ratios of DOX·HCl) for 12 h previously; the mixture was stirred in darkness for 4 h before transferring into dialysis bag (MWCO 3.5 kDa) and was dialyzed against water (pH = 10) for 48 h, respectively. The mixtures were passed through a membrane filter (0.45 μm) and were freeze-dried overnight.The DOX concentration measured using a UV-visible spectrophotometer (UV-Vis, TU-1901 Beijing Spectrum Analysis of General Instrument Co., Ltd) at 480 nm according to the calibration curves of different concentrations of DOX in DMSO were analyzed previously (see Fig. S1 in ESI†). The loading content (LC%) and loading efficiency (LE%) of DOX were calculated using eqn (1) and (2).
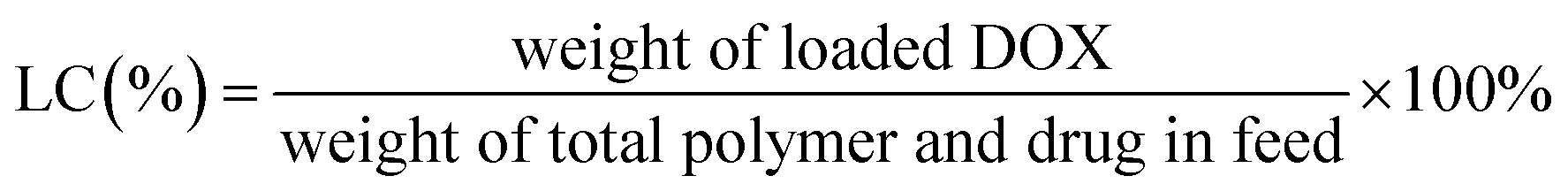 | (1) |
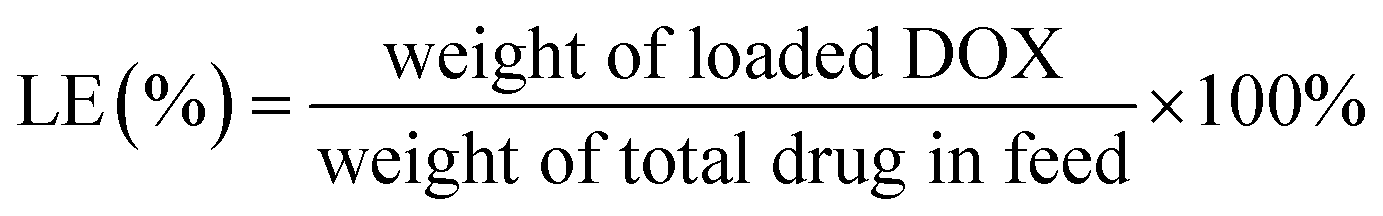 | (2) |
2.7 In vitro drug release
The in vitro release of DOX from three glycopolymer carriers were carried on under pH 5.5 and pH 7.4 PBS (phosphate buffered saline) buffer (0.01 M) medium via diafiltration method. The drug-loaded micelles (5 mg) were dissolved in PBS solutions (5 mL) and were transferred to dialysis bags (MWCO 14 kDa), were dialyzed against PBS solutions (45 mL) accordingly at 37 °C and 100 revolution per minute (rpm) in incubator (TU-1901 Beijing Spectrum Analysis of General Instrument Co., Ltd). The PBS (5 mL) was added after sampling the medium (5 mL) outside the bags at specific time intervals. The DOX concentrations of samples were determined by UV-visible spectrophotometer at 480 nm according to the analyzed calibration curves of different concentrations of DOX in specific PBS buffer previously (see Fig. S2 and S3 in ESI†). Moreover, the accumulative release percentage of DOX was calculated using eqn (3), and all release experiments were carried out in triplicate, and all the results were reported as the mean ± standard deviation (n = 3).
