
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of novel 1,2,3-triazole furocoumarin derivatives via click chemistry and their anti-vitiligo activity
Chao Niuab,
Xueying Luab and
Haji Akber Aisa *ab
*ab
aKey Laboratory of Plant Resources and Chemistry of Arid Zone, Xinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Urumqi 830011, China. E-mail: haji@ms.xjb.ac.cn
bState Key Laboratory Basis of Xinjiang Indigenous Medicinal Plants Resource Utilization, Xinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Urumqi 830011, China
First published on 14th January 2019
Abstract
The extracts of Psoralea corylifolia L. were often used for the repigmentation of leukoderma (vitiligo) in traditional Uygur medicine thousands years ago. Nowadays, its active ingredient, furocoumarins, has been clinically applied since it exhibited strong photosensitivity. Thus, a new series of furocoumarin derivatives (8a–8o) containing 1,2,3-triazole were designed and synthesized based on our previous work. After biological evaluation for melanin contents and tyrosinase activity in B16 murine cells, the SAR was summarized. The results indicated that five compounds (8a, 8j, 8m–8o) were more potent than the positive control (8-MOP) on melanogenesis. Among them, 8a and 8o showed the best stimulating effect on tyrosinase activity as well, and were submitted for further pharmacological study of anti-vitiligo.
1 Introduction
Vitiligo is an acquired pigmentary disorder of the skin characterized by circumscribed depigmented macules due to the destruction of the melanocyte and subsequent obstruction of melanin synthesis (Fig. 1).1,2 The prevalence of vitiligo is often referred to as 0.5–2% of the world's population.3 The origin of vitiligo is thought to be a complex interplay of genetics, environment, oxidative stress, and autoimmunity.4,5 And the autoimmune theory is the leading hypothesis that is supported by strong evidence.6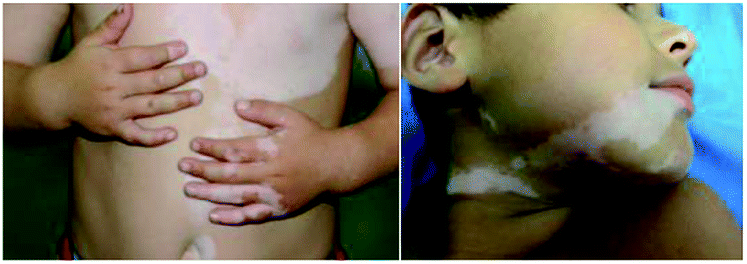 | ||
| Fig. 1 Vitiligo with typical lesions of the face and trunk. | ||
In humans, melanin is the primary determinant of skin color and thought to protect skin cells from UVB radiation damage, reducing the risk of cancer.7 It is produced by melanocytes which are found in the basal layer of the epidermis and regulated by enzymatic cascade, including tyrosinase (TYR), tyrosinase-related protein 1 (TRP-1) and tyrosinase-related protein 2 (TRP-2).8 Among them, TYR is the rate-limiting enzyme and is mainly involved in two distinct reactions of melanin synthesis: firstly, the hydroxylation of a monophenol and secondly, the conversion of an o-diphenol to the corresponding o-quinone.9,10
Presently, there are several drugs used for the treatment of vitiligo including photosensitizer (8-MOP, khellin), topical corticosteroids (dexamethasone), calcineurin inhibitors (tacrolimus), vitamin D3 analogs (tacalcitol),11 and JAK inhibitors (ruxolitinib, baricitinib, tofacitinib) (Fig. 2), which are often used for myelodysplastic disorders, were recently proven to be efficacious for vitiligo based on a large number of clinical cases.12,13
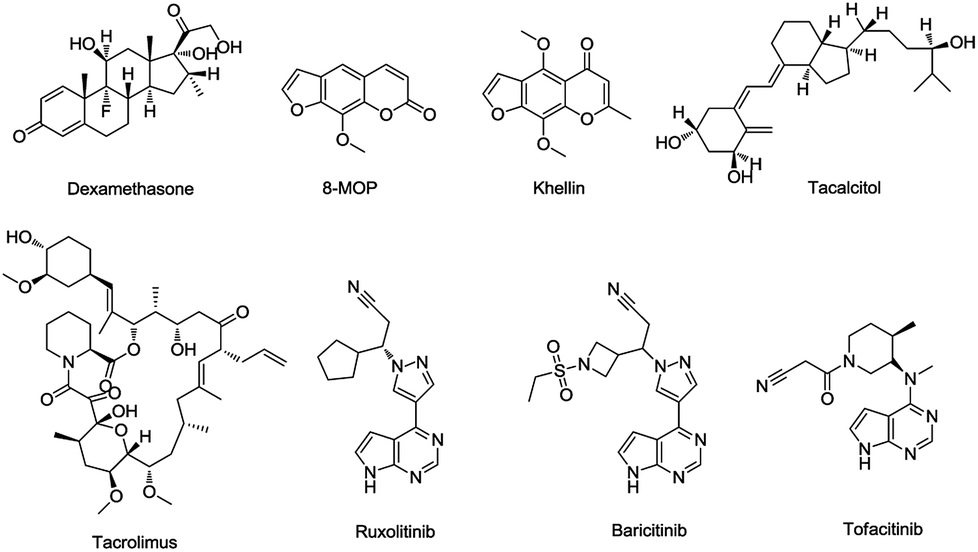 | ||
| Fig. 2 Structures of commonly used drugs for vitiligo. | ||
Traditional Chinese medicine is an important source of modern drugs. In Uygur medicine, the extract of Psoralea corylifolia L. (Fig. 3) was often used for repigmentation of vitiligo with natural sunlight and initially recorded in ‘Yao Yong Zong Ku’ around 300 years ago. Its active ingredient, furocoumarins, has been clinically applied, including 8-methoxypsoralen (8-MOP), 5-methoxypsoralen (5-MOP) and 4,5,8-trimethylpsoralen (TMP) (Fig. 4). Further research proved that these compounds exhibited strong photosensitivity,14 which may be beneficial for the repigmentation of skin with subsequent exposure to long-waved ultraviolet radiation.15 In spite of some undesired side effects,16,17 the therapy is still the most effective way to treat the disease today.
 | ||
| Fig. 3 The plant of the Psoralea corylifolia L. | ||
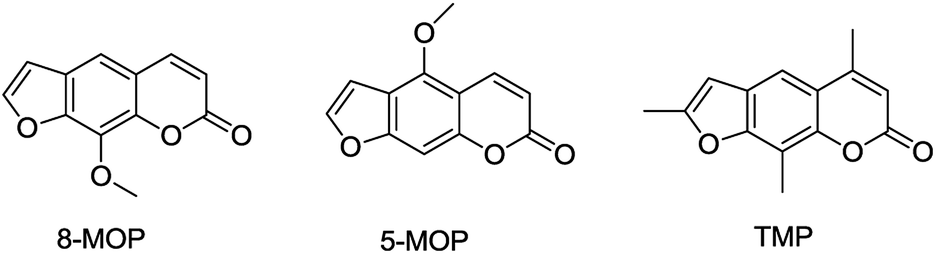 | ||
| Fig. 4 The structures of the furocoumarins used for vitiligo. | ||
Unfortunately, the targets of furocoumarin for the therapies was still unknown due to the complexity of pathogenesis, and few derivatives with anti-vitiligo activity were reported.18–20 During past few years, many molecules with superior effect on melanogenesis and tyrosinase activity were isolated or synthesized by our group.21–24 The preliminary SAR of furocoumarins on vitiligo was outlined in Fig. 5 based on these studies.
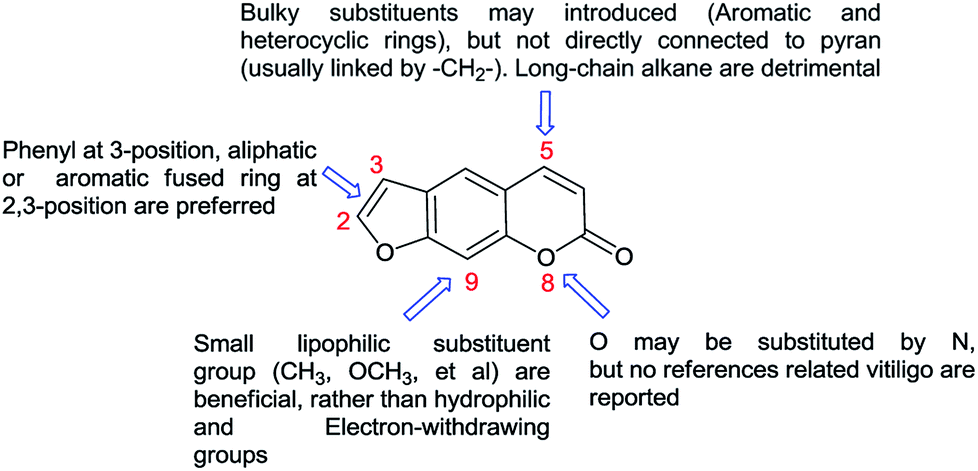 | ||
| Fig. 5 The SAR of furocoumarins on vitiligo. | ||
1,2,3-Triazole and its derivatives showed particular promise as amide bond isosteres, given their diverse biological properties and facile synthesis from readily available azide and alkyne.25–28 The moiety is stable to metabolic degradation and capable of hydrogen bonding, which could be favorable in binding of biomolecular targets and increasing water solubility.29
As depicted in Fig. 5, it is quite clear that more structure modification should be proceed on C-5 position of the furocoumarin to improve this efficiency. Thus, a new series of derivatives containing 1,2,3-triazole group was synthesized, which may be developed as better candidates for the vitiligo.
2 Results and discussion
2.1 Chemistry
The synthetic route for the target compounds is outlined in Scheme 1. First, the commercially available material resorcine was converted to the desired intermediate 4-methylumbelliferone 1 at a 92% yield via Pechmann reaction.30,31 Then, Williamson reaction between 1 and 2-bromoacetophenone in the presence of potassium carbonate afforded intermediate 2, which was cyclized in 4% KOH ethanolic solution to form 3.32 Selenium dioxide was applied in selective oxidation of compound 3 to produce 5-carbaldehyde 4. After that, it was further reduced with NaBH4 to achieve alcohol 5 in 76% yield.33,34 Bromination and azidation of hydroxy with PBr3 and NaN3 successively gave the key intermediate 7. Finally, compound 7 was converted to the target 8a–8o through click chemistry with different alkyne a–o.35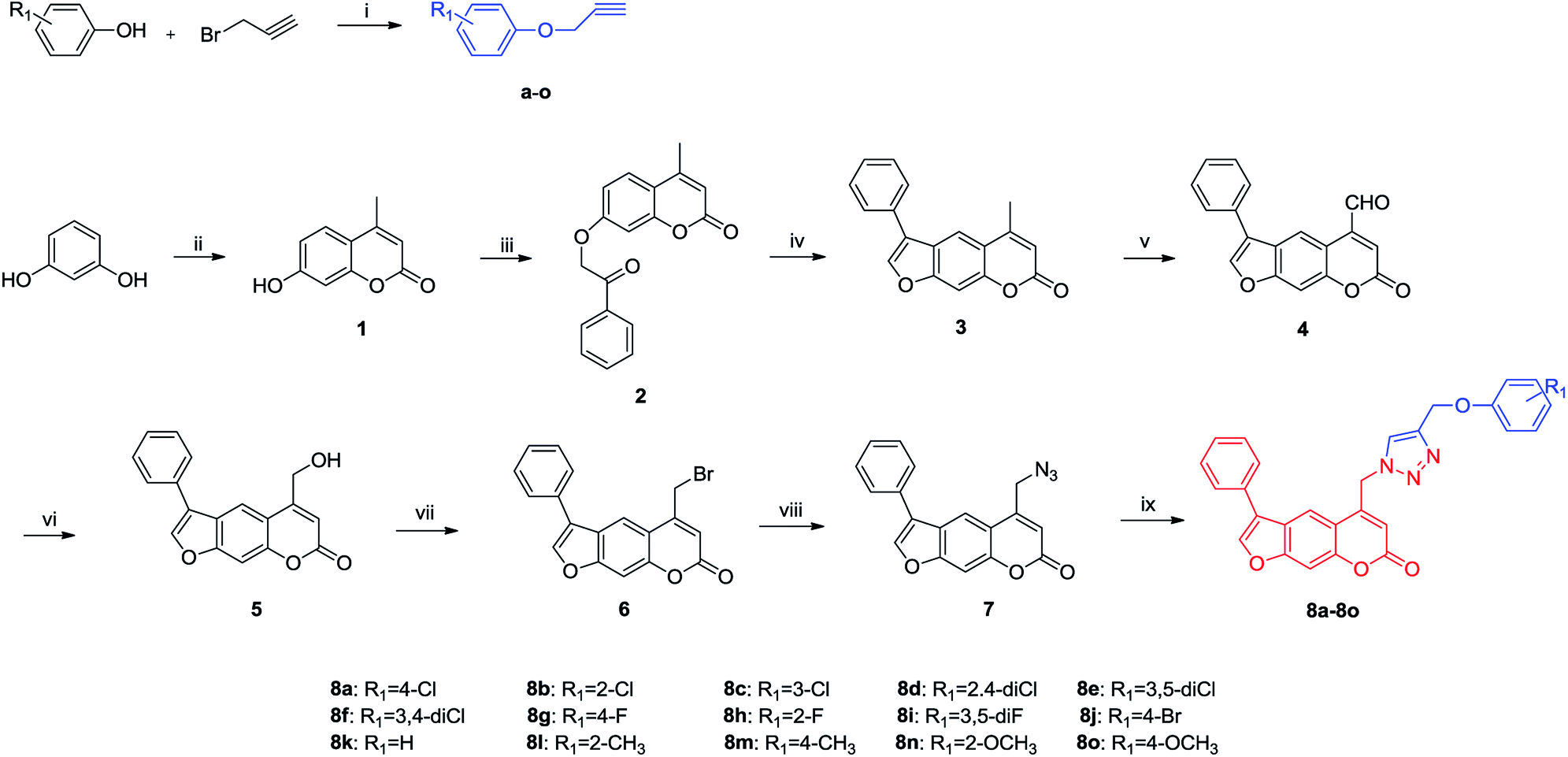 | ||
Scheme 1 Synthesis of furocoumarin derivatives containing 1,2,3-triazole (8a–8o). Reagents and conditions: (i) DMF, K2CO3, 70 °C (ii) ethyl acetoacetate, H2SO4, 60 °C (iii) 2-bromoacetophenone, K2CO3, acetone, reflux (iv) 4% KOH ethanolic solution, reflux (v) SeO2, xylene, reflux (vi): NaBH4, methanol, rt (vii): PBr3, THF, 0 °C-rt (viii): NaN3, CH3CN, reflux (ix) a–o, CuSO4·5H2O, Cu, t-BuOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 1:3, 80 °C. :H2O = 1:3, 80 °C. | ||
2.2 Biological evaluation
Compounds 8a–8o were evaluated for their activity on melanin synthesis in murine B16 cells, with a known method.36 According to the results in Fig. 6, the reference drug 8-MOP enhanced the melanin synthesis by 126.03 ± 2.97% at 50 μm as compared to the blank control. Most of synthesized compounds could increase the melanin content, especially for 8a, 8j, 8m–8o, which were more potent than the positive control (8-MOP) with a value from 134% to 211%. For example, derivatives bearing EDG (8l–8o) on benzene exhibited a stronger effect than unsubstituted 8k. The OCH3 group contributed more than CH3 to the activity, such as 8o > 8m, 8n > 8l. And it seemed that the para-position may be superior for the substituent to improve melanin synthesis (8m > 8l, 8o > 8n). Among these halogenated derivatives (8a–8j), ones with –Cl and –Br showed higher activity compared with –F (8a, 8j > 8g, 8b > 8h). The position of the halogen on benzene also played an important role for their efficacy, the shift of –Cl substituent from the para- (8a, 166%) into the ortho- or meta-position led to 8b (118%) or 8c (106%) separately, which induced a decreased value in melanin content. The similar result was observed in compounds 8g and 8h, substituted with –F group as well. However, introduction of a second –F or –Cl to the benzene may not strengthen this ability (8a > 8d–8f, 8g > 8i), and it is apparent that the presence of a halogen group in the para-position of the phenyl ring was crucial for melanogenesis (8d > 8f > 8e).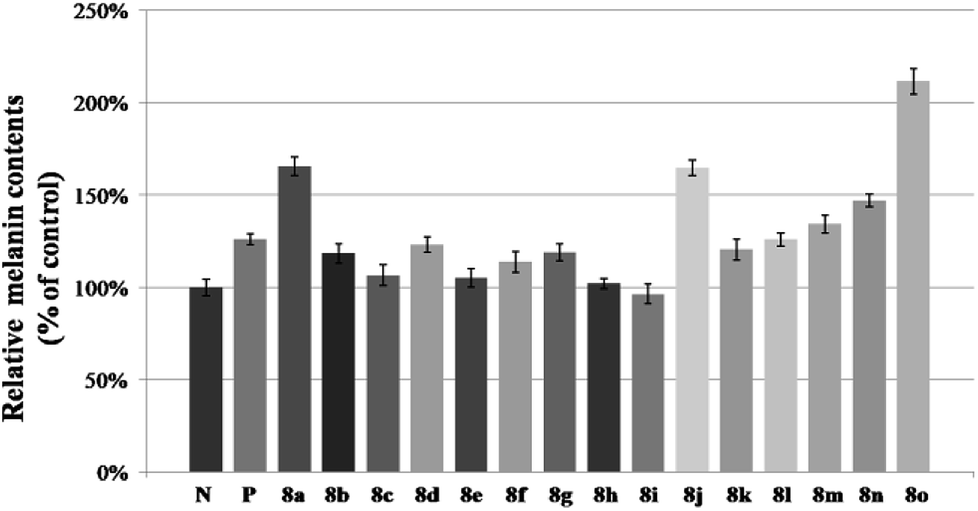 | ||
| Fig. 6 Stimulation of melanin content of B16 cells by 8a–8o. N means negative control; P means positive control (8-MOP); the B16 cells was treated with 50 μM different furocoumarin derivatives for 48 h. After that, melanin content was measured directly. Values are expressed as the mean ± SD of three separate experiments, P < 0.05. | ||
After that, all these derivatives were further studied for their stimulating effect on tyrosinase.37 As shown in Fig. 7, only two compounds (8a, 8o) showed a higher value than 8-MOP. And it can be noticed that the –Cl, and –OCH3 groups might be favorable to enhance the activity among these monosubstituted compounds, since the value of them were all over 110% (8a–8c, 8n–8o > 110%). Changing the position of –Cl on benzene ring weakened stimulatory efficiency (8a > 8b or 8c), indicating that the presence of a substituent in the para-position may be beneficial for the enzyme activation, and it was also applicable for the OCH3 group (8o > 8n). Nevertheless, dihalide derivatives possessed a lower activity than mono- ones. Additionally, the effect of most potential 8a, 8n, on both melanin synthesis and tyrosinase activity, were positively in a concentration dependent manner as depicted in Fig. 8.
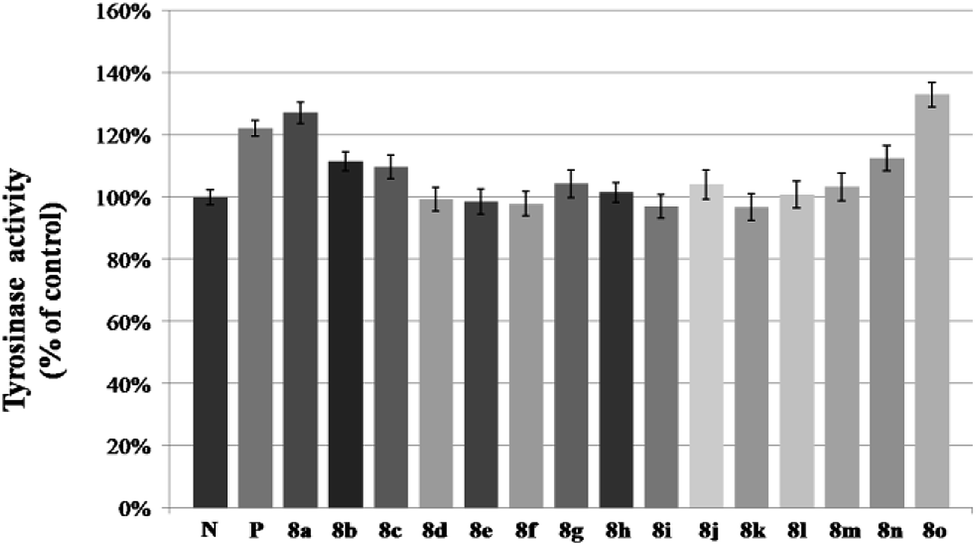 | ||
| Fig. 7 Stimulation of tyrosinase activity of B16 cells by 8a–8o. N means negative control; P means positive control (8-MOP). B16 cells was treated with 50 μM different furocoumarin derivatives for 48 h. After that, tyrosinase activity was measured directly. The data are the mean ± SD of three experiments, P < 0.05. | ||
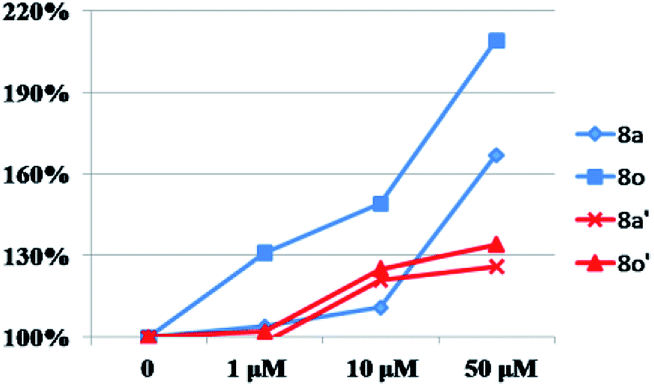 | ||
| Fig. 8 The concentration dependent manner of 8a, 8o on melanin synthesis and tyrosinase activity. “◆”, “■” and “×”, “▲” means the effect of 8a, 8o on melanin synthesis (in blue) and tyrosinase activity (in red) separately at different concentration in B16 cells. | ||
3 Experimental
3.1 Chemistry
Reagents and solvents were purchased from Sigma, and used without further purification. Thin-layer chromatography (TLC) was carried out on glass plates coated with silica gel (Qingdao Haiyang Chemical Co., G60F-254) and visualized by UV light (254 nm). The products were purified by column chromatography over silica gel (Qingdao Haiyang Chemical Co., 200–300 mesh). Melting points were determined on a Buchi B-540 apparatus and uncorrected. All the NMR spectra were recorded with a Varian 400 MHz NMR spectrometer in CDCl3 or acetone-d6, using TMS as an internal standard. High-resolution mass spectra (HRMS) were recorded on AB SCIEX QSTAR Elite quadrupole time-of-flight mass spectrometry. The IR data were recorded on a Thermo Fisher Scientific Nicolet 6700 FT-IR infrared spectrometer (KBr).a: 1H NMR (400 MHz, CDCl3) δ 7.26 (d, J = 8.9 Hz, 2H), 6.92 (d, J = 8.9 Hz, 2H), 4.67 (d, J = 2.4 Hz, 2H), 2.52 (t, J = 2.4 Hz, 1H).
d: 1H NMR (400 MHz, CDCl3) δ 7.38 (d, J = 2.5 Hz, 1H), 7.20 (dd, J = 8.8, 2.5 Hz, 1H), 7.02 (d, J = 8.8 Hz, 1H), 4.76 (d, J = 2.4 Hz, 2H), 2.55 (t, J = 2.3 Hz, 1H).
e: 1H NMR (400 MHz, CDCl3) δ 7.01 (t, J = 1.7 Hz, 1H), 6.88 (d, J = 1.7 Hz, 2H), 4.67 (d, J = 2.4 Hz, 2H), 2.57 (t, J = 2.4 Hz, 1H).
k: 1H NMR (400 MHz, CDCl3) δ 7.37–7.29 (m, 2H), 7.05–6.97 (m, 3H), 4.71 (d, J = 2.4 Hz, 2H), 2.53 (t, J = 2.4 Hz, 1H).
l: 1H NMR (400 MHz, CDCl3) δ 7.26–7.21, (m, 2H), 7.06–6.95 (m, 2H), 4.76 (d, J = 2.4 Hz, 2H), 2.56 (t, J = 2.4 Hz, 1H), 2.34 (s, 3H).
o: 1H NMR (400 MHz, CDCl3) δ 6.93 (d, J = 9.1 Hz, 2H), 6.85 (d, J = 9.1 Hz, 2H), 4.64 (d, J = 2.4 Hz, 2H), 3.77 (s, 3H), 2.51 (t, J = 2.3 Hz, 1H).
:1) were added copper sulfate pentahydrate (0.44 mg, 1.75 mmol) and copper (0.06 g, 1.0 mmol). The reaction was stirred at 80 °C overnight. Then the suspension was extracted with DCM (10 mL × 2), the combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure to give a residue, which was purified by silica gel chromatography to afford final compounds 8a–8o.
3.1.9.1 5-((4-((4-Chlorophenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (8a). Yield 84%, light yellow solid, mp 180–182 °C; 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.86 (s, 1H), 7.70 (s, 1H), 7.58–7.53 (m, 5H), 7.48–7.41 (m, 1H), 7.21 (d, J = 9.0 Hz, 2H), 6.88 (d, J = 9.0 Hz, 2H), 6.05 (s, 1H), 5.80 (s, 2H), 5.21 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 160.06, 157.52, 156.70, 152.07, 148.18, 143.47, 130.56, 129.60, 129.54, 128.48, 127.65, 124.70, 123.40, 122.48, 116.24, 115.20, 114.02, 113.62, 101.06, 62.33, 50.95; IR (KBr) ν: 2925, 1732, 1635, 1576, 1489, 1240, 1151, 823 cm−1; HRMS (ESI) calcd for C27H19ClN3O4[M + H]+ 484.0986, found 484.0993.
3.1.9.2 5-((4-((2-Chlorophenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (8b). Yield 86%, light yellow solid, mp 198–201 °C; 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.86 (s, 1H), 7.77 (s, 1H), 7.59–7.50 (m, 5H), 7.48–7.40 (m, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.22–7.15 (m, 1H), 7.06 (d, J = 8.2 Hz, 1H), 6.94–6.87 (m, 1H), 6.02 (s, 1H), 5.81 (s, 2H), 5.33 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 160.05, 157.51, 153.66, 152.08, 148.24, 145.51, 143.42, 132.60, 131.02, 130.57, 129.53, 128.95, 128.46, 127.98, 127.67, 124.68, 123.49, 122.51, 115.16, 114.57, 113.92, 101.05, 63.52, 50.90; IR (KBr) ν: 2926, 1719, 1634, 1484, 1273, 1152, 1067, 754 cm−1; HRMS (ESI) calcd for C27H19ClN3O4[M + H]+ 484.0986, found 484.0977.
3.1.9.3 5-((4-((3-Chlorophenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (8c). Yield 82%, light yellow solid, mp 110–111 °C; 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.86 (s, 1H), 7.71 (s, 1H), 7.59–7.52 (m, 5H), 7.48–7.41 (m, 1H), 7.22–7.15 (m, 1H), 6.99–6.92 (m, 2H), 6.85 (d, J = 8.6 Hz, 1H), 6.06 (s, 1H), 5.81 (s, 2H), 5.22 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 160.06, 158.84, 157.53, 152.09, 148.17, 143.45, 135.14, 131.06, 130.56, 130.50, 129.54, 128.48, 127.67, 123.36, 121.83, 115.56, 115.21, 114.04, 113.64, 113.22, 110.17, 108.07, 101.08, 62.27, 50.96; IR (KBr) ν: 2924, 1733, 1635, 1457, 1274, 1152 cm−1; HRMS (ESI) calcd for C27H19ClN3O4[M + H]+ 484.0986, found 484.1004.
3.1.9.4 5-((4-((2,4-Dichlorophenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (8d). Yield 86%, light yellow solid, mp 121–123 °C; 1H NMR (400 MHz, CDCl3) δ 8.03 (s, 1H), 7.86 (s, 1H), 7.75 (s, 1H), 7.59–7.50 (m, 5H), 7.48–7.41 (m, 1H), 7.32 (d, J = 2.4 Hz, 1H), 7.14 (dd, J = 8.8, 2.5 Hz, 1H), 7.00 (d, J = 8.8 Hz, 1H), 6.03 (s, 1H), 5.81 (s, 2H), 5.30 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 161.13, 157.53, 155.69, 152.48, 148.16, 143.47, 142.31, 131.06, 130.29, 129.53, 128.99, 128.47, 127.87, 127.65, 125.78, 124.70, 123.58, 115.35, 115.14, 113.98, 110.98, 101.09, 63.68, 50.95; IR (KBr) ν: 2925, 1733, 1635, 1576, 1473, 1271 cm−1; HRMS (ESI) calcd for C27H18Cl2N3O4[M + H]+ 518.0669, found 518.0680.
3.1.9.5 5-((4-((3,5-Dichlorophenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (8e). Yield 88%, light yellow solid, mp 223–224 °C; 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.86 (s, 1H), 7.70 (s, 1H), 7.57–7.51 (m, 5H), 7.48–7.41 (m, 1H), 6.97 (t, J = 1.8 Hz, 1H), 6.87 (d, J = 1.7 Hz, 2H), 6.07 (s, 1H), 5.82 (s, 2H), 5.20 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 160.04, 157.32, 156.52, 152.79, 148.11, 143.48, 131.06, 129.54, 129.41, 128.99, 128.49, 127.66, 124.73, 123.47, 121.98, 115.20, 114.06, 113.77, 109.69, 101.10, 62.44, 50.98; IR (KBr) ν: 2926, 1732, 1634, 1571, 1457, 1273, 1150, 1074 cm−1; HRMS (ESI) calcd for C27H18Cl2N3O4[M + H]+ 518.0669, found 518.0651.
3.1.9.6 5-((4-((3,4-Dichlorophenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (8f). Yield 80%, light yellow solid, mp 159–161 °C; 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.86 (s, 1H), 7.71 (s, 1H), 7.59–7.52 (m, 5H), 7.49–7.41 (m, 1H), 7.30 (d, J = 8.9 Hz, 1H), 7.07 (d, J = 2.9 Hz, 1H), 6.82 (dd, J = 9.0, 2.9 Hz, 1H), 6.06 (s, 1H), 5.81 (s, 2H), 5.20 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 160.05, 157.54, 157.10, 156.31, 152.08, 148.13, 143.49, 130.95, 129.53, 128.48, 127.65, 124.72, 123.47, 122.49, 117.05, 115.19, 114.77, 114.05, 113.60, 110.15, 101.09, 62.46, 50.98; IR (KBr) ν: 2924, 1733, 1653, 1576, 1473, 1274, 1048 cm−1; HRMS (ESI) calcd for C27H18Cl2N3O4[M + H]+ 518.0669, found 518.0682.
3.1.9.7 5-((4-((4-Fluorophenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (8g). Yield 82%, light yellow solid, mp 184–185 °C; 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.86 (s, 1H), 7.71 (s, 1H), 7.59–7.50 (m, 6H), 7.48–7.41(m, 1H), 6.99–6.85 (m, 4H), 6.04 (s, 1H), 5.80 (s, 2H), 5.19 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 160.06, 157.50, 156.54, 154.20, 152.05, 148.24, 143.45, 131.02, 129.53, 128.93, 128.46, 127.65, 124.67, 123.40, 122.48, 116.21, 115.98, 115.20, 113.95, 113.63, 101.03, 62.71, 50.90; IR (KBr) ν: 2926, 1732, 1634, 1576, 1506, 1389, 1202, 1152, 1040 cm−1; HRMS (ESI) calcd for C27H19FN3O4[M + H]+ 468.1354, found 468.1367.
3.1.9.8 5-((4-((2-Fluorophenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (8h). Yield 85%, light yellow solid, mp 166–167 °C; 1H NMR (400 MHz, CDCl3) δ 8.03 (s, 1H), 7.85 (s, 1H), 7.71 (s, 1H), 7.61–7.49 (m, 5H), 7.47–7.40 (m, 1H), 7.12–6.96 (m, 3H), 6.96–6.85 (m, 1H), 6.01 (s, 1H), 5.80 (s, 2H), 5.30 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 160.03, 157.50, 154.29, 152.06, 148.21, 146.00, 143.42, 132.45, 131.05, 130.56, 129.52, 128.97, 128.45, 127.66, 124.58, 122.41, 116.46, 115.19, 113.94, 113.66, 101.03, 63.65, 50.94; IR (KBr) ν: 2925, 1733, 1634, 1506, 1457, 1257, 1152, 1047 cm−1; HRMS (ESI) calcd for C27H19FN3O4[M + H]+ 468.1354, found 468.1371.
3.1.9.9 5-((4-((3,5-Difluorophenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (8i). Yield 83%, light yellow solid, mp 210–212 °C; 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.86 (s, 1H), 7.72 (s, 1H), 7.59–7.52 (m, 5H), 7.47–7.43 (m, 1H), 6.50 (d, J = 8.8 Hz, 2H), 6.43 (t, J = 8.9 Hz, 1H), 6.06 (s, 1H), 5.82 (s, 2H), 5.19 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 160.04, 157.54, 155.60, 152.09, 148.12, 143.48, 131.06, 129.54, 128.99, 128.49, 127.66, 124.74, 123.02, 122.53, 115.20, 114.05, 113.58, 109.19, 101.09, 62.49, 50.98; IR (KBr) ν: 2923, 1732, 1599, 1465, 1275, 1152, 1042, 996 cm−1; HRMS (ESI) calcd for C27H18F2N3O4[M + H]+ 486.1260, found 486.1248.
3.1.9.10 5-((4-((4-Bromophenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (8j). Yield 84%, light yellow solid, mp 104–105 °C; 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.86 (s, 1H), 7.71 (s, 1H), 7.59–7.51 (m, 5H), 7.48–7.41 (m, 1H), 7.35 (d, J = 8.9 Hz, 2H), 6.84 (d, J = 8.9 Hz, 2H), 6.05 (s, 1H), 5.80 (s, 2H), 5.20 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 160.06, 157.52, 157.21, 153.19, 152.07, 148.17, 143.47, 132.54, 129.54, 128.48, 127.65, 124.69, 123.41, 122.49, 116.75, 115.20, 114.03, 113.85, 113.62, 101.07, 62.25, 50.95; IR (KBr) ν: 2926, 1733, 1635, 1576, 1489, 1457, 1276, 1150 cm−1; HRMS (ESI) calcd for C27H19BrN3O4[M + H]+ 528.0553, found 528.0571.
3.1.9.11 5-((4-(Phenoxymethyl)-1H-1,2,3-triazol-1-yl)methyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (8k). Yield 85%, light yellow solid, mp 152–154 °C; 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.86 (s, 1H), 7.72 (s, 1H), 7.61–7.50 (m, 5H), 7.48–7.41 (m, 1H), 7.30–7.24 (m, 2H), 7.00–6.92 (m, 3H), 6.05 (s, 1H), 5.80 (s, 2H), 5.24 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 160.07, 158.12, 157.51, 152.08, 148.24, 143.42, 130.57, 129.72, 129.53, 128.46, 127.67, 124.68, 123.32, 122.52, 121.59, 115.23, 114.91, 114.00, 113.67, 101.03, 62.09, 50.90; IR (KBr) ν: 2925, 1732, 1634, 1490, 1274, 1150, 1047 cm−1; HRMS (ESI) calcd for C27H20N3O4[M + H]+ 450.1448, found 450.1433.
3.1.9.12 5-((4-((o-Tolyloxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)3-phenyl-7H-furo[3,2-g]chromen-7-one (8l). Yield 89%, light yellow solid, mp 160–161 °C; 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.86 (s, 1H), 7.68 (s, 1H), 7.61–7.50 (m, 5H), 7.48–7.41 (m, 1H), 7.08–7.17 (m, 2H), 6.96–6.83 (m, 2H), 6.04 (s, 1H), 5.81 (s, 2H), 5.25 (s, 2H), 2.19 (s, 3H).13C NMR (101 MHz, CDCl3) δ 160.09, 157.51, 156.28, 152.08, 148.31, 143.39, 131.02, 130.57, 129.53, 128.46, 127.67, 127.01, 124.68, 122.52, 121.31, 115.19, 113.89, 113.69, 111.70, 101.07, 62.41, 50.92, 29.86; IR (KBr) ν: 2925, 1733, 1635, 1460, 1273, 1152, 1046 cm−1; HRMS (ESI) calcd for C28H22N3O4[M + H]+ 464.1605, found 464.1616.
3.1.9.13 5-((4-((p-Tolyloxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (8m). Yield 91%, light yellow solid, mp 218–220 °C; 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.86 (s, 1H), 7.72 (s, 1H), 7.60–7.51 (m, 5H), 7.48–7.41 (m, 1H), 7.06 (d, J = 8.1 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 6.05 (s, 1H), 5.79 (s, 2H), 5.20 (s, 2H), 2.27 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 160.07, 157.50, 156.00, 152.08, 148.23, 143.45, 143.37, 130.89, 130.57, 130.15, 129.54, 128.46, 127.67, 124.67, 122.52, 115.28, 115.23, 114.81, 114.06, 113.99, 113.68, 101.05, 62.26, 50.98, 29.85; IR (KBr) ν: 2925, 1733, 1647, 1508, 1457, 1388, 1228, 1151, 1050 cm−1; HRMS (ESI) calcd for C28H22N3O4[M + H]+ 464.1605, found 464.1589.
3.1.9.14 5-((4-((2-Methoxyphenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (8n). Yield 90%, light yellow solid, mp 172–173 °C; 1H NMR (600 MHz, CDCl3) δ 8.04 (s, 1H), 7.86 (s, 1H), 7.71 (s, 1H), 7.59–7.51 (m, 5H), 7.48–7.41 (m, 1H), 6.99 (d, J = 8.0 Hz, 1H), 6.95–6.90 (m, 1H), 6.89–6.81 (m, 2H), 6.05 (s, 1H), 5.78 (s, 2H), 5.32 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 160.06, 157.52, 156.69, 152.11, 148.21, 147.44, 145.97, 143.39, 130.57, 130.18, 129.54, 128.45, 127.68, 124.67, 123.44, 122.39, 121.02, 115.30, 114.94, 114.09, 113.70, 112.08, 101.02, 63.48, 55.96, 50.90; IR (KBr) ν: 2926, 1733, 1635, 1505, 1457, 1388, 1253, 1150, 1074, 1026 cm−1; HRMS (ESI) calcd for C28H22N3O5[M + H]+ 480.1554, found 480.1539.
3.1.9.15 5-((4-((4-Methoxyphenoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (8o). Yield 88%, light yellow solid, mp 195–197 °C; 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.86 (s, 1H), 7.71 (s, 1H), 7.60–7.50 (m, 5H), 7.48–7.41 (m, 1H), 6.93 (d, J = 9.1 Hz, 2H), 6.84 (d, J = 9.1 Hz, 2H), 6.03 (s, 1H), 5.79 (s, 2H), 5.19 (s, 2H), 3.77 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 160.07, 157.50, 154.47, 152.19, 151.82, 148.25, 143.46, 130.57, 129.54, 128.46, 127.67, 124.67, 122.51, 116.29, 116.08, 115.20, 114.84, 114.00, 113.67, 101.06, 62.88, 55.79, 50.98; IR (KBr) ν: 2927, 1717, 1631, 1574, 1439, 1388, 1257, 1151, 1082, 1109 cm−1; HRMS (ESI) calcd for C28H22N3O5[M + H]+ 480.1554, found 480.1570.
3.2 Biological activity
000g for 15 min to obtain the supernatant. After protein quantification and adjustment, 90 μL of the supernatant was incubated in duplicate with 10 μL of freshly prepared substrate solution (10 mM L-DOPA) in a well of a 96-well plate. Then the cells were incubated at 37 °C in dark for 60 min, the absorbance was measured at 490 nm and the samples treated cells was presented as percentage against the untreated cells.4 Conclusions
In summary, a novel series of furocoumarin derivatives bearing 1,2,3-triazole had been prepared via click chemistry. Two of them (8a, 8o) could not only promote the melanin synthesis, but also the tyrosinase activity in vitro than positive control (8-MOP) in a concentration dependent manner, and the SAR was summarized. In light of our findings, it may provide scientific guidance for further structural modification of the furocoumarin, even if without knowing the specific targets of the disease. Further studies on action mechanism and animal experiment on transgenic mouse is under way.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the Xinjiang Key Laboratory of Xinjiang Indigenous Medicinal Plants Resource Utilization (No. 2018D04020); Funds for the Foundation of Director of XTIPC, CAS (2016TP001); Natural Science Foundation of Xinjiang, China (No. 2017D01A76).Notes and references
- K. Ezzedine, V. Eleftheriadou, M. Whitton and N. van Geel, Lancet, 2015, 386, 74–84 CrossRef.
- M. Rodrigues, K. Ezzedine, I. Hamzavi, A. G. Pandya and J. E. Harris, J. Am. Acad. Dermatol., 2017, 77, 1–13 CrossRef CAS PubMed.
- K. Ezzedine, H. W. Lim, T. Suzuki, I. Katayama, I. Hamzavi, C. C. E. Lan, B. K. Goh, T. Anbar, C. Silva de Castro, A. Y. Lee, D. Parsad, N. van Geel, I. C. Le Poole, N. Oiso, L. Benzekri, R. Spritz, Y. Gauthier, S. K. Hann, M. Picardo and A. Taieb, Pigm. Cell Melanoma Res., 2012, 25, E1–E13 CrossRef CAS PubMed.
- N. C. Laddha, M. Dwivedi, M. S. Mansuri, A. R. Gani, M. Ansarullah, A. V. Ramachandran, S. Dalai and R. Begum, Exp. Dermatol., 2013, 22, 245–250 CrossRef CAS PubMed.
- S. Wang, R. Jin, R. Wang, Y. Hu, X. Dong and A. Xu, RSC Adv., 2016, 6, 106308–106315 RSC.
- J. M. Richmond, M. L Frisoli and J. E. Harris, Curr. Opin. Immunol., 2013, 25, 676–682 CrossRef CAS PubMed.
- P. Meredith and J. Riesz, Photochem. Photobiol., 2004, 79, 211–216 CrossRef CAS PubMed.
- M. Sandoval-Cruz, M. García-Carrasco, R. Sánchez-Porras, C. Mendoza-Pinto, M. Jiménez-Hernández, P. Munguía-Realpozo and A. Ruiz-Argüelles, Autoimmun. Rev., 2011, 10, 762–765 CrossRef CAS PubMed.
- M. M. Garcia-Molina, J. L. Muñoz-Muñoz, F. Garcia-Molina, P. A. García-Ruiz and F. Garcia-Canovas, J. Agric. Food Chem., 2012, 60, 6447–6453 CrossRef CAS PubMed.
- Q. Wu, Z. Xu, Y. Duan, Y. Zhu, M. Oua and X. Xu, RSC Adv., 2017, 7, 28114–28123 RSC.
- M. L. Felsten, A. Alikhan and V. Petronic-Rosic, J. Am. Acad. Dermatol., 2011, 65, 493–514 CrossRef PubMed.
- W. Damsky and B. A. King, J. Am. Acad. Dermatol., 2017, 76, 736–744 CrossRef CAS PubMed.
- J. E. Harris, M. Rashighi, N. Nguyen, A. Jabbari, G. Ulerio, R. Clynes, A. M. Christiano and J. Mackay-Wiggan, J. Am. Acad. Dermatol., 2016, 74, 370–371 CrossRef PubMed.
- W. L. Fowucs, D. G. Griffith and E. L. Oginsky, Nature, 1958, 18, 571–572 Search PubMed.
- T. B. Fitzpatrick, J. A. Parrish and M. A. Pathak, in Phototherapy of vitiligo (idiopatic leukodermia) in sunlight and man, Tokyo University Press, 1974, pp. 365–398 Search PubMed.
- L. M. Felsten, A. Alikhan and V. Petronic-Rosic, J. Am. Acad. Dermatol., 2011, 65, 473–491 CrossRef PubMed.
- S. Tippisetty, D. Goudi, A. W. Mohammed and P. Jahan, Toxicol. In Vitro, 2013, 27, 438–440 CrossRef CAS PubMed.
- A. Guiotto, P. Rodighiero, P. Manzini, G. Pastohi, F. Bordin, F. Baccichetti, F. Carlassare, D. Vedaldi, F. Dall'Acqua, M. Tamaro, G. Recchia and M. Cristofolinis, J. Med. Chem., 1984, 27, 959–967 CrossRef CAS PubMed.
- H. Matsuda, N. Hirata, Y. Kawaguchi, M. Yamazaki, S. Naruto, M. Shibano, M. Taniguchi, K. Baba and M. Kubo, Biol. Pharm. Bull., 2005, 28, 1229–1233 CrossRef CAS.
- E. Chodurek, A. Orchel, J. Orchel, S. Kurkiewicz, N. Gawlik, Z. Dzierżewicz and K. Stęppień, Cell. Mol. Biol. Lett., 2012, 17, 616–632 CAS.
- C. Niu, G. X. Pang, G. Li, J. Dou, L. F. Nie, H. Himit, K. Kabas and H. A. Hisa, Bioorg. Med. Chem., 2016, 24, 5960–5968 CrossRef CAS PubMed.
- L. Yin, G. X. Pang, C. Niu, J. Dou and H. A. Aisa, Int. J. Mol. Med., 2018, 41, 3727–3735 CAS.
- C. Niu, D. Zang and H. A. Aisa, Chem. Res. Chin. Univ., 2018, 34, 408–414 CrossRef CAS.
- C. Niu and H. A. Aisa, Molecules, 2017, 22, 1303–1330 CrossRef PubMed.
- P. Yadav, K. Lal, A. Kumar, S. K. Guru, S. Jaglan and S. Bhushan, Eur. J. Med. Chem., 2017, 126, 944–953 CrossRef CAS PubMed.
- N. Pribut, C. G. L. Veale, A. E. Basson, W. A. L. van Otterlo and S. C. Pelly, Bioorg. Med. Chem. Lett., 2016, 26, 3700–3704 CrossRef CAS PubMed.
- S. Maračić, T. G. Kraljević, H. Č. Paljetak, M. Matijašić, D. Verbanac, M. Cetina and S. Raić-Malić, Bioorg. Med. Chem., 2015, 23, 7448–7463 CrossRef PubMed.
- N. Fu, S. Wang, Y. Zhang, C. Zhang, D. Yang, L. Weng, B. Zhao and Li. Wang, Eur. J. Med. Chem., 2017, 136, 596–602 CrossRef CAS PubMed.
- N. S. Vatmurge, B. G. Hazra, V. S. Pore, F. Shirazi, P. S. Chavan and M. V. Deshpande, Bioorg. Med. Chem. Lett., 2008, 18, 2043–2047 CrossRef CAS PubMed.
- B. L. Zhang, C. Q. Fan, L. Dong, F. D. Wang and J. M. Yue, Eur. J. Med. Chem., 2010, 45, 5258–5264 CrossRef CAS PubMed.
- E. C. Gaudino, S. Tagliapietra, K. Martina, G. Palmisanob and G. Cravotto, RSC Adv., 2016, 6, 46394–46405 RSC.
- A. Chilin, P. Manzini, S. Caffieri, P. Rodighiero and A. Guiotto, J. Heterocycl. Chem., 2001, 38, 431–434 CrossRef CAS.
- Q. Shen, Q. Peng, J. L. Sha, X. Liu, Z. Huang, X. Pu, L. Ma, Y. M. Li, A. S. C. Chan and L. Gu, Eur. J. Med. Chem., 2005, 40, 1307–1315 CrossRef CAS PubMed.
- R. O. Schönleber, J. Bendig, V. Hagen and B. Giese, Bioorg. Med. Chem., 2002, 10, 97–101 CrossRef.
- Y. Hu, H. Shen, X. Zhang, Y. Liu and X. Sun, RSC Adv., 2018, 8, 23252–23256 RSC.
- S. Y. Park, M. L. Jin, Y. H. Kim, Y. Kim and S. J. Lee, Arch. Dermatol. Res., 2011, 303, 737–744 CrossRef CAS PubMed.
- H. J. Kim, J. S. Kim, J. T. Woo, J. T. Woo, I. S. Lee and B. Y. Cha, Acta Biochim. Biophys. Sin., 2015, 47, 548–556 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |