
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a hybrid peptide dendrimer micellar carrier system and its application in the reformulation of a hydrophobic therapeutic agent derived from traditional Chinese medicine
Jing Jing†
a,
Karnaker R. Tupally†a,
Ganesh R. Kokila,
Zhi Qua,
Sibao Chen *bc and
Harendra S. Parekh*a
*bc and
Harendra S. Parekh*a
aSchool of Pharmacy, The University of Queensland, 20 Cornwall Street, Woolloongabba, QLD 4012, Australia. E-mail: h.parekh@uq.edu.au
bState Key Laboratory of Chinese Medicine and Molecular Pharmacology (Incubation), The Hong Kong Polytechnic University Shenzhen Research Institute, Shenzhen, 518057, People's Republic of China. E-mail: sibao.chen@polyu.edu.hk
cInstitute of Medicinal Plant Development, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100193, People's Republic of China
First published on 18th January 2019
Abstract
The discovery that a cane toad poison-derived steroid, bufalin can significantly impact cancer cell proliferation supports its potential use in cancer therapy. However, its poor aqueous solubility and tissue deposition characteristics hamper its broader application as an anticancer therapeutic agent in its own right. To address this we developed an amphiphilic dendrimer-based delivery system, which self-assembles into discrete micelles in an aqueous environment. The bufalin–micelle inclusion complex was prepared by the co-precipitation method and their presence was confirmed by dynamic light scattering (DLS), zeta potential and differential scanning calorimetry (DSC) and transmission electron microscopy (TEM) measurements. The self-assembled bufalin-containing micelles were found to form at/above the dendrimer concentration of 105.38 μmol L−1, and showed a more than threefold increase in the aqueous solubility (142.9 μg mL−1) of bufalin, when compared with a saturated bufalin aqueous solution (42.4 μg mL−1), and two non-assembling peptides of similar composition (79.3 and 62.5 μg mL−1 respectively).
1. Introduction
Bufalin, a cardioactive C-24 class of steroid is an active ingredient of a well-known toad-based Traditional Chinese Medicine (TCM) Chan Su, which has long been approved as a treatment for a range of cancers by the Chinese Food & Drug Administration.1 The active ingredients in TCM are widely recognised to have great potential for the development into medicines for cancer therapy, globally. Taking the bioactive bufalin as an example, it has similarities and shortfalls associated with well-known western medicines such as digoxin, digitoxin, and ouabain, each of which is emerging as potential candidate for the treatment of cancer.2 Despite the reported pharmacological action of individual active ingredient in TCM, a range of universal issues remain, which centre around their poor aqueous solubility, limited stability and short biological half-life.3 These features are also common to bufalin, which to-date have limited its broader application in a clinical setting, and an aspect this work attempts to address through rational design and formulation of a hybrid nanoparticulate system.4 Through the self-assembling features of amphiphilic peptide dendrimers, capable of incorporating bufalin in their core, stable, micellar structures of low nm dimensions with enhanced aqueous solubility were formed and characterised using a range of conventional techniques.Peptide dendrimers, derived exclusively from natural amino acids are wedge-like branched macromolecules consisting of a amino acid-based branching core and/or covalently attached surface functional units.5–8 Our extensive earlier studies have shown promise of peptide dendrimers in the delivery of a diverse range of molecules, from small molecules to genetic cargo.9–11 We explored the cell internalisation characteristics of a broad variety of peptide dendrimer designs, providing all-important structure-cell delivery relationship data for rational design of carrier systems.12 To elaborate, we reported on the influence of dendrimer head group chemistry (lysine, histidine, arginine) on cellular entry characteristics, finding that cationic guanidine head groups of arginine were the most efficient at driving cellular internalisation.12 This is in agreement with earlier studies where peptide dendrimers with terminal arginine residues showed enhanced solubility, permeation and cellular internalization for delivery of small molecules.13 To-date very few drug delivery systems (e.g. chitosan, PEG–PLGA–PLL, agglutinin-based) have been trialled for enhancing cellular delivery of bufalin although they are limited by their cytotoxicity (chitosan-induced cell lysis), complex tri-block composition (PEG–PLGA–PLL) and quality control/characterisation issues (wheat germ-based agglutinin).14–16 In contrast low generation amphiphilic peptide dendrimers reported here are well known for their non-cytotoxic and biocompatible nature, their synthesis can be automated and is scalable (mg to kg), while they self-assemble into nanomicelles and readily accommodate poorly soluble drugs, such as bufalin into their core. Based on the evidence from our extensive research in various aspects peptide dendrimer design, synthesis and evaluation we present a first of its kind study reporting on the potential of hybrid-peptide dendrimers to accommodate the steroid bufalin, while simultaneously forming nano micelles, which have inherent properties promoting deposition in tumour tissue.17,18
In this study, arginine-terminated peptide dendrimers with a 4+ (Fig. 1a: non-lipidated form and Fig. 1c: lipidated form) or 8+ surface charge (Fig. 1b), were prepared wholly by solid phase peptide synthesis. Next, each system was assessed for its ability to increase the solubility of bufalin while self-assembly was determined using a range of confirmatory techniques, as is elaborated on below.