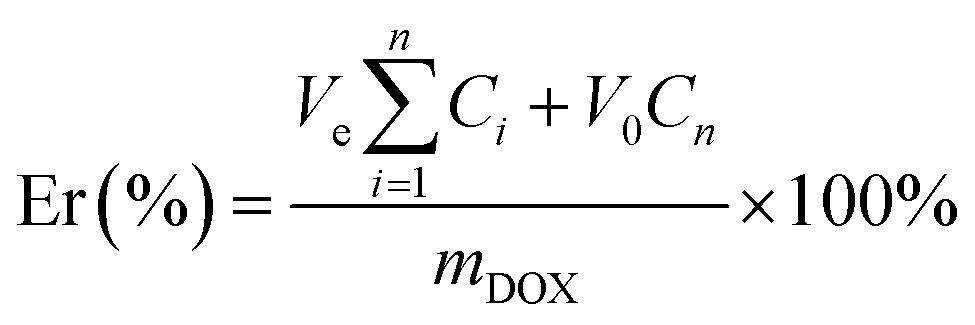 | (3) |
3 Results and discussion
3.1 Synthesis and characterization of pH-sensitive glycopolymers
In order to investigate the release-tunable carriers, three different structures of glycopolymers with the same compositions for self-assembled micelles with three typical constructions were prepared using the macro-initiator PEG-Br for ATRP method of DEA and GAMA. PEG-b-P(DEA-co-GAMA) block glycopolymers were obtained with random architecture of DEA and GAMA chains in the second block by directly polymerized both DEA and GAMA monomers at 60 °C, and triblock glycopolymer, PEG-b-PDEA-b-PGAMA and PEG-b-PGAMA-b-PDEA, were prepared via successive polymerization of DEA or GAMA with high conversion above 90% and then second monomers were added to construct the third block. After polymerization and purification, those block glycopolymers were characterized by 1H NMR as shown in Fig. 1. The specific signals of block glycopolymers could be detected in D2O/DCl at pH 5, as those glycopolymers have same compositions and fixed proportions. However, all of the glycopolymers showed almost the same spectra, and the characteristic peaks at 3.64 ppm (c in PEG), 4.38 ppm (d in PDEA), 4.30–4.15 ppm and 3.9 ppm (d′, i, j in GAMA) were observed. Moreover, the degree of polymerization of DEA and GAMA could be calculated, 40 and 30, 41 and 34, and 32 and 38. Thus the obtained triblock glycopolymers were denoted as PEG113-b-P(DEA40-co-GAMA30), PEG113-b-PDEA41-b-PGAMA34, and PEG113-b-PGAMA32-b-PDEA38 respectively. Furthermore, relatively narrow and unimodal molecular weight distribution with polydispersity Mw/Mn of 1.23, 1.19 and 1.20 showed in the GPC chromatograms (as shown in Table 1) indicates the ideal purity of those glycopolymers.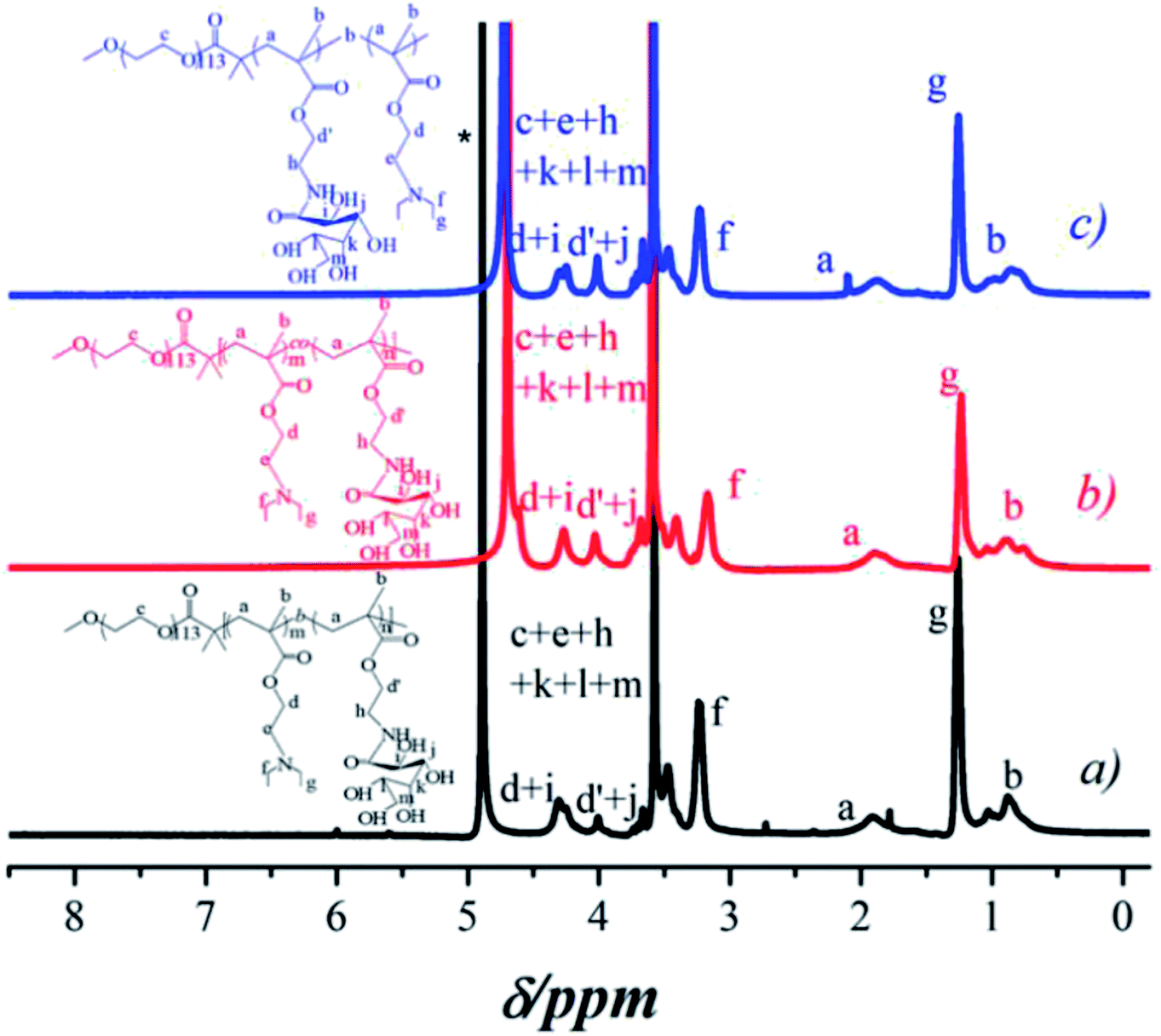 | ||
| Fig. 1 1H NMR spectra of (a) PEG113-b-PDEA41-b-PGAMA34, (b) PEG113-b-P (DEA40-co-GAMA30), and (c) PEG113-b-PGAMA32-b-PDEA38 block glycopolymers. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 008
0083.2 CMCs and assemble transitions of pH-sensitive micelles
The critical micelle concentration (CMC) of pH-sensitive glycopolymers, as a significant parameter to determinate the self-assembly ability, were measured using a fluorescence spectrophotometer with Nile Red as a probe. Theoretically, PDEA block represented as hydrophilic when it protonized below its pKa of 7.3, while it showed hydrophobicity as it reversed above pH 7.3 and the PDEA based copolymers self-assembled into micelles with PDEA cores in this situation. This pH-induced micellization/de-micellization behavior was spontaneous and reversible, which has already been investigated in detail, according to Liu's works.29–33 The transition of DEA covered over the range of pH 6–8 according to the variation of hydrodynamic diameter of assemblies (as shown in Fig. 2) from the DLS in keeping with reported works,29,30 these copolymers micelles demonstrated a reversible micellization behaviors during the mentioned pH values.34 However, where the PEG-b-P(DEA-co-GAMA) assembled micelles showed wider transition range compared with the other two for the mixed core of DEA and GAMA random segment. Thus, the CMCs were measured at pH 10 insuring the micelles were at stable equilibriums. Moreover, it should be noted that all of the glycopolymers had similar DP of each block, meanwhile, their architectures of random and block moieties in the second or third parts were designed to further study the relation between structures of pH-responsive block glycopolymers and their pH induced micellization behaviors. The CMCs of those self-assembled glycopolymer micelles, PEG113-b-P(DEA40-co-GAMA30), PEG113-b-PDEA41-b-PGAMA34, and PEG113-b-PGAMA32-b-PDEA38, were calculated to be 0.10 mg mL−1, 0.012 mg mL−1 and 0.011 mg mL−1 respectively according to the turning points of emission wavelength (λmax) of Nile Red on the logarithmic concentration of glycopolymers of the curve (as shown in Fig. 3).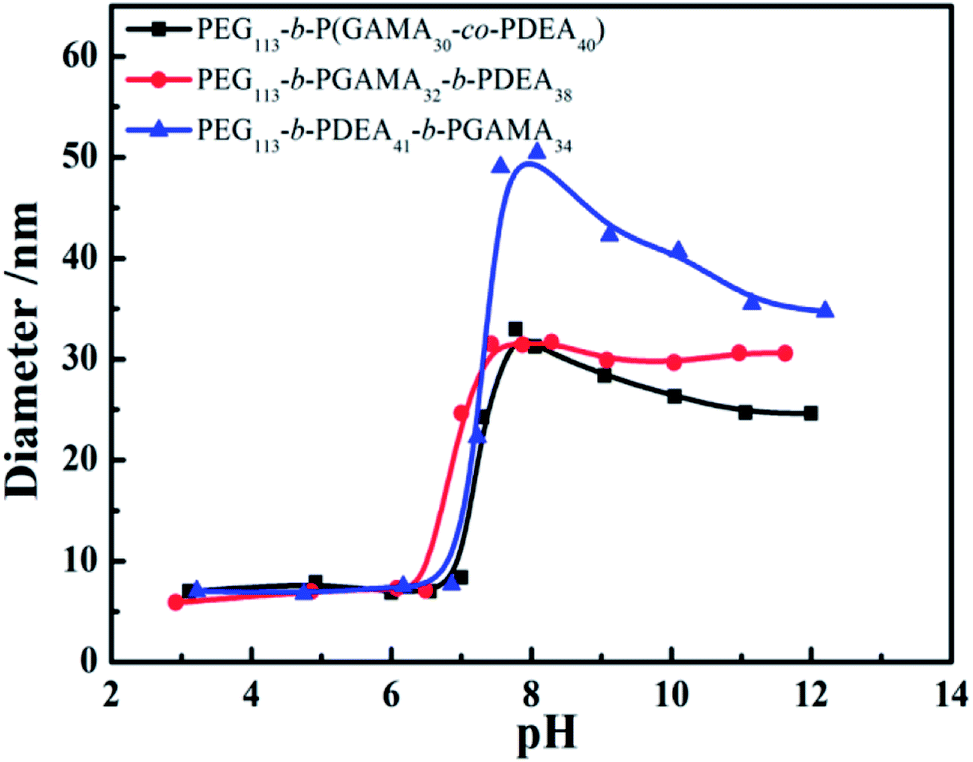 | ||
| Fig. 2 Variation of pH dependence of average hydrodynamic diameters: PEG113-b-P(DEA40-co-GAMA30), PEG113-b-PGAMA32-b-PDEA38, and PEG113-b-PDEA41-b-PGAMA34. | ||
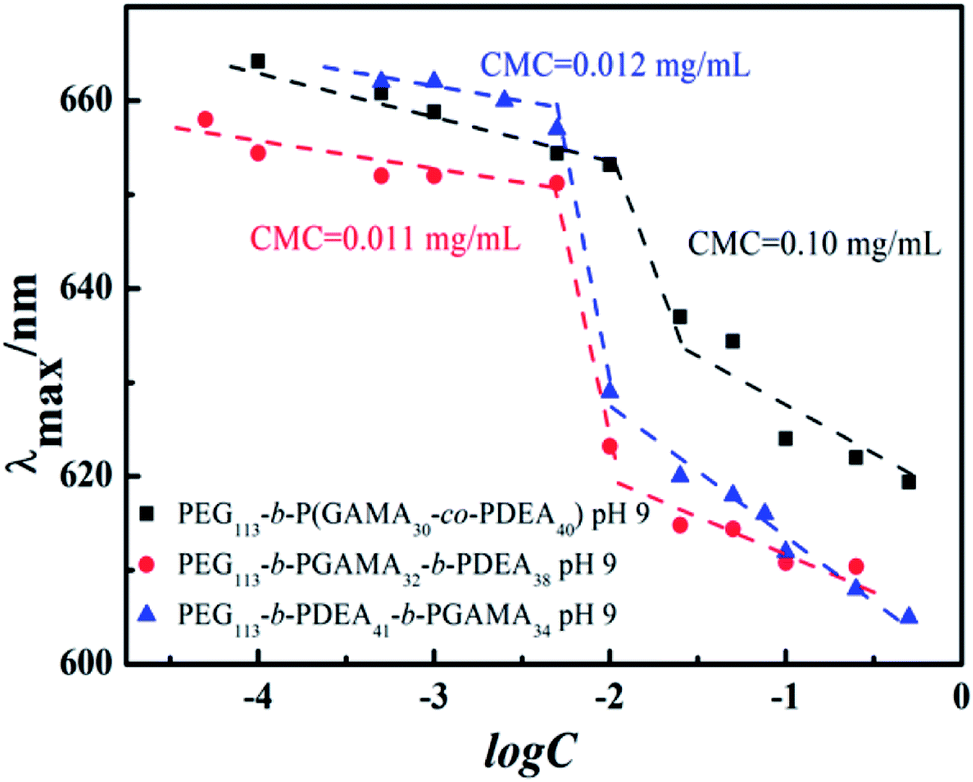 | ||
| Fig. 3 The dependence of maximum emission wavelength (λmax) of Nile Red on the logarithmic concentration of PEG113-b-P(DEA40-co-GAMA30), PEG113-b-PGAMA32-b-PDEA38 and PEG113-b-PDEA41-b-PGAMA34 block glycopolymers. | ||
The CMCs of self-assemblies depended on the hydrophobicity of core segment to a great extent owing to the assembling driving force of similarity-intermiscibility.35 Herein, the PEG-b-P(DEA-co-GAMA) self-assembled micelles represented a quite high CMC compared with the other two triblock-assembled micelles due to the hydrophilic PGAMA segment mixed core, while the core of the other two micelles