DOI:
10.1039/C8RA10184A
(Paper)
RSC Adv., 2019,
9, 1613-1624
Destabilization of amyloid fibrils on interaction with MoS2-based nanomaterials†
Received
11th December 2018
, Accepted 18th December 2018
First published on 11th January 2019
Abstract
The present work is motivated by the established concept that the structure and energetics of biomacromolecules can be modulated by confining their dimensions in the nanoscale. In particular, here we use force-field methods to understand the stability of amyloid fibrils at nanostructured interfaces, which can be useful for the development of new therapeutics for Alzheimer's disease. We explore the binding modes and structural properties of fibrils at the interface of molybdenum disulphide nanotubes and the nanosurface using classical molecular dynamics simulations. We find that in general the MoS2 materials induces disruptions in the structure of the amyloid fibrils where the beta sheet conformation of the fibrils changes to a turned conformation, and it is large in the case of nanotubes in comparison to the nanosurfaces. The intermolecular hydrogen bonds, hydrophilic and hydrophobic contacts between the monomer peptides in the fibril are reduced due to their adsorption onto the MoS2 materials, which results in a destabilization of the fibril. The destabilization of fibril is to some extent compensated for by the van der Waals interactions between the fibril and MoS2. Overall the results indicate that MoS2-based materials can be useful in inhibiting the aggregation of smaller protofibrils to matured fibrils and to bust the already formed fibrils. Therapeutic materials should not exhibit any cross interaction with other off-targets compounds. In order to test whether the MoS2 nanomaterial has any such effect we have studied its interaction with two additional biomacromolecules, the human serum albumin and p53 protein, and we report no significant changes in the secondary structure of these biomolecules. Through molecular docking studies we also established that the drug binding ability of HSA is not altered by its surface binding to MoS2 nanosurface.
1. Introduction
The misfolding and aggregation of proteins including huntingtin, α-synuclein, and amyloid β cause various neurodegenerative diseases such as Huntington's, Parkinson's and Alzheimer's disease.1–8 The formed protein aggregates interrupt normal cell functioning and affect memory and motor activities depending upon the accumulated brain region. In particular, Alzheimer's is the most commonly occurring disease in elder people causing memory loss, mental depression and mortality.9,10 The misfolding of the alpha helical amyloid beta peptides to disordered structures, and the subsequent formation of aggregates which are enriched with a beta sheet conformation are key molecular level events associated with this disease. Initially, the disordered amyloid peptides form oligomers of beta sheet structures, which aggregate and further grow due to the addition of monomers, finally leading to the formation of mature fibrils.11 Recent studies have proven that the oligomers are more toxic than the matured fibrils.12,13 However, the fibrils also contribute to neurotoxicity of Alzheimer's disease. In histological studies, dystrophic neurites have been found around the amyloid plaques, which indicates that fibrils are also neurotoxic similar to oligomers.14,15 Previous studies also proved the cell death through exposure of neurons to preformed fibrils and oxidative stress also may be caused due to fibrils.16 Therefore, it is necessary to destabilize the fibrils and make them soluble so that brain the organelles can clear them. In order to treat Alzheimer's disease the following steps have often been proposed, (i) the clearing of amyloid plaques from the brain, (ii) destruction of formed oligomers and matured fibrils, and (iii) inhibition of the aggregation processes of the amyloid peptides either through stabilization of the native helical structure or the disordered structures. Molecules that inhibit fibril growth are in general referred to as fibril inhibitors while molecules that can disintegrate formed fibrils are referred to as fibril busters. Several small molecules such as methylthioninium chloride, curcumin, resveratrol, epigallocatechin-3-gallate (EGCG), brazilin, tanshinone and orcein have been identified to interact with the amyloid fibrils and to inhibit the aggregation process.17–25 The green tea polyphenol molecule has been shown to directly bind to unfolded proteins and prevent the further formation of rich beta sheet structures.25 D-Peptides, retro-inverso peptides, N-methyl peptides, molecular tweezers, polyphenols and quinone derivatives have also been used to inhibit the aggregation of amyloid beta peptides.26 However, many small molecules failed to provide a permanent solution for Alzheimer's due to unfavourable pharmacokinetic properties as discussed in detail in a recent review.27
In addition to small molecules, nanoparticles have been shown to inhibit the fibrillization of amyloids and have been used to modulate the secondary structure in the amyloid peptide aggregates.28–35 For example, nano-epigallocatechin-3-gallate has been shown to inhibit the aggregation of amyloids and to disintegrate the already formed fibril structures.28 Gold nanoparticles have gained significant attention due to their tunable surface chemistry where the effect of the surface charge of the nanoparticles has been studied for the prevention of fibrillization of the amyloid peptides.32 Polyoxometalates have been identified to prevent self-aggregation of amyloid beta peptides and metal ion induced aggregation.36 In addition, many investigations have shown that carbon based nanomaterials such as fullerenes, carbon nanotubes, graphene and graphene oxide could be useful to inhibit the aggregation of amyloid peptides and to destroy matured fibrils.37–40 The hydrophobic interaction of fullerenes gives a size-dependent destabilization of the fibril structure.41,42 It has been shown that carbon nanotubes inhibit the formation of aggregates from amyloid beta peptides (16–22).43 Carbon nanotubes are also known to induce the formation of beta barrels in amyloid fragments (25–35).44 Experimental and computational studies have shown that graphene sheets can penetrate into the fibril structure and disintegrate peptides through the π–π stacking interactions between the aromatic surface of graphene and phenylalanine residues.45 Experiments prove that graphene oxide can be useful to bust the fibrils into individual peptide fragments and to clear them.45
In addition to carbon based materials, two dimensional nanomaterials, such as tungsten disulphide and molybdenum disulphide, have also found many applications due to their unique properties46–49 semiconductivity, ferromagnetism, photoluminescence and its ability to serve as good template for various adsorbates.50 Recent experiments indicate that tungsten disulphide surfaces can bind to amyloids and inhibit the aggregation process and as well as disintegrate the matured fibrils.49 Tungsten disulphide surfaces can also be used for imaging of fibrils due to their absorption properties.49 Recent experiments have proven that molybdenum disulphide surfaces also are able to modulate the aggregation of amyloid peptides.51,52 Previous computational studies have shown that molybdenum disulphide nanotubes and nanosurfaces can induce changes in the structure of helical and beta sheet peptides.53,54 However, understanding of the interactions of the fibrils with molybdenum disulphide based materials is still elusive and calls for further theoretical scrutiny. In this study, the interaction of amyloid fibrils with molybdenum nanotubes and nanosurfaces was investigated using classical molecular dynamics simulations.
2. Computational details
The NMR structure of amyloid (1–42) fibril was taken from the protein data bank – pdb id: 2BEG.55 The N-terminus of each of the peptide fragments was capped with an acetyl group to mimic the full length peptide and the C-terminal was deprotonated based on the previous report.56 The missing N-terminal residues were not considered in this study. The fibril has five monomers and each one is named as chain A, chain B, chain C, chain D and chain E. The chain A and chain E are growing ends of the fibril. The structures of a molybdenum disulphide (MoS2) nanotube and a nanosurface were considered in this study for their binding to the amyloid fibril and were built using gaussview package.57 The (8, 8) nanotube length was assumed to be 98.5 Å and the dimensions of the nanosurface were set to 105 Å and 99.6 Å. The structures of the nanotube and nanosurface was considered as if they are infinite structures. Force-field molecular dynamics simulations was performed to understand the interaction between the fibril and the MoS2-based nanomaterials. Based on previous reports, the force field parameters for molybdenum were ε = 80.874 kJ mol−1, σ = 0.255 nm and for sulphur were ε = 5.847 kJ mol−1, σ = 0.337 nm. The charges for these atoms were 0.76 and −0.38 in electronic units, respectively, and were based on electronic structure theory calculations.53,54 As can be seen, the Mo potential is very attractive which will influence the melting and transition temperature of the MoS2 crystal or the hybrid system formed with the MoS2 nanomaterials. For describing the interaction of amyloid fibril, we employed the amber99sb-ildn force field.58 The complexes of the amyloid fibril and MoS2 nanomaterials were placed in a box and solvated with the help of the TIP3P water model. The whole system was neutralized using Na+ ions. The solvated structures were relaxed using the steepest descent minimization method. The obtained structures were equilibrated at 293 K temperature and 1 bar of pressure using the velocity rescaling and Parrinello–Rahman algorithms, respectively.59–61 During the equilibration process, the structures of the fibril and MoS2 were position restrained. The equilibrated structures were simulated for 100 ns with a time step of 2 femtoseconds in the NPT ensemble. The particle mesh Ewald method was used to calculate the electrostatic interactions.62 The bonds of hydrogen with heavy atoms were constrained by employing the LINCS algorithm.63 The trajectories obtained from the molecular dynamics simulations were visualized using the VMD package.64 All the simulations were performed with the help of the GROMACS package and the analysis of trajectories was made using tools available in GROMACS.65–67 The number of hydrogen bonds was calculated using the criteria that the distance between donor and acceptor should be 3.5 Å with the angle between the donor, the acceptor and with the hydrogen attached atom set to 180°. The dictionary of protein secondary structure (DSSP) tool which recognizes the hydrogen bond patterns and geometrical features has been used to investigate the secondary structure of the fibril.68
3. Results and discussion
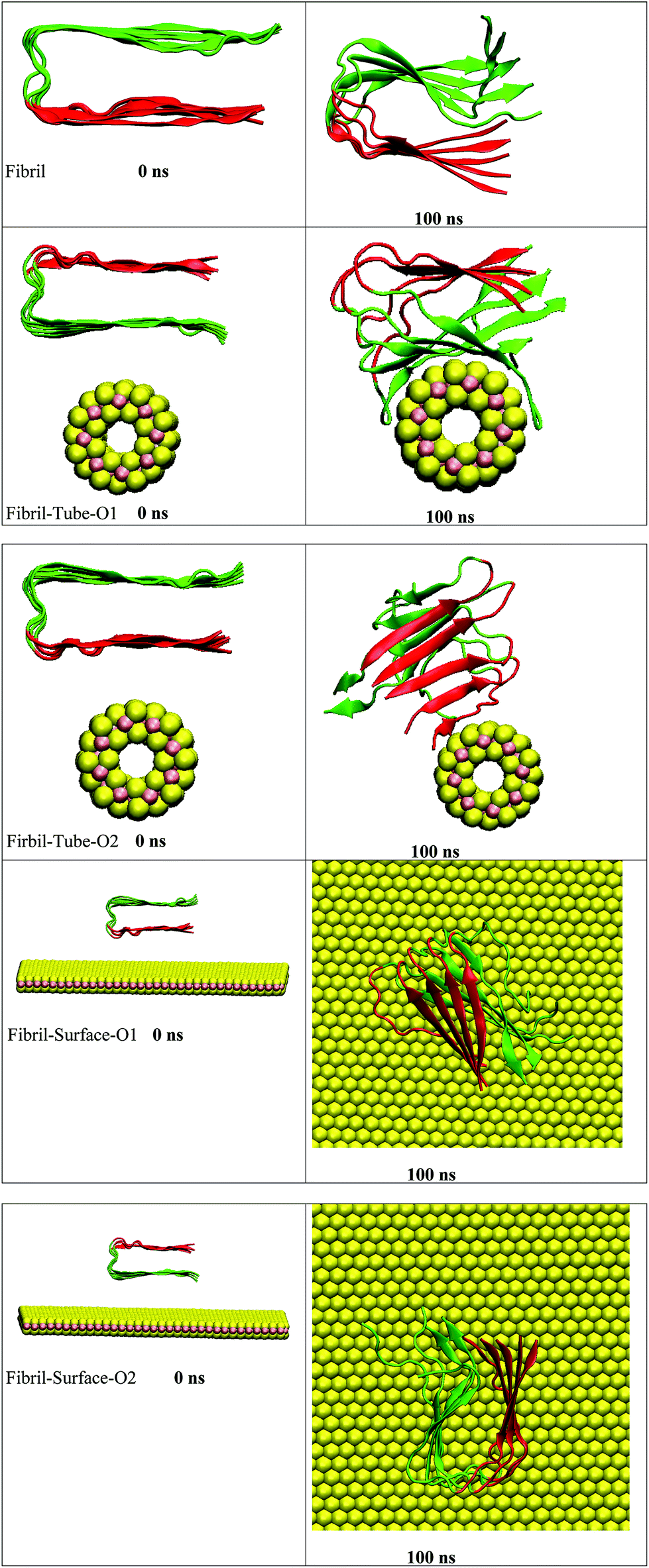
The amyloid fibrils are simulated for 100 ns in aqueous solution and the secondary structure is found intact throughout the simulation. Initially, the amyloid fibrils were placed above the surface of the nanotube, and oriented in two directions, in order to study their interaction, see structures in Fig. 1. Some salient structural features of the interacting complexes can be derived from the simulations: the distance between the fibril and nanotube surface decreases due to the van der Waals interactions. The fibril gets in contact with the sulphur atoms of the nanotube, and, in both orientations, it is adsorbed onto the surface forming stable complexes. In orientation 1 (referred as O1), all chains interact with the nanotube and wrap around the surface of the nanotube adopting its shape. The amino acids (KGAIIGLMVGGVVIA) from 28–42 are in particular involved in the interactions with the nanotube. In the starting structure of orientation 2 (referred as O2), the negatively charged carboxylic acids groups, present in the fibril, are oriented towards the nanotube surface. The partial charges of the sulphur atoms of MoS2 are also negative and due to the repulsion between the negatively charged groups the fibril changes its orientation and interacts through its edge. The chain E is involved in the interaction with the nanotube and this binding pattern is different from orientation 1, indicating that the binding mode of the fibril depends on its orientation. Previous studies have indicated an influence of the curvature of carbon and boron nitride based materials on interaction with peptides and proteins.69,70 In order to disclose the effect of geometry on the adsorption pattern, the amyloid fibrils were allowed to interact with the planar surface of MoS2 in two orientations. Independent of initial structure, in both orientations, the fibril adjusted in such a way that the fibril growth axis is in the perpendicular direction to the surface of MoS2 and forms stable complexes, as can be seen from Fig. 1. In fact, only one chain of the fibril is in contact with the surface. These results clearly show that the binding mode of the fibril with MoS2 is dependent on the curvature, and that the fibril to nanotube binding pattern allows further growth in the case of orientation 1, whereas for orientation 2, the growth can be restricted to one direction rather than from both edges of the fibril. The binding mode of the fibril with the surface is similar to that with the carbon material C60 which has been shown to induce disruptions in the structure of the fibril.41
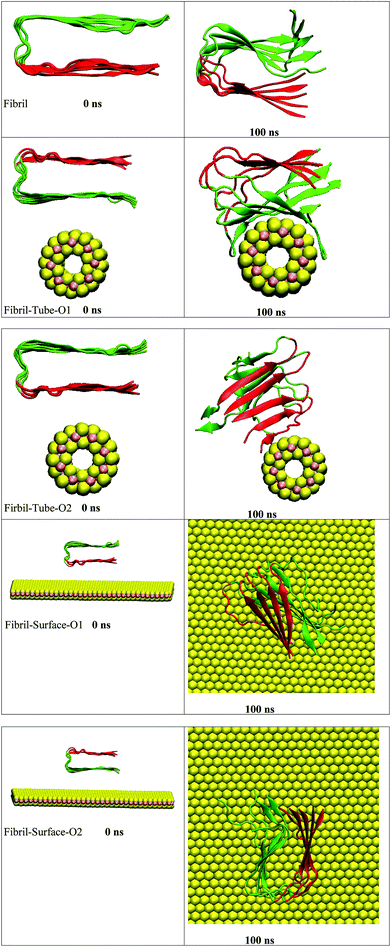 |
| | Fig. 1 The initial and final snap shots of the interaction between amyloid fibril and MoS2 nanomaterials for all the systems. Red and green color in fibril represent upper and lower surface of fibril, respectively. | |
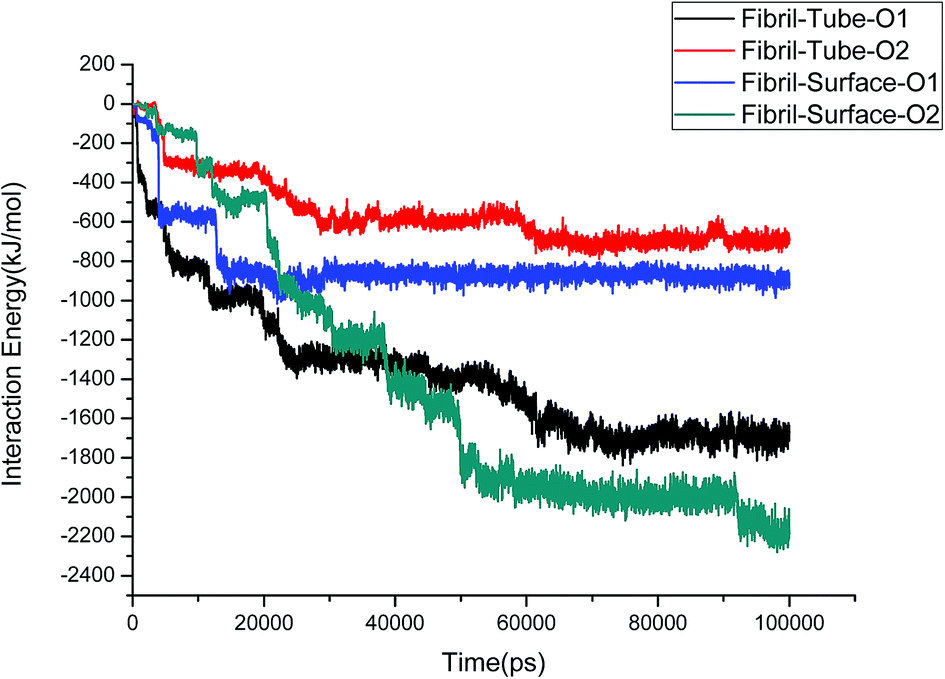
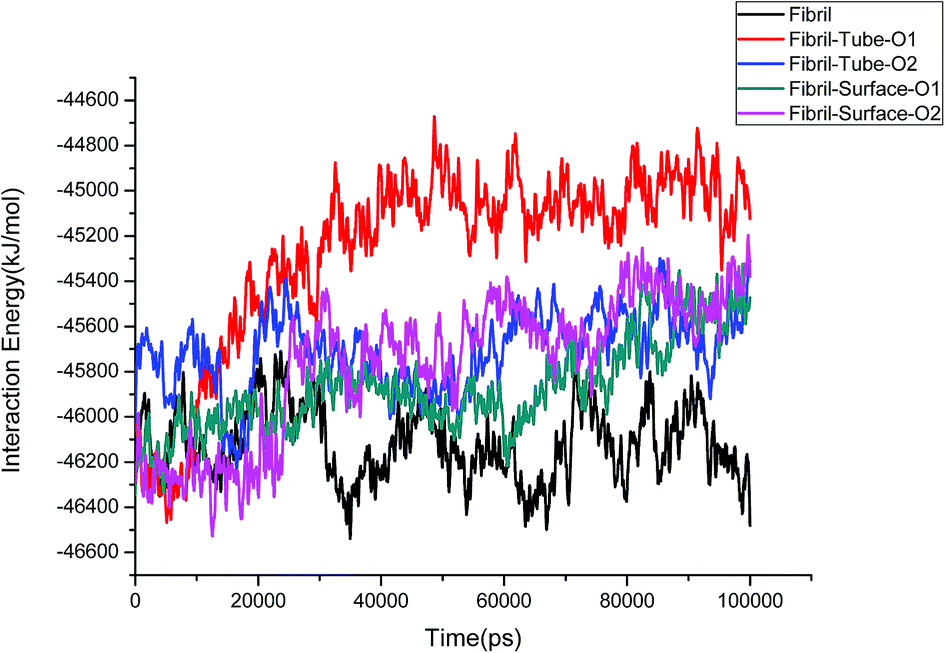
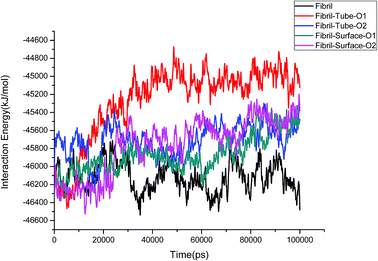
The structural stability of protofibrils is essential for its growth to form plaques. To explore the stability of the complexes of fibril with the MoS2 nanotube and nanosurface, we calculated the interaction energies, see Fig. 2. Interaction energy is defined as the sum of van der Waals and electrostatic energies between amyloid fibril and MoS2. It can be seen that the interaction energy increases with increase in time (we refer to magnitude). The change of the orientation of the fibril on the surface of MoS2 materials maximizes the interaction between them. We have noted that the structural changes in the fibrils are marginal after 50 ns on interaction with MoS2 materials. It can be seen in the interaction energy plots where the energies oscillate around constant values after 50 ns. The interaction energy is high for the nanosurface when compared to the tube in the two orientations. The strength of the interaction seems to be inversely related to the curvature of the MoS2 materials. The calculated average number of contacts (over the last 10 ns) between MoS2 and fibril within 4 Å is 409, 174, 193 and 484 for two orientations in the case of the nanotube and surface, respectively. The number of contacts is proportional to the interaction energy values and is high for the surface owing to the larger available surface area. We have also calculated the contact area between MoS2 and fibril using the solvent accessible surface area (presented in the ESI†). The contact area is found to be in proportion to the interaction energies. The contact area and the number of contacts are thus important quantities to form energetically stable complexes for amyloid fibrils.
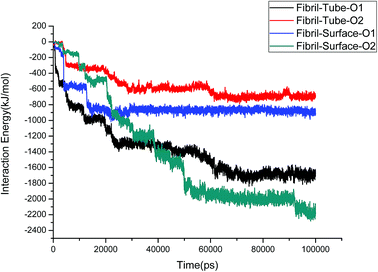 |
| | Fig. 2 The interaction energies between amyloid fibril and MoS2 nanomaterials for all the systems. | |
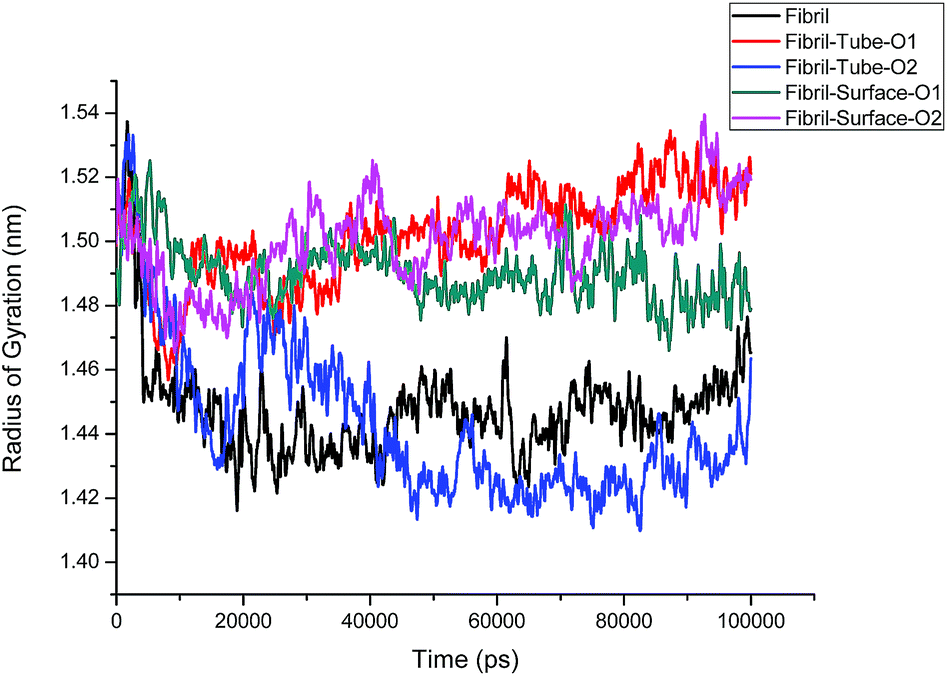
The structural analysis of fibrils can also be discussed using the radius of gyration (Rg) parameter, which gives information about the compactness of the system. The calculated Rg values versus time for all simulated systems are shown in Fig. 3. Initially, Rg decreases and then reaches an equilibrium value for the fibrils in aqueous solution, but grows larger in the case of nanotube-O1, surface-O1 and Surface-O2 when compared to the aqueous solution, indicating the strong interaction with the MoS2 nanotube and surface. It can be noted from Fig. 1 that the chain interacting with MoS2 is separated from the fibrils. The separation of the chain can result in the prevention of fibril growth in one direction in the presence of the nanosurface. The parting of one chain from the pentamer (fibril) decreases the size of the oligomers. This demonstrates the induced destabilization of the fibril upon interaction with MoS2 based materials.
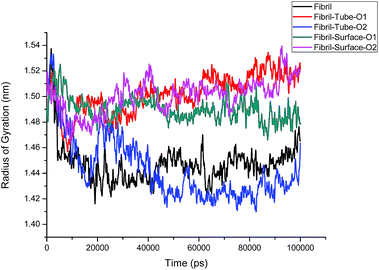 |
| | Fig. 3 Radius of gyration of fibril on interaction with MoS2 based nanomaterials for all the cases. | |
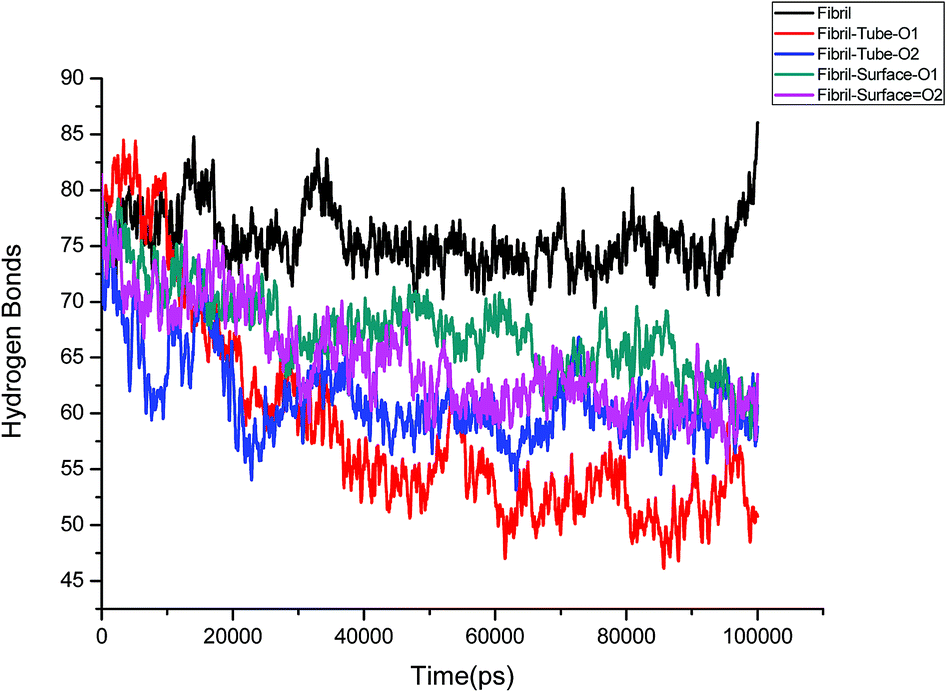
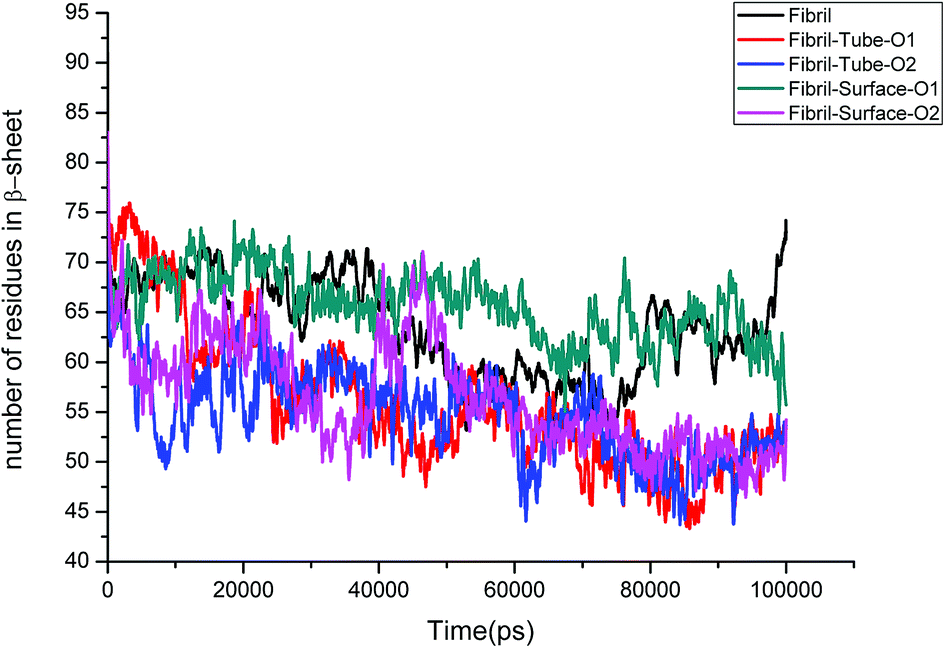
Further, an analysis has been carried out to understand the secondary structure of the fibrils. The beta sheet secondary structure of the oligomer fibrils is important to form larger matured fibrils. The dysfunction of brain components and memory loss is due to the accumulation of insoluble amyloid beta sheet fibrils in the extraneuronal compartments. The destruction of the secondary structure of the fibril may also help to prevent fibril growth and cure the disease. The secondary structures are stabilized through the formation of hydrogen bonds between the monomer peptides. The calculated number of hydrogen bonds in the fibril with respect to simulation time is shown in Fig. 4. It can be clearly seen that a reduction in the number of hydrogen bonds on interaction with MoS2 takes place as compared to fibrils in aqueous solution. The reduction in the number of hydrogen bonds decreases the electrostatic energy between the monomer peptides and leads to a destabilization of the fibrils. More precisely, the calculated average number of hydrogen bonds over the last 10 ns is 76, 52, 51, 62 and 60 for the fibril, tube-O1, tube-O2, surface-O1 and surface-O2, respectively. The reduction of hydrogen bonds is thus larger in the case of tube-O1 and tube-O2 when compared to other cases. The decrease in hydrogen bonds may result in the change of the secondary structure. The calculated number of residues present in the beta sheet conformation of the fibrils is shown in Fig. 5, indicating that the beta sheet content significantly decreases on interaction with MoS2 when compared to fibrils in aqueous solution, except in the case of surface-O1. The average number of amino acids in the beta sheet conformation over last 10 ns are 60, 51, 51, 62 and 50 for the fibril in aqueous conditions for tube-O1, tube-O2, surface-O1 and surface-O2, respectively. The disruption in the secondary structure is more significant for tube-O1, tube-O2 and surface-O2 and correlates with the number of hydrogen bonds in the fibril, except for surface-O2. Previous studies have shown that the loss of helical content of the peptides is inversely proportional to the curvature of the carbon and boron nitride nanomaterials.69,70 In this study, we find that the loss of beta content is more prominent for curved nanotubes than for the planar nanosurface, something that can be derived from the differences in the binding mode of interaction between the tube and the surfaces. In the case of the tube, all five chains of the fibril are involved in the interaction whereas this is not so for the surface. The results confirm that MoS2-based materials induce disruptions in the beta sheet conformations. The destabilization in the structure may increase further on extending the simulation to longer time scales.
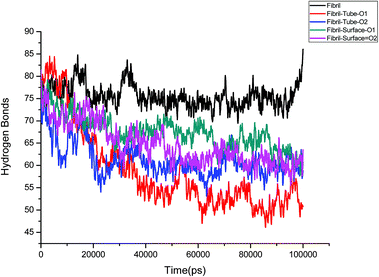 |
| | Fig. 4 Number of hydrogen bonds in amyloid fibril throughout the simulation. | |
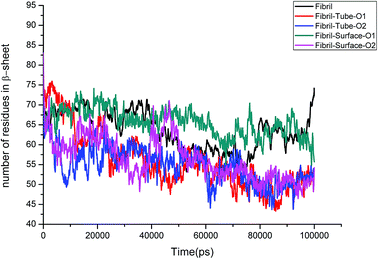 |
| | Fig. 5 Number of amino acids present in beta sheet conformation in amyloid fibril. | |
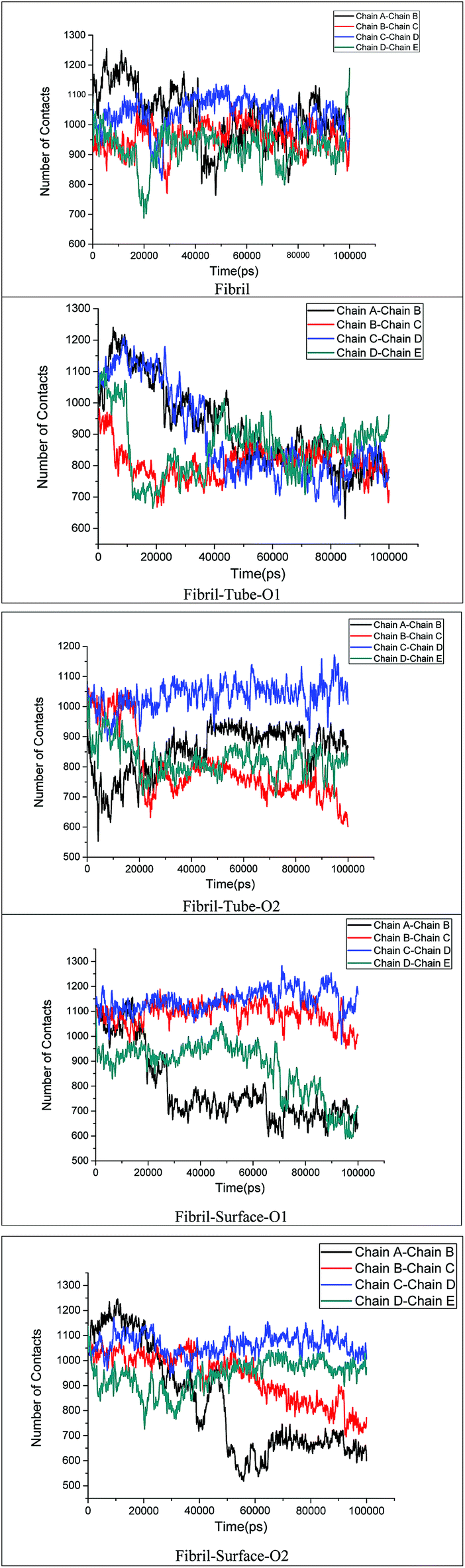
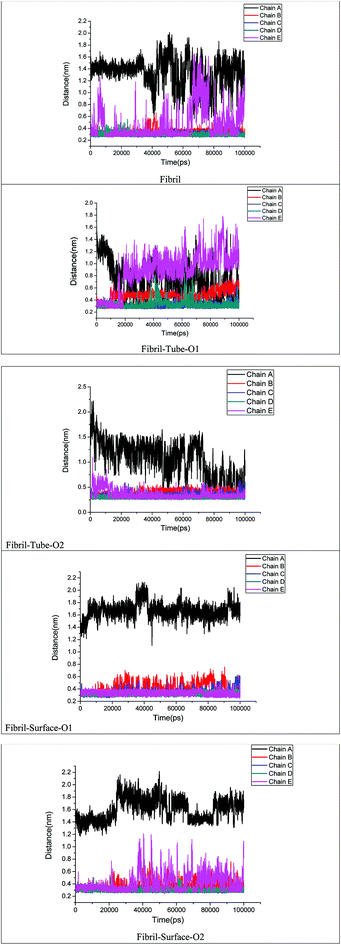
The stability of the fibrils originates from interactions such as hydrogen bonding, hydrophobic forces and salt bridge formations. These interactions change the distances between the fibril chains and influence the strength of the fibril. The contacts between peptides give clear information about the changes in the hydrophobic interactions. The calculated number of contacts between adjacent chains present in the fibrils within 4 Å is shown in Fig. 6. It can be seen that the number of contacts oscillates around a constant value throughout the simulation in the case of the fibril alone in an aqueous solution, showing that the stability of the fibrils is retained in that case. From Fig. 6 the significant changes in the number of contacts between the monomer peptides upon interaction with MoS2 based materials can be noted, and that the number of contacts is reduced when compared to fibrils alone in aqueous solution. The number of contacts between the peptides ranges from 800–1200 for the fibril and after interaction with MoS2 materials it changes to 600–1200. In the case of tube-O1, the number of contacts between all adjacent peptides is drastically decreased compared to the other cases. The reduction in the number of contacts influences the strength of interactions between the monomeric peptides in the fibrils. The interaction energy between all the peptide chains have been calculated and compared with the case of fibril alone in aqueous solution, as shown in Fig. 7. It shows that there is a significant decrease in the interaction energy between the peptide chains on interaction with MoS2 when compared to the case of the fibril alone. The loss of interaction energy is larger in the case of tube-O1 than that of the others. This result is thus in close agreement with the hydrogen bonding and secondary structure analysis given above.
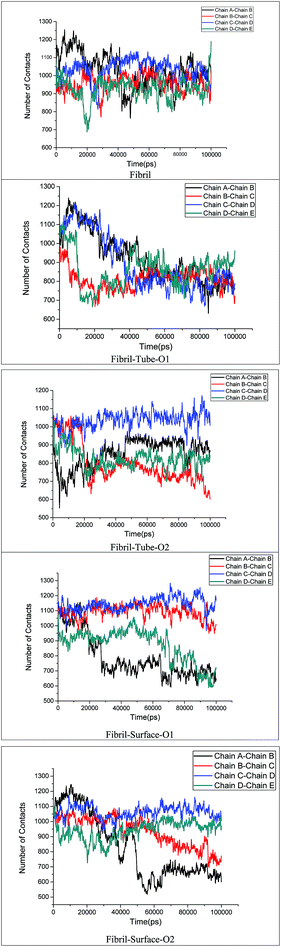 |
| | Fig. 6 Number of contacts the monomer chains present in amyloid fibril. | |
 |
| | Fig. 7 Interaction energy between the monomer chains present in amyloid fibril. | |
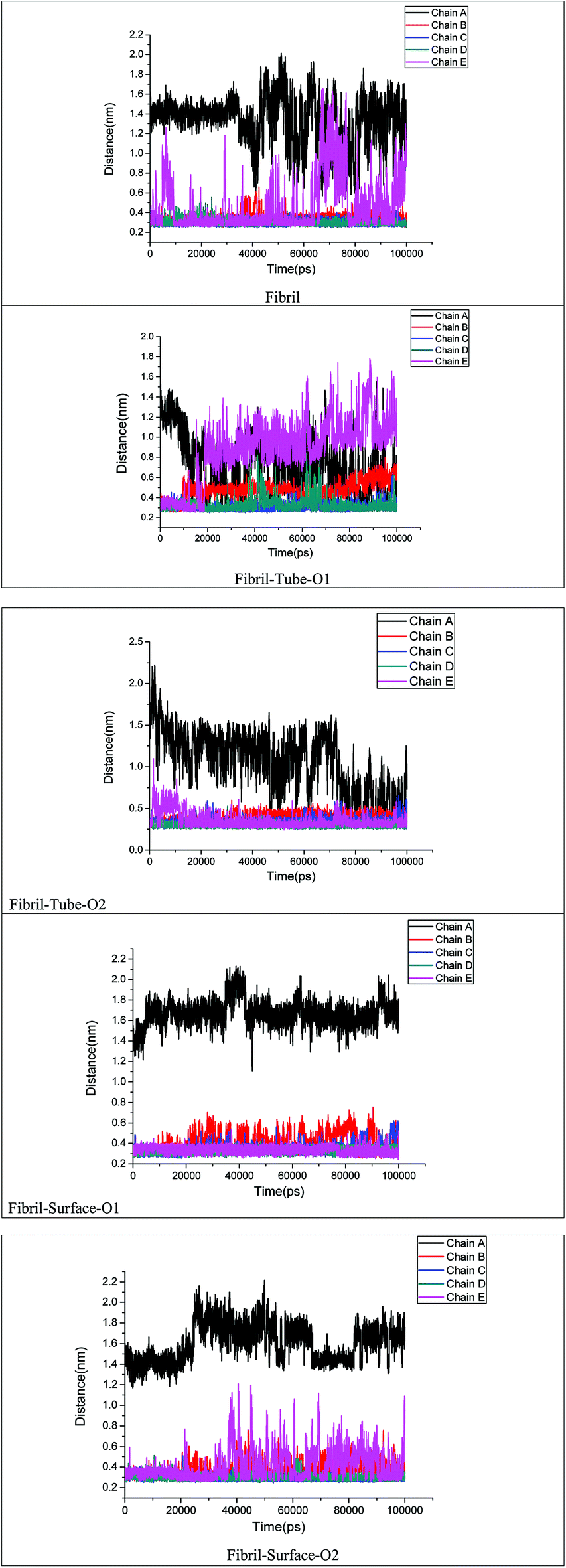
The stability of fibril is also dependent on the presence of inter and intra salt bridges between D23 and K28. We calculated the distance between D23 and K28 in order to analyse the strength of the salt bridge interactions after adsorption onto MoS2 based materials, see Fig. 8. Oscillations between the formation and breaking of the salt bridges present at the end chains of a single fibril in aqueous solution can be seen. In the case of tube-O1, an increase in the distance between the D23 and K28 for chains B and E on interaction with MoS2 tube can be clearly noted. However, there are no significant changes in the salt bridges in other cases except for chain A. Overall, the analysis of the hydrogen bonds, the number of contacts and the salt bridge interactions show that the MoS2 based nanomaterials can be exploited for the fibril destabilization. MoS2 sheets have shown toxicity towards environmental microbial. However, not much is known on their ability to cross the BBB and toxicity in human subjects.71 The technique to disrupt the BBB using pulsed ultrasound may be useful for nanoparticle systems which cannot cross BBB.72
 |
| | Fig. 8 Distance between D23 and K28 amino acids of each monomer chain in amyloid fibril. | |
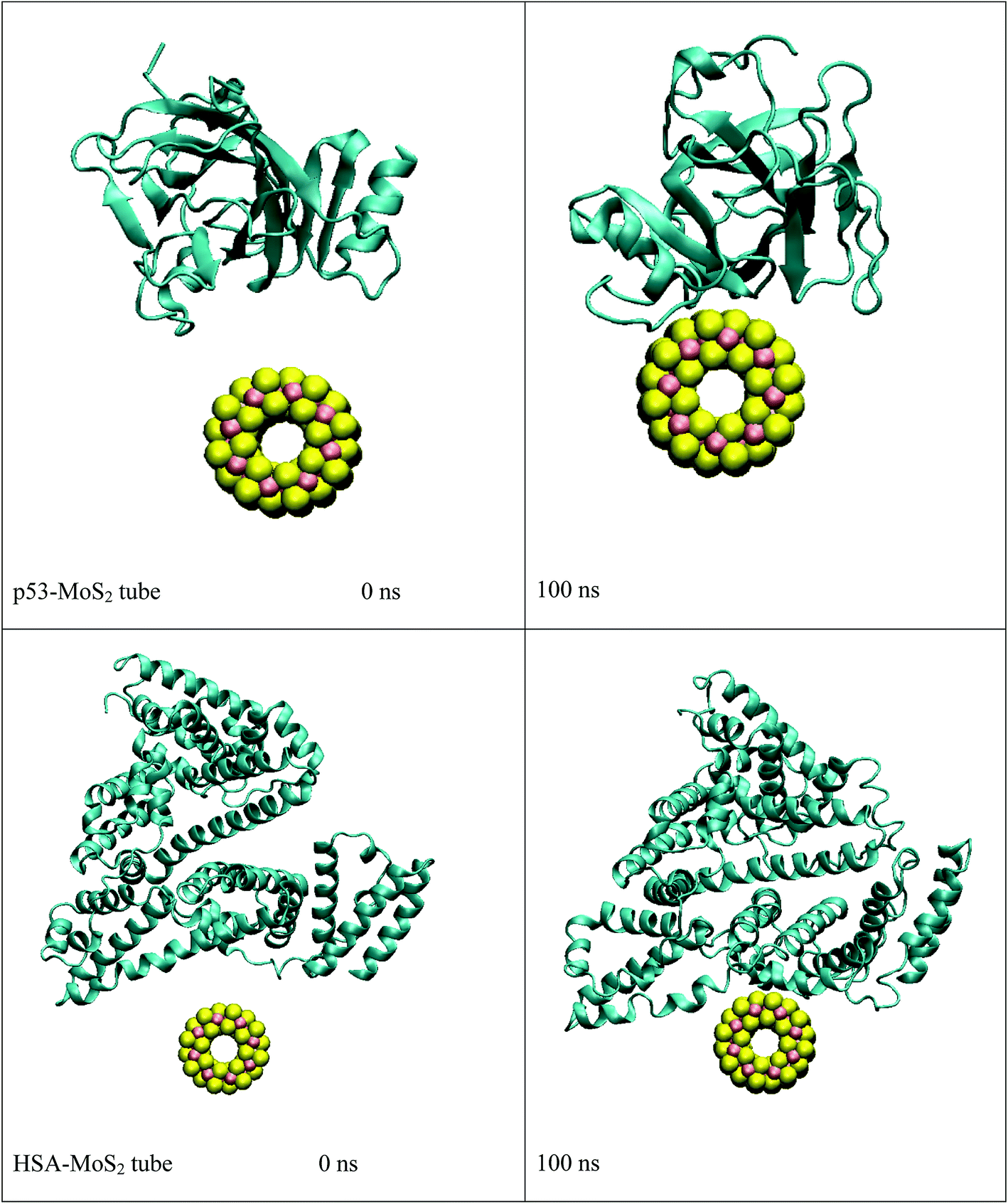
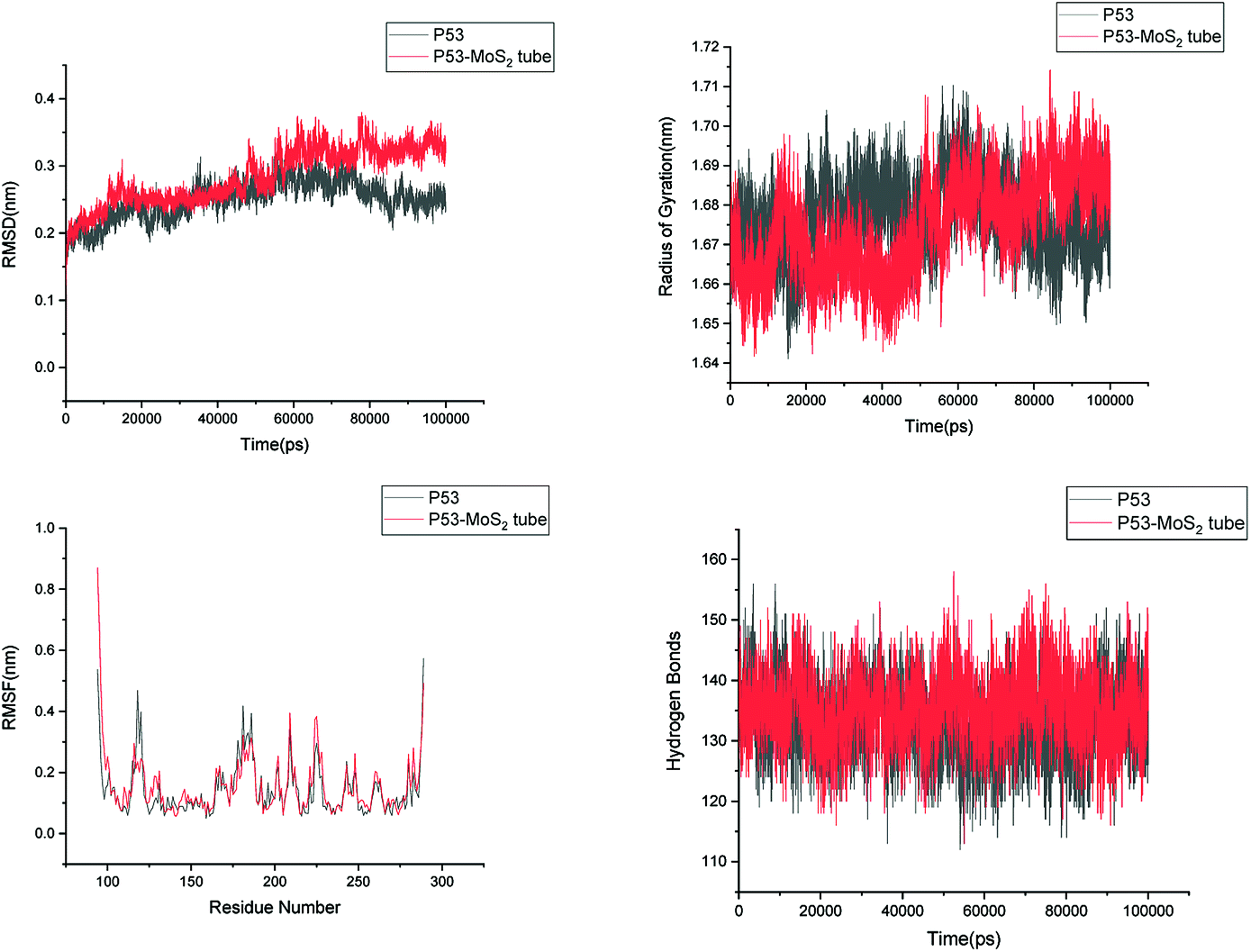
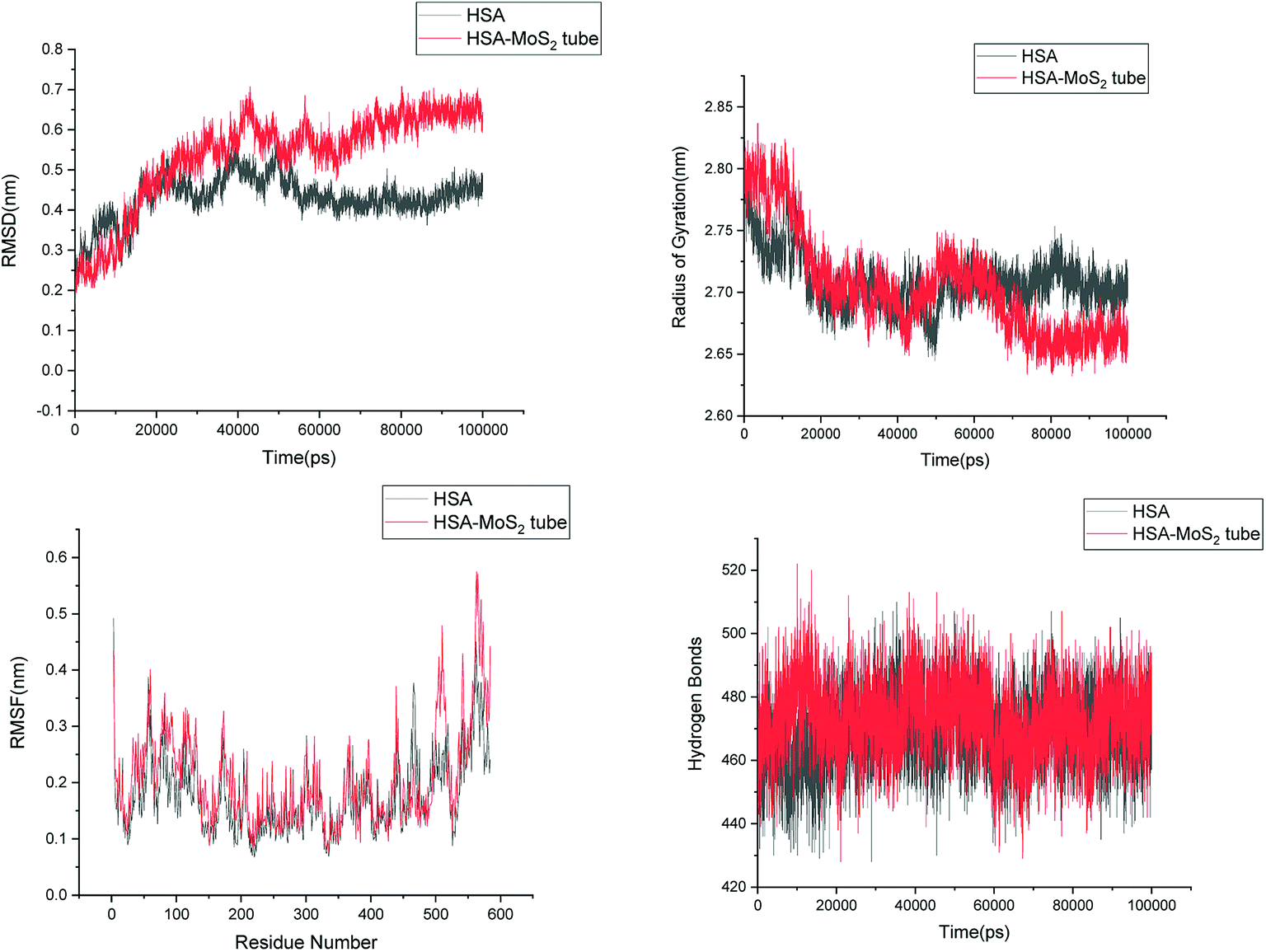
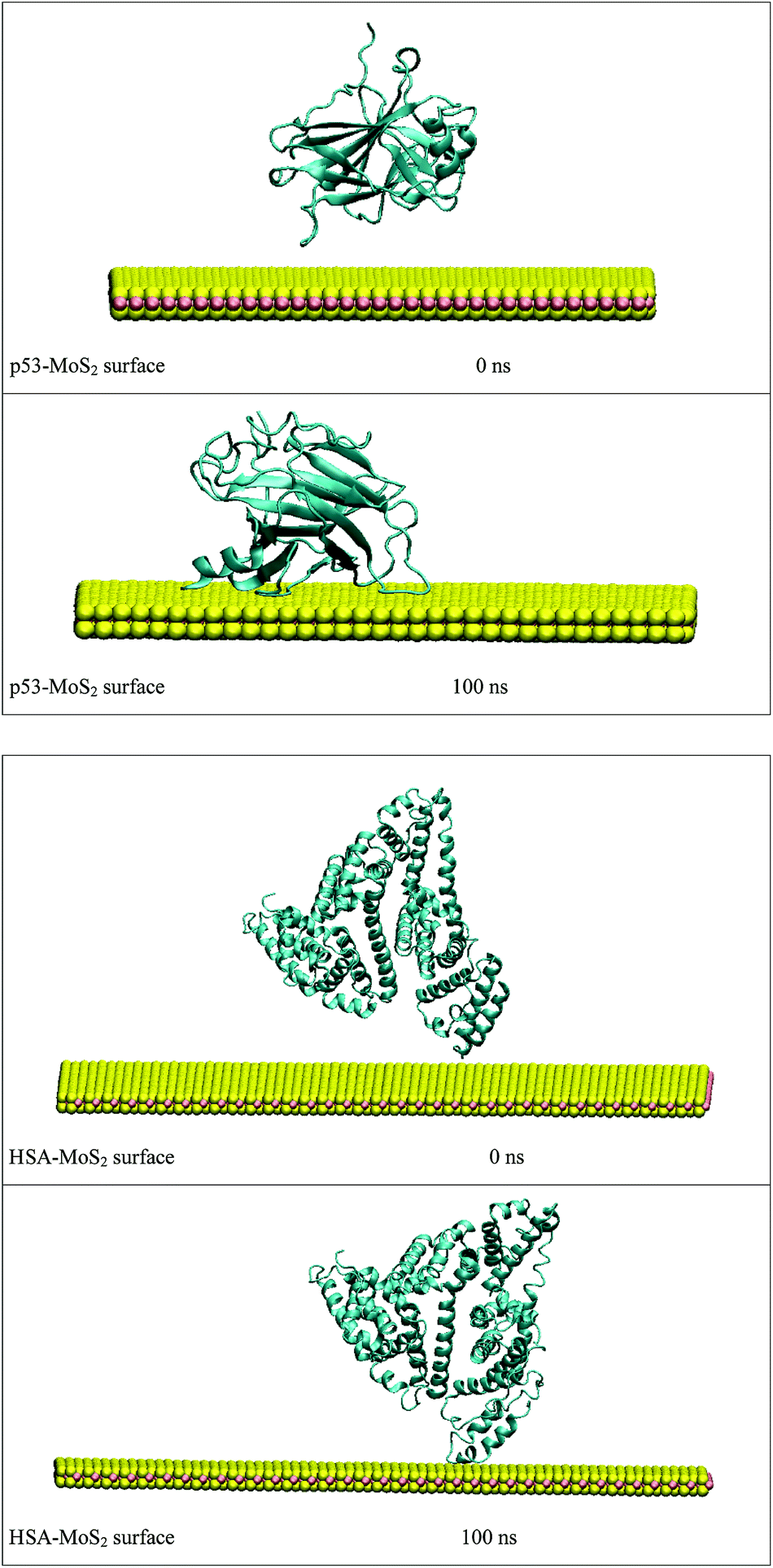
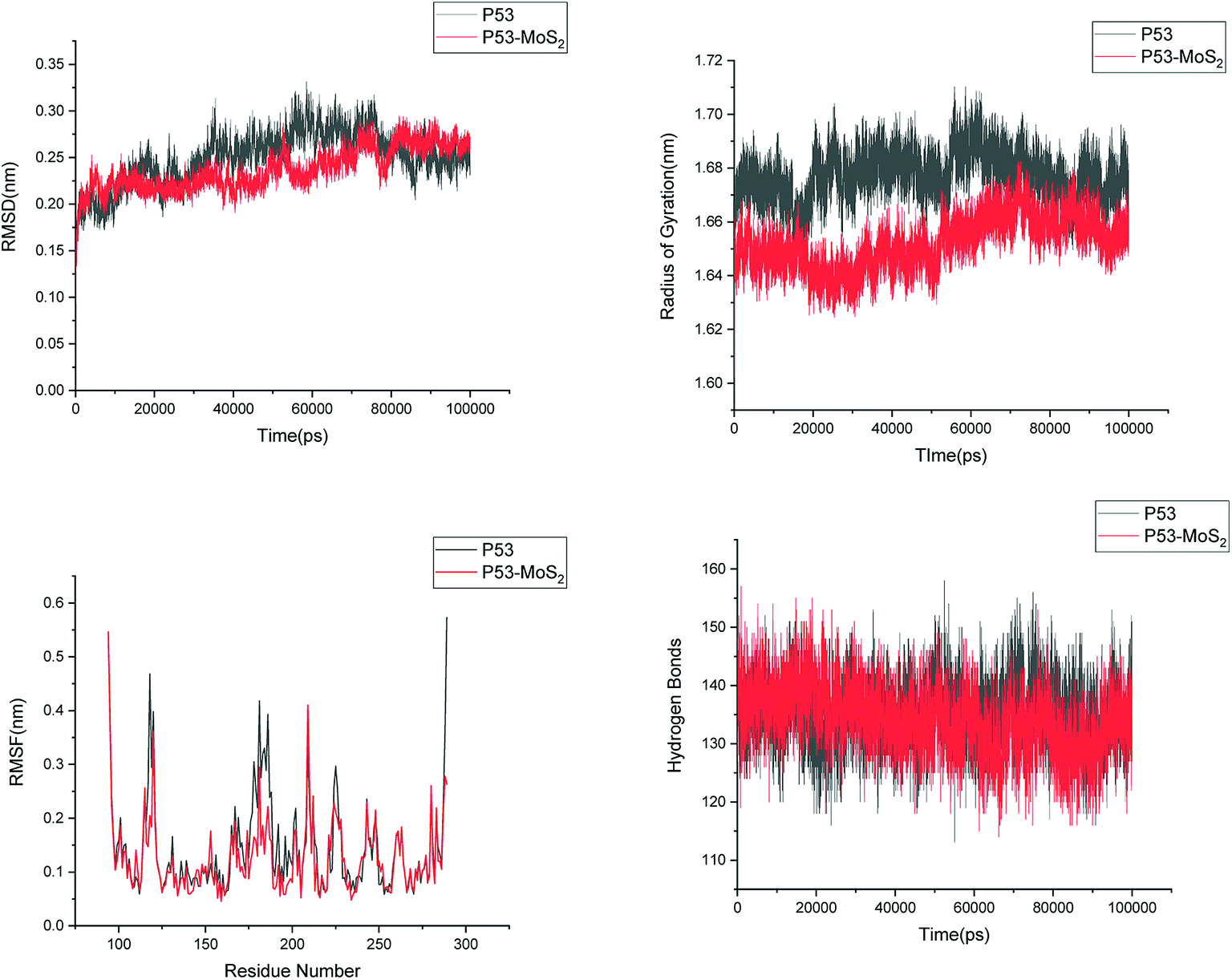
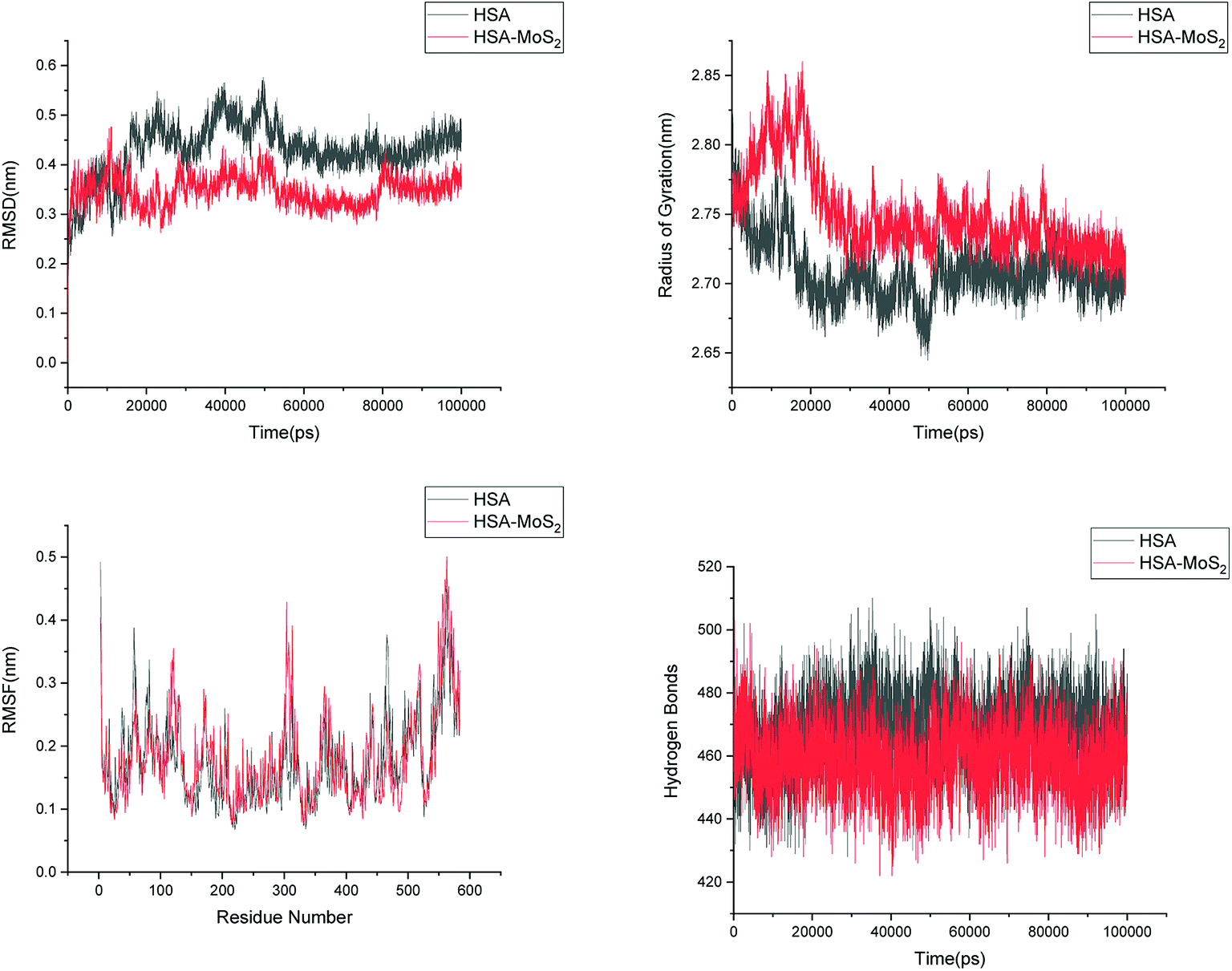
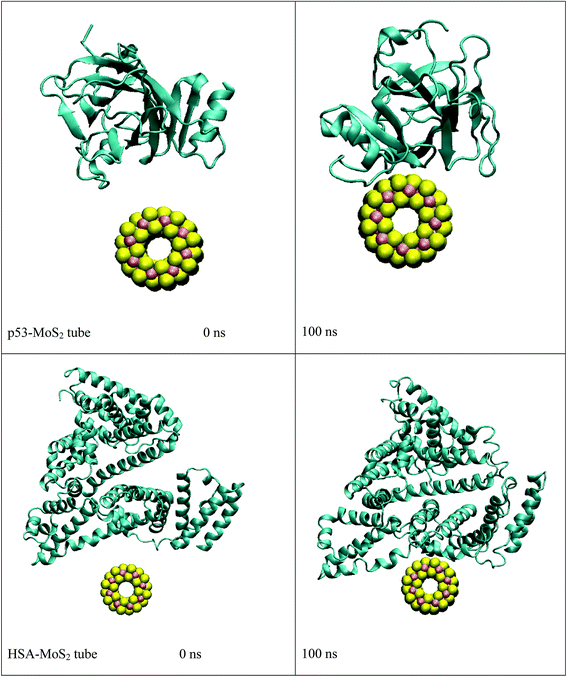
In order to address questions about the toxicity of MoS2 computationally, we studied its off-target interaction with certain vital proteins. In particular, proteins such as HSA (human serum albumin, pdb id: 3B9M) and P53 (pdb id: 1TUB) have been studied for their interaction with MoS2 materials. Several studies have proven that planar surfaces have more impact on the secondary structure of proteins rather than curved ones.69,70 However, we considered both the cases where HSA and P53 proteins interact with MoS2 tube and surface. The dimensions of the MoS2 tube and surface, similar to the fibril case, were used for p53 as it is relatively smaller in size. However, a larger MoS2 sheet was considered for HSA. These proteins were placed above the surface of the MoS2 materials and simulated using similar protocols as in the case of fibril. Two proteins adsorbed onto the surface of MoS2 tube and sheet and form stable complexes. The initial and final structures of the simulated systems in the case of HSA and P53 are given in Fig. 9 and 12. It can be seen clearly that larger part of HSA was exposed to the surface of MoS2 in the initial configuration, whereas the small part of HSA only has significant interaction with MoS2 after 100 ns of simulation. p53 has a larger number of contacts than HSA with MoS2 surface. The structure of a protein is important for performing its activity. Proteins are composed of different secondary structural elements which are responsible for its function. The interaction of the MoS2 materials may change the secondary structure of HSA and P53 and would lead to toxicity. Therefore, the secondary structural details were calculated upon interaction with MoS2 materials and compared with control simulations in water. The calculated structural details are shown in Table 1. The secondary structural elements are retained after interaction with the tube and surface for HSA and p53 in the simulated time. It can be noted that there are marginal changes in the average helical content of p53 on interaction with MoS2 surface. For p53, the total helical content is 8% in aqueous solution. The interaction of MoS2 reduces the helical content from 8% to 6% in the case of p53. However, the decrease in helical content is not significant. The other secondary structures are not affected after interaction with MoS2. The beta sheet content of these proteins does not change due to the interaction with the MoS2 materials. It can be clearly established that HSA and p53 retain their secondary structures on interaction with the MoS2 tube and surface. Further, we have calculated root mean square deviation (RMSD), radius of gyration (Rg), root mean square fluctuation (RMSF) and hydrogen bonds for p53 and HSA after interaction with MoS2 based materials and compared with results from the control simulations (correspond to HSA and p53 in aqueous condition) and the data are presented in Fig. 10, 11, 13 and 14. It can be seen that there are no substantial changes (i.e. in terms of the magnitude there is no significant difference) in RMSD, Rg, RMSF and the hydrogen bonds of these proteins due to MoS2 interaction (except that the conformational flexibility is lowered to some extent as can be seen from RMSF). These results are consistent with the analysis of the secondary structure where we again did not observe any remarkable changes due to the MoS2 tube and surface binding. We have also investigated whether HSA binding to MoS2 leads to any changes in its drug binding ability. For this, we did two independent molecular docking studies using the HSA structure taken from solvent simulation and from the MoS2 surface simulation. In particular, we calculated the binding affinity of three drug molecules and the results are presented in Table 2. As can be seen, the drug binding affinity is not altered for HSA due to the interaction with MoS2 surface. Overall, the analysis of the secondary structures of HSA and p53 shows that these proteins will not aggregate (as there are less significant changes in the beta sheet content) after interaction with the MoS2 surface and the aggregate formation is one of the reasons for the malfunctioning of these proteins.
 |
| | Fig. 9 Initial and final structures of p53 and HSA after interaction with MoS2 tube. | |
Table 1 Secondary structural elements of HSA and p53 on interaction with MoS2 surface
| System |
Coil |
β-Sheet |
β-Bridge |
Bend |
Turn |
α-Helix |
310-Helix |
| p53 |
0.27 |
0.34 |
0.01 |
0.13 |
0.17 |
0.05 |
0.03 |
| p53-MoS2 tube |
0.27 |
0.34 |
0.01 |
0.15 |
0.16 |
0.05 |
0.02 |
| p53-MoS2 surface |
0.29 |
0.33 |
0.01 |
0.13 |
0.18 |
0.04 |
0.02 |
| HSA |
0.13 |
— |
— |
0.08 |
0.15 |
0.59 |
0.05 |
| HSA-MoS2 tube |
0.13 |
— |
— |
0.08 |
0.14 |
0.62 |
0.03 |
| HSA-MoS2 surface |
0.13 |
— |
— |
0.08 |
0.15 |
0.61 |
0.03 |
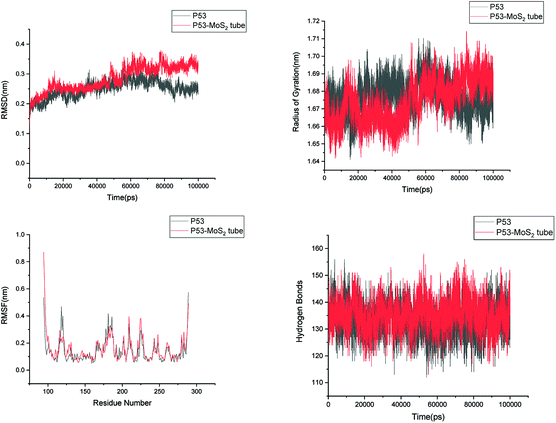 |
| | Fig. 10 Root mean square deviation (RMSD), radius of gyration (Rg), root mean square fluctuation (RMSF) and hydrogen bonds of p53 in aqueous solution and on interaction with MoS2 tube. | |
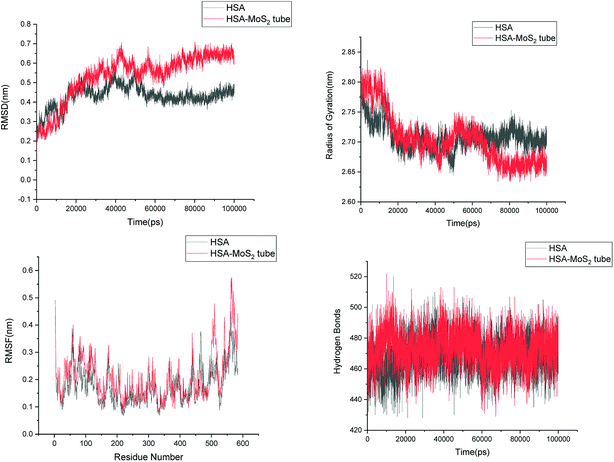 |
| | Fig. 11 Root mean square deviation (RMSD), radius of gyration (Rg), root mean square fluctuation (RMSF) and hydrogen bonds of HSA in aqueous solution and on interaction with MoS2 tube. | |
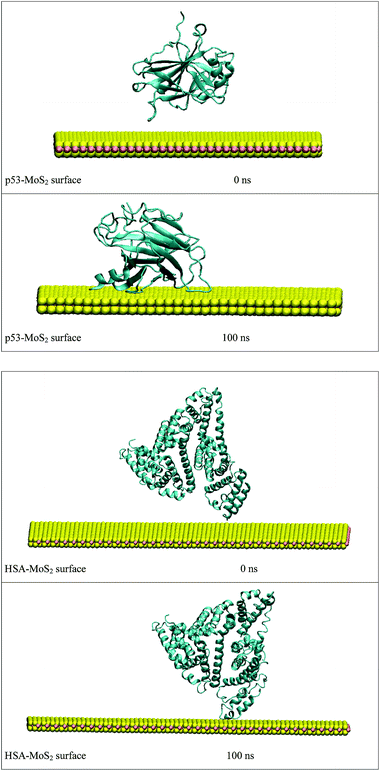 |
| | Fig. 12 Initial and final structures of P53 and HSA after interaction with MoS2 surface. | |
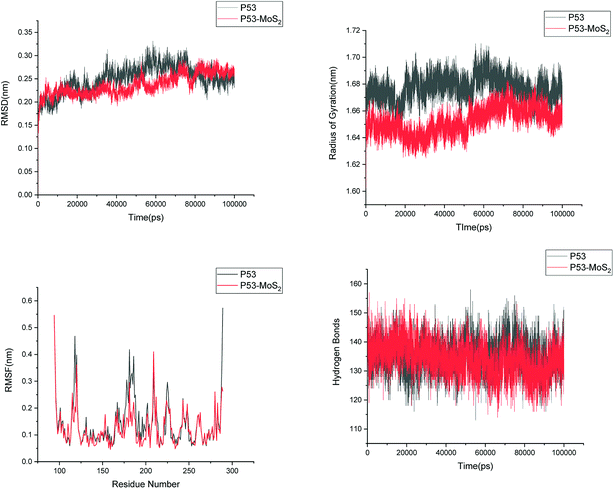 |
| | Fig. 13 Root mean square deviation (RMSD), radius of gyration (Rg), root mean square fluctuation (RMSF) and hydrogen bonds of p53 in aqueous solution and on interaction with MoS2 surface. | |
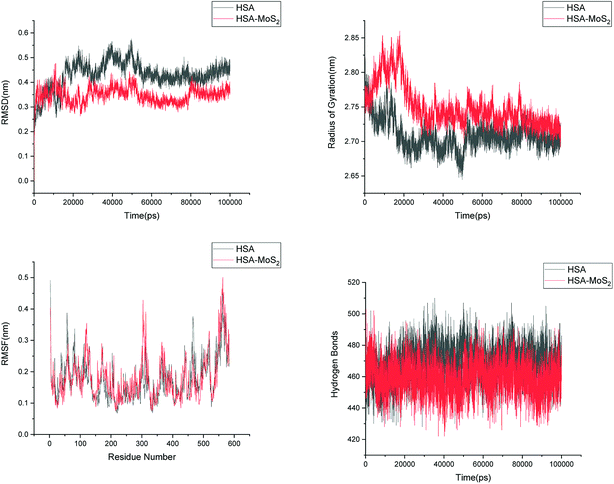 |
| | Fig. 14 Root mean square deviation (RMSD), radius of gyration (Rg), root mean square fluctuation (RMSF) and hydrogen bonds of HSA in aqueous solution and on interaction with MoS2 surface. | |
Table 2 The binding affinity of three drug molecules namely warfarin, ibuprofen, diclofenac with HSA structure taken from solvent simulation and from MoS2 surface simulation. The binding free energies and inhibition constants were computed from molecular docking using autodock4 software. The binding free energies are given in kcal mol−1
| Drug |
Warfarin |
Ibuprofen |
Diclofenac |
| HSA in water |
−8.42 (671.9 nM) |
−7.02 (7.10 μM) |
−8.23 (920.7 nM) |
| HSA on MoS2 surface |
−9.63(87.40 nM) |
−7.01 (7.25 μM) |
−9.19 (182.9 nM) |
4. Conclusions
The interaction between amyloid fibril and MoS2-based materials has been investigated in this study using classical molecular dynamics simulations. It is found that MoS2 forms stable complexes with the fibrils which tend to wrap around the surface of the nanotubes. Fibril interacts with the MoS2 surface through its edges rather than with the upper or lower surface. The structural properties such as the secondary structure, hydrogen bonding, hydrophobic contacts and salt bridges, which are important for the stabilization of the fibrils were analysed upon interaction with MoS2-based materials. MoS2 based materials induce disruptions in secondary structural elements and change the beta sheet conformation to a turned form. The hydrophobic and hydrophilic interactions in the fibrils are also significantly decreased on interaction with the nanotube and the nanosurface. The destabilization of the fibril is more pronounced in the case of the nanotubes when compared to the nanosurfaces due to the difference in binding modes. However, both the nanotube and the nanosurface induces destabilization in the fibril structure.
Therefore, the overall results clearly indicate the destabilization of the amyloids fibril upon interaction with MoS2 nanotube, and that the MoS2 surface could be useful in inhibiting further growth of smaller protofibrils into matured fibrils as well as to destabilize the fibrils. We have also studied the off-target interaction of MoS2 materials with certain vital proteins such as human serum albumin and p53 protein and the secondary structural analysis do not show any significant changes in beta sheet contents of these proteins, ruling out any MoS2 material induced coagulation/aggregation to make these proteins non-functional. These results could be useful as guidance at a molecular level to design therapeutics for Alzheimer's disease, which would, however, would require a considerable efforts for in vivo testing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge support from the Swedish Foundation for Strategic Research (SSF) through the project “New imaging biomarkers in early diagnosis and treatment of Alzheimer's disease” and the support from SLL through the project “Biomolecular pro-filing for early diagnosis of Alzheimer's disease”. This work was supported by the grants from the Swedish Infrastructure Committee (SNIC) for the projects “Multiphysics Modeling of Molecular Materials” (SNIC2017-12-49) and “In silico Diagnostic Probes Design” (SNIC2018-3-3). Authors NAM, VS and HÅ acknowledge VR-DST for funding the Indo-Swedish joint research program entitled “Use of Nanoparticles as Protein Aggregation Modulator for Healthcare and Pharmaceutical Applications” (Project ID: 2015-06726) and for supporting the exchange visit to India. One of the authors (SKM) wishes to thank DST-VR (DST/NTSWD/VR/P-03/2016) for the support to the Indian partners.
References
- D. J. Selkoe, Nature, 1999, 399, A23–A31 CrossRef CAS PubMed.
- D. J. Selkoe, Nature, 2003, 426, 900–904 CrossRef CAS PubMed.
- N. R. Jana and N. Nukina, J. Chem. Neuroanat., 2003, 26, 95–101 CrossRef CAS PubMed.
- A. L. Fink, Acc. Chem. Res., 2006, 39, 628–634 CrossRef CAS PubMed.
- T. Ban, K. Yamaguchi and Y. Goto, Acc. Chem. Res., 2006, 39, 663–670 CrossRef CAS PubMed.
- D. Eisenberg, R. Nelson, M. R. Sawaya, M. Balbirnie, S. Sambashivan, M. I. Ivanova, A. Ø. Madsen and C. Rieke, Acc. Chem. Res., 2006, 39, 568–575 CrossRef CAS PubMed.
- K. A. Jellinger, J. Neural Transm., 2006, 113, 1603–1623 CrossRef CAS PubMed.
- R. N. Rambaran and L. C. Serpell, Prion, 2008, 2, 112–117 CrossRef PubMed.
- Alzheimer's Association, Alzheimer's Dementia, 2016, 12, 459–509 CrossRef.
- R. E. Tanzi and L. Bertram, Cell, 2005, 120, 545–555 CrossRef CAS PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- R. Sultana and D. A. Butterfield, Mol. BioSyst., 2008, 4, 36–41 RSC.
- J. Kang, H. G. Lemaire and A. Unterbeck, et al., Nature, 1987, 325, 733–736 CrossRef CAS PubMed.
- D. J. Selkoe, Nature, 1999, 399, A23–A31 CrossRef CAS.
- T. Luhrs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Dobeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef CAS PubMed.
- A. I. Bush, Trends Neurosci., 2003, 26, 207 CrossRef CAS PubMed.
- K. Rajasekhar and T. Govindaraju, RSC Adv., 2018, 8, 23780–23804 RSC.
- M. Biancalana and S. Koide, Biochim. Biophys. Acta, 2010, 1804, 1405–1412 CrossRef CAS PubMed.
- M. G. Savelieff, A. S. DeToma, J. S. Derrick and M. H. Lim, Acc. Chem. Res., 2014, 47, 2475–2482 CrossRef CAS PubMed.
- M. Tanaka, Y. Machida, S. Niu, T. Ikeda, N. R. Jana, H. Doi, M. Kurosawa, M. Nekooki and M. Nukina, Nat. Med., 2004, 10, 148–154 CrossRef CAS PubMed.
- J. S. Derrick, R. A. Kerr, Y. Nam, S. B. Oh, H. J. Lee, K. G. Earnest, N. Suh, K. L. Peck, M. Ozbil, K. J. Korshavn, A. Ramamoorthy, R. Prabhakar, E. J. Merino, J. Shearer, J.-Y. Lee, B. T. Ruotolo and M. H. Lim, J. Am. Chem. Soc., 2015, 137, 14785–14797 CrossRef CAS PubMed.
- J. Bieschke, J. Russ, R. P. Friedrich, D. E. Ehrnhoefer, H. Wobst, K. Neugebauer and E. E. Wanker, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7710–7715 CrossRef CAS PubMed.
- F. L. Palhano, J. Lee, N. P. Grimster and J. W. Kelly, J. Am. Chem. Soc., 2013, 135, 7503–7510 CrossRef CAS PubMed.
- L. Fonseca-Ornelas, S. E. Eisbach, M. Paulat, K. Giller, C. O. Fernández, T. F. Outeiro, S. Becker and M. Zweckstetter, Nat. Commun., 2014, 5, 5857 CrossRef CAS PubMed.
- L. M. Young, P. Cao, D. P. Raleigh, A. E. Ashcroft and S. E. Radford, J. Am. Chem. Soc., 2014, 136, 660–670 CrossRef CAS PubMed.
- A. J. Doig and P. Derreumaux, Curr. Opin. Struct. Biol., 2015, 30, 50–56 CrossRef CAS PubMed.
- A. J. Doig, M. P. Del Castillo-Frias, O. Berthoumieu, B. Tarus, J. Nasica-Labouze, F. Sterpone, P. H. Nguyen, N. M. Hooper, P. Faller and P. Derreumaux, ACS Chem. Neurosci., 2017, 8, 1435–1437 CrossRef CAS PubMed.
- K. Debnath, S. Shekhar, V. Kumar, N. R. Jana and N. R. Jana, ACS Appl. Mater. Interfaces, 2016, 8, 20309–20318 CrossRef CAS PubMed.
- S. I. Yoo, M. Yang, J. R. Brender, V. Subramanian, K. Sun, N. E. Joo, S.-H. Jeong, A. Ramamoorthy and N. A. Kotov, Angew. Chem., Int. Ed., 2011, 50, 5110–5115 CrossRef CAS PubMed.
- M. Richman, S. Wilk, N. Skirtenko, A. Perelman and S. Rahimipour, Chem.–Eur. J., 2011, 17, 11171–11177 CrossRef CAS PubMed.
- S. Palmal, A. R. Maity, B. K. Singh, S. Basu, N. R. Jana and N. R. Jana, Chem.–Eur. J., 2014, 20, 6184–6191 CrossRef CAS PubMed.
- S. Palmal, N. R. Jana and N. R. Jana, J. Phys. Chem. C, 2014, 118, 21630–21638 CrossRef CAS.
- S. Li, L. Wang, C. C. Chusuei, V. M. Suarez, P. L. Blackwelder, M. Micic, J. Orbulescu and R. M. Leblanc, Chem. Mater., 2015, 27, 1764–1771 CrossRef CAS.
- J. Zhang, X. Zhou, Q. Yu, L. Yang, D. Sun, Y. Zhou and J. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 8475–8487 CrossRef CAS PubMed.
- M. J. Kogan, N. G. Bastus, R. Amigo, D. Grillo-Bosch, E. Araya, A. Turiel, A. Labarta, E. Giralt and V. F. Puntes, Nano Lett., 2006, 6, 110–115 CrossRef CAS PubMed.
- Q. Chen, L. Yang, C. Zheng, W. Zheng, J. Zhang, Y. Zhou and J. Liu, Nanoscale, 2014, 6, 6886–6897 RSC.
- Z. Wang, C. Zhu, S. V. Bortolini, A. Hoffmann, H. X. Amari, Z. L. Liu and M. D. Dong, Nanotechnology, 2016, 27, 304001 CrossRef PubMed.
- S. Bag, R. Mitra, S. Dasgupta and S. J. Dasgupta, J. Phys. Chem. B, 2017, 121, 5474–5482 CrossRef CAS PubMed.
- J. Luo, S. K. Wärmländer, C. H. Yu, K. Muhammad, A. Gräslund and J. Pieter Abrahams, Nanoscale, 2014, 6, 6720–6726 RSC.
- Y. Sun, Z. Qian and G. Wei, Phys. Chem. Chem. Phys., 2016, 18, 12582–12591 RSC.
- A. Sebastián Andujar, L. Francesca, H. Siegfried, R. D. Enriz and F. Zerbetto, Phys. Chem. Chem. Phys., 2012, 14, 8599–8607 RSC.
- P. D. Q. Huy and M. Suan Li, Phys. Chem. Chem. Phys., 2014, 16, 20030–20040 RSC.
- H. Li, Y. Luo, P. Derreumaux and G. Wei, Biophys. J., 2011, 101, 2267–2276 CrossRef CAS PubMed.
- Z. Fu, Y. Luo, P. Derreumaux and G. Wei, Biophys. J., 2009, 97, 1795–1803 CrossRef CAS PubMed.
- Z. Yang, C. Ge, J. Liu, Y. Chong, Z. Gu, C. A. Jimenez-Cruz, Z. Chaia and R. Zhou, Nanoscale, 2015, 7, 18725–18737 RSC.
- N. Kapil, A. Singh, M. Singh and D. Das, Angew. Chem., Int. Ed., 2016, 128, 7772–7776 CrossRef PubMed.
- S. G. Wang, Y. Chen, X. Li, W. Gao, L. L. Zhang, J. Liu, Y. Y. Zheng, H. R. Chen and J. L. Shi, Adv. Mater., 2015, 27, 7117–7122 CrossRef CAS PubMed.
- S. G. Wang, X. Li, Y. Chen, X. J. Cai, H. L. Yao, W. Gao, Y. Y. Zheng, X. An, J. L. Shi and H. R. Chen, Adv. Mater., 2015, 27, 2775–2782 CrossRef CAS PubMed.
- M. Li, A. Zhao, K. Dong, W. Li, J. Ren and X. Qu, Nano Res., 2015, 8, 3216–3227 CrossRef CAS.
- W. Zhongying and Mi. Baoxia, Environ. Sci. Technol., 2017, 51, 8229–8244 CrossRef PubMed.
- Q. Han, S. Cai, L. Yang, X. Wang, C. Qi, R. Yang and C. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 21116–21123 CrossRef CAS PubMed.
- J. Wang, L. Liu, D. H. Ged, H. X. Zhang, Y. H. Feng, Y. B. Zhang, M. Chen and M. D. Dong, Chemistry, 2018, 24, 3397–3402 CrossRef CAS PubMed.
- Z. Gu, Z. Yang, S. G. Kang, J. R. Yang, J. Luo and R. Zhou, Sci. Rep., 2016, 6, 28252 CrossRef CAS PubMed.
- Z. Gu, P. De Luna, Z. Yang and R. Zhou, Phys. Chem. Chem. Phys., 2017, 19, 3039–3045 RSC.
- T. Luhrs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Dobeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef CAS PubMed.
- J. A. Lemkul and D. R. Bevan, J. Phys. Chem. B, 2010, 114, 1652–1660 CrossRef CAS PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E Shaw, Proteins, 2010, 78, 1950–1958 CAS.
- S. Nose and M. L. Klein, Mol. Phys., 1983, 50, 1055–1076 CrossRef CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 14101–14107 CrossRef PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Bendersen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1473 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model., 2001, 7, 306–317 CrossRef CAS.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- S. K. Mudedla, K. Balamurugan and V. Subramanian, J. Phys. Chem. C, 2016, 120, 28246–28260 CrossRef CAS.
- K. Balamurugan, E. R. A. Singam and V. Subramanian, J. Phys. Chem. C, 2011, 115, 8886–8892 CrossRef CAS.
- N. Wu, Y. Yu, T. Li, X. Ji, L. Jiang, J. Zong and H. Huang, PLoS One, 2016, 11, e0167245 CrossRef PubMed.
- A. Carpentier and M. Canney, et al., Sci. Transl. Med., 2016, 8, 343re2 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The contact areas are shown in Fig. S1. See DOI: 10.1039/c8ra10184a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Natarajan Arul Murugan
a,
Natarajan Arul Murugan