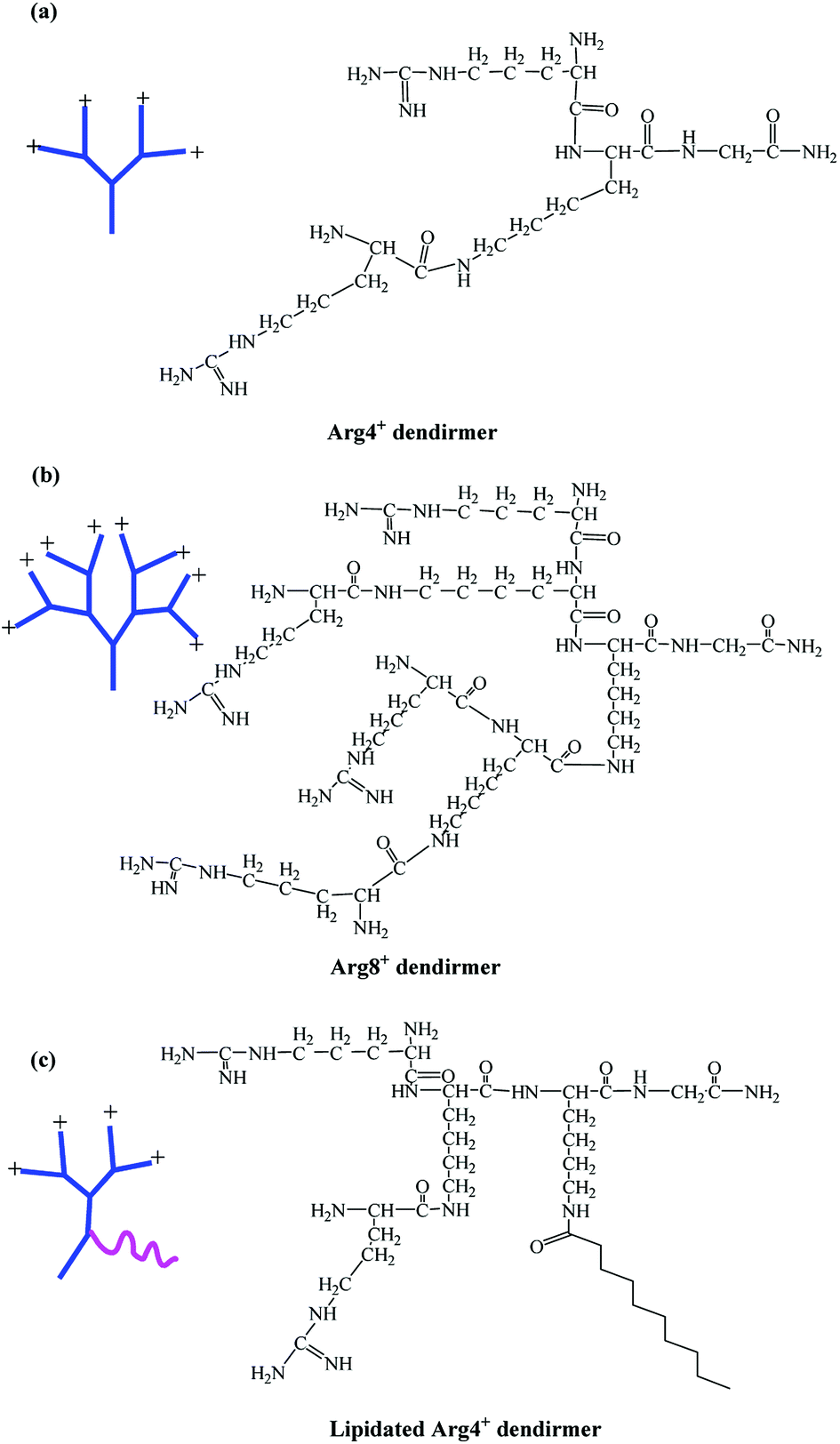 | ||
| Fig. 1 Chemical structures of peptide dendrimers. (a) 4+ charge; (b) 8+ charge; (c) lipidated 4+ charge. | ||
2. Materials and methods
2.1 Materials
Fmoc-Gly–OH, Fmoc-Lys (Fmoc)–OH, Fmoc-Lys (Boc)–OH, Fmoc-Arg (Pbf)–OH, O-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were purchased from GL Biochem (Shanghai) Ltd, China. Rink amide resin (200–400 mesh, loading capacity 0.34 g mmol−1) was purchased from Chem-impex International, Inc (USA). The solvent used throughout the synthesis was peptide grade N,N-dimethylformamide (DMF; Merck Australia), trifluoroacetic acid (TFA), N,N-diisopropylethylamine (DIEA), dichloromethane (DCM), triisopropylsilane (TIPS), piperidine, were purchased from Sigma-Aldrich (Australia).2.2 Synthesis of Fmoc-Lys (Mtt)–OH
Fmoc-Lys-NH (Boc)–OH (0.46, 1 mmol) was dissolved in 5 mL of dichloromethane (DCM). To this solution, 5 mL of trifluoroacetic acid (TFA) was added slowly and reaction stirred for 1 h at room temperature. The reaction was monitored with TLC to confirm the complete disappearance of Fmoc-Lys (Boc)–OH in the reaction mixture. DCM and TFA were removed using high vacuum and further toluene was added to the crude to remove any traces of TFA using high vacuo. The dried product was collected and again dissolved in 10 mL of DCM. To this solution 2 equivalence of triethylamine (TEA, 0.202 g, 2 mmol) and methyl trityl chloride (Mtt, 0.356 g, 1.2 mmol) were added and the reaction continued for 24 h by monitoring with TLC to make sure complete disappearance Fmoc-Lys (NH2)–OH spot on TLC plate. The mixture was extracted with DCM (2 × 20 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography using n-hexane and ethyl acetate (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 v/v). Yield (74%, 0.461 g), TLC: Rf value 0.55 (n-hexane:ethyl acetate = 3:2, UV active, illuminates under UV light).