were pH-sensitive PDEA segment only. Due to the respectable characteristics of the PGAMA, it was extensively applied in drug delivery system to supply hydrophilicity, the biocompatibility, and functional components.16 Moreover, the micelles assembled by PEG-b-PDEA-b-PGAMA and PEG-b-PGAMA-b-PDEA showed similar and desirable low CMCs, which increased the stability in physical conditions.36
3.3 Morphology and size of pH-sensitive micelles
The TEM images of self-assemblies prepared from glycopolymers were obtained to clarify the morphologies of micelles at a structural stable pH and to evidence the hydrodynamic diameter of micelles detected by DLS, and then to provide the additional analysis of micelles structures (as shown in Fig. 4). | ||
| Fig. 4 Typical TEM image of micelles (2 mg mL−1) prepared from the glycopolymers at pH 10: (a) PEG113-b-P(DEA40-co-GAMA30) (diameter 15 nm) (b) PEG113-b-PGAMA32-b-PDEA38 (diameter 25 nm) and (c) PEG113-b-PDEA41-b-PGAMA34 (diameter 40 nm). | ||
The isoelectric points (pIs) of PEG113-b-P(DEA40-co-GAMA30), PEG113-b-PGAMA32-b-PDEA38 and PEG113-b-PDEA41-b-PGAMA34 glycopolymers are 9.3, 9.1 and 9.0, respectively, which considered by zeta potential at various solution pHs as shown in Fig. 5. The aggregates of PEG-b-P(DEA-co-GAMA) showed the smallest size, while the two layers micelles assembled from PEG-b-PDEA-b-PGAMA showed the biggest both in TEM and DLS results (as shown in Table 2). Compared with PEG-b-PGAMA-b-PDEA triblock assembled three layers micelles, the micelles assembled from PEG-b-PDEA-b-PGAMA triblock owned a bigger size in spite of less layer than micelles from PEG-b-PGAMA-b-PDEA and same size of hydrophobic cores according to the similar DP of PDEA of both triblock.37 Herein, three-layer micelles possessed most compact shell construction as the large density of shell for the PEG and PGAMA shell component compared with PEG shelled PEG-b-P(DEA-co-GAMA) aggregates and same density of shell but smaller size compared with PEG-b-PDEA-b-PGAMA aggregates. It should be noted that the whole micellization/de-micellization behaviors are reversible upon pH changes.
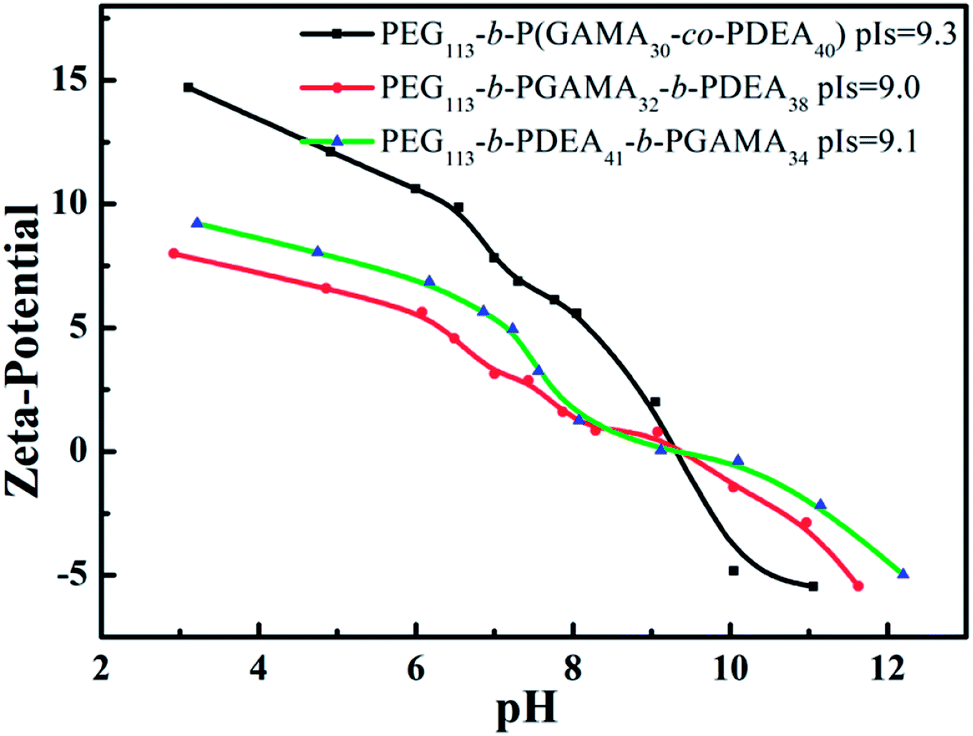 | ||
| Fig. 5 Zeta potentials of PEG113-b-P(DEA40-co-GAMA30), PEG113-b-PGAMA32-b-PDEA38 and PEG113-b-PDEA41-b-PGAMA34 copolymers at versus solution pH. | ||
| Micelles form | Dh/nm | DTEM/nm | pIs | LE (%) | LC (%) |
|---|---|---|---|---|---|
| PEG113-b-P(DEA40-co-GAMA30) | 23 | 15 | 9.3 | 47.3 | 9.5 |
| PEG113-b-PDEA41-b-PGAMA34 | 35 | 40 | 9.0 | 47.0 | 9.3 |
| PEG113-b-PGAMA32-b-PDEA38 | 32 | 25 | 9.1 | 58.0 | 12 |
3.4 Encapsulation and pH-responsive release of DOX
DOX-loaded micelles were prepared by diafiltration method based on the pH-responsive glycopolymers at pH 10. Under the circumstances, the PEG and PGAMA or PEG block self-assembled into hydrophilic shells with good biocompatibility to provide the stability of micelles, while the PDEA block or PDEA and PGAMA random block formed the cores of micelles and encapsulated deprotonated hydrophobic DOX to deliver the drugs. In theory, the LC and LE increase with the increase of hydrophobic contents of block copolymers due to the hydrophobic interaction among the drugs and PDEA segment at the same feed ratio of drug and polymers.38 However, the hydrophilic shell entrapped the drugs to certain extent and was influenced by the thickness and density of polymer chains of shell, according to the calculated LC and LE results as shown in Table 2. The three-layer PEG-b-PGAMA-b-PDEA assembled micelle showed the highest loading content (12 wt%) compared with the other two (9.5 wt% and 9.3 wt%) for the additional interlayer and more compact shell structure, while the two layers micelle showed similar loading content as the similar amount of DEA segments the core contained.The release profiles of hydrophobic DOX from the pH-responsive micelles were studied using diafiltration method in PBS (10 mM, pH 7.4 and 10 mM, pH 5.5) at 37 °C and 100 rpm in the incubator. DOX release profile of PDEA core micelle in reported works showed the suitable difference in different pH values, more at acidic, less at basic conditions.39 The tertiary amino groups contained PDEA blocks with pKa of 7.3 were deprotonated hydrophobic blocks at pH 7.4 and maintained the shell–core structure in theory, thus, the hydrophobic drugs were held in the core and showed a low release amount at this condition, while they represented hydrophilic for protonation at pH 5.5 and released drugs instantly for the de-micellization of carriers. Nevertheless, PEG-b-P(DEA-co-GAMA) self-assembled micelles with mixed cores showed a relative high accumulated drug release up to 50% in 250 h at pH 7.4 while the other two stayed at a lower level about 25% and 35% (as shown in Fig. 6). Promoted protonation of PDEA by hydrophilic segment mixed core and shortened release behaviors by the small size of micelles were the two main reasons for the highest release amount of the PEG-b-P(DEA-co-GAMA) self-assembled micelles. Furthermore, PEG-b-PGAMA-b-PDEA aggregates with more compact three-layer structure had the lowest release amount below 25% in 250 h for its high density of polymer chains delayed the release progress. On the other hand, under the acidic conditions of pH 5.5, both of two-layer micelles showed high release amount of drugs in 250 h, while the three-layer micelles held almost a half of drugs in the structure after 250 h releasing due to the hindering of both inward diffusion of proton and outward diffusion of drugs by the compact shell. It is hard to release a similar amount of drug from multilayers micelle compared with the other different micelles' structures within same periodic time; it needs longer time to release equal or more amount drug component as in other different structures' micelles. Moreover, the shorter distance with relatively loose structure from core to the medium as well as the promoted proton diffusion by hydrophilic GAMA in the core, PEG-b-P(DEA-co-GAMA) self-assembled micelles showed the highest release of drugs approaching 100% in 250 h, and PEG-b-PDEA-b-PGAMA aggregates showed moderate release amount of drugs up to 90% in 250 h owing to the longer distance with relatively loose structure. Besides, two-layer micelles assembled from PEG-b-P(DEA-co-GAMA) and PEG-b-PDEA-b-PGAMA showed quick release in an earlier stage compared with three-layer micelles due to the difference of constructions.
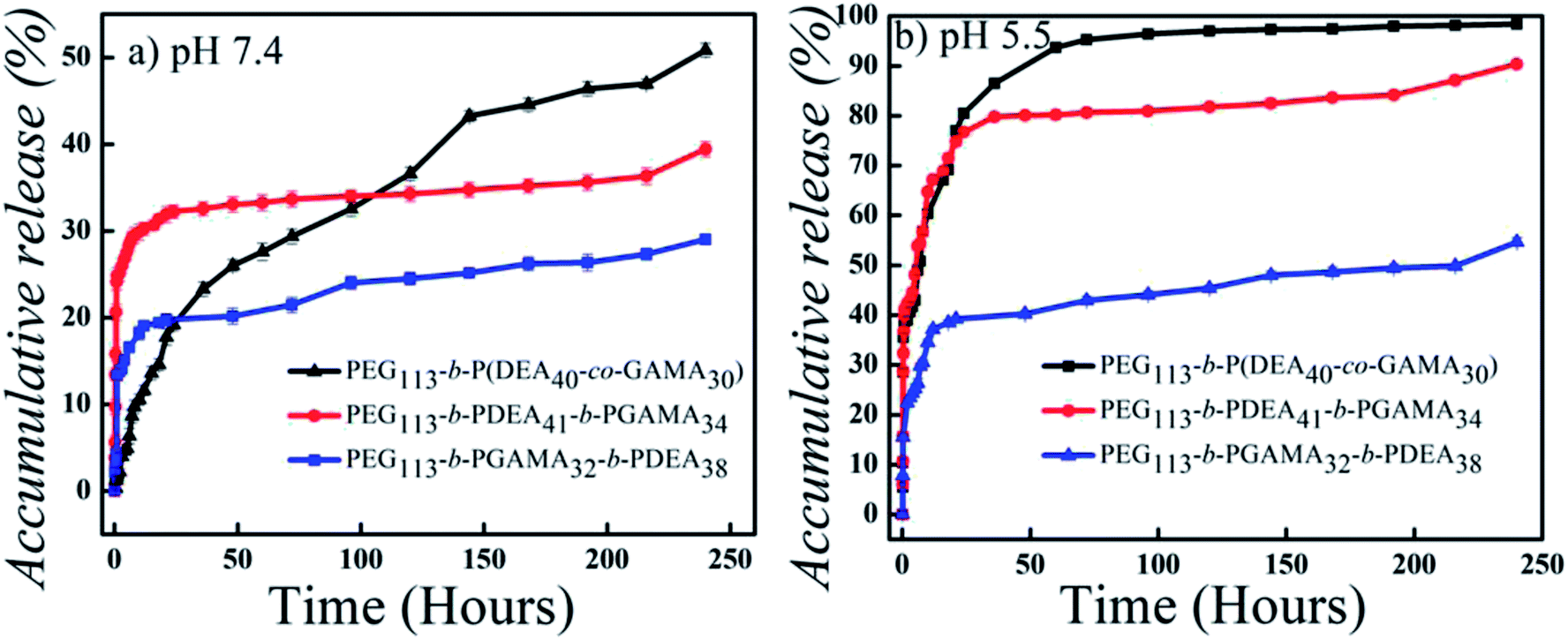 | ||
| Fig. 6 In vitro drug-release profiles of DOX-loaded micelles self-assembled from pH-responsive PEG113-b-P(DEA40-co-GAMA30), PEG113-b-PGAMA32-b-PDEA38 and PEG113-b-PDEA41-b-PGAMA34 block glycopolymers at (a) pH 7.4 and (b) pH 5.5 (mean ± SD; n = 3). | ||
Therefore, the pH-sensitive micelles formed by PEG-b-PDEA-b-PGAMA exhibited desirable properties in comparison with the other two pH-responsive assemblies, as the two-layer structure had a relatively low density of polymer chains in the shell but the drug has to travel longer distance; as well as the compact pure PDEA core permits suitable rate of drug release.
4 Conclusions
Three types of pH-responsive block glycopolymers with similar compositions but different structures were successfully synthesized via ATRP method. The aggregates of these glycopolymers presented obvious pH responsibility as forming micelles at base condition and dissociate to unimers at the acidic condition. The difference of constructions between three types of micelles played an important role in hydrophobic drug release in vitro. Micelles formed by PEG-b-P(DEA-co-GAMA) containing hydrophilic PGAMA mixed PDEA core with a small size presented high release amount both at pH 5.5 and pH 7.4, due to hydrophilic PGAMA decreased the hydrophobicity of core and promoted protonation of PDEA. More interestingly, PEG-b-PDEA-b-PGAMA self-assembled micelles have the biggest size, bigger even compared with three-layer PEG-b-PGAMA-b-PDEA formed micelles, which indicates the shell construction of PEG-b-PDEA-b-PGAMA formed micelles were looser than PEG-b-PGAMA-b-PDEA formed micelles, thus, leading to the drug loading and releasing behaviors in optimal level. Furthermore, the results investigated the influence of micelles structure on drug loading and releasing behaviors and provided an interesting design solution for desirable drug carriers aiming at controlled release.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledged the financial support of the National Natural Science Foundation of China (No. 51473035 and No. 21204010), the Research Program of Shanghai Science and Technology Commission (No. 13NM1400102 and No. 14521100600), and the Fundamental Research Funds for the Central Universities (No. 2232014D3-35).References
- S. Manchun, C. R. Dass and P. Sriamornsak, Life Sci., 2007, 80, 381–387 Search PubMed.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- A. S. Mikhail and C. Allen, J. Controlled Release, 2009, 138, 214–223 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64, 37–48 CrossRef.
- A. K. Bajpai, S. K. Shukla, S. Bhanu and S. Kankane, Prog. Polym. Sci., 2008, 33, 1088–1118 CrossRef CAS.
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174–211 CrossRef CAS.
- J. Zhuang, M. R. Gordon, J. Ventura, L. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421–7435 RSC.
- G. Kocak, C. Tuncer and V. Bütün, Polym. Chem., 2016, 8, 144–176 RSC.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- R. J. Christie and D. W. Grainger, Adv. Drug Delivery Rev., 2003, 55, 421–437 CrossRef CAS PubMed.
- C. J. Rijcken, C. J. Snel, R. M. Schiffelers, C. F. van Nostrum and W. E. Hennink, Biomaterials, 2007, 28, 5581–5593 CrossRef CAS PubMed.
- T. H. Tran, T. Ramasamy, J. Y. Choi, H. T. Nguyen, T. T. Pham, J. H. Jeong, S. K. Ku, H. G. Choi, C. S. Yong and J. O. Kim, Asian J. Pharm. Sci., 2015, 10, 5249–5262 CAS.
- G. Pasparakis, A. Cockayne and C. Alexander, J. Am. Chem. Soc., 2007, 129, 11014–11015 CrossRef CAS.
- X. Li and G. Chen, Polym. Chem., 2015, 6, 1417–1430 RSC.
- R. Narain and S. P. Armes, Biomacromolecules, 2003, 4, 1746 CrossRef CAS PubMed.
- X. H. Dai, Z. M. Wang, Y. F. Huang, J. M. Pan, Y. S. Yan, D. M. Liu and L. Sun, RSC Adv., 2014, 4, 42486–42493 RSC.
- C. F. V. Nostrum, Soft Matter, 2011, 7, 3246–3259 RSC.
- X. Xu, A. E. Smith and C. L. Mccormick, Aust. J. Chem., 2009, 62, 1520–1527 CrossRef CAS.
- K. Wang, Y. Liu, W. J. Yi, C. Li, Y. Y. Li, R. X. Zhuo and X. Z. Zhang, Soft Matter, 2012, 9, 692–699 RSC.
- X. Hu, H. Li, S. Luo, T. Liu, Y. Jiang and S. Liu, Polym. Chem., 2013, 4, 695–706 RSC.
- F. Jia, Y. Wang, H. Wang, Q. Jin, T. Cai, Y. Chen and J. Ji, Polym. Chem., 2015, 6, 2069–2075 RSC.
- X. Hu, J. Tian, T. Liu, G. Zhang and S. Liu, Macromolecules, 2013, 46, 6243–6256 CrossRef CAS.
- A. Das and P. Theato, Chem. Rev., 2016, 116, 56–60 CrossRef PubMed.
- X. Jiang, S. Luo, S. P. Armes, W. Shi and S. Liu, Macromolecules, 2006, 39, 5987–5994 CrossRef CAS.
- S. K. Jin and H. Y. Ji, Polymer, 2009, 50, 2204–2208 CrossRef.
- R. Narain and S. P. Armes, Chem. Commun., 2002, 23, 2776–2777 RSC.
- R. N. And and S. P. Armes, Macromolecules, 2003, 36, 4675–4678 CrossRef.
- Z. Deng, M. Ahmed and R. Narain, J. Polym. Sci., Part A: Polym. Chem., 2010, 47, 614–627 CrossRef.
- W. J. Lin, S. Y. Nie, Q. Chen, Y. Qian, X. F. Wen and L. J. Zhang, AIChE J., 2015, 60, 3634–3646 CrossRef.
- W. Lin, S. Nie, Q. Zhong, Y. Yang, C. Cai, J. Wang and L. Zhang, J. Mater. Chem. A, 2014, 2, 4008–4020 CAS.
- C. Zhang, W. Wu, N. Yao, B. Zhao and L. Zhang, RSC Adv., 2014, 4, 40232–40240 RSC.
- X. Jiang, G. Zhang, R. Narain and S. Liu, Langmuir, 2009, 25, 2046–2054 CrossRef CAS.
- S. Y. Liu, W. Jvm, Y. Q. Tang, N. C. Billingham, S. P. Armes and K. Tribe, Macromolecules, 2002, 35, 6121–6131 CrossRef CAS.
- S. Liu, J. V. M. Weaver, A. Maud Save and S. P. Armes, Langmuir, 2002, 18, 1347–1353 Search PubMed.
- M. A. Rub, N. Azum, A. M. Asiri, S. Y. Alfaifi and S. S. Alharthi, J. Mol. Liq., 2017, 227, 1–14 CrossRef CAS.
- J. Huang, H. Zhang, Y. Yu, Y. Chen, D. Wang, G. Zhang, G. Zhou, J. Liu, Z. Sun and D. Sun, Biomaterials, 2014, 35, 550–566 CrossRef CAS.
- Q. Y. Yu, L. B. Lin, X. Y. Xing, H. L. Dong, X. Z. Jiang and M. F. Zhu, Mater. Sci. Forum, 2015, 815, 359–366 Search PubMed.
- K. Y. Peng, M. Y. Hua and R. S. Lee, Carbohydr. Polym., 2014, 99, 710–719 CrossRef CAS PubMed.
- Y. Wang, J. Fang, D. Cheng, Y. Wang and X. Shuai, Polymer, 2014, 55, 3217–3226 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Loading calibration curve in water of DOX, release calibration curve in PBS media pH 7.4 of DOX, and release calibration curve in PBS media pH 5.5 of DOX. See DOI: 10.1039/c8ra09475f |
| This journal is © The Royal Society of Chemistry 2019 |