
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of tetragonal to cubic phase transition on the upconversion luminescence properties of A/B site erbium-doped perovskite BaTiO3†
Hyeongyu Bae and
Kang Taek Lee*
and
Kang Taek Lee*
Department of Chemistry, Gwangju Institute of Science and Technology, Cheomdangwagi-ro 123, Buk-gu, Gwangju, Republic of Korea. E-mail: ktlee@gist.ac.kr
First published on 18th January 2019
Abstract
With the increasing number of applications for upconversion materials, a more detailed understanding of the intrinsic mechanisms of their optical processes is required. Thus far, various lanthanide-doped host materials or nanoparticle systems have been investigated as representative upconversion systems owing to their major advantage as optical probes. As for the energetics of upconversion and the associated upconversion pathways, the role of the host material is very important because it provides a unique microscopic environment; for example, a unique local lattice structure in the case of crystalline samples. In general, the upconversion luminescence intensity decreases as a function of temperature owing to thermally accelerated multiphonon relaxation. Here, we report that the temperature dependence of the upconversion luminescence efficiency is affected differently in an Er3+-doped perovskite material, barium titanate (BaTiO3, BT), than in a general system. We show that Er3+ doped at the A (Ba2+) and B (Ti4+) sites of tetragonal phase BT, referred to as A-BT and B-BT, respectively, show different upconversion behaviors. The slope of the plot of the upconversion emission intensity as a function of temperature changed significantly in case of B-BT, but not for A-BT. This anomalous behavior of Er3+-doped BT is attributed to the phase transition (at ∼120 °C) of BT from tetragonal to cubic phase. Essentially, the temperature-dependent upconversion luminescence trend depends on the doping sites of Er3+, i.e., at A or B sites in BT, which is explained by the difference in the symmetry of the crystalline structure with different crystal phase surrounding the Er3+ ions.
Introduction
Upconversion (UC) is a useful photophysical phenomenon and has been applied in various fields, such as the solar cell industry,1–3 biological imaging4,5 or pH sensing,6–8 and temperature sensing.9–11 The range of spectral emissions of upconversion luminescence (UCL) from upconversion materials (e.g. upconverting nanoparticles, UCNPs) is wide and extends throughout the UV, visible, and IR regions, depending on the type of lanthanide ion doped in the host material. UCNPs increase the energy of the incident photons through the upconversion process. Although various mechanisms have been suggested, every mechanism is based on multiphoton absorption aided by the lanthanide electronic states with long lifetimes ranging from microsecond to millisecond timescale. Moreover, the ladder-like evenly spaced electronic states enable efficient absorption of photons of a single wavelength. The relevant transitions generally involve forbidden 4f–4f transitions, which are extremely stable because of the shielding effect by 5s or 5p electrons. The shielding effect also produces sharp 4f–4f transition bands due to weak coupling of the lattice and weak crystal field.12 Despite these advantageous features, the relatively low UC quantum yield of UC materials limits their application in many fields, especially in biomedical pharmacology, where a small dose of photons is preferred. Therefore, numerous research efforts are underway to enhance the efficiency of UCL by various methods such as doping of different sized ions13,14 or making a core–shell structure.15,16The intensity of the 4f–4f transition was first described by Judd17 and Ofelt using the electric dipole mechanism.18 Because the intra-4f transitions are parity forbidden, which results in weak transition dipole moment, the lanthanide ions show low upconversion efficiency and weak absorption or emission.19 Judd–Ofelt's theory is based on the assumption that mixing of the ground 4fN state with higher electronic configurations has odd terms in the crystal field Hamiltonian.17,18,20 The luminescence intensities of lanthanide ions are significantly influenced by their site symmetry.21 In general, a local crystal structure with low symmetry around the activator (emitting ion) leads to enhanced mixing between the 4f state and higher electronic configuration; consequently, the upconversion efficiency is enhanced.12 Various studies have been published concerning the problem of low upconversion efficiency. Wisser et al. showed that site-specific doping of gadolinium (Gd) and lutetium (Lu) at Y sites in NaYF4 UCNPs leads to a local symmetry-distorted lattice around the activator (Er3+).14 Furthermore, the radiative efficiency of transitions from the excited states to the ground state (4I15/2) have been enhanced substantially. With an increasing odd-parity interaction of the Er3+ ion with the host crystal, the f orbitals and higher electronic configurations are mixed significantly, leading to enhanced emission intensity.12 Thus, in the absence of such non-symmetrical crystal fields, the degenerate f orbitals of lanthanide ion would not produce any radiative transition.
BaTiO3 (BT) and related advanced ABO3 perovskite materials have been widely investigated as host materials for upconversion.22–24 BT is a versatile perovskite material characterized by a high dielectric constant, high charge storage capacity, electrical insulating property, and optical transparency in the visible range.25–28 The cubic and tetragonal crystal structures of BT are shown in Fig. 1. In the cubic phase, Ba2+ is located at the corner of the cube (A site), O2− is located at the center of the face (X site), and Ti4+ is located at the center of the body (B site).29 In the tetragonal phase structure, the unit cell is stretched along one axis (z-axis), and the position of the Ti4+ ion at the center of the body is slightly shifted upward or downward in the same axis (z-axis).30–32 This phenomenon leads to the ferroelectricity of BaTiO3, and the unit cell shows polarization. The Curie temperature of BT is ∼120 °C,31,33 above which BT undergoes a transition from the tetragonal phase to the cubic phase. In addition, as shown in the infrared spectrum of BT (Fig. S1†), BT is confirmed to be an appropriate host with a low maximum phonon energy. In this study, we prepared two site-specific Er3+-doped BT samples, that is, Er3+ doped at A sites of BT (A-BT), and Er3+ doped at B sites of BT (B-BT), separately via solid-state reactions. In addition, we investigated the temperature dependent upconversion luminescence properties of the powders (A-BT and B-BT). We found that the UCL changed similarly as a function of temperature, but A-BT and B-BT show different behaviors at the Curie temperature. This is the first demonstration of the relationship between UCL and the phase transition of the BT host.
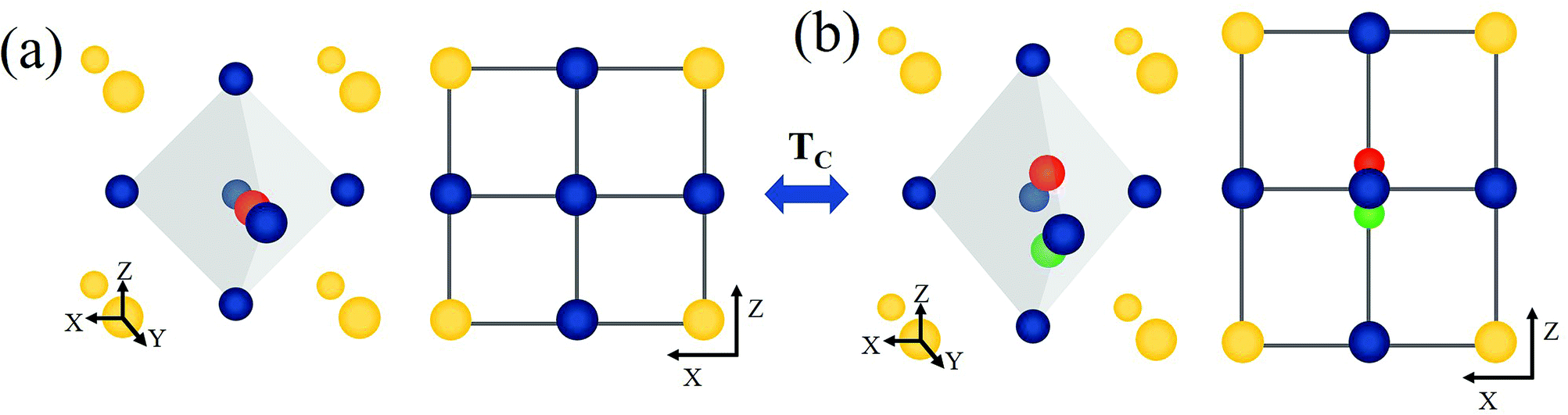 | ||
| Fig. 1 Schematic of a BaTiO3 unit cell (A site Ba2+ ion: yellow, B site Ti4+ ion: red or green, O2− ion: blue): (a) BaTiO3 unit cell in the cubic phase, 3D view (left) and (001) plane view (right). (b) BaTiO3 in the tetragonal phase with different orientations of polarization (green and red Ti4+), 3D view (left) and (001) plane view (right). In case of tetragonal phase, the centered Ti4+ ion moves along the [001]-direction. Tc represents the Curie temperature, the temperature at which reversible phase transition occurs between the two phases. | ||
Purely tetragonal Er3+-doped BT powders were synthesized via solid-state reactions. Since the solid-state reaction is an ion-size-dependent doping method that uses accurately calculated stoichiometric quantities of reagents, the solid-state reaction is suitable for doping the ion at a specific site in the crystal.34 In this study, Ba0.98TiO3:Er0.02 (A-BT) and BaTi0.98O3:Er0.02 (B-BT) were synthesized, because the fraction of Er3+, 2%, turned out to yield the highest emission intensity, clearly at 525 and 550 nm as shown in Fig. S2.† This results is the same result as the previous study.35 We anticipate that if 1% and 3% Er3+ ions are site specifically doped at A site and B site of BT, the phase transition dependent upconversion luminescence intensities decay tendency will be similar with 2% Er3+ ion doped BT (A-BT, B-BT). To increase the surface area of the reaction, sodium chloride (99.5%, TCI) with a melting point of 801 °C was used as the flux. Barium carbonate (≥99%, Sigma-Aldrich) and titanium(IV) oxide (99.8%, Sigma-Aldrich) were used as barium and titanium source, respectively. Erbium oxide (99.9%, Sigma-Aldrich) was used as the Er3+ source. The molar ratios of the reagents vary depending on the concentration of the doped ions (see Table 1). The amount of sodium chloride was adjusted at NaCl/BaTiO3 = 1. A stoichiometric amount of the precursor powder material was ground for 1 h with an agate mortar and pestle. After that, the mixed reagents were loaded in a furnace and sintered for 5 h at 1100 °C (heating rate: 5 °C min−1) The product, tetragonal BT, is a white delicate powder that turns slightly pinkish after Er3+ doping. The structures of the as-prepared A-BT and B-BT powders were characterized by X-ray diffraction (XRD, X'Pert PRO Multi-Purpose X-ray Diffractometer, PANalytical) using Cu Kα radiation in the 2θ range of 20° to 80°. Further, X-ray diffraction (D/MAX-2500, RIGAKU) between 65 and 155 °C (at intervals of 10 °C) under vacuum conditions was also performed using Cu Kα radiation in the same 2θ range.
| Material | Dopant ions | Dopant concentration | Concentrations of species (mol%) | |||
|---|---|---|---|---|---|---|
| Ba | Ti | Er | NaCl | |||
| t-BaTiO3 | Er3+ | 2% (on Ba2+ site) | 0.98 | 1 | 0.02 | 1 |
| t-BaTiO3 | Er3+ | 2% (on Ti4+ site) | 1 | 0.98 | 0.02 | 1 |
The room-temperature XRD patterns of A-BT and B-BT are shown in Fig. 2a. The blue and pink diffraction patterns are from A-BT and B-BT, respectively. The XRD patterns of BT with reflections from the (200) and (002) planes are the fingerprint patterns of the tetragonal phase BT. In addition, the positions of these peaks are related to the size of the lattice. Fig. 2b shows an enlargement of the 44–46.5° range of the pattern shown in Fig. 2a. If we define the lattice constants in a conventional way, the relationship between them in the tetragonal phase is as follows:
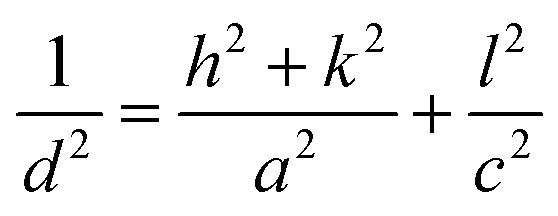 | (1) |
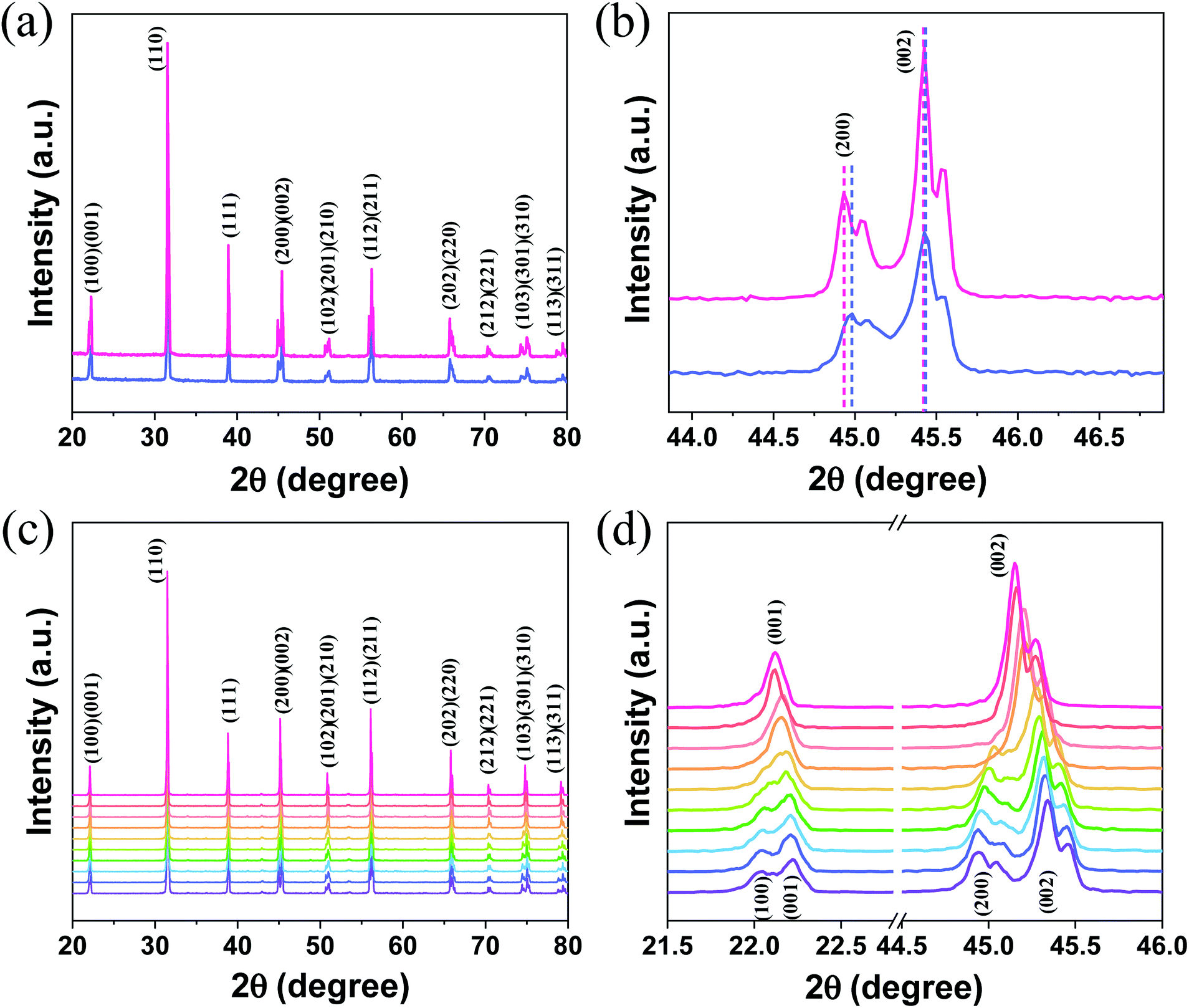 | ||
| Fig. 2 (a) Room-temperature XRD patterns of tetragonal A-BT (blue) and B-BT (pink) and (b) magnification of the 44.0–46.5° region. (c) High-temperature XRD patterns of B-BT and (d) magnification of the 21.5–23° and 44.5–46° regions. Purple to pink: 65–155 °C with an interval of 10 °C. | ||
In Fig. 2b, a slightly different angle (2θ) of the (002) plane diffraction was observed because of the different site substitution of Er3+ ions into the BT host. The inter-planar spacing (d spacing), the lattice constants (a and c) (Å), volume of the unit cell (106 pm3), and density (g cm−3) were calculated using the tetragonal phase equation (eqn (1)). The calculated parameters for A-BT and B-BT are listed in Table 2.
| Material | Dopant ions | Ion concentration | Lattice constant and density | |||
|---|---|---|---|---|---|---|
| a (Å) | c (Å) | Vcal (106 pm3) | dcal (g cm−3) | |||
| t-BaTiO3 | Er3+ | 2% (on Ba2+ site) | 3.9799 | 4.0256 | 63.7640 | 6.0532 |
| t-BaTiO3 | Er3+ | 2% (on Ti4+ site) | 3.9845 | 4.0301 | 63.9811 | 6.0738 |
Upon comparison, the lattice constant of B-BT is larger than that of A-BT, indicating the expansion of the unit cell in B-BT, as mentioned above. This expansion arises from the differences in the ionic sizes of Er3+, Ba2+, and Ti4+ (Er3+: 0.89 Å (with 6 coordination), Ba2+: 1.61 Å, Ti4+: 0.605 Å, O2−: 1.35 Å).36–38 In the case of doping at site A (Ba2+), the size of Ba2+ is larger than the size of Er3+; therefore, the lattice constant depends on the size of the O2− ion rather than the size of Er3+. When we substitute Er3+ at site B (Ti4+), the size of Er3+ is larger than the size of Ti4+, and this causes the expansion of the unit cell, despite the low doping concentration of Er3+. Therefore, when comparing the diffraction patterns of A-BT with B-BT, a small peak shift is observed (44.9 vs. 45.0) corresponding to the (200) peak (Fig. 2b).
Fig. 2c shows the high-temperature XRD patterns of B-BT obtained at various temperatures (65–155 °C) (purple to pink, the baselines of the patterns were arbitrarily set for better visualization). We found that (a) the doublet (200)/(002) peak converged into a singlet (002) peak and (b) the doublet (100)/(001) peak merged into a singlet (001) peak. We attributed this to the phase transition from the tetragonal phase to cubic one, with an increase in temperature to ∼120 °C, the Curie temperature of the phase transition of BaTiO3. The transition from the tetragonal to cubic phase induces a change in the UCL intensity of Er3+.
The log(I) versus log(P) plots (here, I denotes the UC intensity and P denotes the laser power) with slopes of nearly 2 (Fig. S3†) indicate that the 2H11/2 → 4I15/2, 4S3/2 → 4I15/2, and 4F9/2 → 4I15/2 transitions of Er3+ in both A-BT and B-BT are all two-photon upconversion processes, as depicted in Fig. S4.† In Fig. 3a and b, the upconversion emission spectra of B-BT and A-BT under 980 nm CW lasers are shown in the same temperature range (65–155 °C) as used in the high-temperature XRD experiments. Here, we focus only on the green emission (540–556 nm) because our samples show very low signal intensity in the red spectral region (∼660 nm). The temperature-dependent UC spectra of A-BT and B-BT under 980 nm CW laser irradiation were acquired at intervals of 10 °C between 65 and 155 °C. The optical setup for the temperature-dependent UCL measurement is shown in Fig. S5.† The sample powder was evenly spread on the sample holder placed on a hot plate. The focal point of the 980 nm laser was directed on the sample with the power of the laser being uniform at 340 mW. The spectra were obtained using spectrometer (HR2000+, Ocean Optics) after maintaining the temperature of the hot plate at a given temperature for 5 min to facilitate thermal equilibrium between the sample and hot plate. The reliability of the optical setup was tested by acquiring the intensity data from different positions of the samples, typically 40 times. As shown in Fig. S6b,† despite the possible heterogeneity of the powder density, standard deviation errors were not significant when the emission was measured by irradiating the laser at various locations on the sample.
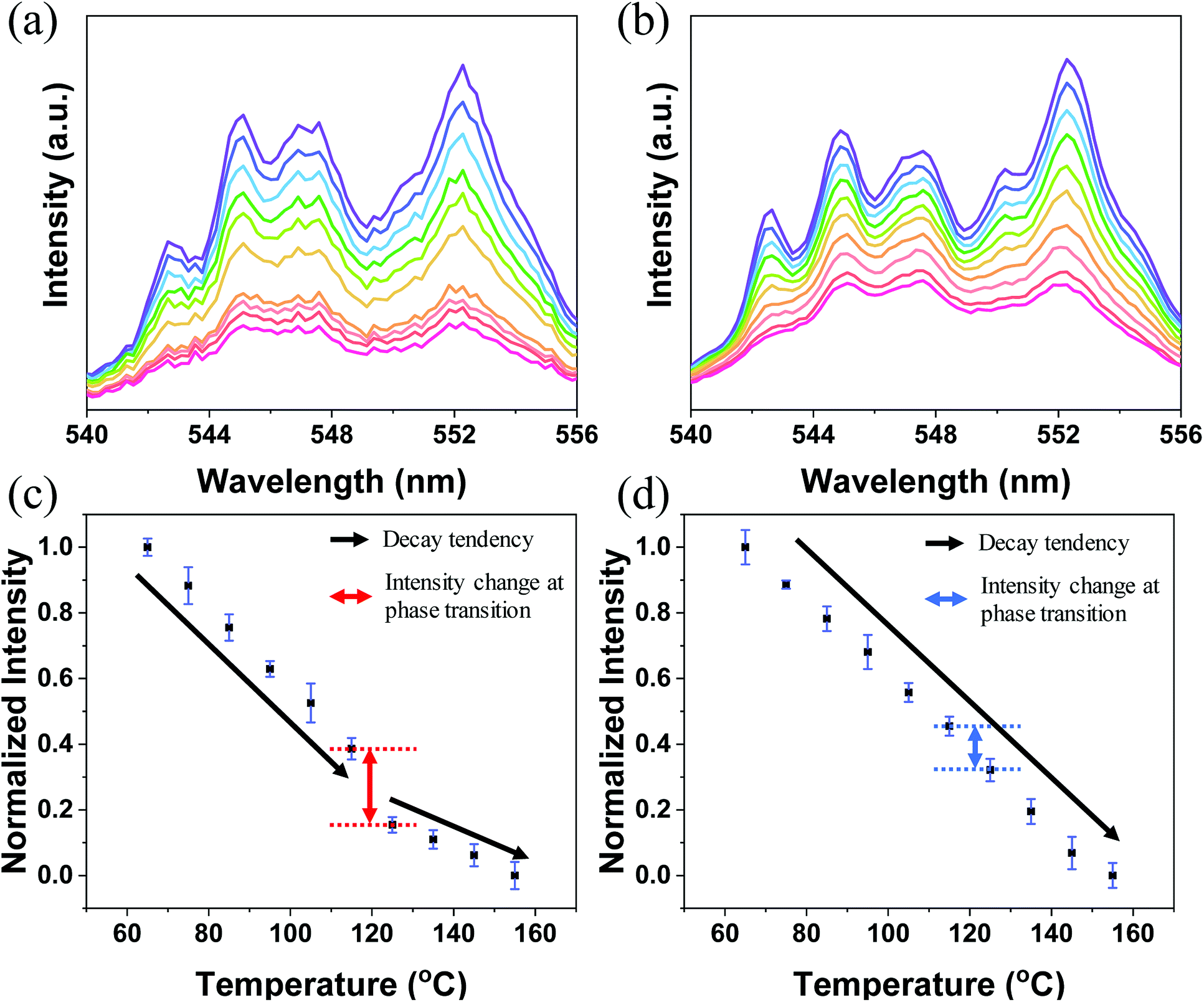 | ||
| Fig. 3 Upconversion spectra of (a) B-BT and (b) A-BT in the 540 to 556 nm range (purple to pink: 65 to 155 °C with an interval of 10 °C) and the normalized 4S3/2 → 4I15/2 emission intensity of (c) B-BT and (d) A-BT. The standard deviation error bar with the point of average normalized intensity is shown. Ten spectra were recorded at each temperature. | ||
Fig. 3a and c show the emission spectrum of B-BT in the 540–556 nm region and the normalized intensity of 4S3/2 → 4I15/2 emission (see Fig. S4† for the energy level diagram of Er3+) versus temperature, obtained by repeating the experiments 10 times. Fig. 3c and d are obtained by integrating the emission spectra of the region of 4S3/2 → 4I15/2 and normalizing the area to 1 for the data at 65 °C. We easily found that, in general, the intensity decreased linearly with increasing temperature. However, interestingly, we observed an abrupt attenuation of the intensity at 125 °C and a nearly linear decrease in intensity with further increase in the temperature beyond 125 °C for B-BT. As shown in Fig. S7,† the reversibility of the phase transition of B-BT was observed. The temperature, 125 °C, is higher than the Curie temperature determined by XRD analyses. In clear contrast, the UCL of A-BT (Fig. 3d) did not show any anomalous temperature dependence at high temperatures, but simply attenuated with a single slope. Therefore, we may conclude that, as B-BT undergoes phase transition from the tetragonal to cubic phase, the UCL is also affected by the phase transition. In other words, the Er3+ ion at the B site experiences interaction with the surrounding ions centro-symmetrically during the phase transition and deviates from the linear attenuation of UCL at lower temperatures. Subsequently, the intensity decreases linearly with a smaller slope at higher temperatures up to 155 °C.
Fig. 4a and b illustrate the unit cells of B-BT and A-BT crystals, respectively. Fig. 4c and d represent the interaction of Er3+ surrounded by the ions of B-BT and A-BT, respectively. In Fig. 4c and d, regions 1 and 2 are the temperature ranges below and above the Curie temperature. Further, in Fig. 4c, the tetragonal structure of B-BT with (001) plane view is shown. Because of the structure of the tetragonal phase of BT, with a body-centered ion that deviates from the center, the interaction between the emitter Er3+ ion and the surrounding ions (face-centered O2− and unit cell edge Ba2+) is not centro-symmetric. In this case, despite the parity-forbidden 4f–4f transition nature of Er3+, the intensity of the UCL tends to be high. In contrast, in the cubic state of B-BT at region 2, Er3+ occupies the center of the cubic unit cell. Thus, the interaction becomes centro-symmetric, which reduces the intensity of UCL. In other words, the system with distorted centro-symmetry, wherein the Laporte forbidden selection rule is relaxed, leads to the enhancement of the emission intensity. However, when the system transitions to the cubic phase, the luminescence intensity decreases substantially. In Fig. 4d, the (001) plane view of the tetragonal phase of A-BT is shown. In this case, the biased Ti4+ and Er3+ have symmetry-distorted interactions and the interaction of O2− with Er3+ is centro-symmetric. The temperature-dependent UCL, which tends to decrease in region 1 (65–115 °C) with the tetragonal phase, shows the same linear attenuation for both A-BT and B-BT. However, the UCL of A-BT and B-BT are different at the Curie temperature: the slope of intensity attenuation of A-BT remains constant at regions 1 and 2. In Fig. 4d, the interactions of Er3+ with surrounding ions in tetragonal A-BT are shown. Compared to B-BT, A-BT has a larger number of surrounding ions (12 O2− and 8 Ti4+). However, O2− and Ti4+ interact with Er3+ in different ways: O2− and Er3+ interact centro-symmetrically, whereas Ti4+ and Er3+ interact non-centro-symmetrically. In this way, while there are two different interaction factors in A-BT, the ionic radius of Ti4+ (0.605 Å) is significantly smaller than that of Ba2+ (1.61 Å) or O2− (1.35 Å). Therefore, regarding symmetry with one ion at the center, the effects of Ba2+ and O2− on the surrounding ions are greater than those of Ti4+ affecting the surrounding ions. In A-BT, the centro-symmetry distorted interaction between Er3+ and Ti4+ is insufficient to induce the crystal structure effect on the UCL, whereas, in the case of B-BT, Ba2+ and O2− with large ionic sizes have a significant distorted centro-symmetric interaction with Er3+. Consequently, in tetragonal A-BT, only the Er3+ ion and Ti4+ ion with a relatively small ionic radius undergo a distorted centro-symmetric interaction through some translocation of Ti4+, and this does not lead to any significant change in the UCL.
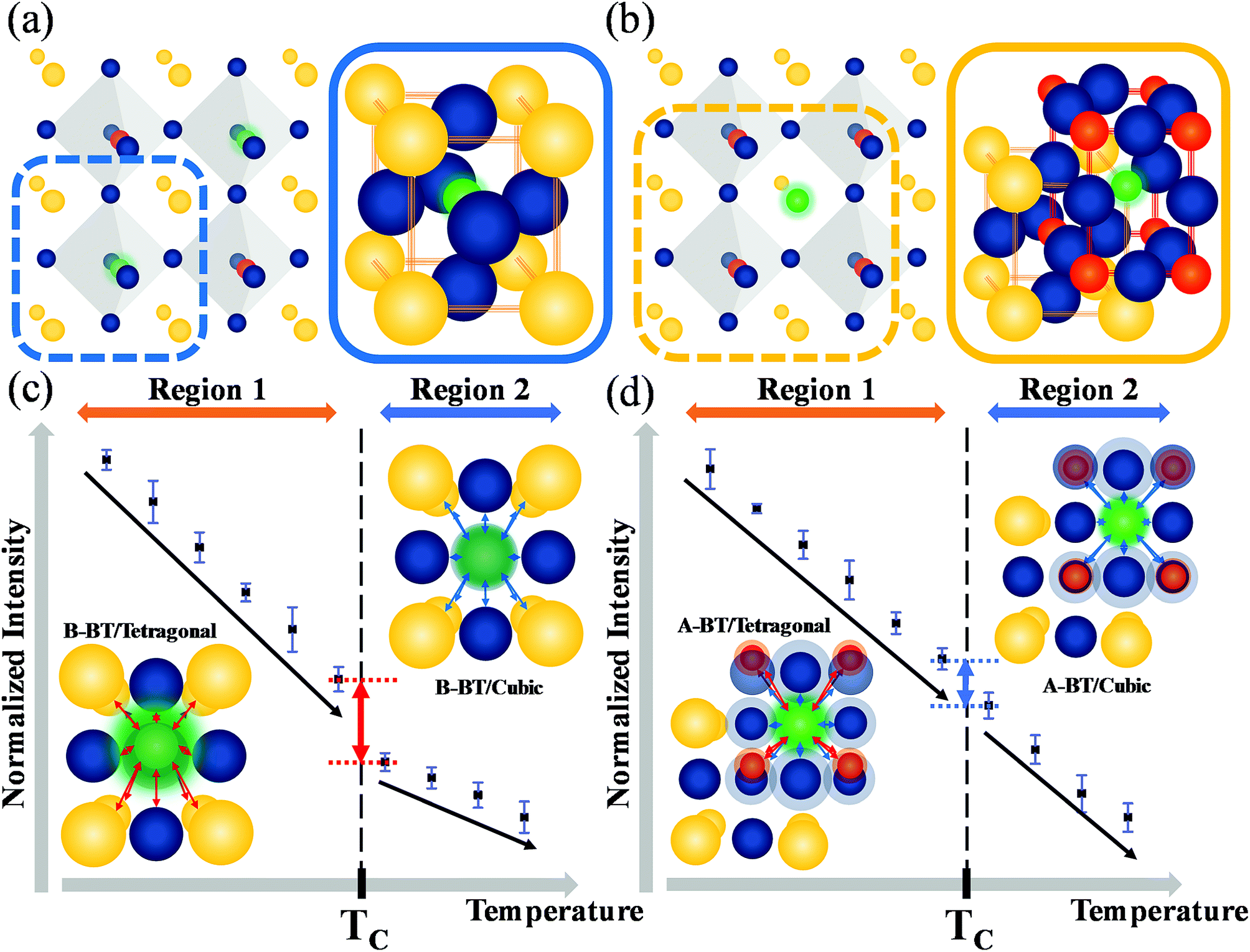 | ||
| Fig. 4 Illustrations of the unit cells of (a) B-BT and (b) A-BT along with the crystal structure (yellow: Ba2+, blue: O2−, green: Er3+, red: Ti4+) and temperature-dependent normalized upconversion intensity data with the (001) plane view of the interaction between Er3+ with surrounding ions in (c) B-BT and (d) A-BT. (c) Centro-symmetry distorted interaction of Er3+ with the surrounding ions of tetragonal B-BT at region 1 and centro-symmetric interaction of Er3+ with the surrounding ions of cubic B-BT at region 2. (d) Centro-symmetry distorted interaction of Er3+ with Ti4+ ions and centro-symmetric interaction of Er3+ with O2− ions of tetragonal A-BT at region 1 and centro-symmetric interaction of Er3+ with the surrounding ions of cubic A-BT at region 2. Arrows represent the interaction between the ions with red for centro-symmetry distorted interaction and blue for the centro-symmetric interaction. | ||
Conclusions
In summary, we investigated the upconversion luminescence of Er3+ ion-doped BT based on the idea that the symmetry-distorted interaction between Er3+ and the surrounding ions enhances the probability of a radiative transition (4f → 4f) in the Er3+ ions. The Er3+-doped BTs (A-BT, B-BT) were synthesized with solid-state reaction, where A- and B- represent the doping at “A site” and “B site” of the general perovskite structure, respectively. The UCL of Er3+ was measured over the temperature range of 65–155 °C to observe the phase-transition effects at ∼120 °C. UCL is generally less intense as the temperature increases because the rate of non-radiative phonon relaxation increases with increasing thermal energy. B-BT also showed such behavior up to the Curie temperature (∼120 °C) of BT; however, in the vicinity of the Curie temperature, the linearly attenuating intensity showed an abrupt change in the rate of decrease. On the contrary, the other sample, tetragonal A-BT, maintained its linear dependency on temperature. This distinct behavior indicates that the change in the upconversion intensities is due to the symmetry of the local crystal structure around emitter lanthanides, despite the fact that lanthanides are not affected significantly by the surrounding environment. This research gives significant insights into mechanisms for increasing the quantum efficiency of lanthanide-doped UC materials and how local crystal structure symmetry works at upconversion intensities of lanthanides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. T. Lee was supported by a GIST Research Institute (GRI) grant funded by the GIST in 2018, and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2017R1A2B3005414).Notes and references
- J. Roh, H. Yu and J. Jang, ACS Appl. Mater. Interfaces, 2016, 8(31), 19847 CrossRef CAS PubMed.
- F.-L. Meng, J.-J. Wu, E.-F. Zhao, Y.-Z. Zheng, M.-L. Huang, L.-M. Dai, X. Tao and J.-F. Chen, Nanoscale, 2017, 9, 18535 RSC.
- M. He, X. Pang, X. Liu, B. Jiang, Y. He, H. Snaith and Z. Lin, Angew. Chem., Int. Ed., 2016, 55(13), 4280 CrossRef CAS PubMed.
- W. Hou, Y. Liu, Y. Jiang, Y. Wu, C. Cui, Y. Wang, L. Zhang, I.-T. Teng and W. Tan, Nanoscale, 2018, 10, 10986 RSC.
- Y. Goh, Y. H. Song, G. Lee, H. Bae, M. K. Mahata and K. T. Lee, Phys. Chem. Chem. Phys., 2018, 20(16), 11359 RSC.
- R. Arppe, T. Nareoja, S. Nylund, L. Mattsson, S. Koho, J. M. Rosenholm, T. Soukka and M. Schaferling, Nanoscale, 2014, 6, 6837 RSC.
- T. Ma, Y. Ma, S. Liu, L. Zhang, T. Yang, H.-R. Yang, W. Lv, Q. Yu, W. Xu, Q. Zhao and W. Huang, J. Mater. Chem. C, 2015, 3, 6616 RSC.
- Y.-X. Wu, X.-B. Zhang, D.-L. Zhang, C.-C. Zhang, J.-B. Li, Y. Wu, Z.-L. Song, R.-Q. Yu and W. Tan, Anal. Chem., 2016, 88(3), 1639 CrossRef CAS PubMed.
- C. Ye, J. Ma, S. Chen, J. Ge, W. Yang, Q. Zheng, X. Wang, Z. Liang and Y. Zhou, J. Phys. Chem. C, 2017, 121(37), 20158 CrossRef CAS.
- M. K. Mahata, T. Koppe, T. Mondal, C. Brüsewitz, K. Kumar, V. Kumar Rai, H. Hofsäss and U. Vetter, Phys. Chem. Chem. Phys., 2015, 17(32), 20741 RSC.
- Y. Hao, S. Lv, Z. Ma and J. Qiu, RSC Adv., 2018, 8(22), 12165 RSC.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114(10), 5161 CrossRef CAS PubMed.
- Q. Sun, X. Chen, Z. Liu, F. Wang, Z. Jiang and C. Wang, J. Alloys Compd., 2011, 509(17), 5336 CrossRef CAS.
- M. D. Wisser, S. Fischer, P. C. Maurer, N. D. Bronstein, S. Chu, A. P. Alivisatos, A. Salleo and J. A. Dionne, ACS Photonics, 2016, 3(8), 1523 CrossRef CAS.
- X. Chen, D. Peng, Q. Ju and F. Wang, Chem. Soc. Rev., 2015, 44, 1318 RSC.
- H.-S. Qian and Y. Zhang, Langmuir, 2008, 24(21), 12123 CrossRef CAS PubMed.
- B. R. Judd, Phys. Rev., 1962, 127(3), 750 CrossRef CAS.
- G. S. Ofelt, J. Chem. Phys., 1962, 37(3), 511 CrossRef CAS.
- O. L. Malta and L. D. Carlos, Quim. Nova, 2003, 26(6), 889 CrossRef CAS.
- O. L. Malta, S. J. L. Rebeiro, M. Faucher and P. Porcher, J. Phys. Chem. Solids, 1991, 52(4), 587 CrossRef CAS.
- W. You, D. Tu, W. Zheng, P. Huang and X. Chen, J. Lumin., 2018, 201, 255 CrossRef CAS.
- A. Siaï, K. Horchani-Naifer, P. Haro-González and M. Férid, J. Lumin., 2016, 172, 65 CrossRef.
- S. Gong, M. Li, Z. Ren, X. Yang, X. Li, G. Shen and G. Han, J. Phys. Chem. C, 2015, 119(30), 17326 CrossRef CAS.
- X. Wu, K. W. Kwok and F. Li, J. Alloys Compd., 2013, 580, 88 CrossRef CAS.
- V. Petrovsky, T. Petrovsky, S. Kamlapurkar and F. Dogan, J. Am. Ceram. Soc., 2008, 91(11), 3590 CrossRef CAS.
- G. C. Manika and G. C. Psarras, High Voltage, 2016, 1(4), 151 CrossRef.
- V. Swaminathan, S. S. Pramana, T. J. White, L. Chen, R. Chukka and R. V. Ramanujan, ACS Appl. Mater. Interfaces, 2010, 2(11), 3037 CrossRef CAS PubMed.
- W. Cai, C. Fu, J. Gao, Q. Guo, X. Deng and C. Zhang, Phys. B, 2011, 406(19), 3583 CrossRef CAS.
- K. Itoh, L. Z. Zeng, E. Nakamura and N. Mishima, Ferroelectrics, 1985, 63, 29 CrossRef CAS.
- H. D. Megaw, Nature, 1945, 155, 484 CAS.
- S. Nayak, B. Sahoo, T. K. Chaki and D. Khastgir, RSC Adv., 2014, 4(3), 1212 RSC.
- J. J. Wang, F. Y. Meng, X. Q. Ma, M. X. Xu and L. Q. Chen, J. Appl. Phys., 2010, 108, 034107 CrossRef.
- K. Sakayori, Y. Matsui, H. Abe, E. Nakamura, M. Kenmoku, T. Hara, D. Ishikawa, A. Kokubu, K. Hirota and T. Ikeda, Jpn. J. Appl. Phys., 1995, 34, 5443 CrossRef CAS.
- Y. Zhang, J. Hao, C. L. Mak and X. Wei, Opt. Express, 2011, 19(3), 1824 CrossRef CAS PubMed.
- L. Chen and X. H. Wei, Integr. Ferroelectr., 2012, 140(1), 187 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32(5), 751 CrossRef.
- R. L. Moreira and A. Dias, J. Phys. Chem. Solids, 2007, 68(8), 1617 CrossRef CAS.
- A. S. Verma and V. K. Jindal, J. Alloys Compd., 2009, 485(1–2), 514 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectrum, pump power dependence, optical setup. See DOI: 10.1039/c8ra09783f |
| This journal is © The Royal Society of Chemistry 2019 |