:20 v/v). Yield (74%, 0.461 g), TLC: Rf value 0.55 (n-hexane:ethyl acetate = 3:2, UV active, illuminates under UV light).
2.3 Synthesis of (lipidated)-peptide dendrimers
A panel of arginine-terminated lipidated dendrimers (4+, C-to-N sequence: Gly-Lys (C10)-Lys-(Arg)2) were synthesized by Fmoc-solid phase chemistry purified and characterized as described in our earlier publications. Briefly, Rink amide resin was swollen with N,N-dimethylformamide (DMF) for 2 h and deprotected with piperidine solution (20% v/v in DMF, 2 × 8 min). After washing with DMF, first amino acid (pre-activated Fmoc-Gly–OH with 0.5 M HBTU and DIEA) was coupled to the Rink amide resin and then repeat the deprotection and coupling of Fmoc-Lys (Mtt) OH. The resulting product was treated with TFA solution (3% v/v in DCM) and washed with DMF. Then the lipid pre-activated decanoic acid with 0.5 M HBTU and DIEA was coupled. Sequential deprotection and coupling was performed until the desired dendrimer construct formed by adding Fmoc-Lys (Fmoc)–OH and Fmoc-Arg (Pbf)–OH. Ninhydrin test was performed to confirm the coupling efficiency (≥99.8% coupling) of amino acid coupling. After the final deprotection, resin was thoroughly washed with DMF and dry DCM. The dried resin was collected into the round bottom flask and added with cleavage cocktail (TFA, DCM, water and TIPS). TFA was removed using high vacuo and residual TFA azeotroped with toluene. The crude peptide was triturated with cold diethyl ether. Further, the crude peptide dissolved in water and lyophilized. Synthesized dendrimers were purified by using a preparative RP-HPLC system (Agilent 1200)2.4 MS and HPLC condition
HPLC analysis was carried out using a Shimadzu Prominence liquid chromatography system, equipped with a binary mobile phase pump with an auto-sampler and a DAD detector. The stationary phase comprised a GraceVydac® C18 protein analytical column (5 μm, 4.6 mm × 250 mm). The detection wavelength of interest was 210 nm and the flow rate was maintained at 1.0 mL min−1. The gradient elution of the mobile phase consisted of (A) 0.1% TFA and (B) 100% acetonitrile with the gradient program optimised as follows: 0–30 min, linear gradient 1–100% B; 30–35 min, isocratic at 100% B, then 35–50 min 100–2% B.Mass spectrometric electrospray ionization (ESI+-MS) analysis was performed on an Applied Biosystem/MDS Sciex Q TRAP LC/MS/MS system. Declustering potential (DP) and entrance potential (EP) were set at 200 and 10 mV respectively.
2.5 Solubility studies
Saturation solubility of bufalin was determined in aqueous solutions of at neutral pH. An excess amount of bufalin was added to deionized water and aqueous dendrimer solutions (Arg4+ & 8+ dendrimer, and Arg4+ lipidated dendrimer) and stirred for 24 h at room temperature. The mixture was then filtered through a 0.45 μm syringe filter, and the filtrate directly analysed by HPLC and bufalin concentration determined.The HPLC analysis of bufalin was undertaken using mobile phases comprising 0.1% TFA in MilliQ water (solvent A) and acetonitrile (solvent B) equilibrated, under isocratic (2:3 v/v) conditions. Working solutions of 20, 50, 100, 200, 500 nmol L−1 of bufalin were prepared by appropriate dilution of the stock solution (1000 mg L−1) with methanol. The stationary phase comprised a GraceVydac® C18 analytical column (5 μm, 4.6 mm × 250 mm). The detection wavelength of interest was 296 nm and the flow rate was maintained at 1.0 mL min−1.
