
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of carbon quantum dots/single crystal TiO2 nanosheets with exposed {001} and {101} facets and their visible light driven catalytic activity
Hongqin Huang,
Hao Ouyang,
Tiehu Han,
Huigang Wang * and
Xuming Zheng
* and
Xuming Zheng
Department of Chemistry, Key Laboratory of Advanced Textiles Materials, Manufacture Technology of the Ministry of Education, Engineering Research Center for Eco-Dyeing, Finishing of Textiles of the Ministry of Education, Zhejiang Sci-Tech University, Davis, Hangzhou 310018, P. R. China. E-mail: zdwhg@163.com; hugwang@ucdavis.edu; Tel: +86-571-8684-3627 Tel: +86-571-8665-2179
First published on 25th January 2019
Abstract
Carbon quantum dots were successfully doped into anatase TiO2 single crystal nanosheets (TNS) with exposed {001} and {101} reactive facets by a facile solvothermal process. SEM and TEM confirmed the as-prepared TiO2 nanosheet structure and that the dominant exposed face is the {001} facet, and the loaded N-CDs are nearly spherical with an average size of about 3 nm. XPS results confirmed that the deposited N-CDs were chemically integrated into the TiO2 nanosheets. UV-vis DRS spectroscopy shows that with the dotting of N-CDs, the absorption edge of N-CDs/TNS has been extended into the visible light region. The ability of the N-CDs/TNS to degrade Rhodamine B (RhB) in aqueous solution under visible light irradiation (λ ≥ 400 nm) was investigated. The results show that the photocatalytic performance of N-CDs/TNS was substantially improved relative to pure TNS. The photodegradation efficiency reached its maximum value with 6 mL of N-CDs/TNS, showing a 9.3-fold improvement in photocatalytic activity over TNS. Fluorescence spectroscopy (PL) and electron paramagnetic resonance (EPR) studies were conducted to characterize the active species during the degradation period, based on which the possible photodegradation mechanism of N-CDs/TNS by visible light irradiation was given.
1 Introduction
In 1972 Fujishima and Honda first reported that titanium dioxide (TiO2) can split water into hydrogen and oxygen by a photo-assisted electrochemical reaction.1 This work triggered enormous interest by scientists into semiconductor photocatalysis investigation. Photocatalytic degradation of pollutants using TiO2 was first reported in 1977 by Frank and Bard,2 since when a dramatic improvement has taken place in this area because of the urgent need for water and air purification.The photocatalysis process generally begins with the absorption of a photon with energy greater than the band gap of the semiconductor, which interacts with an electron in the valence band (VB), and then excites the electron to the conduction band (CB).3–5 Only a photon with energy greater than the band gap of the semiconductor can excite the electron to the CB; in the case of anatase TiO2, the band gap is 3.2 eV, therefore only light with a wavelength shorter than 387 nm satisfies the condition.6,7 However, there are some strategies to make anatase TiO2 active in the visible light region. Hetero-element structures such as doping,8–11 p–n heterojunctions,12,13 metal/semiconductor Schottky junctions14,15 and dye sensitization15–17 are proven to be effective ways to extend the absorption range into the visible light region. For example, N-doped TiO2 creates a hybrid orbital above the VB of TiO2 by imbedding an N atom into the lattice of TiO2 (ref. 10) or C-doped TiO2 can create a hybrid orbital above the VB of TiO2 by replacing a lattice Ti atom or lattice O atom by C atoms.11,18,19 They all successfully expand the catalyst's photoresponse to the visible light region with an enhanced visible light photocatalytic performance.20
Nowadays homo-structures, including surface hetero facet junctions21–23 and Ti3+ self-doped black TiO2−x strategies, arouse great attention in the solar cell and pollution treatment fields.24 Anatase TiO2 single crystal nanosheets (TNS) with highly exposed {001} and {101} facets boast enhanced photocatalytic activity as a result of forming a surface heterojunction that induces photogenerated e− and h+ to move to {101} and {001} facets, respectively.25–27 Tachikawa et al.28 also reported a study on the electron transfer between {101} and {001} facets on the TiO2 surface. Moreover, TNS provide perfect single crystal facets for an investigation of the mechanism of facet-dependent catalytic activity.
Carbon quantum dots are nanoparticles with intriguing fast charge mobility and large surface area properties.29 The effective doping of TiO2 with carbon quantum dots is expected to be beneficial to increasing the electron transfer rate and the adsorption amount of dye molecules to be degraded. Therefore, it is believed that TNS nanosheets combined with carbon quantum dots and N atoms to form N-CDs/TNS nanosheets could be an ideal designed photocatalyst with superior catalytic performance.30,31
Herein, in this work, novel anatase N-CDs/TNS nanosheets with exposed {101} and {001} facets are prepared for the first time using HF as a morphology control agent through a solvothermal method. The morphology, chemical structure, and the influence of N-CDs content in the composites on catalytic performance were systematically investigated. The prepared N-CDs/TNS nanosheets with exposed {001} facets and a narrow band gap exhibit a much higher photocatalytic activity relative to pure TNS under visible-light irradiation. Furthermore, the photocatalytic efficiency exhibits good stability after five cycles. A possible photocatalytic mechanism of the as-prepared catalyst has been proposed.
2 Experimental section
2.1 Preparation of N-CDs
0.768 g of citric acid and 0.72 g of urea were dissolved into 20 mL of water, and stirred to form a clear solution. Then the solution was transferred into a 100 mL Teflon lined stainless autoclave. The sealed autoclave was heated to 200 °C in an electric oven and kept for an additional 8 hours. It was cooled to obtain a suspension, followed by adjustment of the pH to 7 with acetic acid. The suspension was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min to remove large carbon residues, yielding a dark yellowish-brown solution of N-CDs. Further purification of the N-CDs was conducted through a dialysis tube (kept molecular weight: 500–3500 Da) for 4 days in the dark (changing the deionized water every 8 h). The liquid was collected and concentrated to 20 mL by rotary evaporation.
000 rpm for 5 min to remove large carbon residues, yielding a dark yellowish-brown solution of N-CDs. Further purification of the N-CDs was conducted through a dialysis tube (kept molecular weight: 500–3500 Da) for 4 days in the dark (changing the deionized water every 8 h). The liquid was collected and concentrated to 20 mL by rotary evaporation.
2.2 Preparation of TNS
TiO2 nanosheets with dominant {001} crystal planes were prepared by a facile one-pot solvothermal reaction of TBT-HAc solution: a precursor solution containing 1 mL of tetrabutyl titanate and 3 mL of ethylene glycol was added dropwise to 50 mL of HAc with continuous stirring. 60 min later 0.18 mL of HF was added and stirred for 5 min more. The obtained white suspension was transferred to a dried Teflon-lined autoclave at 200 °C for 24 h. After being cooled to room temperature, the product was collected by high-speed centrifugation, washed with ethanol and water several times, dried at 60 °C overnight, and finally calcined at 600 °C for 90 min to remove the residual organics and F−.2.3 Preparations of N-CDs/TNS photocatalysts
N-CDs/TNS were prepared by a hydrothermal deposition method. In the standard preparation conditions, an appropriate volume of N-CDs and 0.2 g of TNS were dispersed in 40 mL of distilled water, and stirred for 1 h. The suspension was put into a dried Teflon-lined autoclave at 200 °C for 6 h. After being cooled to room temperature, the product was collected by high-speed centrifugation. According to the added volume of N-CDs, we named the samples (2 mL) N-CDs/TN, (4 mL) N-CDs/TNS, (6 mL) N-CDs/TNS, (8 mL) N-CDs/TNS and (10 mL) N-CDs/TNS.2.4 Photocatalysis experiments
The photocatalytic activities of the materials were examined by degrading Rhodamine B (RhB) in aqueous solution under visible light irradiation using a 300 W xenon lamp (CEL-HXUV300, Beijing CEAULIGHT) with a 400 nm cutoff filter as the light source. In each experiment, a total of 50 mg of catalyst was added to a 100 mL solution of 10 mg L−1 RhB in a 120 mL quartz reactor with a circulating water system to maintain a constant temperature, and the distance between the light and the surface of the solution was set at about 10 cm. Before irradiation, the suspension was stirred in the dark for 30 min to ensure the adsorption–desorption equilibrium of the solution. After the start of the photocatalytic reaction, approximately 3 mL of the mixture was taken at regular time intervals, and then centrifuged to separate the photocatalyst particles. The concentration was analyzed by measuring the maximum absorbance at 554 nm for RhB using a UV-vis spectrometer (Varian Cary 50).2.5 Characterization
Crystalline phases of the prepared samples were characterized by X-ray diffraction (XRD) on a DX-2700 diffractometer (Dandong Hanyuan Instrument Co. Ltd, China) using Cu Kα radiation (λ = 0.15418 nm). Transmission electron microscope (TEM) images were taken on a JEOL-2100 electron microscope operating at an accelerating voltage of 200 kV. The morphology of the materials was analyzed by a Hitachi S-4800 field emission scanning electron microscope (FE-SEM). UV-vis diffuse-reflectance spectroscopy (DRS) was obtained from a Shimadzu UV-2450 spectrophotometer using BaSO4 as the reference. X-ray photoelectron spectroscopy (XPS) was performed on a Kratos AXIS Ultra DLD instrument with an Al Kα monochromatic source. All binding energies were referenced to the C 1s peak (284.6 eV) arising from adventitious hydrocarbons.3 Results and discussion
The preparation procedure for the catalysts N-CDs/TNS is illustrated in Scheme 1. N-doped CDs were carbonized with a solvothermal process and purified through centrifugation and dialysis. TNS nanosheets were prepared from tetrabutyl titanate using a solvothermal method, and HF was used as a crystal facet control agent. In this work, the mixture of as-prepared N-CDs and TNS goes through a hydrothermal deposition process, and Ti–OH can be easily dehydration condensed with the abundant oxygen functional groups on the N-CDs surfaces, which is more favourable to the formation of the {001} surfaces. That is, N-CDs as a stabilizer plays an important role in helping to maintain a large single crystal of TNS with exposed {001} facets.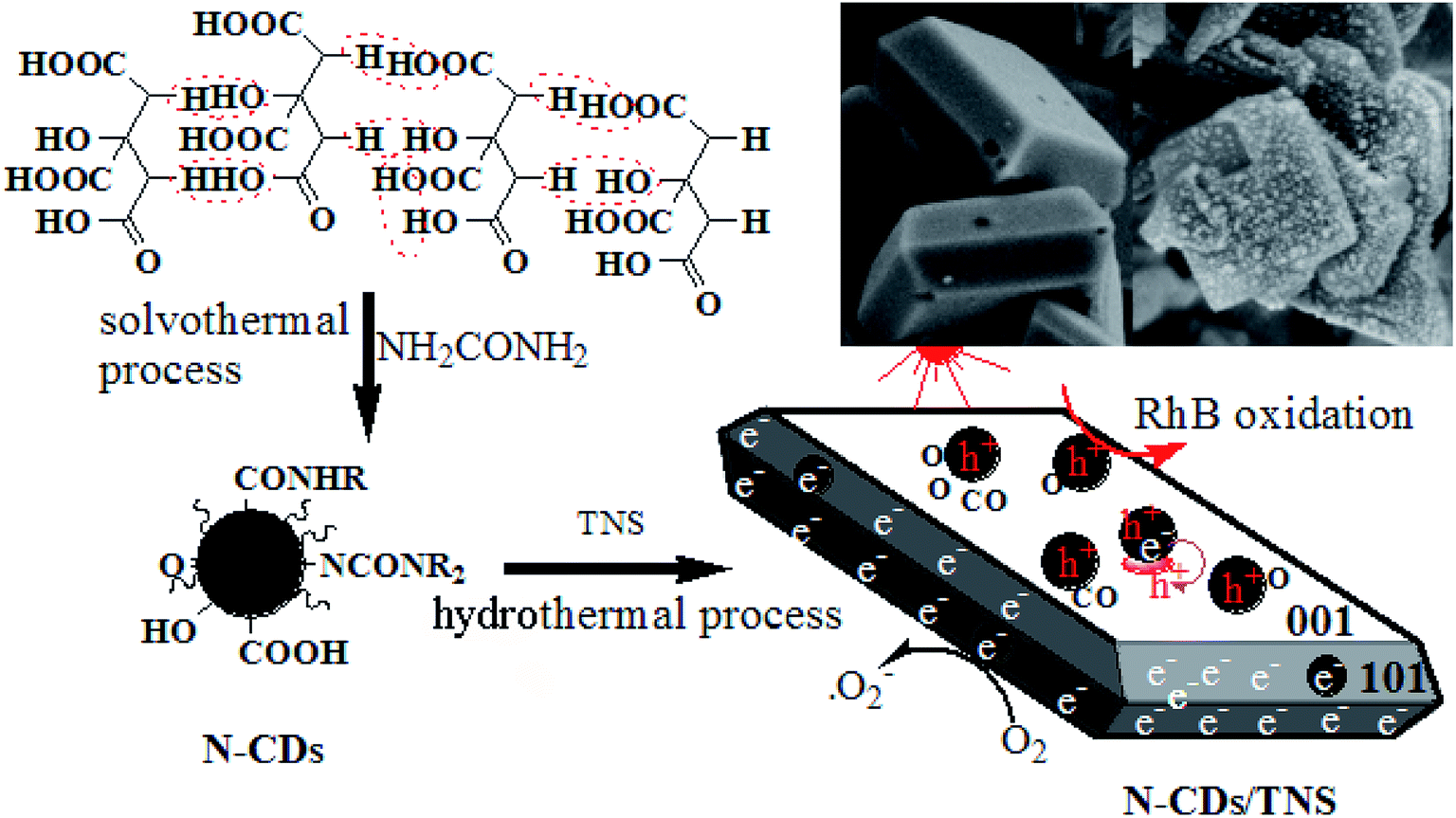 | ||
| Scheme 1 Synthetic route to the photocatalyst N-CDs/TNS from citric acid and tetrabutyl titanate through hydrothermal deposition. | ||
X-ray diffraction (XRD) measurements were used to identify the crystallographic phase structures of the synthesized products. XRD patterns of as-prepared N-CDs, pure TNS, and N-CDs/TNS are shown in Fig. 1. Fig. 1 (bottom) shows a typical broad noisy peak around 21°, assigning a graphitic structure to the spectrum for the N-CDs, corresponding to the (002) planes of graphite. The interlayer spacing is 0.35 nm, which is broader than that of graphite, indicating that N-CDs had a bigger interlayer spacing than that of graphite. This result could be attributed to the nitrogen-containing groups introduced in the dehydrolysis and hydrothermal process, which enhanced the interlayer distance. From TNS and (6 mL) N-CDs/TNS in Fig. 1, it can be seen that no peak ascribed to N-CDs can be observed in the (6 mL) N-CDs/TNS composites; this is due to the fact that the N-CDs are highly dispersed in the TiO2 nanosheet structures and the low loading quantity of N-CDs is beyond the XRD detection limit. Simultaneously, all the diffraction peaks for (6 mL) N-CDs/TNS in Fig. 1 are the same as for the crystal structure of pure TNS, and match well with the crystal structure of anatase-phase TiO2 (space group: I41/amd, JCPDS no. 21-1272), indicating the formation of (6 mL) N-CDs/TNS nanocomposites.
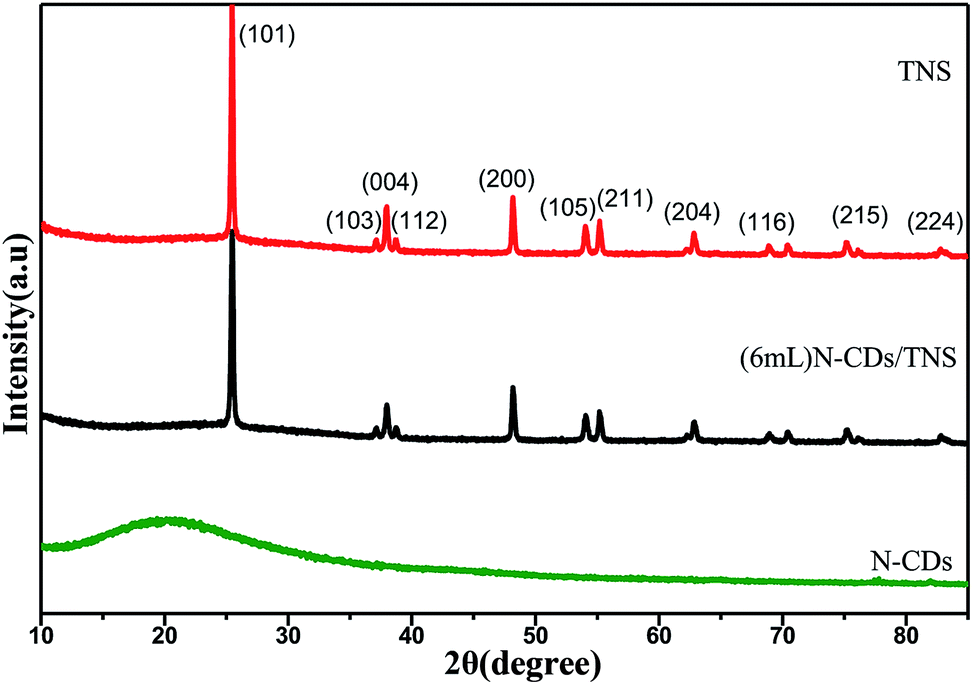 | ||
| Fig. 1 XRD pattern of N-CDs, TNS and N-CDs/TNS. | ||
The TEM image in Fig. 2a shows that the N-CDs nanoparticles are uniform and monodispersed, with an average diameter of about 3 nm. The high resolution TEM (HRTEM) image (inset of Fig. 2a) shows that the N-CDs have crystallinity with lattices of 0.242 nm. These lattices may be sp2 clusters in N-CDs or carbon quantum dots obtained in the solvothermal route.32 Fig. 2b and c show the SEM image of pure single crystal TNS nanosheets; the rectangular TNS nanosheets have a typical dimension (length × width × height) of 150 × 120 × 20 nm. The HRTEM image shows the continuous {001} atomic planes of anatase TiO2 single crystals with a lattice spacing of 0.235 nm (inset of Fig. 2b). The percentage of the {001} facets can be statistically estimated to be 75%, based on SEM and TEM measurements. A previous study demonstrated that the {001} facets and {101} facets can form a surface heterojunction due to the more negative flat-band potential of anatase {001} facets, which is favorable for the separation of e− and h+.33 Therefore, the as-synthesized TNS nanosheets composed of {001} and {101} facets are particularly beneficial to photocatalytic applications. Fig. 2d and e show the TEM and SEM images of the as-prepared catalysis N-CDs/TNS composites; they demonstrate that the N-CDs were uniformly distributed on the exposed surfaces of TNS. The main size distribution of N-CDs, obtained from the statistical result in Fig. 2d, was around 3 nm. The uniform distribution of N-CDs on all exposed surfaces of TNS derived from the crucial advantage of our hydrothermal deposition process: the homogeneous high-pressure deposition of the N-CDs precursor on the spatial locationally confined TNS.
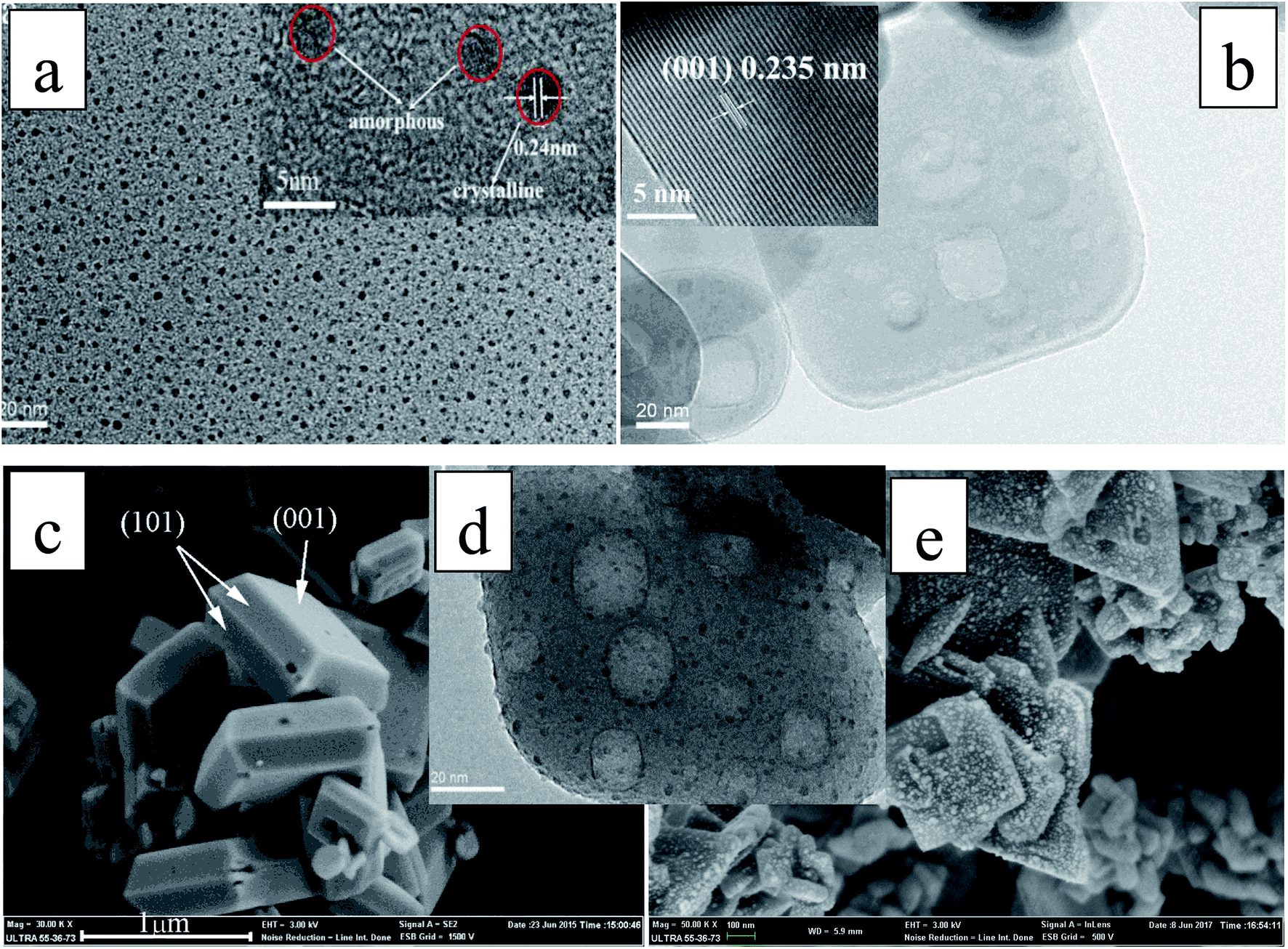 | ||
| Fig. 2 (a) TEM image of N-CDs (inset shows the HRTEM image); (b) TEM image of single crystal TNS nanosheets (inset shows the HRTEM image); (c) SEM image of single crystal TNS nanosheets; (d) TEM image of N-CDs/TNS; (e) SEM image of N-CDs/TNS. | ||
To investigate the surface composition and the chemical states of the elements in as-prepared N-CDs/TNS and the interactions between TNS nanosheets and N-CDs, XPS studies were conducted over N-CDs before and after loading over TNS. As shown in Fig. 3a, the wide-scan survey spectra of N-CDs and N-CDs/TNS all contain O, N and C elements. Compared with N-CDs, the survey spectrum of N-CDs/TNS shows the presence of Ti 2p originating from TiO2. In addition, the oxidation state of the Ti in N-CDs/TNS is shown (in inset of Fig. 3a). Two bands located at 458.94 eV and 464.71 eV can be recognized, and assigned to the distinct Ti 2p3/2 and Ti 2p1/2 signals in the Ti4+ chemical state.34 In contrast, the peak intensities of the O 1s substantially decreased in the XPS survey spectrum (Fig. 3a), confirming the increasing Ti percentage in the N-CDs/TNS composites. It should be taken into consideration that for such small-sized carbon nanodots, less than 3 nm, most of the XPS signal comes from the whole particles, both core and surface, rather than being from the surface alone. The reason is attributed to the fact that the size of the CDs (below 3 nm) is smaller than the depth of the XPS (which is about 15 nm).
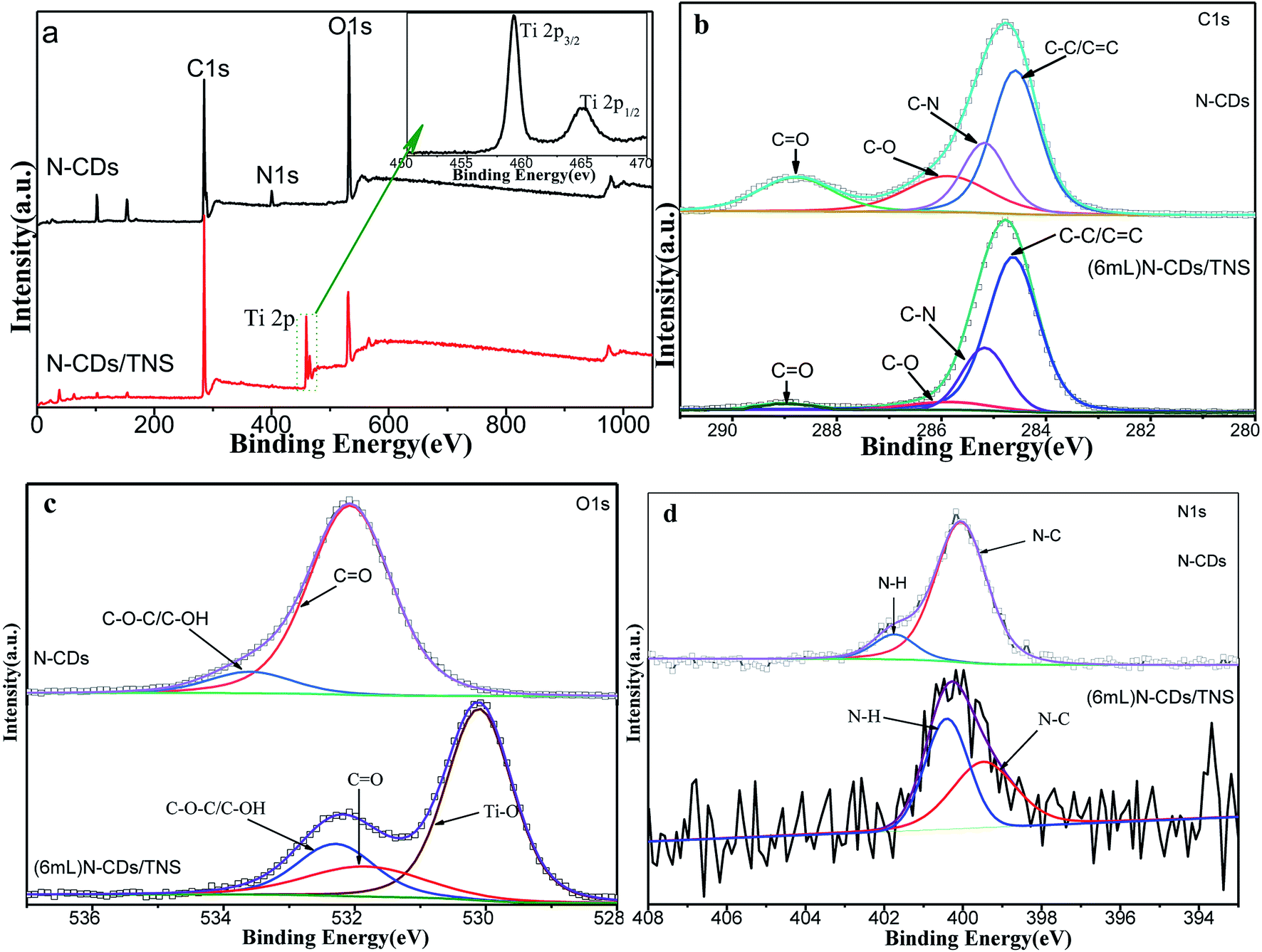 | ||
| Fig. 3 XPS spectral comparison of N-CDs and N-CDs/TNS. (a) XPS survey spectrum, (b) high-resolution C 1s spectrum, (c) O 1s spectrum and (d) N 1s spectrum. | ||
The high resolution deconvoluted spectra of each XPS peak are shown in Fig. 3b–d. In Fig. 3b the C 1s XPS spectrum is composed of four peaks based on peak fitting. The peak at a binding energy of 285.63 eV is assigned to C–O and C–O–C, the signal at 288.90 eV is attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxygen-containing carbonaceous band and the peak at 285.17 eV is coming from C–N. In addition, the deconvoluted peak centered at a binding energy of 284.63 eV is assigned to the C–C, CC and C–H bonds (sp2). These results suggest that there are relatively high amounts of oxygen groups on the N-CDs or the surface of the N-CDs/TNS, and there is a nitrogen group bonded to the N-CDs and N-CDs/TNS as well. In contrast, the peak intensities of the oxygen functional groups substantially decreased in the C 1s XPS spectrum of N-CDs/TNS (Fig. 3b), confirming that N-CDs has been covered by TiO2 nanosheets. It is worth noting that no peak assigned to C–F is detected, indicating no F-doped TNS was produced during the solvothermal process. In addition, the O 1s XPS spectra of N-CDs and N-CDs/TNS nanosheets are presented in Fig. 3c, which exhibit a different peak shape. For N-CDs, the O 1s peak at 533.59 eV is closely related to the significant C–O–C and hydroxyl groups, and the peak at 532.07 eV was assigned to CO. For N-CDs/TNS nanosheets the three peaks fitted with O 1s peaks were assigned to three different groups: 530.12 eV to Ti–O, 531.88 eV to CO and 532.27 eV to C–O–C/C–OH. All these results further confirm the successful integration between TNS and N-CDs and the presence of a strong interaction between the Ti, O, and C after the solvothermal treatment.
O oxygen-containing carbonaceous band and the peak at 285.17 eV is coming from C–N. In addition, the deconvoluted peak centered at a binding energy of 284.63 eV is assigned to the C–C, CC and C–H bonds (sp2). These results suggest that there are relatively high amounts of oxygen groups on the N-CDs or the surface of the N-CDs/TNS, and there is a nitrogen group bonded to the N-CDs and N-CDs/TNS as well. In contrast, the peak intensities of the oxygen functional groups substantially decreased in the C 1s XPS spectrum of N-CDs/TNS (Fig. 3b), confirming that N-CDs has been covered by TiO2 nanosheets. It is worth noting that no peak assigned to C–F is detected, indicating no F-doped TNS was produced during the solvothermal process. In addition, the O 1s XPS spectra of N-CDs and N-CDs/TNS nanosheets are presented in Fig. 3c, which exhibit a different peak shape. For N-CDs, the O 1s peak at 533.59 eV is closely related to the significant C–O–C and hydroxyl groups, and the peak at 532.07 eV was assigned to CO. For N-CDs/TNS nanosheets the three peaks fitted with O 1s peaks were assigned to three different groups: 530.12 eV to Ti–O, 531.88 eV to CO and 532.27 eV to C–O–C/C–OH. All these results further confirm the successful integration between TNS and N-CDs and the presence of a strong interaction between the Ti, O, and C after the solvothermal treatment.
The N 1s peaks at 398.20 and 400.11 eV shown in Fig. 3d indicate that nitrogen existed mostly in the form of N–C (400.05 eV) or N–H (401.74 eV). It is worth noting that no peak assigned to C–Ti is detected, indicating that N-CDs were not directly bonded to Ti atoms, and most probably were bonded to TNS through oxygen functional groups during the solvothermal process.
The FTIR technique was used to identify the organic functional groups on the surface of the N-CDs. As shown in Fig. 4, the amide group is clearly identified through the 1661 cm−1 CO stretching vibration. Absorptions at 3200 cm−1 and 1100 cm−1 are ascribed to O–H symmetric stretching vibration and C–O stretching vibration, respectively. The peaks at 1575 cm−1 and 1406 cm−1 are assigned to CC stretching and C–N bending respectively. The peak at 1207 cm−1 is described as C–O–C stretching vibration. The strong absorption peaks of C–O, C–N and O–H in N-CDs suggest that the N-CDs have an abundance of amide groups and hydroxyl groups, thus guaranteeing their good solubility in water. In addition, the absorption peaks between 2800 cm−1 and 3000 cm−1 are attributed to the C–H stretching vibration and the peaks within the range 1300–1500 cm−1 represent the C–H bending vibration.
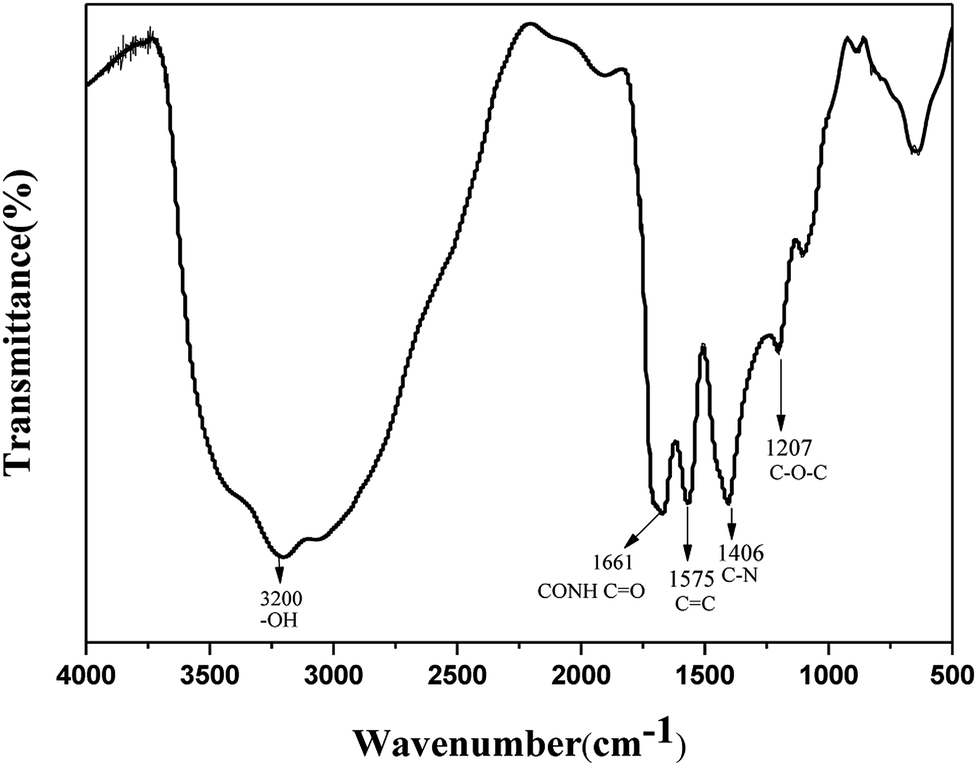 | ||
| Fig. 4 FTIR spectra of N-CDs. | ||
Fig. 5a shows the UV-vis absorption spectrum of N-CDs, with inset photographs of N-CDs under different light. A clear adsorption feature is observed at about 228 nm, which is assigned to the π–π* transition of CC bonds. In addition, N-CDs have a broad absorption band ranging from 280 nm to 500 nm with decreasing intensity, which is ascribed to the typical n to π* transitions of CO and CN groups. Such a wide absorption band ensures the photoluminescent emission of the N-CDs under excitation from UV light to red light. The phenomenon of emission wavelength dependence on the excitation wavelength is a common property observed in carbon quantum dot-based materials. The solution of N-CDs presents blue light under UV beam excitation but a yellow color under visible light excitation (Fig. 5a inset).
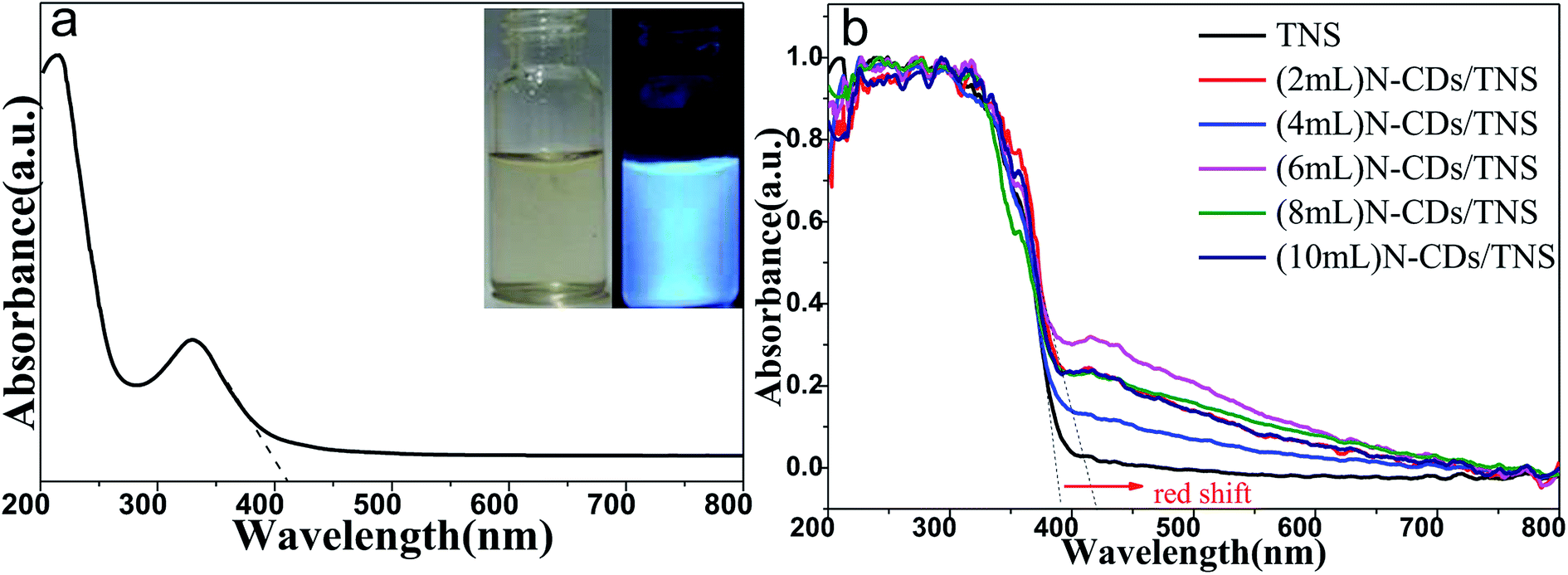 | ||
| Fig. 5 UV-vis absorption spectrum (a) of the as-prepared N-CDs solution (inset is a photograph of N-CDs solution under visible light and UV light) and (b) UV-vis diffuse reflectance spectra (UV-vis DRS) of N-CDs/TNS. | ||
Fig. 5b shows the UV-vis DRS of as-prepared TNS and N-CDs/TNS photocatalysts. In the UV region no absorption difference can be discerned among all the catalysts while in the visible light region a red shift in the absorption edge could be observed for N-CDs/TNS relative to TNS. This red shift could be ascribed to the deposition of carbon quantum dots in the composites, reducing the reflection of light. (6 mL) N-CDs/TNS have the highest visible light absorption.
Fig. 6a shows the fluorescence emission spectra of water solutions of N-CDs excited by a series of laser wavelengths. It shows that the emission spectra of N-CDs are broad, ranging from 350 nm to 550 nm; moreover, the emission spectra are wavelength dependent on the excitation laser wavelengths. This indicates that the band structures of N-CDs are complicated and disordered. The emission property of the C dots is highly related to the CO and CN functional groups in N-CDs, which can affect the electronic structures of core sp2 clusters and introduce new energy levels into their electronic structures, consequently resulting in multiple electronic transition possibilities. Multiple electronic transitions mix together, broadening the absorption band range. The weight of each transition is different and changes with the excitation wavelength. The excited electrons will be deactivated by radiative recombination to their own ground state, leading to multiple fluorescence emissions. A specific excitation wavelength corresponds to specific weight multiple electronic transitions, resulting in tailored backward fluorescence wavelength emissions. The excitation wavelength changes, the weight of each electronic transition changes, and the weight of each fluorescence emission changes, shifting with the emission peak wavelength, presenting wavelength-dependent properties of N-CDs. Our N-CDs show the strongest blue fluorescence under an excitation wavelength of 330 nm. It was reported that doping with nitrogen element moves the conduction band edge of carbon down so as to decrease the band gap. Moreover, the nitrogen bonding to carbon may cause disorder in the carbon hexagonal rings and create new luminescent centers by trapping the radiative electron–hole pairs, so prolonging the lifetime of electron–hole pairs and making room for electron transfer to another semiconductor. Fig. 6b shows that doping of N-CDs into TNS changes the electron radiative solution: it may trap the excited electrons and cause quenching of fluorescence.
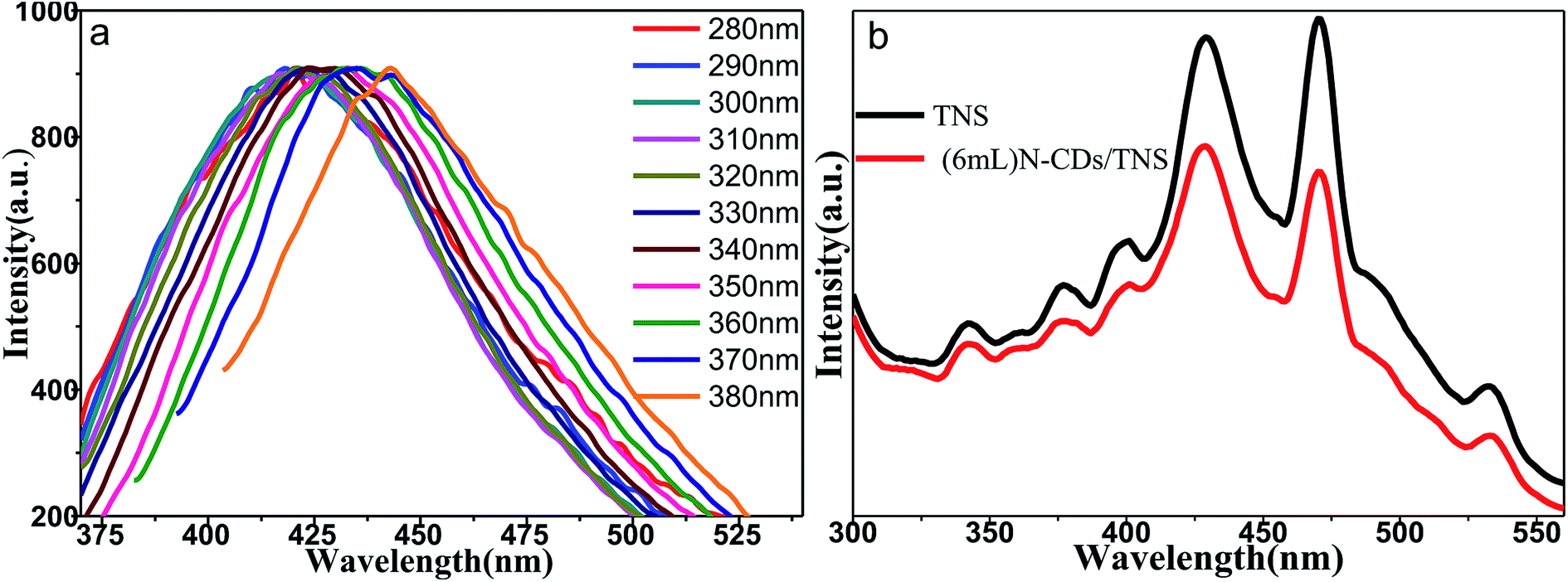 | ||
| Fig. 6 (a) Normalized fluorescence emission spectra of water solutions of N-CDs acquired using various excitation wavelengths. (b) PL spectra of TNS and (6 mL) N-CDs/TNS. | ||
Rhodamine B (RhB) is a representative organic dye and has been widely used in industrial applications, contaminating environments. Thus the photocatalytic degradation of RhB in aqueous solution was selected as a probe reaction in this work to evaluate the photocatalytic performance of N-CDs/TNS photocatalysts, together with those on N-CDs and pure TNS for a comparison. The concentration change in RhB vs. photo illumination time is shown in Fig. 7. Under visible light irradiation (400–780 nm), no RhB self-degradation or no obvious concentration decrease was observed over N-CD catalysts, and only 25% degradation was observed over TNS within 180 minutes, as shown in Fig. 7. Whereas, the N-CDs/TNS with different N-CDs contents demonstrated higher photodegradation efficiency: 92% RhB degraded within 180 minutes over (6 mL) N-CDs/TNS. Through careful observation, the RhB degradation efficiency continuously increases with N-CDs loading content increasing from 2 mL to 6 mL. Further increasing N-CDs content, however, decreases catalytic activity. On the basis of a simplified Langmuir–Hinshelwood model, the linear relationship of ln(C/C0) versus time (see Fig. 8a) upon visible light irradiation indicates that RhB degradation follows pseudo first order kinetics, and the apparent rate constant (k) shown in Fig. 8b was calculated from the plot of ln(C/C0) vs. time. The highest apparent rate constant, obtained for (6 mL) N-CDs/TNS, is 1.282 × 10−2 min−1, which shows a 9.29, 1.69, 2.29, 1.66 and 2.54-fold photocatalytic activity improvement over TNS (k = 0.00138 min−1), (2 mL) N-CDs/TNS (k = 0.0076 min−1), (4 mL) N-CDs/TNS (k = 0.0056 min−1), (8 mL) N-CDs/TNS (k = 0.00772 min−1) and (10 mL) N-CDs/TNS (k = 0.00504 min−1), respectively. The experiments show that a suitable loading content of N-CDs on TNS is a key factor for improving the photocatalytic activity of N-CDs/TNS nanocomposites. The doping of N-CDs into the matrix of TNS endows the N-CDs/TNS photocatalysts with several outstanding characteristics: extended catalytic adaptability, enhanced adsorption of pollutants, extended light absorption range into the visible region and improved separation of photogenerated electron–hole pairs.
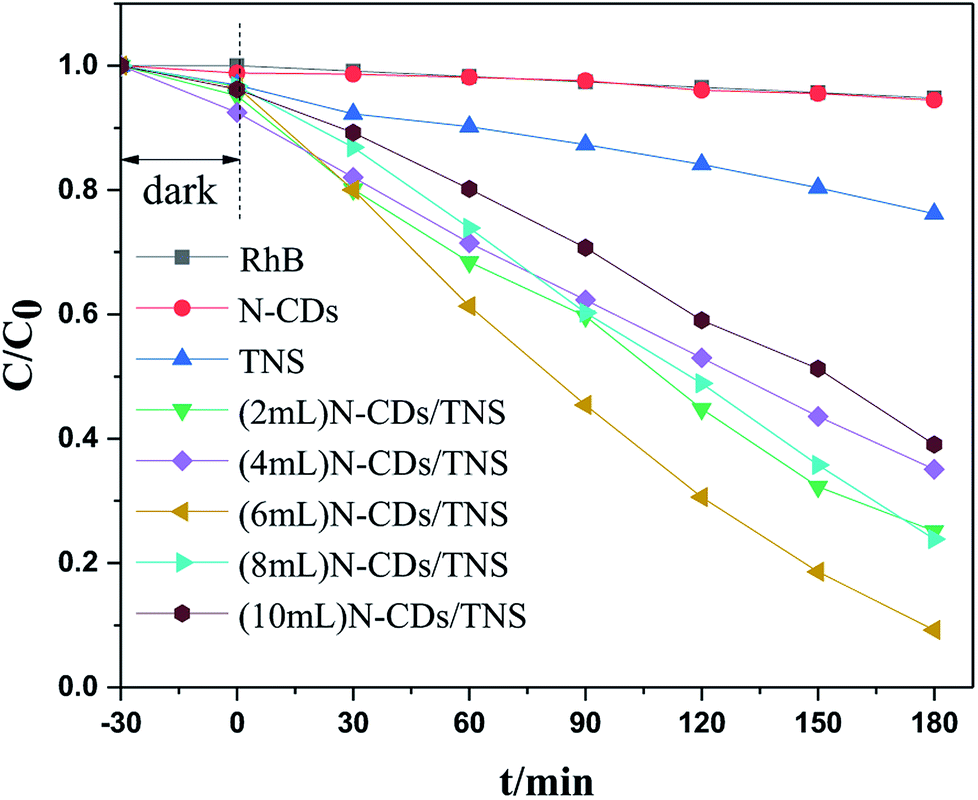 | ||
| Fig. 7 Photocatalytic degradation rate curves of RhB using N-CDs, TNS, and N-CDs/TNS as photocatalysts under visible light. | ||
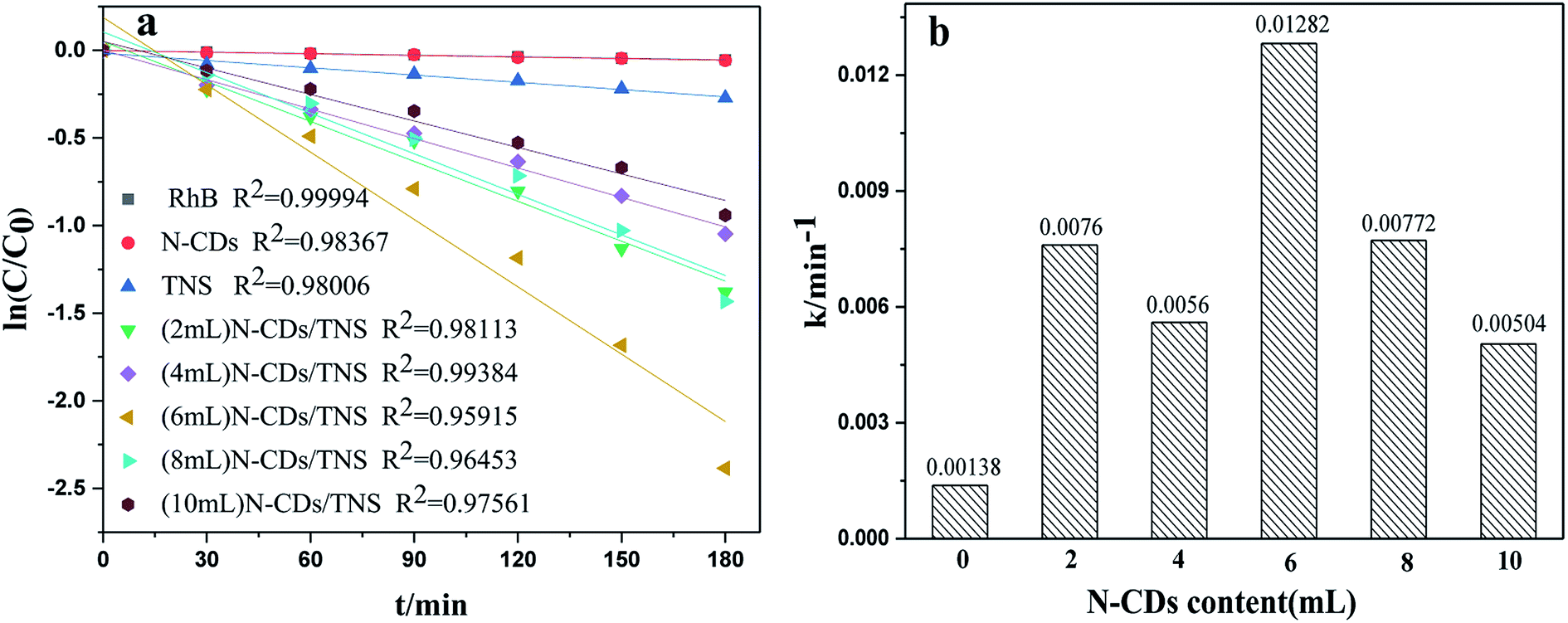 | ||
| Fig. 8 (a) Kinetic linear fitting curves and (b) the apparent rate constant for liquid-phase photocatalytic degradation of RhB over different samples under visible light irradiation. | ||
To investigate the catalytic mechanism, especially the capture of the main active species that trigger the visible light photodegradation, an additional examination was carried out by adding specific radical trapping agents to the reactant before light irradiation. It is well known that tert-butanol (TBA) is an ·OH scavenger, benzoquinone (BQ) is a scavenger for ·O2− radicals, and EDTA–2Na and AgNO3 can trap holes and electrons respectively. Under visible light irradiation, as shown in Fig. 9a, RhB degradation was significantly suppressed when benzoquinone (BQ) or EDTA–2Na were added to the reaction system. However, other radical scavengers had no influence on photoactivity. This indicated that the ·O2− radical and h+ were the main active species during the photodegradation process.
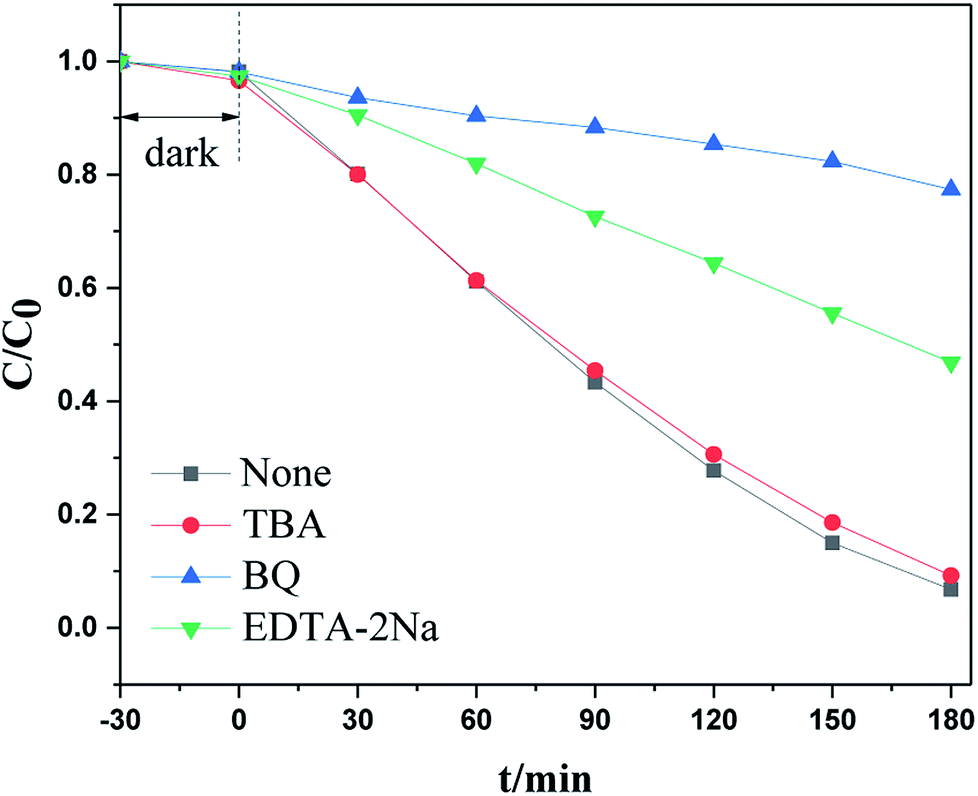 | ||
| Fig. 9 The photocatalytic activity of N-CDs/TNSs with the addition of different scavengers. | ||
An EPR spin-trapping study was conducted in order to verify the presence of radicals and to identify radical species which may be responsible for the degradation of RhB. The EPR spectrum of N-CDs/TNS dispersed in water under visible light irradiation is shown in Fig. 10a and the spectrum dispersed in methanol is shown in Fig. 10b. No signal is present in Fig. 9a, which means there is no DMPO–·OH adduct in solution.35 The EPR spectrum monitored upon continuous irradiation of N-CDs/TNS suspension dispersed in methanol (Fig. 10b) demonstrates the formation of DMPO–·O2− simultaneously with a further four characteristic paramagnetic signals with an intensity ratio of 1:1:1:1, which are typical of the DMPO oxidation product 5,5-dimethyl-1-pyrrolidone-2-oxyl (·DMPO–X, i.e. O2−˙/·OOH36). This experiment verifies that ·O2− is the active species that is produced during light irradiation and which is responsible for the photodegradation of RhB, which is consistent with the experimental trapping results shown in Fig. 9.
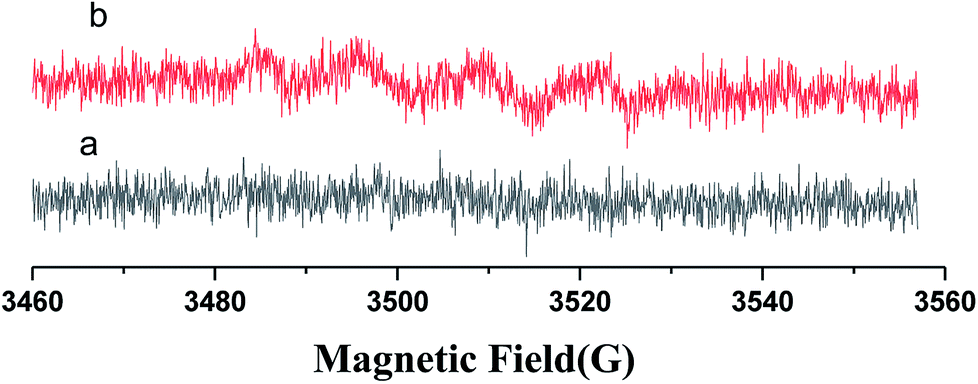 | ||
| Fig. 10 DMPO spin-trapping EPR spectra for (6 mL) N-CDs/TNS. | ||
Fig. 11a presents the cyclic voltammograms (CVs) of N-CDs. It is clear that the electrochemical behavior is reversible, and the reduction potential is approximately at −0.61 V, and according to the equation ELUMO = −e(ERED + 4.4), we can obtain the LUMO energy as −3.79 V, which is very close to that reported in the literature.37 On the basis of the UV-vis DRS spectra of N-CDs and ELUMO, we obtain EHOMO = −6.81 V. Based on the energy data and trapping detection results above, Z-scheme N-CDs/TNS heterojunction structures were proposed to explain the potential mechanisms of the visible light irradiation catalytic activity (Fig. 11b). The visible light irradiation excited the HOMO electron of N-CDs to the excited LUMO orbit. At the same time, the anatase TNS can also absorb visible light due to the weak absorption “tail” around a wavelength of 400 nm, which excites the electrons in the VB of TNS to the CB of TNS. The {001} and {101} facets of TNS can form a surface heterojunction and facilitate the electron preferentially jumping to {101} facets. That is to say, both of the electrons on TNS and N-CDs can be excited to yield electron–hole pairs under visible light illumination. Photogenerated electrons from the LUMO of N-CDs can substantially combine with the photogenerated holes from the TNS accumulated on {001} facets and, as a result, the electron and hole carriers are spatially separated: that is, holes accumulate on N-CDs and electrons accumulate on {101} facets of TNS. The interfacial Z-scheme combination also helps the excited electrons accumulate on the {101} of TNS, preventing them from traveling back to the VB. Since no holes remain in TNS, the hot electrons on the CB should have much longer lifetimes, offering more probabilities to reduce the O2 adsorbed on the {101} facets of TNS and the consequent RhB degradation. On the other hand, the h+ remaining in the HOMO of the N-CDs might be transferred to the surface of the photocatalysts, attacking target organic compounds, or reacting with H2O to form ·OH. Thus, the Z-scheme structure could efficiently separate the photoinduced electron–hole pairs, to obtain enhanced photocatalytic performance for N-CDs/TNS.
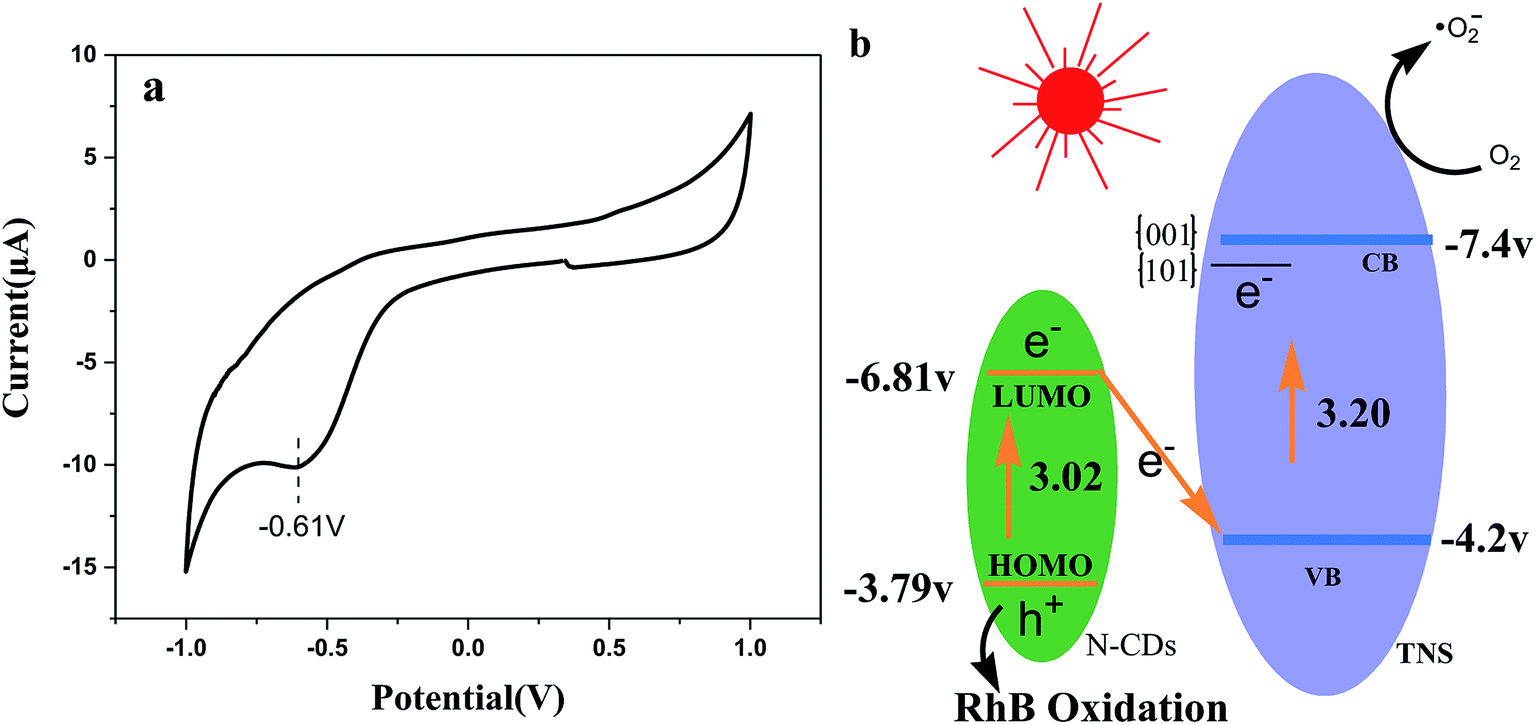 | ||
| Fig. 11 CV curve of N-CDs and (b) the possible photocatalytic mechanism of N-CDs/TNS under visible light irradiation. | ||
Additional experiments were performed to evaluate the stability and reusability of the N-CDs/TNS. As shown in Fig. 12, the RhB photodegradation efficiency exhibits only a slight decrease after 10 successive cycles of experiment under identical conditions, indicating that our as-prepared N-CDs/TNS photocatalysts have superior photocatalytic stability. This can be attributed to a synergistic effect between the high intrinsic single-crystal-like nature of the anatase phase of TNS, the stability of the N-CDs, as well as the strong interaction between the N-CDs and TNS.
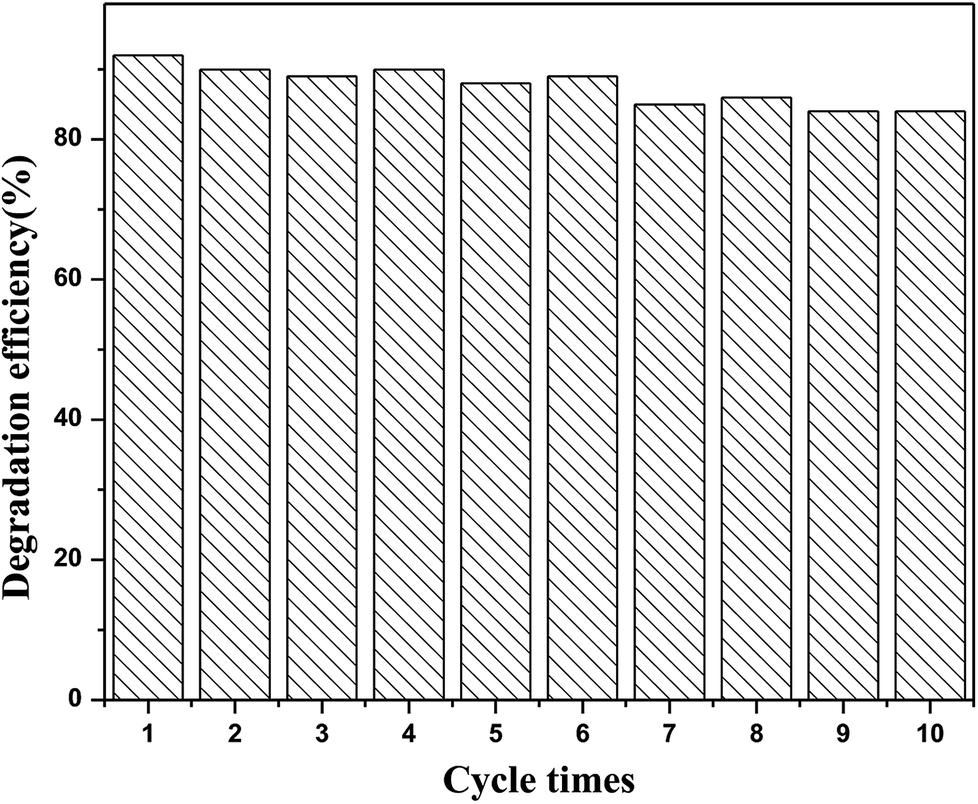 | ||
| Fig. 12 The reusability performance of (6 mL) N-CDs/TNS for the removal of RhB under visible light irradiation. | ||
4 Conclusions
In summary, the incorporation of N-CDs into anatase TNS, denoted as N-CDs/TNS, was performed using a solvothermal method to modify the photoresponse properties of TiO2 particles and improve their photocatalytic stability. Experiments demonstrated that the exposed active {001} facets coupled with the loaded N-CDs dramatically enhanced the visible light photocatalytic performance of TiO2, and the highest photocatalytic activity was found to be 9.3 times that of pure TNS. SEM and TEM confirmed the as-prepared TiO2 nanosheet structure and that the dominant exposed face is the {001} facet, and the loaded N-CDs are nearly spherical with an average size of about 3 nm. XPS results confirmed that the deposited N-CDs were chemically integrated into TiO2 nanosheets. UV-vis DRS spectroscopy shows that with the dotting of N-CDs, the absorption edge of N-CDs/TNS has been extended into the visible light region. The structure of a Z-scheme heterojunction and the {101} {001} facet surface heterojunction, the absorption edge extension, the high charge separation, and the effective exposure of highly reactive {001} facets are the main contributions to the high photocatalytic activity. The prepared N-CDs/TNS catalyst is expected to be applied to photocatalytic degradation, photocatalytic water splitting, solar cells, sensors, etc. The findings of our work provide a new way to steadily improve photocatalytic efficiency through the loading of stable N-CDs onto a single-crystal-like TiO2 nanosheet.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 21473161 and 21873084), the Zhejiang Provincial Natural Science Foundation of China (LZ17B030001), the Alexander von Humboldt Foundation, and Zhejiang SCI-TECH University for 521 Distinguished Scholar's Scheme.References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- S. N. Frank and A. J. Bard, J. Am. Chem. Soc., 1977, 99, 303–304 CrossRef CAS.
- Y. Feng, L. Ling, Y. Wang, Z. Xu, F. Cao, H. Li and Z. Bian, Nano Energy, 2017, 40, 481–486 CrossRef CAS.
- X. Qian, M. Ren, D. Yue, Y. Zhu, Y. Han, Z. Bian and Y. Zhao, Appl. Catal., B, 2017, 212, 1–6 CrossRef CAS.
- L. Ling, L. Liu, Y. Feng, J. Zhu and Z. Bian, Chin. J. Catal., 2018, 39, 639–645 CrossRef CAS.
- V. Etacheri, C. Di Valentin, J. Schneider, D. Bahnemann and S. C. Pillai, J. Photochem. Photobiol., C, 2015, 25, 1–29 CrossRef CAS.
- T. H. Han, H. G. Wang and X. M. Zheng, RSC Adv., 2016, 6, 7829–7837 RSC.
- A. Aghamali, M. Khosravi, H. Hamishehkar, N. Modirshahla and M. A. Behnajady, Mater. Sci. Semicond. Process., 2018, 87, 142–154 CrossRef CAS.
- Z. Duan, Y. Zhu, P. Ren, J. Jia, S. Yang, G. Zhao, Y. Xie and J. Zhang, Appl. Surf. Sci., 2018, 452, 165–173 CrossRef CAS.
- X. Tang, Z. Wang, W. Huang, Q. Jing and N. Liu, Mater. Res. Bull., 2018, 105, 126–132 CrossRef CAS.
- Y. Zhang, M. Xu, H. Li, H. Ge and Z. Bian, Appl. Catal., B, 2018, 226, 213–219 CrossRef CAS.
- Q. Li, H. Qin, H. Zhao, X. Zhao, X. Cheng and W. Fan, Appl. Surf. Sci., 2018, 457, 59–68 CrossRef CAS.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed.
- G. Shen, L. Pan, Z. Lu, C. Wang, A. Fazal e, X. Zhang and J.-J. Zou, Chin. J. Catal., 2018, 39, 920–928 CrossRef CAS.
- P. Chowdhury, S. Athapaththu, A. Elkamel and A. K. Ray, Sep. Purif. Technol., 2017, 174, 109–115 CrossRef CAS.
- W. Wei, D. Liu, Z. Wei and Y. Zhu, ACS Catal., 2017, 7, 652–663 CrossRef CAS.
- H. Wang, D. Zhou, Z. Wu, J. Wan, X. Zheng, L. Yu and D. L. Phillips, Mater. Res. Bull., 2014, 57, 311–319 CrossRef CAS.
- W. Zhao, Y. Guo, Y. Faiz, W.-T. Yuan, C. Sun, S.-M. Wang, Y.-H. Deng, Y. Zhuang, Y. Li, X.-M. Wang, H. He and S.-G. Yang, Appl. Catal., B, 2015, 163, 288–297 CrossRef CAS.
- X. Wu, M. Li, J. Li, G. Zhang and S. Yin, Appl. Catal., B, 2017, 219, 132–141 CrossRef CAS.
- Y.-C. Zhang, Z. Li, L. Zhang, L. Pan, X. Zhang, L. Wang, A. Fazal e and J.-J. Zou, Appl. Catal., B, 2018, 224, 101–108 CrossRef CAS.
- R. Liu, Z. Ji, J. Wang and J. Zhang, Appl. Surf. Sci., 2018, 441, 29–39 CrossRef CAS.
- X. Yan, Z. Xing, Y. Cao, M. Hu, Z. Li, X. Wu, Q. Zhu, S. Yang and W. Zhou, Appl. Catal., B, 2017, 219, 572–579 CrossRef CAS.
- J. Zhang, L. Zhang, Y. Shi, G. Xu, E. Zhang, H. Wang, Z. Kong, J. Xi and Z. Ji, Appl. Surf. Sci., 2017, 420, 839–848 CrossRef CAS.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- M. M. Maitani, K. Tanaka, H. Satou, T. Oshima, A. Ohtomo and Y. Wada, Crystengcomm, 2017, 19, 4734–4741 RSC.
- H. Wang, W. Zhang, B. Liu, S. Shen, X. Zheng, K. Pei and L. Chen, J. Phys. Chem. A, 2011, 115, 14282–14291 CrossRef CAS PubMed.
- J. Yu, J. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS PubMed.
- T. Tachikawa, N. Wang, S. Yamashita, S.-C. Cui and T. Majima, Angew. Chem. Int. Ed., 2010, 49, 8593–8597 CrossRef CAS PubMed.
- Y.-Q. Dang, S.-Z. Ren, G. Liu, J. Cai, Y. Zhang and J. Qiu, Nanomaterials, 2016, 6, 212 CrossRef PubMed.
- X. Hao, Z. Jin, J. Xu, S. Min and G. Lu, Superlattices Microstruct., 2016, 94, 237–244 CrossRef CAS.
- F. W. Low and C. W. Lai, Renewable Sustainable Energy Rev., 2018, 82, 103–125 CrossRef CAS.
- S. J. Zhu, J. H. Zhang, C. Y. Qiao, S. J. Tang, Y. F. Li, W. J. Yuan, B. Li, L. Tian, F. Liu, R. Hu, H. N. Gao, H. T. Wei, H. Zhang, H. C. Sun and B. Yang, Chem. Commun., 2011, 47, 6858–6860 RSC.
- Z. F. Bian, T. Tachikawa, P. Zhang, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2014, 136, 458–465 CrossRef CAS PubMed.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- J. M. Fontmorin, R. C. B. Castillo, W. Z. Tang and M. Sillanpaa, Water Res., 2016, 99, 24–32 CrossRef CAS PubMed.
- A. S. W. Li, K. B. Cummings, H. P. Rothling, G. R. Buettner and C. F. Chignell, J. Magn. Reson., 1988, 79, 140 Search PubMed.
- X. Yan, X. Cui, B. Li and L.-s. Li, Nano Lett., 2010, 10, 1869–1873 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |