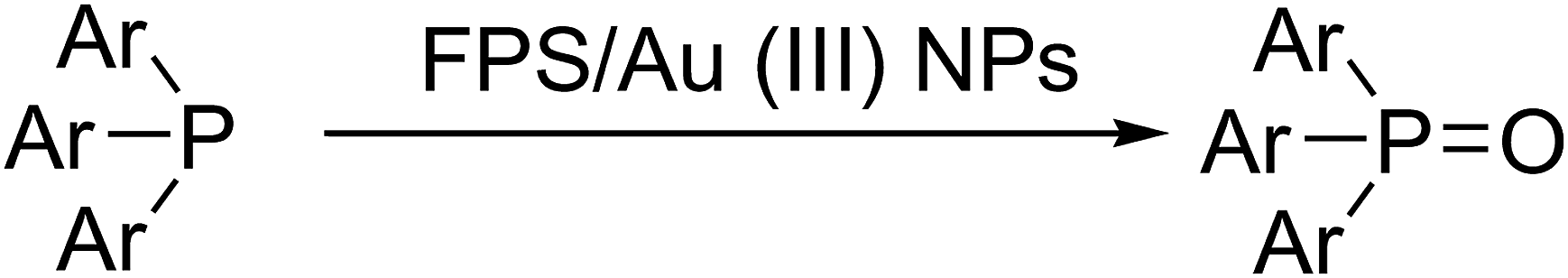DOI:
10.1039/C8RA09721F
(Paper)
RSC Adv., 2019,
9, 1509-1516
Photooxidation of triarylphosphines under aerobic conditions in the presence of a gold(III) complex on cellulose extracted from Carthamus tinctorius immobilized on nanofibrous phosphosilicate
Received
26th November 2018
, Accepted 14th December 2018
First published on 11th January 2019
Abstract
Triarylphosphines were converted to the corresponding oxides via photooxidation as a novel method. In this study, cellulose was extracted from the Carthamus tinctorius plant and then oxidized by sodium metaperiodate. A gold complex was supported on this natural cellulose. Then, a gold complex on natural cellulose supported on FPS (FPS/Au(III)) was synthesized for the reduction of phosphine oxides to corresponding phosphines with remarkable chemoselectivity. The morphology of FPS led to higher catalytic activity. FPS/Au(III) NPs were thoroughly characterized using TEM, FESEM, FTIR, TGA, and BET.
Introduction
Lately, the performance of cellulose has been considered via its hybridization with various ligands and metal nanoparticles. These compounds have attracted much interest in different aspects such as in flexible electronics, biocompatible drug delivery, biosensors, and disposable sensor applications.1,2 Micro-biocomposites are defined as natural compounds with at least one microscale substance scattered in a matrix, which can afford micromaterials with usage in industrial chemicals. In these types of materials, cellulose works as a bio-friendly substrate and is ubiquitous for novel catalytic applications.3,4 Reproducible natural sources are considered as natural raw materials for the development of biodegradable plastics and eco-friendly composites as they can substitute artificial polymers, reduce global affiliation on fossil fuel resources and provide simplified end-of-life access.5–7 Celluloses are the most abundant natural biopolymers providing exclusive mechanical stability to microcomposites.8,9 Various aldehydes can transform into polysaccharides with Schiff base ligands and primary amines in the oxidation of glycolic groups. Usually, for the conversion of polysaccharides to react with primary amines, several aldehyde groups are introduced into polysaccharides.10,11 Schiff bases build dialdehyde cellulose as a precious intermediate for cellulose-based substances for drug deliveries.12 Molybdenum complexes present great potential for their applications in medicinal chemistry due to their various antitumor and biological activities. On the other hand, biomimetic modeling and the design of magnetic molecules have attained much significance. Schiff bases are also used as corrosion inhibitors, pigments, dyes, polymer stabilizers, intermediates in organic synthesis, and catalysts.13
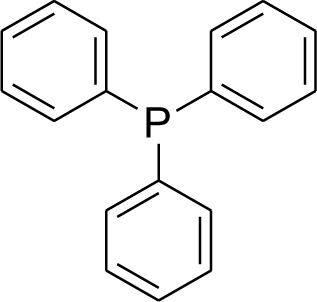
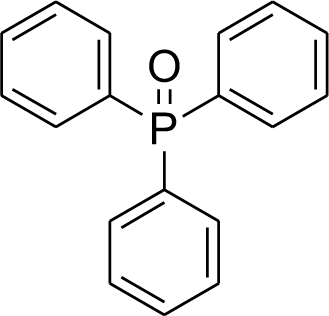
The oxidation of phosphines to phosphine oxides is an important subject for industrial reactions, especially in the reduction of contamination.14 Therefore, the oxidation of phosphines to phosphine oxides has been an important issue in chemistry. Given the importance of this issue, many methods have been extended in these topics.15–29 Despite their significant results, phosphine oxidation reactions have some problems. A catalytic or stoichiometric amount of additives must be added to initiate or catalyze this reaction.17,22,23 Because of this, a large amount of toxic material or organic waste is produced. Also, the formation of by-products cannot be avoided;21,29 this results in problems with the purification and waste products perhaps formed in the workup of purification. The issues listed above show that considerable waste is produced, which is specifically not environmentally friendly. Thus, more efficient reactions that can be performed under ambient conditions are in urgent need considering the increasing environmental concerns. Photooxidation23–29 provides an efficient way to realize this reaction, which normally uses oxygen as the oxidant.25–27
Gold catalysis has quickly become a hot topic in Chemistry in the past decade.30 Gold species are equally impressive as heterogeneous or homogeneous catalysts,31,32 showing excellent results in diversified reactions.33,34 Gold complexes have been employed as a highly efficient catalyst for the formation of C–O, C–C, C–S, C–N, C–P, and C–F bonds starting from alkenes and alkynes.35–37 From a viewpoint of reactivity, Au complexes involving phosphorus ligands are one of the most reactive classes of Au catalysts. Supporting these ligands on a recyclable support is one of the most important approaches to improve their applicability in organic reactions.38 In recent years, it has been revealed that the use of gold complexes grafted on solid supports plays an important role in preventing the aggregation of Au.39
Recently, our research team presented fibrous phosphosilicate (FPS) NPs that have special morphology, and their pore sizes gradually increase from the center to the surface.40 FPS NPs showed a high specific surface area due to the pores in the structures, and the approachability of the active sites significantly increased as a result of the special morphology and structure. Additionally, the 3D architectures generated hierarchical pore structures with macropores, which can also improve the mass transfer of the reactant. The FPS-based sorbents may have several advantages over conventional silica-based sorbents, including (i) high catalyst loading, (ii) minimal reduction in the surface area after functionalization and (iii) greater accessibility of the catalyst sites to enhance the reaction, due to the fibrous structure and highly accessible surface area of FPS. Given our continued interest in nanocatalysis and catalyst development for organic reactions,41–45 a novel strategy was reported for the preparation of desired nanocomposites by the modification of cellulose as a natural resource along with extracting solution of natural plants for cellulose industrial applications. Herein, oxidized cellulose was extracted from Carthamus tinctorius, and we investigated a novel immobilized gold Schiff base complex on natural cellulose and supported on fibrous FPS as a catalyst, catalyzing the photooxidation of phosphines under aerobic conditions. In this study, we found that under simple photoirradiation, triarylphosphines can be easily oxidized into corresponding oxides. This reaction uses oxygen in the air as the oxidant, which is green and abundantly available. Very importantly, after complete conversion, stoichiometric amounts of the oxides were afforded without the formation of any by-products, which indicated that no waste was generated during the workup step since simply recycling the solvent under vacuum is sufficient for purification (Scheme 1). Herein, we wish to report our recent observations on the catalyst photooxidation of phosphines under aerobic conditions at an ambient temperature.
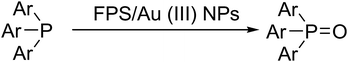 |
| | Scheme 1 Aerobic photooxidation of triarylphosphines. | |
Experimental
General procedure for the preparation of FPS
In a stirred solution of 30 mL of cyclohexane and 1.5 mL of 1-pentanol, 2.0 g of tetraethyl orthosilicate (TEOS) and 3.7 g of tripolyphosphate were dissolved. A solution of 1 g of CPB and 0.5 g of urea in 30 mL of water was added to the top mixture. The obtained mixture was continually stirred for 45 min at r.t. and then placed in a reactor and heated at 120 °C for 5 h. FPS was isolated by centrifugation, washed with deionized water and acetone, and dried in a drying oven.
Extracted cellulose from Carthamus tinctorius
Cellulose obtained from Carthamus tinctorius was treated with NaClO2 salt at pH = 4.0 at 75 °C for 2 h and filtered. The residue was then washed with distilled water and ethanol (95%) and dried in an oven at 50 °C for 13 h. Next, dried residues were extracted with KOH (10%) at room temperature for 8 h. After filtration, the residue was washed until neutral and then washed with ethanol (95%) 5 times. Finally, the samples were dried in an oven at 50 °C for 20 h.
General procedure for the preparation of cellulose-FPS NPs
First, 2 mmol of FPS in 20 mL of water was mixed together. Then, into the mixture, 0.75 mL of acetic acid was dispersed by ultrasonication; 250 mg of cellulose was added at r.t. and stirred for another 16 h at 70 °C. The products were washed with ethanol and deionized water and then dried under vacuum at 50 °C for 3 h.
Preparation of dialdehyde cellulose
First, 0.27 g of sodium metaperiodate was added to 0.5 g of cellulose-FPS suspended in 60 mL of distilled water. The mixture was stirred in the dark at 60 °C for 10 h. Hereinafter, the remaining NaIO4 was decomposed by adding glycerol. Finally, the product was washed with deionized water and dried at r.t. for 12 h.
Preparation of Schiff base supported on dialdehyde cellulose
Here, 0.35 g of DAMC was added to 20 mL of ethanol and then, 0.78 g of 9-aminoacridine was added to the mixture. The mixture was heated under reflux for 36 h. The product was washed with deionized water and ethanol.
General procedure for the preparation of FPS/Au(III) NPs
In a stirred solution of 0.15 mmol of NaAuCl4 in 50 mL of DMF, 1 g of FPS/ligand was added and refluxed under argon for 12 h. The obtained solid was washed with 25 mL of THF and 25 mL of acetone. Further, the product was Soxhlet-extracted with dichloromethane (CH2Cl2) for 24 h to remove unreacted NaAuCl4 and organic impurities. The resulting product was dried using vacuum at 75 °C to furnish FPS/Au(III) NPs.
General procedure for photooxidations
Here, 8 mg of FPS/Au(III) NPs, 1 mmol of triarylphosphine and 10 mL of dimethyl carbonate were added to a dry quartz reaction flask, which was equipped with a magnetic stirrer. The mixture was irradiated by a compact fluorescent lamp (CFL) (20 W) at r.t. under an air atmosphere. Next, the photoreaction was completed as monitored by TLC. The solvent was directly recycled under vacuum to afford triarylphosphine oxides as a solid.
Results and discussion
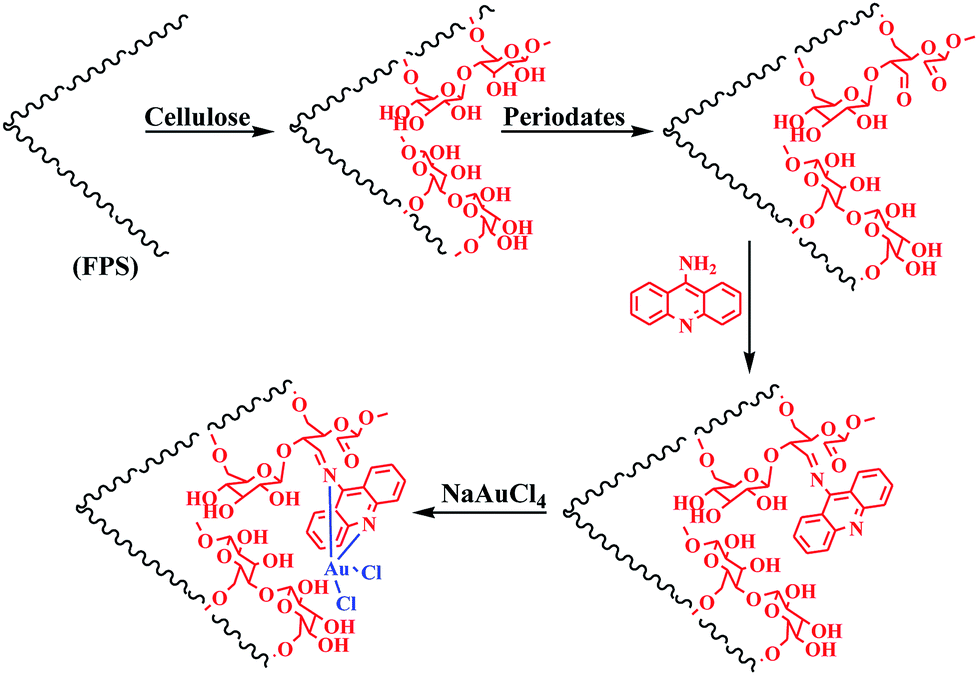
The first stage in designing FPS/Au(III) NPs was the functionalization of FPS with cellulose groups, which could then act as pseudo chelators or ligands to control metal leaching during the reaction. In the next step, the aldehyde group of natural oxidized cellulose reacted with the amine group of 9-aminoacridine through the Schiff base ligand. The immobilized Schiff base ligand was then allowed to react with an appropriate concentration of ammonium heptamolybdate to provide a gold complex. Finally, FPS/Au(III) NPs were characterized by different methods such as TEM, FESEM, FTIR, TGA, and BET (Scheme 2).
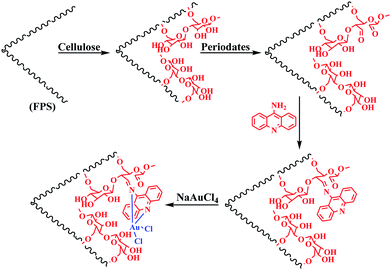 |
| | Scheme 2 Schematic illustration of the synthesis of FPS/Au(III) NPs. | |
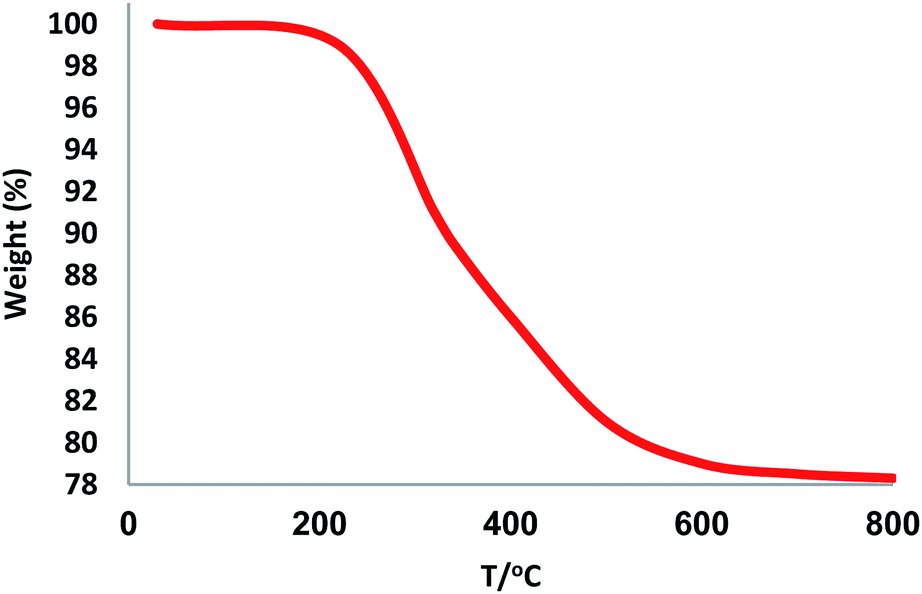
The structure and morphology of FPS/Au(III) NPs were studied with TEM and FESEM (Fig. 1). The FPS sample comprised wall-like domains, and the sizes of walls were stable (Fig. 1a and c). The study of TEM and FESEM revealed that FPS/Au(III) has dendrimeric fibers (thicknesses of 10–12 nm) arranged in three dimensions to form walls, which can allow easy access to the available high level. From FESEM and TEM images of FPS/Au(III) NPs, it can be seen that no significant change was observed after the modification of the morphology of NPs (Fig. 1b and d). Fig. 2 shows the TGA analysis of FPS/Au(III) NPs: the elimination of the physisorbed and chemisorbed solvent on the surface of the FPS/Au(III) material causes weight loss below 150 °C. Also, weight loss in the range of 250–450 °C is about 11.5 wt%, which is related to the organic group derivatives.
 |
| | Fig. 1 TEM images of FPS NPs (a) and FPS/Au(III) NPs (b); FESEM images of FPS NPs (c) and FPS/Au(III) NPs (d). | |
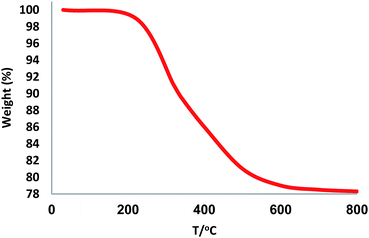 |
| | Fig. 2 TGA diagram of FPS/Au(III) NPs. | |
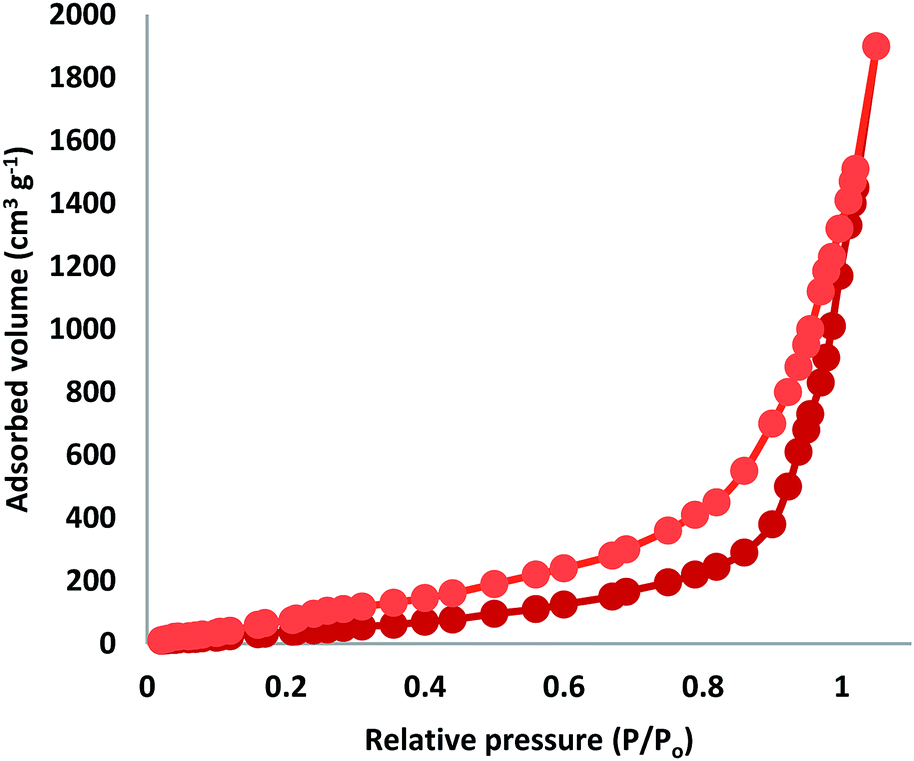
The N2 adsorption–desorption isotherms of FPS/Au(III) NPs showed characteristic type IV curves (Fig. 3). The BET surface area for FPS, the total pore volume and the BJH pore diameter were determined to be 34 m2 g−1, 0.24 cm3 g−1, and 2.43 nm, respectively; also, the corresponding parameters of FPS/Au(III) NPs decreased to 30 m2 g−1, 0.20 cm3 g−1, and 2.21 nm. The nitrogen sorption analysis of FPS/Au(III) NPs also confirmed a regular and uniform mesostructure with decrease in surface area, pore diameter and pore volume in comparison with the observations of pristine FPS. The corresponding pore volumes were drastically reduced with the functionalization of ligand/Au(III)–P. This can be attributed to increased loading with the sensing probe, which occupies a large volume inside the fibers (Fig. 3 and Table 1).
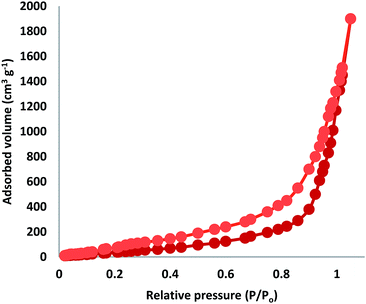 |
| | Fig. 3 Adsorption–desorption isotherms of FPS/Au(III) NPs. | |
Table 1 Structural parameters of FPS and FPS/Au(III) NP materials determined from nitrogen sorption experiments
| Catalysts |
SBET (m2 g−1) |
Va (cm3 g−1) |
DBJH (nm) |
| FPS |
34 |
0.24 |
2.43 |
| FPS/Au(III) |
30 |
0.20 |
2.21 |
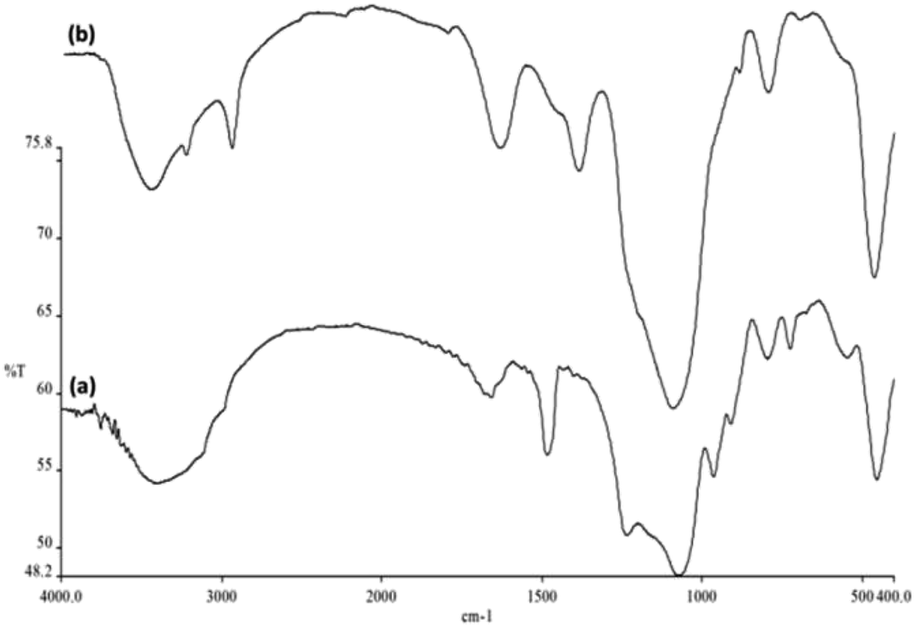
FTIR spectra proved the presence of surface silanol, hydroxyl and phosphate groups as well as anchored complex [HAuCl4] in (a) FPS and (b) FPS/Au NPs (Fig. 4). For FPS, the broad absorption bands at 1092 cm−1 and 3649 cm−1 were attributed to Si–O–Si asymmetric stretching and OH, respectively. Two peaks at 793 cm−1 and 469 cm−1 were assigned to Si–O–Si symmetric stretching and bending vibrations, respectively. The pure FPS sample showed a typical broad peak at around 3399 cm−1 associated with the presence of hydroxyl groups, and the intensity of this peak increased, which could be due to the presence of the immobilized phosphate group on the framework of FPS. Also, the phosphate of FPS showed a peak at around 1483 cm−1 due to TPP.46 The main peak in the spectrum of FPS is the additional peak appearing at 1232 cm−1, which can be assigned to the –P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, indicating the presence of a phosphate group.47,48 The band at 963 cm−1 and the shoulder at 1108 cm−1 were due to the TO and LO modes of the asymmetric stretching of Si–O–P bonds, respectively.49,50 The bands at about 724 and 795 cm−1 were assigned to the asymmetric stretching of the bridging oxygen atoms bonded to a phosphorus atom.51 These peaks suggested the successful reaction between TEOS and TPP (Fig. 4a), clearly indicating the grafting of organic groups on the surface of FPS. The organic groups-FPS composite showed bands at around 1091, 793 and 462 cm−1. A strong and broad absorption band at 3000–3550 cm−1 was related to the –OH and –NH stretching vibrations. The peaks appearing at 3100 cm−1 and 2930 cm−1 are due to the stretching of the C–H aromatic group and the C–H aliphatic group (Fig. 4b).
O stretching vibration, indicating the presence of a phosphate group.47,48 The band at 963 cm−1 and the shoulder at 1108 cm−1 were due to the TO and LO modes of the asymmetric stretching of Si–O–P bonds, respectively.49,50 The bands at about 724 and 795 cm−1 were assigned to the asymmetric stretching of the bridging oxygen atoms bonded to a phosphorus atom.51 These peaks suggested the successful reaction between TEOS and TPP (Fig. 4a), clearly indicating the grafting of organic groups on the surface of FPS. The organic groups-FPS composite showed bands at around 1091, 793 and 462 cm−1. A strong and broad absorption band at 3000–3550 cm−1 was related to the –OH and –NH stretching vibrations. The peaks appearing at 3100 cm−1 and 2930 cm−1 are due to the stretching of the C–H aromatic group and the C–H aliphatic group (Fig. 4b).
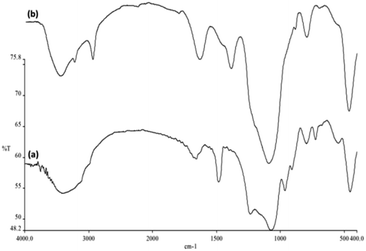 |
| | Fig. 4 FTIR spectra of (a) FPS NPs and (b) FPS/Au NPs. | |
In the first step, a series of conventional solvents were tested (Table 2, entries 1–18). In all the tested solvents, a stoichiometric yield of the desired tris(4-methoxyphenyl)phosphine oxide was provided. The biggest difference is that dimethyl carbonate (DMC) increased the rate of the oxidation process, which may be due to the higher energy transfer efficiency and faster single-electron transfer speed (Table 2, entry 8). Therefore, DMC exhibited the best performance among the solvents. Next, the effect of temperature on the reaction was investigated. Increasing the temperature to 80 °C did not increase the yield. Hence, it was found that the best result was obtained at room temperature for the desired transformation. The catalyst quantity is another important issue in the photooxidation of triarylphosphine, which should be examined. The reaction was performed in the presence of 4 mg, 6 mg, 8 mg, and 10 mg of FPS/Au(II) NPs at r.t. for 2 h (Table 2, entry 25). As expected, using FPS/Au(II) NPs as a catalyst resulted in distinctive change in product performance. The reaction utilizing 4 mg of catalyst offered 52% yield, whereas 81% yield was obtained after using 6 mg of catalyst after 2 h. Product performance increased to 99% when 8 mg of catalyst was used. The influence of time on the photooxidation of triarylphosphine is summarized in Table 1. It is clear that the product performance increased to 99% for 1 h, but an increase in the time did not result in improvement in the product yield. Hence, the desirable time for the photooxidation of triarylphosphine is 1 h. We also examined the reaction in the presence of visible light at different intensities. FPS/Au(II) NPs were screened under lights of different intensities (8, 15, 20, 22 and 32 W), and the other parameters were maintained constant to acquire the respective yields, i.e., 43%, 80%, 99%, 99%, and 99% (Table 2, entries 28–31). The outcomes suggest that the photooxidation of triarylphosphine increases gradually as the light intensity increases from 8 to 20 W and stays constant at 20 W. The use of CFL of 22 and 32 W did not have any noticeable effect on the yield or reaction time.
Table 2 Optimization of the reaction conditions
| Entry |
Solvent |
Temperature (°C) |
Catalyst (mg) |
Time (h) |
Visible light intensity (W) |
Yielda (%) |
| Isolated yield. |
| 1 |
EtOH |
r.t. |
10 |
2 |
22 |
79 |
| 2 |
MeOH |
r.t. |
10 |
2 |
22 |
82 |
| 3 |
i-PrOH |
r.t. |
10 |
2 |
22 |
86 |
| 4 |
CH2Cl2 |
r.t. |
10 |
2 |
22 |
43 |
| 5 |
CH3CN |
r.t. |
10 |
2 |
22 |
41 |
| 6 |
CHCl3 |
r.t. |
10 |
2 |
22 |
38 |
| 7 |
CCl4 |
r.t. |
10 |
2 |
22 |
14 |
| 8 |
DMC |
r.t. |
10 |
2 |
22 |
99 |
| 9 |
DMA |
r.t. |
10 |
2 |
22 |
86 |
| 10 |
1,4-Dioxane |
r.t. |
10 |
2 |
22 |
45 |
| 11 |
EtOAc |
r.t. |
10 |
2 |
22 |
62 |
| 12 |
THF |
r.t. |
10 |
2 |
22 |
59 |
| 13 |
DMSO |
r.t. |
10 |
2 |
22 |
36 |
| 14 |
H2O |
r.t. |
10 |
2 |
22 |
25 |
| 15 |
Toluene |
r.t. |
10 |
2 |
22 |
— |
| 16 |
n-Hexane |
r.t. |
10 |
2 |
22 |
— |
| 17 |
Cyclohexane |
r.t. |
10 |
2 |
22 |
— |
| 18 |
Anisole |
r.t. |
10 |
2 |
22 |
32 |
| 19 |
Solvent-free |
r.t. |
10 |
2 |
22 |
— |
| 20 |
DMC |
60 |
10 |
2 |
22 |
99 |
| 21 |
DMC |
80 |
10 |
2 |
22 |
99 |
| 22 |
DMC |
r.t. |
8 |
2 |
22 |
99 |
| 23 |
DMC |
r.t. |
6 |
2 |
22 |
81 |
| 24 |
DMC |
r.t. |
4 |
2 |
22 |
52 |
| 25 |
DMC |
r.t. |
— |
2 |
22 |
— |
| 26 |
DMC |
r.t. |
5 |
1 |
22 |
99 |
| 27 |
DMC |
r.t. |
5 |
0.5 |
22 |
51 |
| 28 |
DMC |
r.t. |
5 |
1 |
32 |
99 |
| 29 |
DMC |
r.t. |
5 |
1 |
20 |
99 |
| 30 |
DMC |
r.t. |
5 |
1 |
15 |
80 |
| 31 |
DMC |
r.t. |
5 |
1 |
8 |
43 |
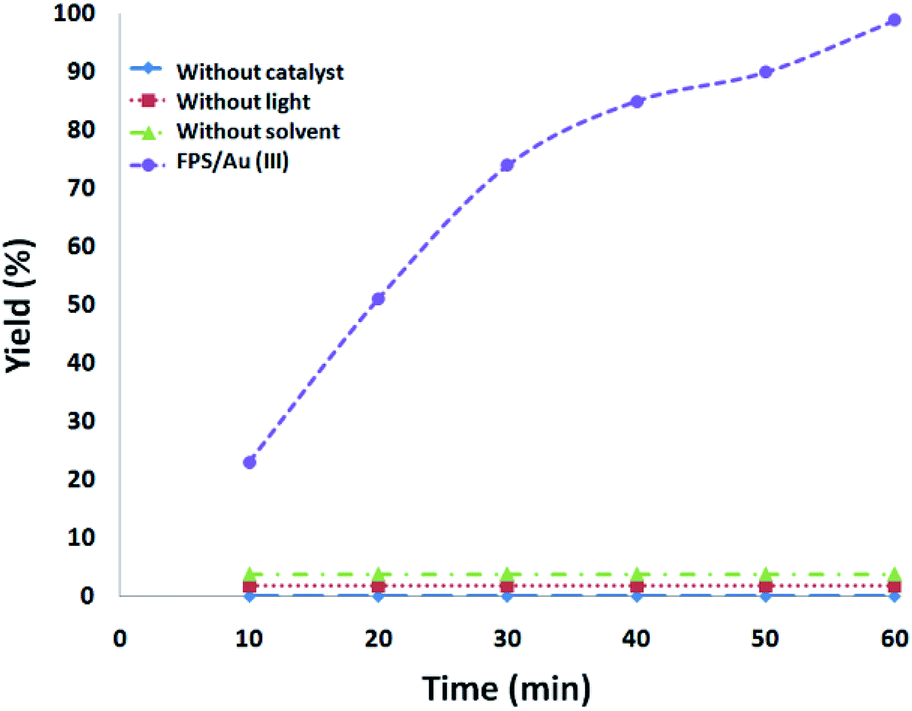
In addition, a series of comparative tests were conducted. In a dark environment, no products were formed, showing that the reaction is photocatalytic. In the absence of FPS/Au(III) NPs or solvent, the reaction also showed that no product was produced under visible-light irradiation. We used N2 instead of O2 to confirm the source of the reduction of phosphine oxides to preprocess FPS/Au(III) NPs. There was only a trace of the reduction of phosphine oxides under 1 h irradiation with FPS/Au(III) NPs as the photocatalyst, indicating that the reduction of phosphine oxides resulted from O2 rather than decomposition of ligands (Fig. 5).
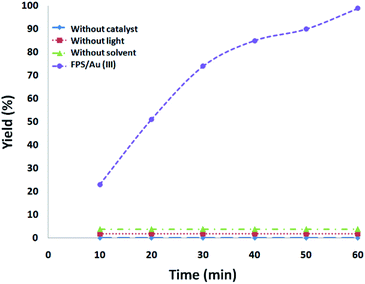 |
| | Fig. 5 The amount of the reduction of phosphine oxides as a function of irradiation time under different conditions. | |
For further review of catalyst performance, various control experiments were conducted, and the information is shown in Table 3. At first, a standard reaction performed using FPS showed that any quantity of the product was not formed after 2 h (Table 3, entry 1). Also, when FPS/ligand was used as the catalyst, there was no reaction (Table 3, entry 2). The organic groups could not give acceptable catalytic activity under optimized conditions. Because these results were disappointing, we did more research into performance improvements of the product by adding Au(III). This research proved that the progression of the reaction is exclusively catalyzed by Au(III) species complexed on FPS. The nano-sized particles increased the exposed surface area of the active sites of the catalyst, thereby dramatically enhancing the contact between reactants and catalyst and mimicking the homogeneous catalysts. As a result, FPS/Au(III) NPs were used in the subsequent investigations because of their high reactivity, high selectivity and easy separation. Furthermore, the activity and selectivity of the nanocatalyst could be manipulated by tailoring physical and chemical properties such as morphology, shape, composition and size. For evaluating the accurate impact of the presence of FPS in the catalyst, FPS/Au(III) NPs were compared with MCM-41/Au(III), SBA-15/Au(III), and nano-SiO2/Au(III). The yields for the reduction of phosphine oxides were good when nano-SiO2/Au(III), MCM-41/Au(III) or SBA-15/Au(III) was used as the catalyst, whereas the yield for FPS/Au(III) was excellent. The large spaces between the fibers could greatly increase the access of the surface of FPS. These results indicated that FPS exhibited better performance than SBA-15, MCM-41, and nano-SiO2 (Table 3, entries 3–6). These results justified the use of FPS NPs in subsequent investigations because of their easy separation, high selectivity and high reactivity (Table 3).
Table 3 Influence of different catalysts for the reduction of phosphine oxidesa
| Entry |
Catalyst |
Yielda (%) |
| Isolated yield. |
| 1 |
FPS |
— |
| 2 |
FPS/ligand |
— |
| 3 |
FPS/Au(III) |
99 |
| 4 |
Nano-SiO2/Au(III) |
47 |
| 5 |
MCM-41/Au(III) |
65 |
| 6 |
SBA-15/Au(III) |
73 |
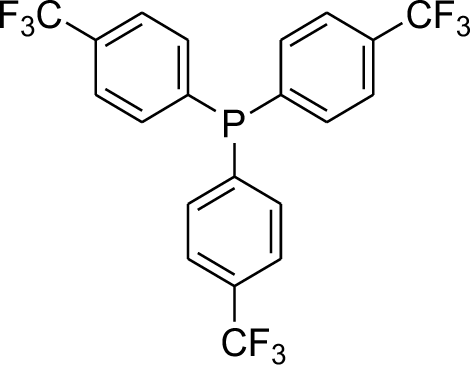
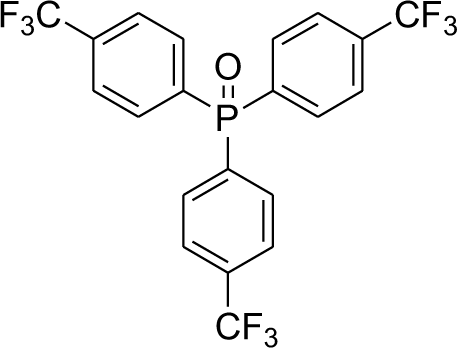
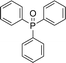
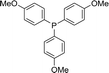
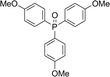
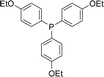
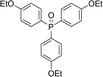
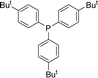
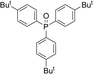
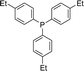
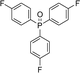
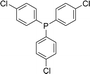
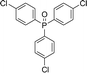
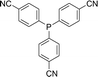
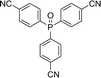
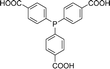
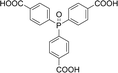
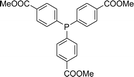
With the above-optimized conditions in hand, the substrate scope of this photooxidation with a series of triarylphosphines under optimal conditions was investigated. The results are summarized in Table 4. It was observed that the reactions of both electron-rich and deficient phenylated phosphine derivatives proceeded in high efficiency, affording the corresponding oxides. No clear electronic effect on the phenyl ring was observed.
Table 4 Photooxidation of triarylphosphines under optimized conditions
| Entry |
Phosphines |
Product |
Yielda (%) |
| Isolated yield. |
| 1 |
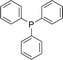 |
 |
99 |
| 2 |
 |
 |
98 |
| 3 |
 |
 |
99 |
| 4 |
 |
 |
96 |
| 5 |
 |
 |
99 |
| 6 |
 |
 |
100 |
| 7 |
 |
 |
97 |
| 8 |
 |
 |
95 |
| 9 |
 |
 |
98 |
| 10 |
 |
 |
96 |
| 11 |
 |
 |
97 |
| 12 |
 |
 |
99 |
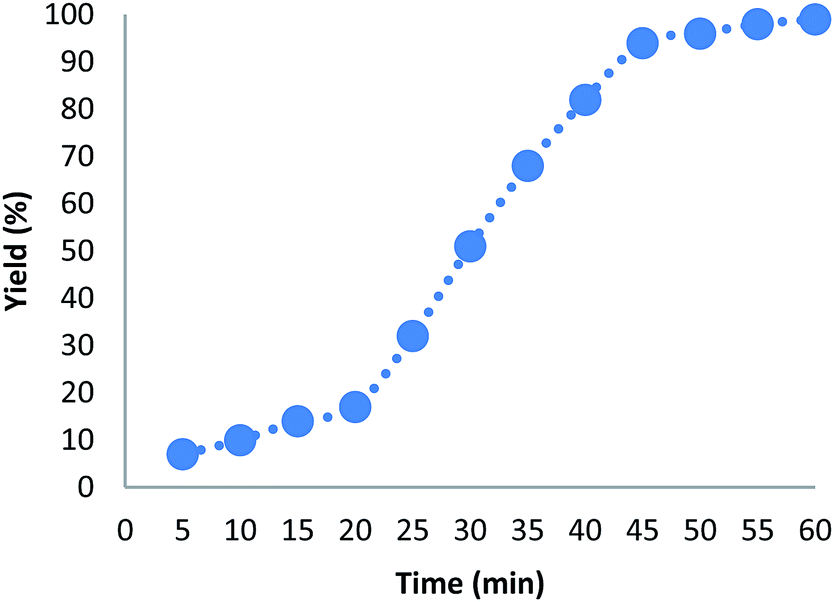
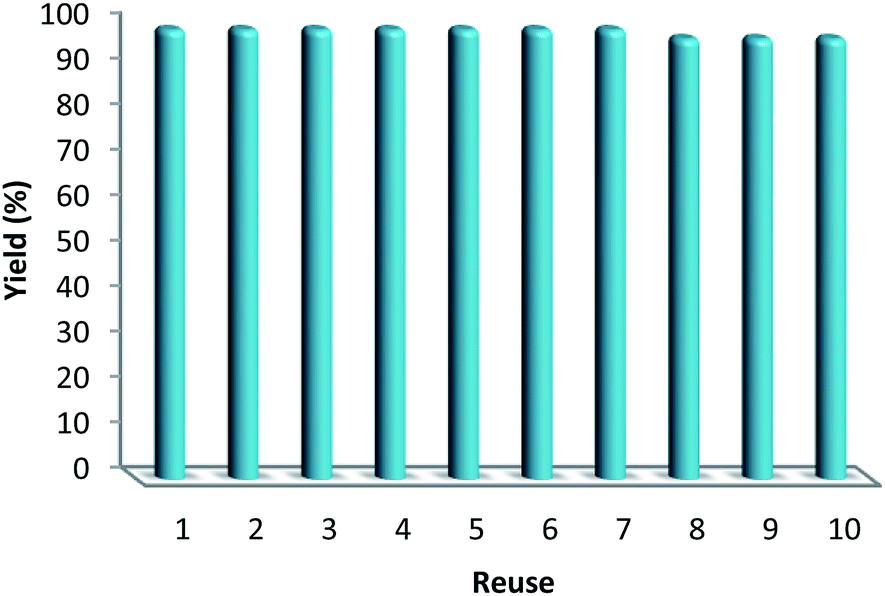
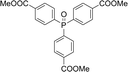
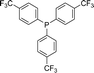
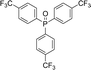
Hot filtration and mercury poisoning tests were performed to obtain information about the heterogeneity or homogeneity of the reaction. At first, the relationship between the progress of reaction and time reached a maximum after 20–45 min. Thus, we chose the reaction time of 45 min for the following experiments. The catalyst was filtered out at above 100 °C after the reaction proceeded for 45 min, and the yield of product was 94%, as determined by GC. The obtained filtrate with additional 1.0 mmol of triarylphosphine was continually stirred under the reaction conditions. After 45 min, the conversion was determined to be 94% (Fig. 6). The same approach was applied to the mercury test. After the first stage, the yield of the product was 93%. In the second stage, after mercury (0.3 mL) was added into the filtrate, the filtrate was stirred for another 45 min under the reaction conditions. The yield of the product remained at 94%. These phenomena showed that a small amount of catalytically active Au(III) species may have leached into the reaction mixture. However, the contents of Au(III) in catalyst before and after the reaction were 4.9% and 4.8%, respectively, as determined by ICP-MS (Table 5). This indicates that most Au(III) species leaching into solution are recaptured onto the fibers of FPS after completion of the reaction. It is important to note that the heterogeneous property of FPS/Au(III) NPs facilitates their efficient recovery from the reaction mixture after the completion of reaction. The amount of Au(III) in FPS/Au(III) NPs was similar after reuse for ten consecutive periods of catalysts. Notably, the catalyst could be recycled up to ten times with a marginal decrease in activity (Fig. 7). A major concern is the plausible presence of trace amounts of other active metals (such as Pd, Cu, Fe, Ni, Cd, and Co) in any component of the reaction. To solve this problem, it was necessary to carry out ICP analysis of the reaction components. It can be seen in Table 6 that the concentrations of Pd, Cu, Fe, Ni, Cd, and Co were very low, which eliminated the slightest doubt.
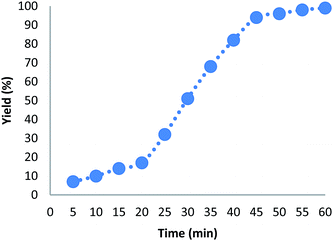 |
| | Fig. 6 Effect of time on the yield of photooxidation of triarylphosphines. | |
Table 5 The loading amount of Au(III) in FPS/Au(III) NPs
| Entry |
Catalyst |
wt% |
| 1 |
FPS/Au(III) NPs |
4.9 |
| 2 |
FPS/Au(III) NPs after ten reuses |
4.8 |
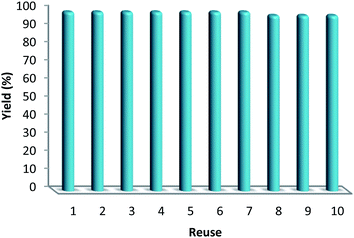 |
| | Fig. 7 Reuse performance of the catalysts. | |
Table 6 Chemical composition of the reaction components using ICP
| Entry |
Element |
Weight percent (%) |
| 1 |
Pd |
— |
| 2 |
Cu |
<0.01 |
| 3 |
Fe |
— |
| 4 |
Ni |
<0.01 |
| 5 |
Cd |
— |
| 6 |
Co |
— |
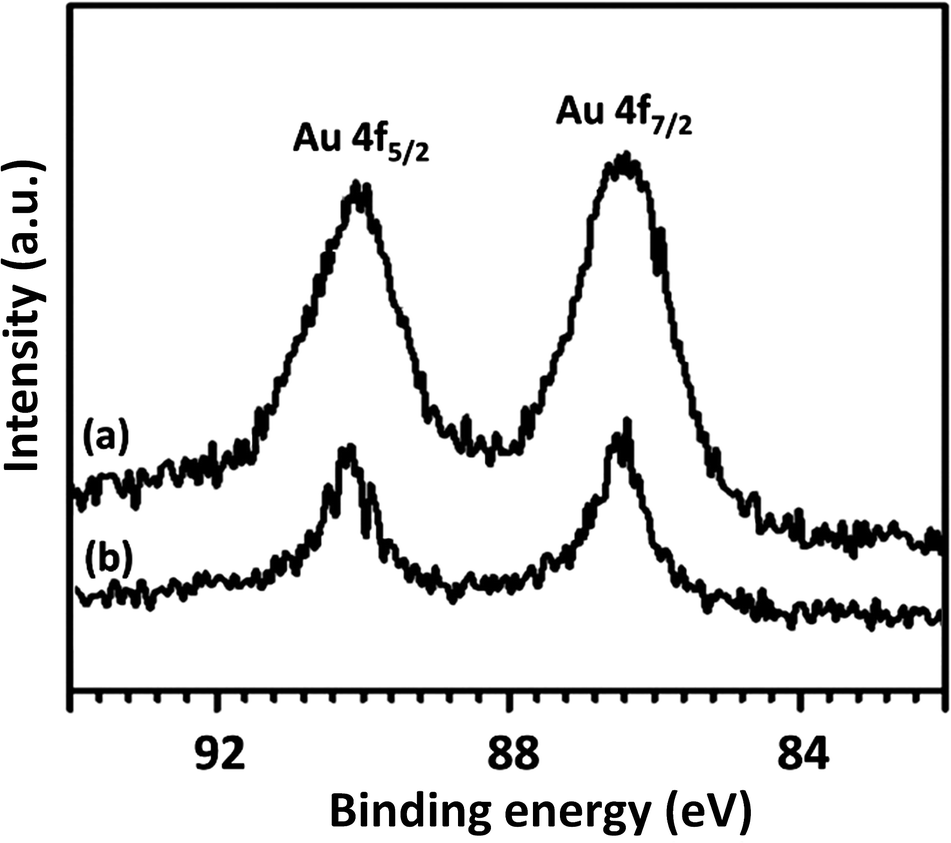
To further understand the underlying reason for the significant difference in recyclabilities, XPS and SEM were employed to characterize fresh and reused FPS/Au(III) NPs. The XPS spectra are shown in Fig. 8. For fresh FPS/Au(III), the Au 4f5/2 and Au 4f7/2 binding energies were determined to be 89.4 and 85.7 eV, respectively. After being reused ten times, 89.4 eV (4f5/2) and 85.7 eV (4f7/2) binding energies were fixed, corresponding to the Au(III) binding energy. SEM images provided further information about FPS/Au(III) NPs. SEM images for fresh FPS/Au(III) NPs and FPS/Au(III) NPs reused ten times are displayed in Fig. 9. After being reused ten times, the wall-like structure of the catalyst could still be observed. Structural similarities between fresh FPS/Au(III) NPs and FPS/Au(III) NPs obtained after ten reuses accounted for high power in recyclability.
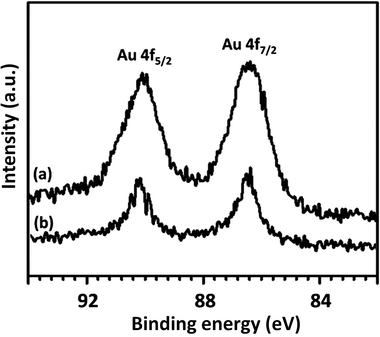 |
| | Fig. 8 XPS spectra of fresh FPS/Au(III) NPs (a) and FPS/Au(III) NPs after ten reuses (b). | |
 |
| | Fig. 9 SEM images of fresh HPG@KCC-1/PPh2/AuNPs (a) and HPG@KCC-1/PPh2/AuNPs after ten reuses (b). | |
Conclusions
In conclusion, visible light-induced oxidation of triarylphosphine was developed under aerobic conditions. This reaction provided a cheaper and greener method for the preparation of phosphine oxide derivatives. Considering that this method is environmentally friendly and that the substrate scope is broad, this protocol may be widely used in the oxidation of phosphine derivatives.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- A. Codou, N. Guigo, L. Heux and N. Sbirrazzuoli, Compos. Sci. Technol., 2015, 117, 54–61 CrossRef CAS.
- H. Liimatainen, M. Visanko, J. A. Sirvio, H. Hormi and J. Niinimaki, Biomacromolecules, 2012, 13, 1592–1597 CrossRef CAS PubMed.
- S. Coseri, G. Biliuta, C. Simionescu, K. Stana-Kleinschek and V. Harabagiu, Carbohydr. Polym., 2013, 93, 207–215 CrossRef CAS PubMed.
- T. Shui, S. Feng, G. Chen, A. Li, Z. Yuan, H. Shui, X. Kuboki and C. Charles, Biomass Bioenergy, 2017, 105, 51–58 CrossRef CAS.
- J. Yongkim and H. Choi, Cellul. Chem. Technol., 2014, 48, 25–32 Search PubMed.
- M. Bansal, G. S. Chauhan, A. Kaushik and A. Sharma, Int. J. Biol. Macromol., 2016, 91, 887–894 CrossRef CAS PubMed.
- P. Zugenmaier, Pure Appl. Chem., 2006, 78, 1843–1855 CAS.
- E. C. S. Filho, J. C. Melo, M. G. Fonseca and C. Airoldi, J. Colloid Interface Sci., 2009, 340, 8–15 CrossRef PubMed.
- U. J. Kim and S. Kuga, J. Chromatogr. A, 2001, 919, 29–37 CrossRef CAS PubMed.
- S. M. A. S. Keshk, A. M. Ramadan and S. Bondock, Carbohydr. Polym., 2015, 127, 246–251 CrossRef CAS PubMed.
- U. J. Kim, S. Kuga, M. Wada, T. Okano and T. Kondo, Biomacromolecules, 2000, 1, 488–492 CrossRef CAS PubMed.
- X. Liu, C. Manzur, N. Novoa, S. Celedón and D. Clloarri, Coord. Chem. Rev., 2018, 357, 144–172 CrossRef CAS.
- J. Lindh, C. Carlsson, M. Strømme and A. Mihranyan, Biomacromolecules, 2014, 15, 1928–1932 CrossRef CAS PubMed.
-
(a) J.-L. Montchamp, Acc. Chem. Res., 2014, 47, 77–87 CrossRef CAS PubMed;
(b) M. Hashiguchi, K. Nagata, K. Tanaka and Y. Matsuo, Org. Process Res. Dev., 2012, 16, 643–646 CrossRef CAS;
(c) E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed;
(d) C.-J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68–82 RSC;
(e) J. I. G. Gadogan and R. K. Mackie, Chem. Soc. Rev., 1974, 3, 81–137 Search PubMed.
- R. Dobrovetsky and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 2516–2519 CrossRef CAS PubMed.
- A. G. Majouga, E. K. Beloglazkina, A. A. Moiseeva, O. V. Shilova, E. A. Manzheliy, M. A. Lebedeva, E. S. Davies, A. N. Khlobystov and N. V. Zyk, Dalton Trans., 2013, 42, 6290–6293 RSC.
- W. Peng and J. M. Shreeve, J. Fluorine Chem., 2005, 126, 1054–1056 CrossRef CAS.
- W. Levason, R. Patel and G. Reid, J. Organomet. Chem., 2003, 688, 280–282 CrossRef CAS.
- B. G. Jacobi, D. S. Laitar, L. Pu, M. F. Wargocki, A. G. DiPasquale, K. C. Fortner, S. M. Schuck and S. Brown, Inorg. Chem., 2002, 41, 4815–4823 CrossRef CAS PubMed.
- B. R. Travis, B. P. Ciaramitaro and B. Borhan, Eur. J. Org. Chem., 2002, 3429–3434 CrossRef CAS.
- L. A. Woźniak and W. J. Stec, Tetrahedron Lett., 1999, 40, 2637–2640 CrossRef.
- Y. Futami, H. Nishino and K. Kurosawa, Bull. Chem. Soc. Jpn., 1989, 62, 3567–3571 CrossRef CAS.
- L. A. Wickramasinghe and P. R. Sharp, Inorg. Chem., 2014, 53, 1430–1442 CrossRef CAS PubMed.
- F. Avenier, C. Herrero, W. Leibl, A. Desbois, R. Guillot, J.-P. M. Mahy and A. Aukauloo, Angew. Chem., Int. Ed., 2013, 52, 3634–3637 CrossRef CAS PubMed.
- K. A. Prokop and D. P. Goldberg, J. Am. Chem. Soc., 2012, 134, 8014–8017 CrossRef CAS PubMed.
- K. Ohkubo, T. Nanjo and S. Fukuzumi, Bull. Chem. Soc. Jpn., 2006, 79, 1489–1500 CrossRef CAS.
- A. Ding, Y. Wang, R. Rios, J. Sun, H. Li and H. Guo, J. Saudi Chem. Soc., 2015, 19, 706–709 CrossRef.
- Y. Zhang, C. Ye, S. Li, A. Ding, G. Gu and H. Guo, RSC Adv., 2017, 7, 13240–13243 RSC.
- A. Ding, S. Li, Y. Chen, R. Jin, C. Ye, J. Hu and H. Guo, Tetrahedron Lett., 2018, 59, 3880–3883 CrossRef CAS.
- L. P. Liu and G. B. Hammond, Chem. Soc. Rev., 2012, 41, 3129–3139 RSC.
- A. S. K. Hashmi, L. Schwarz, J. H. Choi and T. M. Frost, Angew. Chem., Int. Ed., 2000, 39, 2285–2288 CrossRef CAS PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- W. B. Wang, G. B. Hammond and B. Xu, J. Am. Chem. Soc., 2012, 134, 5697 CrossRef CAS PubMed.
- L. Guczi, A. Beck and Z. Paszti, Catal. Today, 2012, 181, 26–32 CrossRef CAS.
- A. S. K. Hashmi, A. M. Schuster, S. Litters, F. Rominger and M. Pernpointner, Chem.–Eur. J., 2011, 17, 5661–5667 CrossRef CAS PubMed.
- M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem.–Eur. J., 2011, 17, 8248–8262 CrossRef CAS PubMed.
- Y. Zhang and C. Zhu, Catal. Commun., 2012, 28, 134–137 CrossRef CAS.
-
(a) T. Yamamoto, T. Yamada, Y. Nagata and M. Suginome, J. Am. Chem. Soc., 2010, 132, 7899–7901 CrossRef CAS PubMed;
(b) X. Zhang, P. Li, Y. Ji, L. Zhang and L. Wang, Synthesis, 2011, 2975–2983 CAS.
- M. S. Sadeghzadeh, RSC Adv., 2015, 5, 68947–68952 RSC.
- R. Zhiania, M. Khoobi and S. M. Sadeghzadeh, Microporous Mesoporous Mater., 2019, 275, 76–86 CrossRef.
- S. M. Sadeghzadeh, RSC Adv., 2015, 5, 17319–17324 RSC.
- S. M. Sadeghzadeh, Appl. Organomet. Chem., 2016, 30, 835–842 CrossRef CAS.
- S. M. Sadeghzadeh, Microporous Mesoporous Mater., 2016, 234, 310–316 CrossRef CAS.
- M. A. Nasseri and S. M. Sadeghzadeh, J. Iran. Chem. Soc., 2014, 11, 27–33 CrossRef CAS.
- S. M. Sadeghzadeh and M. A. Nasseri, Catal. Today, 2013, 217, 80–85 CrossRef CAS.
- Ph. Massiot, M. A. Centeno, I. Carrizosa and J. A. Odriozola, J. Non-Cryst. Solids, 2001, 292, 158–166 CrossRef CAS.
- M. Stan, A. Vasdilescu, S. Moscu and M. Zaharescu, Rev. Roum. Chim., 1998, 43, 425 CAS.
- H. S. Liu, T. S. Chin and S. W. Yung, Mater. Chem. Phys., 1997, 50, 1 CrossRef CAS.
- I. N. Chakraborty and R. A. Condrate, Phys. Chem. Glasses, 1985, 26, 68–73 CAS.
- Y. K. Kim and R. E. Tressler, J. Mater. Sci., 1994, 29, 2531–2535 CrossRef CAS.
- G. Lakshminarayana and M. Nogami, Solid State Ionics, 2010, 181, 760–766 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab and
Rahele Zhianiab
*ab and
Rahele Zhianiab