2.6 Dynamic light scattering (DLS) measurements
The CMC was determined by using a Malvern Zetasizer, NANO ZS (Malvern Instruments Limited, UK) equipped with a 4 mW He–Ne laser operating at a wavelength of 633 nm. The scattered light is detected at an angle of 169°, an optical arrangement known as non-invasive back scatter (NIBS) optic arrangement that maximizes the detection of scattered light while maintaining signal quality. This provides the exceptional sensitivity that is required for measuring the size of entities such as nanoparticles and polymer micelles, at low concentrations. Measurements were carried out in a polystyrene cell at 25 °C. Data processing was carried out with a computer attached to the instrument. The measurements were repeated three times in order to check their reproducibility.2.7 Dendrimer–bufalin complexation
To obtain the complex, bufalin was dissolved in methanol, then evaporated using rotary evaporator to remove methanol completely. Following which the dendrimer in deionized water was added. The reaction mixture was stirred for 24 h in the dark. The dendrimer–drug suspensions were filtered through a 0.2 μm nylon syringe filter to remove excess solid drug and then lyophilized to remove water. The drug–dendrimer complex obtained was in the form of a white powder.Bufalin, dendrimer, bufalin/dendrimer mixture and the bufalin–dendrimer complex were subjected to analysis using TGA/DSC 2 STARe System (Thermal, USA) up to 300 °C at a heating rate of 5 °C min−1. Empty aluminina pan was used as a blank reference. The heat flow as a function of temperature was measured for the samples.
2.8 Zeta potential measurements
Zeta potential of dendrimers, bufalin–dendrimer mixtures was determined using a NanoZS (Malvern Instrument Limited, UK) for the solutions of bufalin, dendrimer and the bufalin loaded dendrimer in pH 7.4 phosphate buffer.2.9 Transmission electron microscopy measurements
Dendrimer were analysed using Cryo-TEM, where in the copper grids were first dipped into a sample solution and immediately transferred into liquid nitrogen and allowed to stay in for 10 min, later the copper grid was analysed using transmission electron microscope using JEOL 1010.3. Results
3.1 Synthesis of peptide dendrimers
A panel of peptide dendrimer were synthesized using in-house protocols as shown in the Fig. 2. We synthesized orthogonally protected Fmoc-Lys (Mtt)–OH and used for on-resin introduction of lipid on to the peptide dendrimers. Synthesized dendrimers were analysed and characterized by using RP-HPLC and ESI+-MS. The yield of crude dendrimers was found no less than 70%. Further preparative RP-HPLC performed to achieve purity no less than 95%. The purified dendrimer showed single peak profile in analytical RP-HPLC (Fig. 3a) and desired molecular ion ([M + Na]+, m/z = 819.5) under ESI+-MS characterization (Fig. 3b), thus confirming the formation of target peptide dendrimer.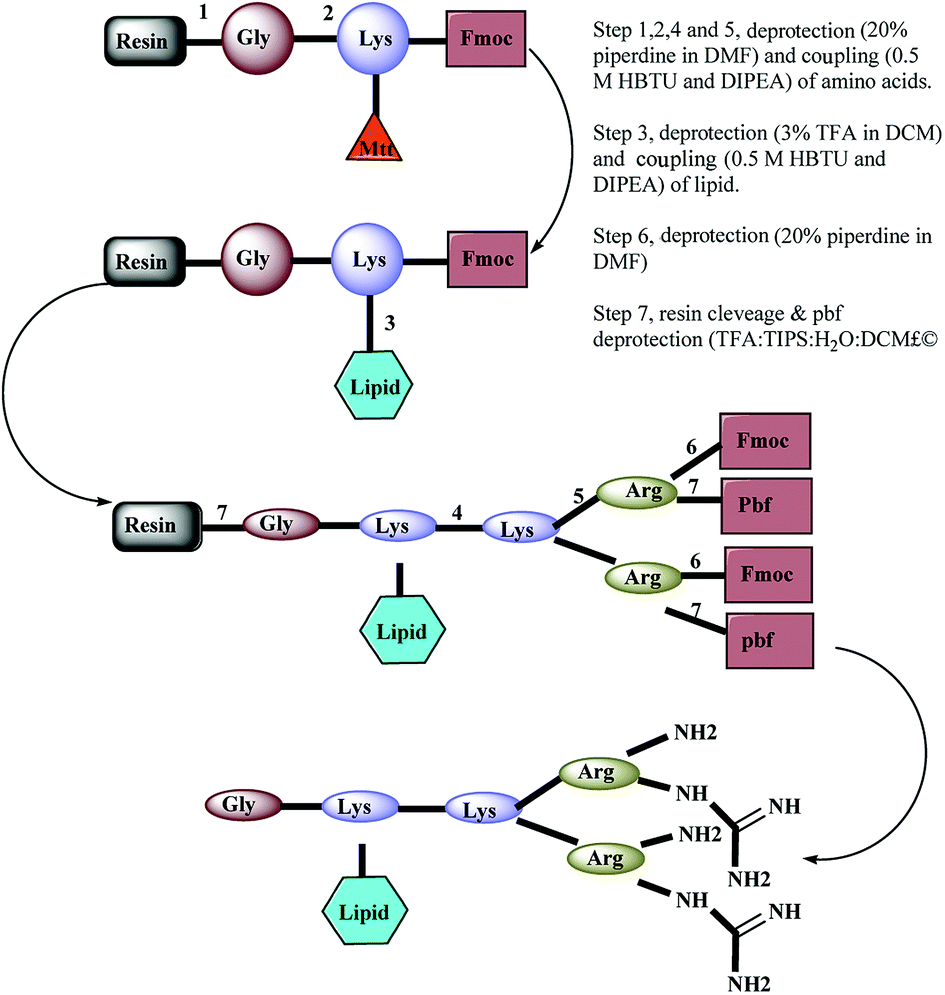 | ||
| Fig. 2 Schematic representation of synthesis pathway of designed lipidated peptide dendrimer. | ||
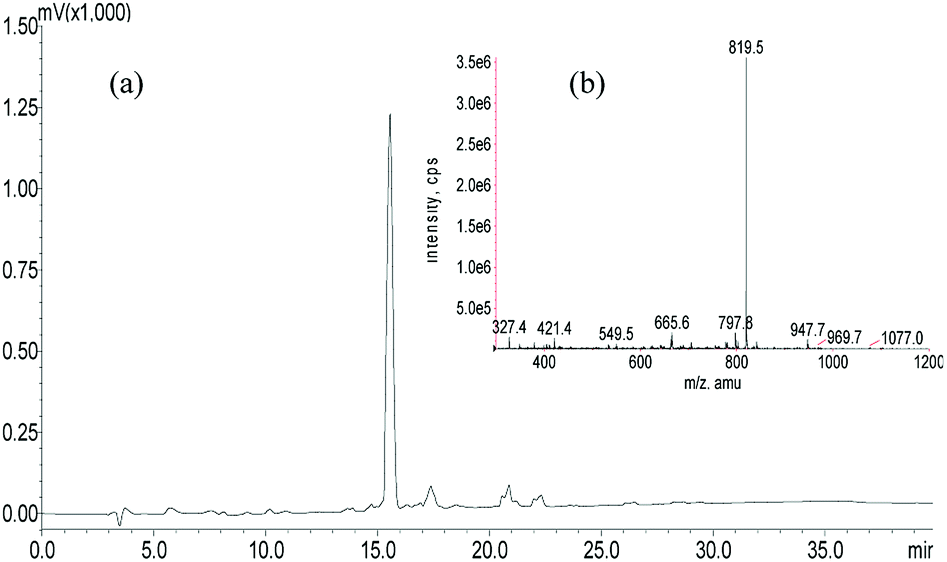 | ||
| Fig. 3 HPLC and MS verification of lipidated dendrimers. (a) HPLC; (b) MS. | ||
3.2 Critical micelle concentration determination for micelles
In this study, self-assembling properties (micellar) of lipidated dendrimers were investigated. Hydrophobic core (decanoic acid) and hydrophilic branching resulted in surfactant like behaviour of lipidated dendrimers tend to self-assemble into micelles in diluted aqueous solution.19 The amphiphilic nature of lipidated dendrimers (monomers) at certain concentration or at critical micelle concentration (CMC) aggregate to form micelles as shown in the Fig. 4. Above this concentration lipidated dendrimer exist as both micelles and monomers, whereas below the concentration only exist as monomers.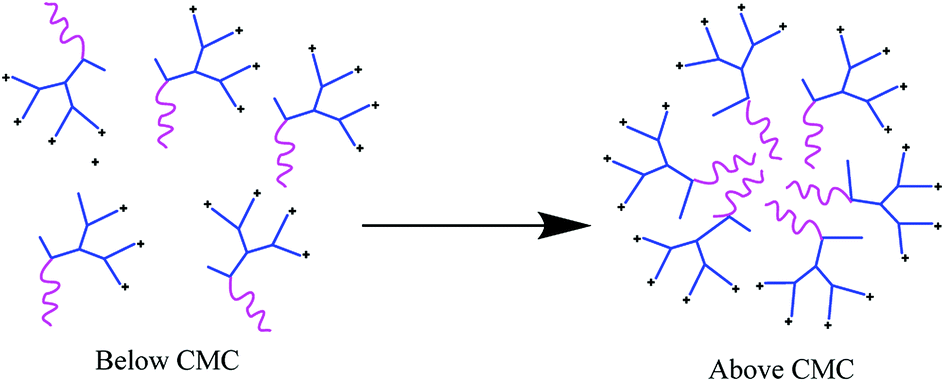 | ||
| Fig. 4 Schematic representation of lipidated dendrimer micelle being formed above its CMC. | ||
To determine the CMC by using DLS technique, aqueous solutions of lipidated dendrimers was prepared within the concentration range of 1–500 μmol L−1. The intensity values of scattered light as a function of concentration of dendrimers are shown in Fig. 5. The scattering intensities detected for dendrimers concentrations below CMC have an approximately constant value corresponding to that of deionized water. As shown in the Fig. 5, the intensity (kcps) starts to show a linear increase with the concentration when it is close CMC, since the number of micelles increases in the solution. However, size of micelles (hydrodynamic diameter) were constant while the concentration approaching to CMC and remain consistent even increase in the concentration. This is an indication for the formation stable micelle of lipidated dendrimers. The intersection of best fit lines drawn through the data point corresponds to 105.38 μmol L−1 is the CMC value of lipidated dendrimers. TEM results showed that the size of the micelles was around 130 nm (Fig. 6).
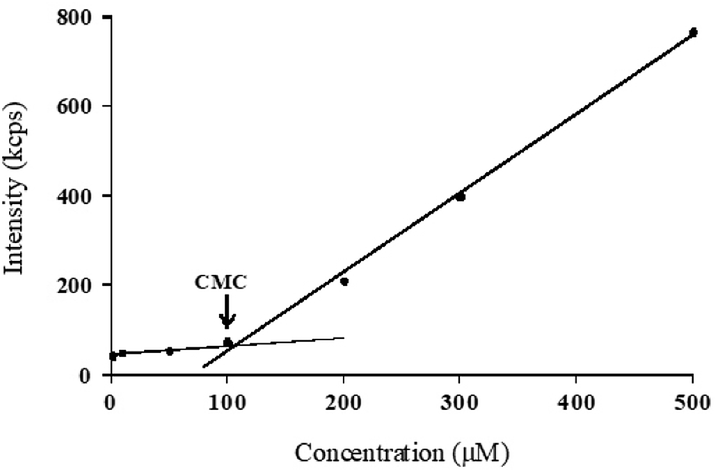 | ||
| Fig. 5 CMC determination: scattered intensity (kcps) as a function of lipidated dendrimers concentration. | ||
 | ||
| Fig. 6 Transmission electron microscopy (TEM) results showed that the size of the micelles was around 130 nm. | ||
3.3 Bufalin–dendrimer micellization
DSC studies were performed separately on dendrimer, drug, and drug–dendrimer complexes. The DSC thermogram Fig. 7d shows the bufalin–dendrimer complex was entirely different from the physical mixture (Fig. 7c) and also bufalin and dendrimers (Fig. 7a and b). Endothermic peak of dendrimer was completely disappeared in the complex. And melting temperature was reduced as shown in the Fig. 7d. Further, zeta potentials were measured to confirm the micellization. The zeta potential was found to be −0.55 mV for bufalin and +18.9 mV for dendrimers. On the other hand, the zeta potential of dendrimer–drug complexes (1:0.5) was +3.03 mV, clearly suggesting that complexes were formed, due to a fall in the net cationic charge, most likely being offset (partial neutralization) by bufalin.
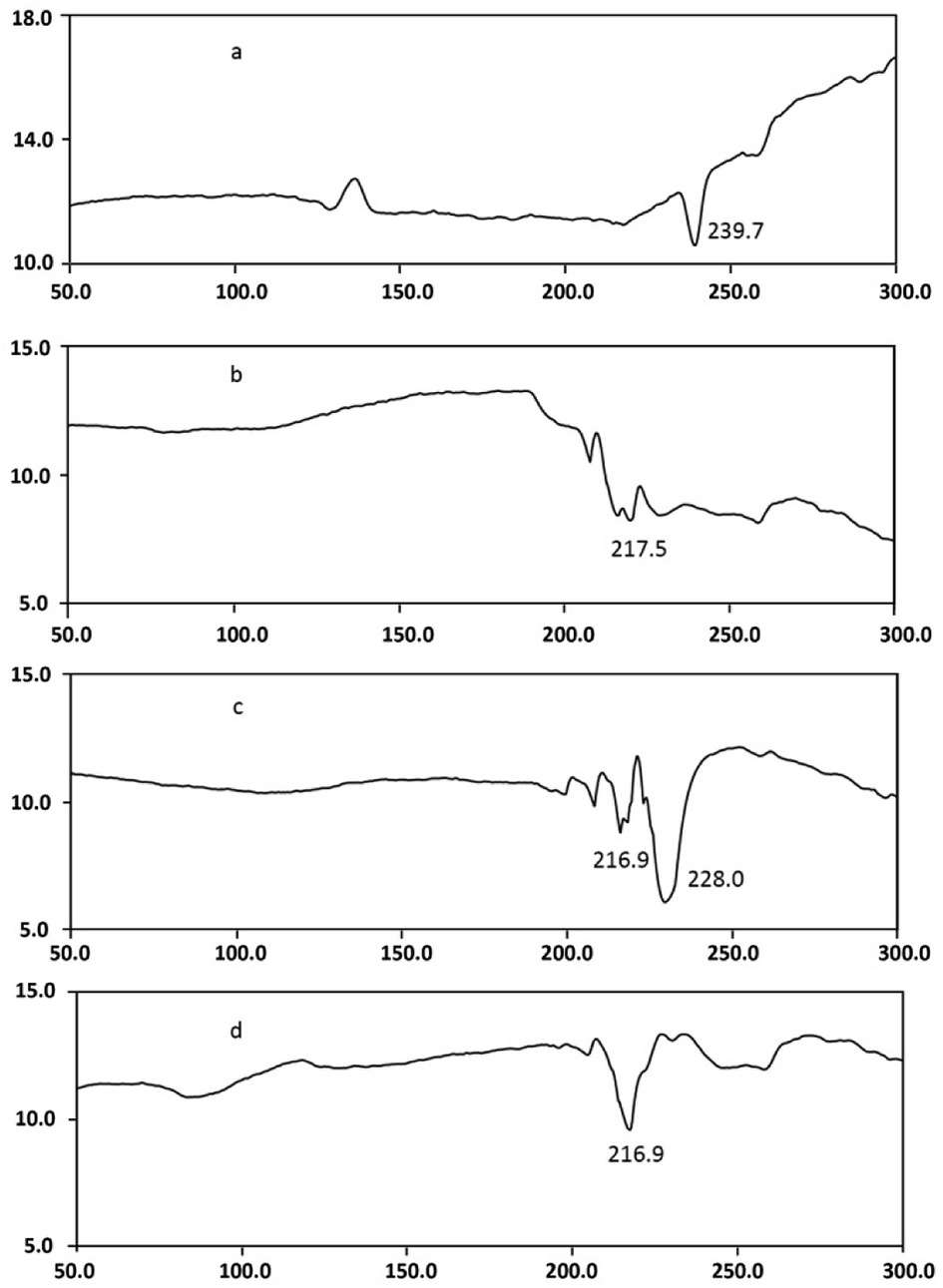 | ||
| Fig. 7 HF-time curves (DSC) of (a) bufalin (melting point 232–241 °C); (b) dendrimer (melting point of L-arginine 216–218 °C); (c) bufalin and dendrimer mixture (1:0.5 molar ratio); (d) bufalin–dendrimer complex. | ||
3.4 Bufalin solubility studies
The concentration of bufalin in water as well as it in different dendrimer (Arg4+ and, 8+ dendrimer and Arg4+ lipidated dendrimer) solutions was determined as an index of the water solubility of bufalin. The solubility of bufalin in different dendrimer solution was determined using HPLC method. Linear least-squares regression analysis of the calibration graph demonstrated linearity in the 20–500 μg mL−1 concentration range. A typical standard relationship (r2 = 0.9999) was described by the equation y = 6363.8 x + 13779. Upon adopting this equation and the peak area obtained for bufalin, the concentration of bufalin in the saturated solution was calculated to be 42.4 μg mL−1, which is close to the reported maximum apparent aqueous solubility of bufalin (32.76 μg mL−1 at 37 °C, pH 7.0). The solubility of bufalin in Arg4+ and Arg8+ dendrimer solution was 62.5 and 79.3 μg mL−1, respectively. Interestingly the solubility of bufalin in Arg4+ lipidated dendrimer significantly increased to 142.9 μg mL−1. The results showed that the solubility of bufalin has been increased in dendrimer solutions, and lipidated dendrimer showed more than twice increased solubility compared non-lipidated counter parts.
4. Discussion
Significant evidences have been proved that the effect Chan Su and its active ingredient bufalin on cancers such as leukaemia,20 breast cancer,21 prostate cancer,22 melanoma and other cancers.23 However, expected clinical outcomes can be achieved only when the drug (bufalin) formulated with right delivery system. The major setback for bufalin is its poor aqueous solubility and drug delivery system to deliver the molecule to the targeted disease. Dendrimers, relatively a new class of carrier systems with hyper-branched structure exist in variable sizes and flexible to change surface functionalities and also for conjugation of targeting ligands and drug molecules. Furthermore, dendrimers also showed enhancing the solubility of hydrophobic drug molecules.23 These combined approach offered to develop amino-acid based peptide dendrimers to enhance the solubility and delivery of bufalin. Earlier, our group published application of asymmetric peptide-dendrimers drug/gene delivery. Now, the first time successfully introduced decanoic acid onto the core by maintaining the terminal guanidine chemistry. Both lipidated and non-lipidated dendrimers were used for the complexation with bufalin. However, lipidated dendrimers being an amphiphilic monomers quickly assembled into micelles at and above the CMC and result in significant enhancement in the solubility compared to non-lipidated dendrimers. DSC studies showed the interaction between bufalin and dendrimer and their inclusion-complex. DSC thermogram of bufalin–drug inclusion complex showed significantly different endothermic melting temperature compare to dendrimer and bufalin. This result is in agreement with zeta potential of bufalin–dendrimer complex, +ve charge of dendrimers was neutralized by bufalin −ve charge. The aqueous solubility data showed bufalin in lipidated peptide dendrimer solution was increased more than 3 folds compare to bufalin alone and 2 folds to non-lipidated counterparts.5. Conclusions
In conclusion, we synthesized and characterised a hybrid lipidated-peptide dendrimer using Fmoc-SPPS. The methodology to incorporate lipid into asymmetric dendrimers is expected to generate self-assembled micelles with greater enhanced solubility of hydrophobic drugs, while retaining arginine terminal can increase the cellular uptake and intracellular delivery of the drug. The micellar formation was clear with enhanced solubility of the guest molecule bufalin and further in vitro studies warranted with optimised formulation. This study here also provides a platform for effective and versatile delivery of other moieties of therapeutic interests.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was co-supported financially by the Shenzhen Science and Technology Innovation Committee (JCYJ20151030164022389; GJHZ20140422173104241) and the National Natural Science Foundation of China (81872769).References
- Z. Meng, P. Yang, Y. Shen, W. Bei, Y. Zhang, Y. Ge, R. A. Newman, L. Cohen, L. Liu and B. Thornton, Cancer, 2009, 115, 5309–5318 CrossRef PubMed.
- R. A. Newman, P. Yang, A. D. Pawlus and K. I. Block, Mol. Interventions, 2008, 8, 36–49 CrossRef CAS PubMed.
- Y. Liu and N. Feng, Adv. Colloid Interface Sci., 2015, 221, 60–76 CrossRef CAS PubMed.
- P. Liu, M. Zhu, Y. Li, H. Wang and Y. Duan, The research on cytotoxicity and cellular uptake of mPEG-PLGA-PLL nanoparticles, The International Conference on Advanced Fibers and Polymer Materials, Shanghai, China, 2009, pp. 1190–1192 Search PubMed.
- S. Mutalik, A. Hewavitharana, P. Shaw, Y. G. Anissimov, M. Roberts and H. S. Parekh, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 3556–3562 CrossRef CAS PubMed.
- H. S. Parekh, Curr. Pharm. Des., 2007, 13, 2837–2850 CrossRef CAS PubMed.
- K. Sadler and J. P. Tam, Rev. Mol. Biotechnol., 2002, 90, 195–229 CrossRef CAS PubMed.
- N. Shah, R. J. Steptoe and H. S. Parekh, J. Pept. Sci., 2011, 17, 470–478 CrossRef CAS PubMed.
- A. R. Hegde, P. V. Rewatkar, J. Manikkath, K. Tupally, H. S. Parekh and S. Mutalik, Eur. J. Pharm. Sci., 2017, 102, 237–249 CrossRef CAS PubMed.
- P. V. Rewatkar, D. P. Sester, H. S. Parekh and M. O. Parat, ACS Biomater. Sci. Eng., 2016, 2, 438–445 CrossRef CAS.
- P. K. Shetty, J. Manikkath, K. Tupally, G. Kokil, A. R. Hegde, S. Y. Raut, H. S. Parekh and S. Mutalik, AAPS PharmSciTech, 2017, 18, 2346–2357 CrossRef CAS PubMed.
- P. V. Rewatkar, H. S. Parekh and M. O. Parat, PLoS One, 2016, 11, e0147491 CrossRef PubMed.
- S. Mutalik, P. K. Shetty, A. Kumar, R. Kalra and H. S. Parekh, Drug Delivery, 2014, 21, 44–54 CrossRef CAS PubMed.
- Y. Liu, P. Wang, C. Sun, J. Zhao, Y. Du, F. Shi and N. Feng, Int. J. Pharm., 2011, 419, 260–265 CrossRef CAS PubMed.
- X. Tian, H. Yin, S. Zhang, Y. Luo, K. Xu, P. Ma, C. Sui, F. Meng, Y. Liu and Y. Jiang, Eur. J. Pharm. Biopharm., 2014, 87, 445–453 CrossRef CAS PubMed.
- P. Yin, Y. Wang, Y. Qiu, L. Hou, X. Liu, J. Qin, Y. Duan, P. Liu, M. Qiu and Q. Li, Int. J. Nanomed., 2012, 7, 3961–3969 CAS.
- C. Oerlemans, W. Bult, M. Bos, G. Storm, J. F. W. Nijsen and W. E. Hennink, Pharm. Res., 2010, 27, 2569–2589 CrossRef CAS PubMed.
- Y. Yang, R. M. Pearson, O. Lee, C. W. Lee, R. T. Chatterton, S. A. Khan and S. Hong, Adv. Funct. Mater., 2014, 24, 2442–2449 CrossRef CAS.
- C. Hüttl, C. Hettrich, R. Miller, B. R. Paulke, P. Henklein, H. Rawel and F. F. Bier, BMC Biotechnol., 2013, 13, e51 CrossRef PubMed.
- X. B. Xie, J. Q. Yin, L. L. Wen, Z. H. Gao, C. Y. Zou, J. Wang, G. Huang, Q. L. Tang, C. Colombo and W. L. He, PLoS One, 2012, 7, e47375 CrossRef CAS PubMed.
- S. Yan, X. Qu, Z. Zhu, L. Zhang, L. Xu, N. Song, Y. Teng and Y. Liu, J. Cancer Res. Clin. Oncol., 2012, 138, 1279–1289 CrossRef CAS PubMed.
- C. H. Yu, S. F. Kan, H. F. Pu, E. Jea Chien and P. S. Wang, Cancer Sci., 2008, 99, 2467–2476 CrossRef CAS PubMed.
- Y. P. Hsiao, C. S. Yu, C. C. Yu, J. S. Yang, J. H. Chiang, C. C. Lu, H. Y. Huang, N. Y. Tang, J. H. Yang and A. C. Huang, J. Evidence-Based Complementary Altern. Med., 2012, 2012, e591241 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |