
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural characterization and anti-inflammatory effect in hepatocytes of a galactoglucan from Antrodia camphorata mycelium†
Huiling Tang‡
ab,
Wenbing Nie‡a,
Jinna Xiaoa,
Zhengqi Zhaa,
Qiuli Chenc and
Hongping Yin *a
*a
aSchool of Life Science and Technology, State Key Laboratory of Natural Medicines, China Pharmaceutical University, No. 24 Tongjiaxiang Road, Nanjing 210009, Jiangsu, People's Republic of China. E-mail: yinhongping63@163.com; Fax: +86 025 83271249; Tel: +86 025 83271108
bDepartment of Pharmacy, Jiangsu Food & Pharmaceutical Science College, Huaian 223003, People's Republic of China
cSchool of Pharmacy, Nanjing University of Chinese Medicine, Nanjing 210029, People's Republic of China
First published on 8th March 2019
Abstract
The galactoglucan ACP2 was isolated from cultured Antrodia camphorata mycelium through anion-exchange column chromatography and Sephadex G-100 chromatography and shown to exhibit hepatoprotective function in L02 cells. Based on monosaccharide composition analysis, ACP2 was mainly composed of glucose, galactose, and 6-deoxyglucose in a molar ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1. The average molecular weight of ACP2 was 1.93 × 104 Da. The primary structure of ACP2 was elucidated with Fourier-transform infrared spectroscopy, gas chromatography-mass spectrometry, and nuclear magnetic resonance spectroscopy. The results indicated the following composition: →6)-linked-β-D-Galp-(1→, →6)-linked-α-D-Glcp-(1→, →3)-linked-α-D-Glcp-(1→, and →2,4)-linked-β-D-Glcp-(1→, with terminal 6-deoxy-α-D-Glcp and α-D-Glcp. ACP2 alleviated lipopolysaccharide-induced hepatocyte inflammation by down-regulating the expressions of COX-2, IL-1β, TNF-α and IL-6. The decreased expressions of TLR4, MyD88, NF-κB, and phosphorylated p38 in ACP2-treated L02 cells indicated that ACP2 might ameliorate inflammation through the TLR4 and p38/NF-κB signaling pathways.
:2:1. The average molecular weight of ACP2 was 1.93 × 104 Da. The primary structure of ACP2 was elucidated with Fourier-transform infrared spectroscopy, gas chromatography-mass spectrometry, and nuclear magnetic resonance spectroscopy. The results indicated the following composition: →6)-linked-β-D-Galp-(1→, →6)-linked-α-D-Glcp-(1→, →3)-linked-α-D-Glcp-(1→, and →2,4)-linked-β-D-Glcp-(1→, with terminal 6-deoxy-α-D-Glcp and α-D-Glcp. ACP2 alleviated lipopolysaccharide-induced hepatocyte inflammation by down-regulating the expressions of COX-2, IL-1β, TNF-α and IL-6. The decreased expressions of TLR4, MyD88, NF-κB, and phosphorylated p38 in ACP2-treated L02 cells indicated that ACP2 might ameliorate inflammation through the TLR4 and p38/NF-κB signaling pathways.
1. Introduction
Hepatitis, which is caused by various etiological factors such as viral infections, alcoholism, autoimmune disorders and cholestasis, is a complex process that threatens human health and has high incidence. Some studies have found that the pivotal etiology of hepatitis is related to acute liver injury.1,2 Some liver injury may be related to lipopolysaccharides (LPS), which are the main component of the cell walls of Gram-negative bacteria and are widely present in the digestive tracts of humans and other animals.3 LPS can trigger a series of inflammatory responses and septic shock, eventually leading to multiple organ disorders and liver injury in particular.4,5Toll-like receptor 4 (TLR4) is a pattern recognition receptor that specially recognizes LPS and initiates signaling cascades, leading to the up-regulation of the expression of pro-inflammatory cytokines.6,7 Myeloid differentiation factor 88 (MyD88) is an important adapter protein of TLR4 that can activate mitogen-activated protein kinases (MAPKs) and nuclear factor-κB (NF-κB) signaling pathways.8–10 P38 is a member of the MAPK family that is closely associated with liver inflammation.11 NF-κB is also a pivotal transcription factor that binds with inhibitors of κB (IκB) in the cytoplasm, preventing the transport of NF-κB into the nucleus. Inflammatory stimuli such as LPS can phosphorylate IκB followed by ubiquitination and proteosomal degradation. This leads to the nuclear translocation of NF-κB, which promotes the transcription of pro-inflammatory genes.6 In turn, a series of inflammatory factors such as cyclooxygenase-2 (COX-2), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) are up-regulated, leading to liver inflammation.12
Antrodia camphorata, which is referred to as niu-chang-chih in China, is a parasitic fungus that lives only on the inner cavity walls of the local evergreen Cinnamomum kanchirai Hay in Taiwan.13 Among Chinese people, A. camphorata is a valuable traditional functional food that is known as a food supplement or health food for the treatment of hypohepatia and hepatitis. A. camphorata exhibits significant hepatoprotection due to its bioactive constituents, which include triterpenoids and ergostane.14,15 However, to the best of our knowledge, the primary structures and hepatoprotective activities of polysaccharides isolated from A. camphorata have not been fully investigated.16,17 A member of our research group isolated a water-soluble β-D-glucan from A. camphorata and found that it possessed antioxidant activity in human hepatocytes.18
Based on a preliminary study of the structures and molecular mechanisms of polysaccharides in our laboratory, we isolated an undescribed galactoglucan termed ACP2 from the mycelium of A. camphorata and evaluated its anti-inflammation effect in L02 cells. ACP2 is special because of the presence of 6-deoxyglucose in its sugar chain, which has rarely been reported in the previously papers. We hypothesized that ACP2 attenuates LPS-induced hepatic injury by inhibiting the TLR4-mediated inflammatory pathway and ameliorating the production of downstream pro-inflammatory molecules. The role of 6-deoxyglucan in the anti-inflammatory activity of ACP2 in hepatocytes, if any, requires further study.
2. Materials and methods
2.1. Materials
Cultured A. camphorata powder was obtained from Jiangsu Shenhua Pharmaceutical Co., Ltd. LPS from Escherichia coli (O55:B5, L2880) and standard monosaccharides were provided by Sigma. A series of glucan standards with different molecular weights were obtained from Shodex. Methyl-thiazolyl-tetrazolium (MTT), which was used for coloration, was purchased from Invitrogen Corp. (Carlsbad, CA, USA). Dulbecco's modified eagle medium (DMEM) and fetal bovine serum were purchased from Gibco (Rockville, MD, USA), and polyvinylidene difluoride (PVDF) membranes were purchased from Bio-Rad (Hercules, CA, USA). All other chemicals and solvents were analytical reagent grade.2.2. Extraction and purification
ACP2 was extracted and purified according to the methods of Wang.19 Briefly, after a series of steps involving dissolving, decolorizing, purifying and drying, we obtained decolorized polysaccharide (dACP). The dACP was separated through a column (300 × 26 mm) packed with diethylaminoethyl (DEAE)-cellulose 52 and eluted with distilled water followed by 1 mol L−1 NaCl. Subsequently, the eluate was collected and purified using a Sephadex G-100 column (1.0 × 80 cm) followed by dialysis and lyophilization in a vacuum freeze-dryer. The resulting depurated polysaccharide fraction was assigned as ACP2. The purity of ACP2 was preliminarily analyzed using ultraviolet (UV) spectroscopy.2.3. Determination of molecular weight
The molecular weight of ACP2 was estimated by high-performance gel-permeation chromatography (HPGPC) with a refractive index detector (detection temperature = 35 °C) and Shodex SB 803 and Shodex SB 805 chromatographic columns. The detailed experimental conditions were as follows: the sample solution (100 μL) was injected and eluted with 0.1 M NaNO3 solution at a flow rate of 0.35 mL min−1.20 The calibration curve was established using GPC software.2.4. Monosaccharide composition analysis of ACP2
High-performance liquid chromatography (HPLC) was used to identify the monosaccharide composition of ACP2 based on 1-phenyl-3-methyl-5-pyrazolone (PMP) labeling.21 Briefly, ACP2 solution (5 mg mL−1) was hydrolyzed by 2 M trifluoroacetic acid (TFA) for 2 h at 100 °C. After removing TFA, the hydrolysate was derivatized via the addition of 0.6 M NaOH (25 μL) and 0.4 M PMP (50 μL). Subsequently, the solution was neutralized with 50 μL of 0.3 M HCl and extracted three times with chloroform. The aqueous phase was then loaded onto an HPLC using an Agilent 1260 system (UV detector VWD, wave length 250 nm) equipped with a ZORBAX Eclipse XDB-C18 (250 mm × 4.6 mm) column and eluted at 0.8 mL min−1 with acetonitrile and phosphate buffer (pH 6.7) in a ratio of 17:83 (v/v).
2.5. Fourier-transform infrared (FT-IR) spectroscopy
ACP2 was analyzed on a FT-IR spectrophotometer (Bruker Tensor 27, Bruker, Germany). Briefly, the sample was ground with dried KBr powder and then compressed into a 1 mm pellet for analysis in the frequency range of 4000–400 cm−1.2.6. Nuclear magnetic resonance (NMR) spectroscopy
ACP2 samples were dissolved in 1.0 mL D2O. NMR spectra were recorded at 25 °C using a JEOL JNM ECP 600 NMR spectrometer (JEOL, Tokyo, Japan). 1D (1H and 13C NMR) and 2D NMR spectra, 1H-1H correlated (1H-1H COSY) spectra, heteronuclear single quantum correlation (HSQC) spectra, and heteronuclear multiple bond correlation (HMBC) spectra were collected.2.7. Methylation analysis
ACP2 was methylated repeatedly in accordance with Ciucanu's method.22 The absolute methylated products were hydrolyzed with 2 M TFA and converted into the partially methylated alditol acetates (PMAAs) that were analyzed by using a gas chromatography-mass spectrometry (GC-MS; HP 6890 II, Agilent USA) equipped with a DB-225MS fused-silica capillary column (Agilent Technologies Co. Ltd., USA).2.8. Biological activity
2.9. Statistical analysis
All experimental data are presented as means ± standard deviations. Statistical comparisons were made using Graphpad Prism 5.0 software. Two levels of statistical significance were considered: *p < 0.05 and **p < 0.01.3. Results and discussion
3.1. Extraction, purification, and characterization of ACP2
ACP2 was obtained from the mycelium of A. camphorata in a single symmetrical chromatographic peak after a series of purification steps using DEAE-52 and Sephadex G-100 columns. Hydrolyzed ACP2 had no absorption at either 260 nm or 280 nm in the UV spectrum, which indicated the absence of proteins and nucleic acids. The HPGPC profile of ACP2 exhibited a single, symmetrical sharp peak (Fig. 1A), which is characteristic of a homogeneous polysaccharide. Based on standard curve and retention time of the polysaccharide (Fig. 1B), the average molecular weight of ACP2 was estimated to be 1.93 × 104 Da.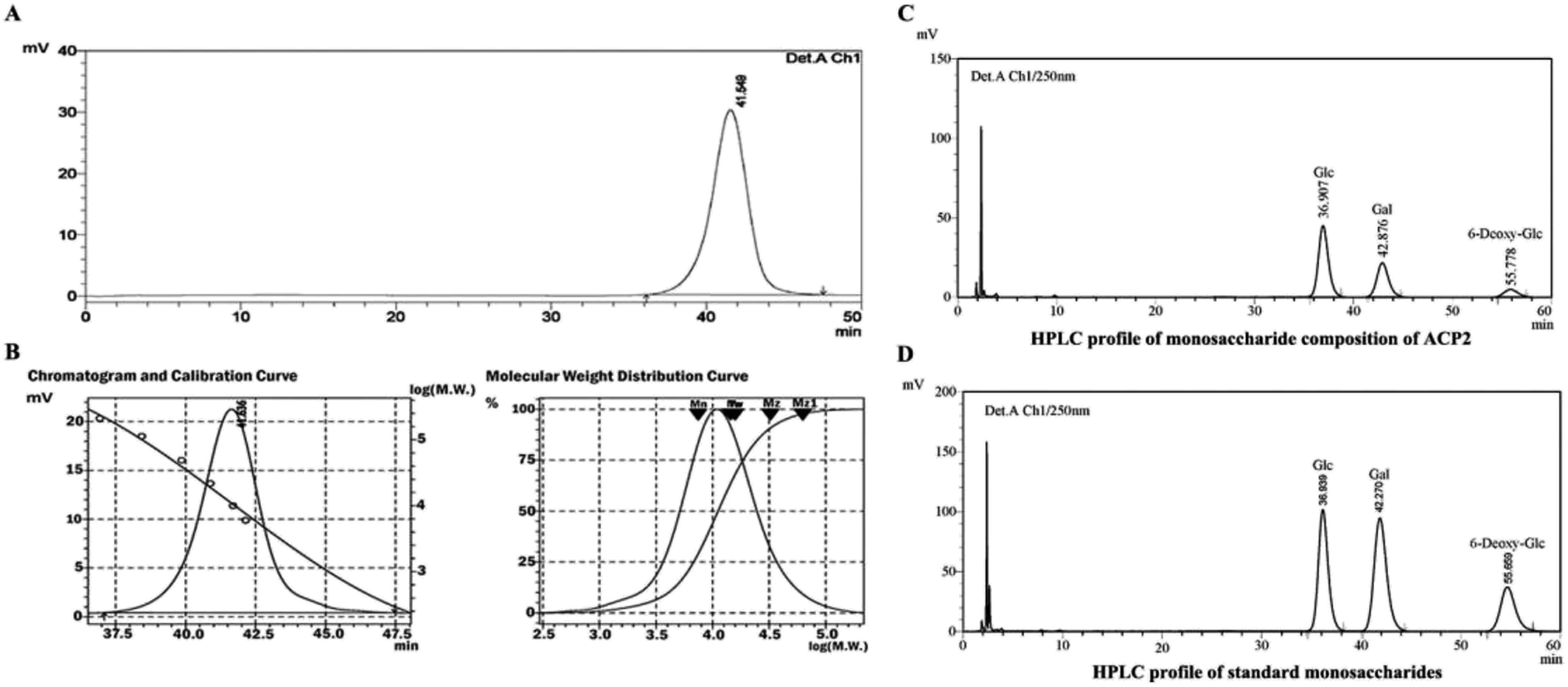 | ||
| Fig. 1 Elution profile of ACP2 in HPGPC with 0.1 M NaNO3 at a flow rate of 0.35 mL min−1 on Shodex SB805 and 802 columns. (A) ACP2 was identified as a single symmetrical sharp peak, indicating that it was a homogeneous polysaccharide. (B) Chromatogram and calibration curve of Dextran standards plotted with GPC software and molecular weight (Mw) on a log scale versus the retention time of ACP2. (C) Monosaccharide composition of ACP2. (D) HPLC profile of standard monosaccharides. | ||
3.2. Structural characterization
:2:1 (Fig. 1D).| Methylated sugars (as alditol acetates) | Type of linkage | Mass fragments (m/z) (relative abundance, %) |
|---|---|---|
| a 2,3,4,6-Me4-Glcp = 1,5-di-O-acetyl-2,3,4,6-tetra-O-methyl-glucopyranose, etc. | ||
| 2,3,4-Me3-6-deoxy-Glcp | T-6-deoxy-Glc | 43,59,71,72,89,101,115,117,131,161,175 |
| 2,3,4,6-Me4-Glcpa | T-Glc | 43,58,59,71,75,87,101,117,129,145,161,205 |
| 2,4,6-Me3-Glcp | 1,3-Linked-Glc | 43,45,71,87,101,117,129,161,189,233 |
| 2,3,4-Me3-Glcp | 1,6-Linked-Glc | 43,45,58,71,87,101,117,129,161,173,189,233 |
| 2,3,4-Me3-Galp | 1,6-Linked-Gal | 43,45,58,71,87,101,117,129,161,173,189,233 |
| 3,6-Me2-Glcp | 1,2,4-Linked-Glc | 43,45,71,87,99,113,129,159,189,233 |
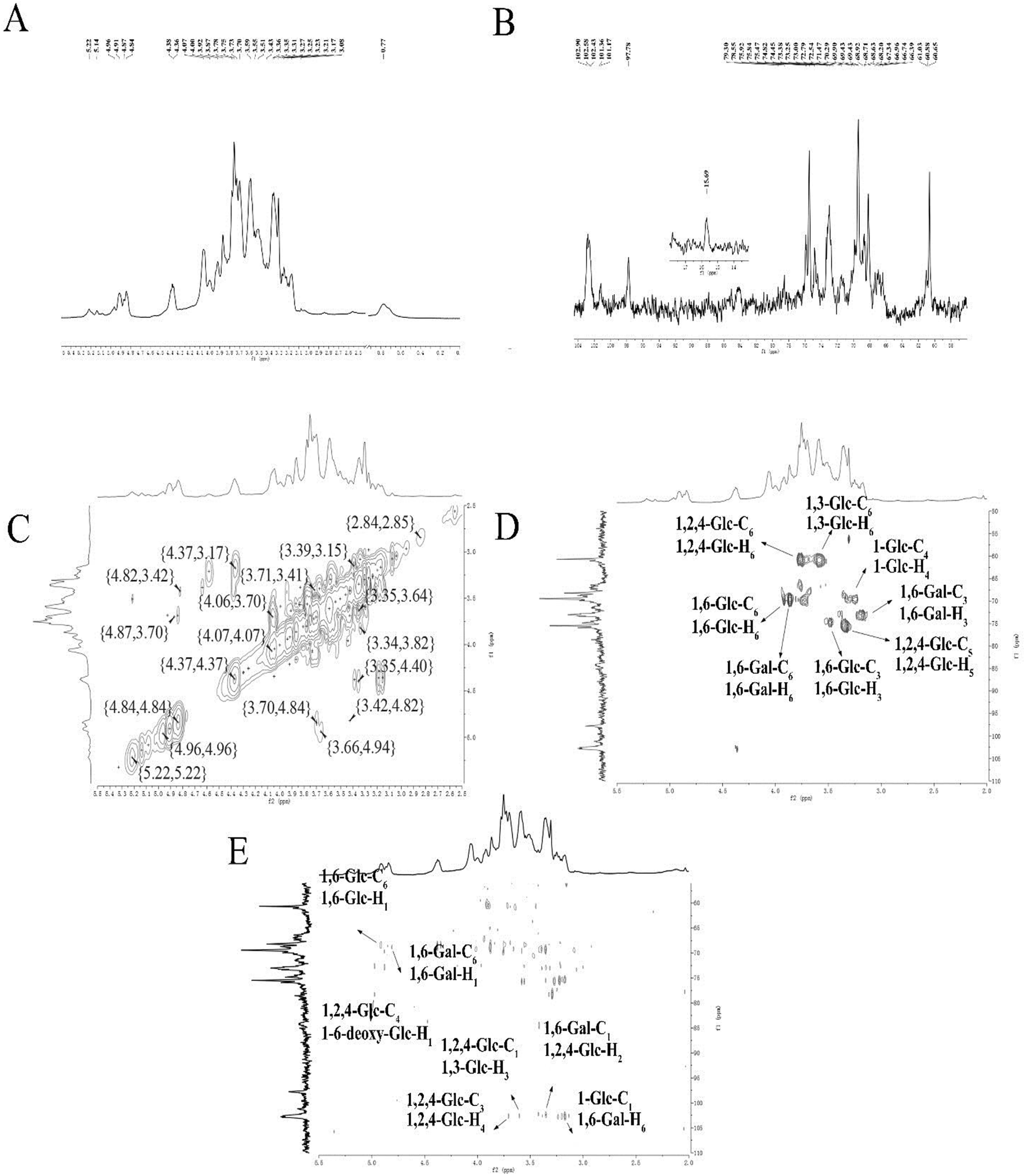 | ||
| Fig. 2 NMR spectra of ACP2 in D2O. (A) 1H NMR spectrum. (B) 13C NMR spectrum. (C) 1H-1H COSY spectrum. (D) HSQC spectrum. (E) HMBC spectrum. | ||
| Sugar residues | δ 13C/1H (ppm) | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| 6-Deoxy-α-D-Glcp-(1→ | 102.90 | 71.47 | 73.25 | 68.97 | 75.47 | 15.69 |
| 4.96 | 3.35 | 3.51 | 3.08 | 3.75 | 0.77 | |
| α-D-Glcp-(1→ | 101.36 | 74.82 | 73.38 | 69.90 | 75.84 | 60.88 |
| 5.22 | 3.36 | 3.70 | 3.59 | 3.73 | 3.78/3.87 | |
| →3)-α-D-Glcp-(1→ | 97.78 | 74.82 | 73.38 | 69.43 | 75.84 | 60.88 |
| 5.14 | 3.78 | 3.59 | 3.31 | 3.75 | 3.78/3.87 | |
| →6)-α-D-Glcp-(1→ | 102.43 | 74.82 | 74.45 | 69.90 | 75.84 | 68.63 |
| 4.91 | 3.78 | 3.43 | 3.25 | 3.70 | 3.78/3.92 | |
| →6)-β-D-Galp-(1→ | 102.43 | 72.54 | 72.79 | 68.71 | 68.92 | 68.71 |
| 4.84 | 3.17 | 3.87 | 4.07 | 4.38 | 3.87/3.78 | |
| →2,4)-β-D-Glcp-(1→ | 102.58 | 79.30 | 75.47 | 78.55 | 75.47 | 60.65 |
| 4.87 | 3.35 | 3.87 | 3.70 | 3.35 | 3.78/4.36 | |
The correlations between hydrogen and carbon atoms were determined from the 1H-1H COSY and HSQC spectra (Fig. 2C and D, respectively).27 The signals at δ H 3.78, 4.36/δ C 60.65 were attributed to H-6/C-6 of →2, 4)-β-D-Glcp-(1→, and the peaks at δ H 3.78, 3.92/δ C 68.63 corresponded to H-6/C-6 of →6)-α-D-Glcp-(1→. The signals at δ H 3.87, 3.78/δ C 68.71 were attributed to H-6/C-6 of →6)-β-D-Galp-(1→. The peaks at δ H 3.43/δ C 74.45, δ H 3.35/δ C 74.47, and δ H 3.87/δ C 73.25 were assigned to H-3/C-3 of →6)-β-D-Glcp-(1→, H-5/C-5 of →2,4)-β-D-Glcp-(1→, and H-3/C-3 of →6)-β-D-Galp-(1→, respectively. The peaks crossing at δ H 3.59/δ C 69.90 and δ H 3.78, 3.87/δ C 60.880 were attributed to H-4/C-4 of α-D-Glcp-(1→ and H-3/C-3 of →3)-α-D-Glcp-(1→.
The HMBC spectrum indicates both the remote coupling correlation between carbon and hydrogen along with the corresponding position of the glycosidic linkage. As shown in Fig. 2E, the signals at δ H 4.91/δ C 68.63 ppm were attributed to H-1 of →6)-α-D-Glcp-(1→ and C-6 of →6)-α-D-Glcp-(1→, and the peaks at δ H 4.81/δ C 68.71 ppm were speculated to correspond to H-1 of →6)-β-D-Galp-(1→ and C-6 of →6)-β-D-Galp-(1→. The signals at δ H 4.96/δ C 78.55 ppm were assigned to H-1 of 6-deoxy-α-D-Glcp-(1→ and C-4 of →2,4)-β-D-Glcp-(1→, while the peaks at δ H 3.70/δ C 102.58 ppm attributed to H-4 of →2,4)-β-D-Glcp-(1→ and C-1 of →2,4)-β-D-Glcp-(1→. The signals at δ H 3.59/δ C 102.58 ppm were assigned to H-3 of →3)-α-D-Glcp-(1→ and C-1 of →2,4)-β-D-Glcp-(1→, whereas the signals at δ H 3.35/δ C 102.43 ppm were attributed to H-2 of →2,4)-β-D-Glcp-(1→ and C-1 of →6)-β-D-Galp-(1→. The signals at δ H 3.78/δ C 101.36 ppm corresponded to H-6 of →6)-β-D-Galp-(1→ and C-1 of α-D-Glcp-(1→. All 13C NMR chemical shifts are shown in Table 2.
In consideration of the monosaccharide composition, FT-IR spectroscopy, GC-MS, and 1H/13C NMR spectroscopy analyses, the likely backbone of ACP2 is as follows: →6)-linked-β-D-Galp-(1→, →6)-linked-α-D-Gclp-(1→, →3)-linked-α-D-Glcp-(1→, and →2,4)-linked-β-D-Glcp-(1→, with terminal 6-deoxy-α-D-Glcp and α-D-Glcp.
3.3. Protective effect of ACP2 on L02 cells
Prior to determining the anti-inflammatory activity of ACP2, we examined the cytotoxicity of LPS, GSH and ACP2 in L02 cells using MTT assay. As shown in Fig. 3A, LPS significantly inhibited the proliferation of L02 cells at the concentration of 10 μg mL−1 (##p < 0.01). GSH, a common reducing substance with good hepatoprotection, was used as a positive control in this study. However, GSH inhibited the growth of L02 cells when the concentration reached 15 mM (Fig. 3B); the optimal concentration of GSH was 10 mM. As shown in Fig. 3C, ACP2 had no obvious toxic effect on L02 cells until its concentration reached 200 μg mL−1 (##p < 0.01). The data also indicated a protective effect of ACP2 compared to LPS as concentrations of 25, 50 and 100 μg mL−1. Therefore, ACP2 promoted the proliferation of L02 cells. In subsequent tests, we evaluated ACP2 concentrations of 25 and 100 μg mL−1, the GSH concentration of 10 mM, and the LPS concentration of 10 μg mL−1 (Fig. 3D).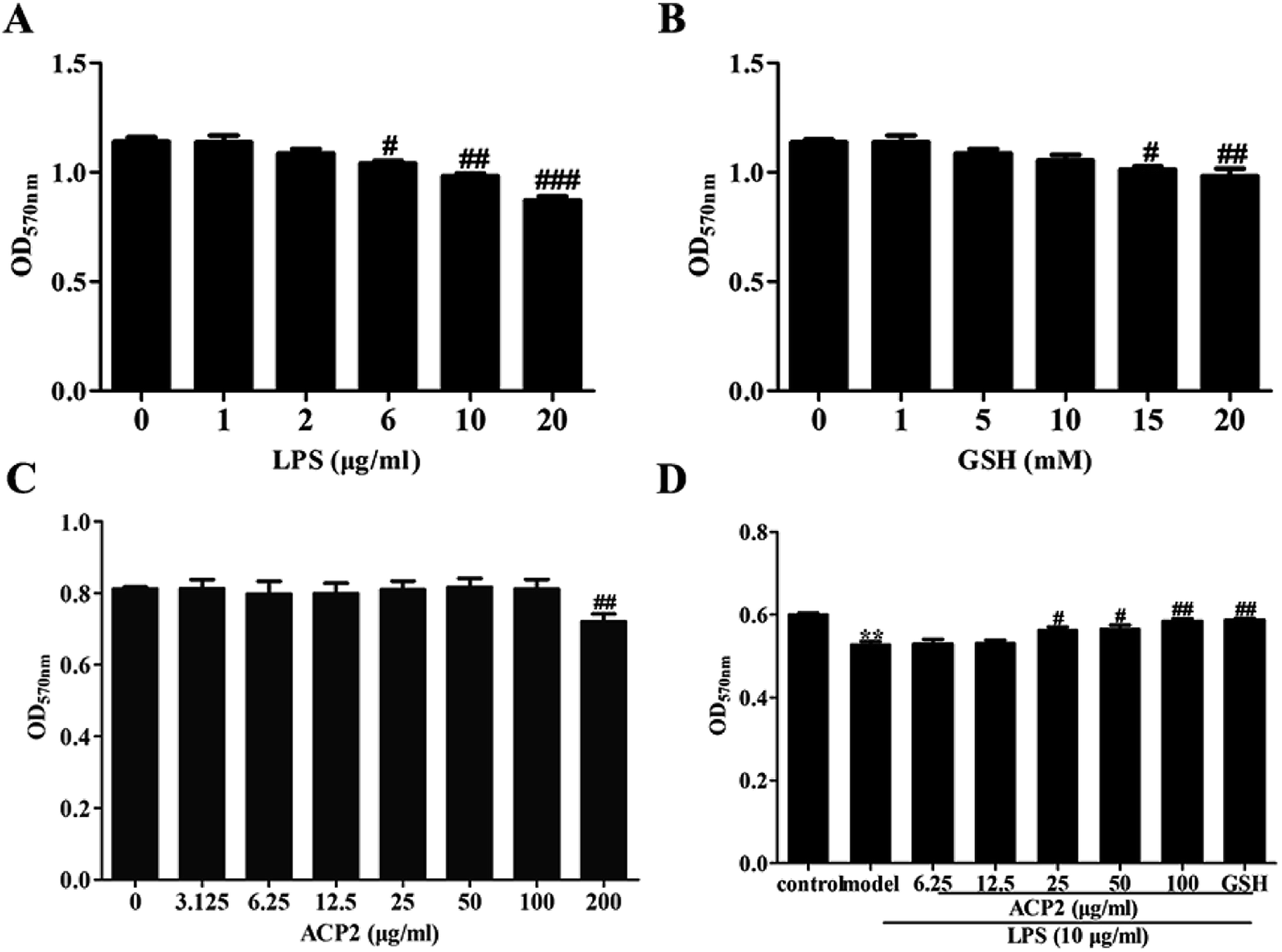 | ||
| Fig. 3 Cell viability of ACP2 on L02 cells induced by LPS. MTT assay was conducted on L02 cells which treated with different concentrations of ACP2 for 24 h. (A) Effects of various concentrations of LPS on L02 cells. (B) Effects of various concentrations of GSH on L02 cells. (C) Effects of various concentrations of ACP2 on L02 cells. (D) The proliferation of L02 cells when treated with ACP2 and LPS (n = 6, **p < 0.01 vs. the control, #p < 0.05 and ##p < 0.01 vs. the model). | ||
3.4. ACP2 inhibited TLR4/NF-κB signal pathway
Inflammation is an intricate pathophysiological phenomenon that is thought to depend on some pathogen recognition receptors such as TLRs,28 which play a pivotal role in liver injury.5 Recent studies have demonstrated the presence of TLRs in different cell types, including macrophages, dendritic cells and hepatocytes.29 Given that the mass of hepatic cells in the liver, therefore, TLRs can be responsive to various pathogens. MyD88, a signaling adaptor that can be activated by TLRs, plays a crucial role in various biological and pathological processes in hepatocytes. MyD88 triggers the secretion of inflammatory factors that regulate and amplify inflammatory response through TLR signals.30 To determine if ACP2 regulates the TLR4-MyD88 signaling pathway in hepatic cells, we evaluated the protein levels of TLR4 and MyD88 in L02 cells. As shown in Fig. 4A–C, compared to the control group, the expressions of TLR4 and MyD88 were significantly up-regulated by treatment with LPS (##p < 0.01), while ACP2 treatment clearly down-regulated TLR4 and MyD88 at the concentration of 100 μg mL−1 (##p < 0.01). These results imply that the anti-inflammatory effect of ACP2 is related to the TLR4-MyD88 signaling pathway.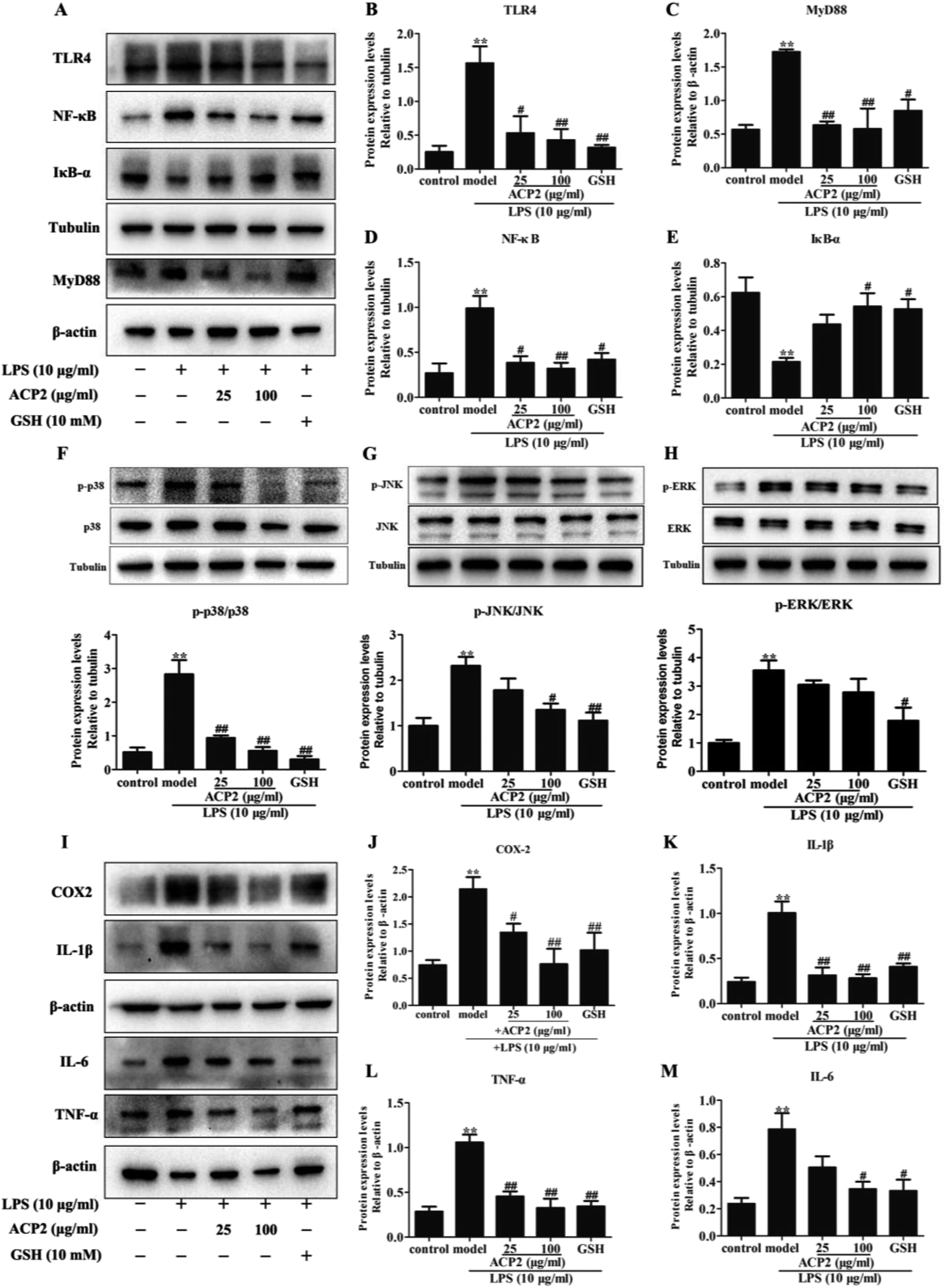 | ||
| Fig. 4 (A) ACP2 attenuates the up-regulation of protein expression by LPS in L02 cells. Columns of the protein expression levels of TLR4 (B), MyD88 (C), NF-κB (D), IκB-α (E). ACP2 down-regulated the expression of protein associated with MAPKs. The p-p38 (F), p-JNK (G), p-ERK (H) expression levels which was standardized by tubulin. (I) The protein levels of COX-2 (J), IL-1β (K), TNF-α (L), IL-6 (M). (n = 3, *p < 0.05 and **p < 0.01 vs. the control, #p < 0.05 and ##p < 0.01 vs. the model). | ||
Numerous studies have shown that NF-κB and MAPKs are two downstream effectors of TLR4-MyD88 signaling that regulate the expression of pro-inflammatory cytokines.31,32 NF-κB, which is one of the most pivotal downstream targets of MyD88, it activated by two signaling pathways: the NF-κB translocation-dependent pathway and the MAPK phosphorylation pathway. NF-κB is a heterodimer of p50 and p65 held in the cytosol with an inactive state by the inhibitor IκB-α. NF-κB is activated and translocated to the nucleus via phosphorylation followed by the degradation of IκB-α.33 p38, a member of MAPK family, regulates the transcription of NF-κB and contributes to the up-regulation of some pro-inflammatory factors due to considered as an upstream mediators kinase of NF-κB.34 Therefore, we examined whether ACP2 regulated the expression of proteins related to MAPKs and the NF-κB signaling pathway. First, we assessed the protein levels of IκB-α and NF-κB. The results indicated that LPS dramatically inhibited the level of IκB-α and increased the level of NF-κB. Meanwhile, ACP2 at the concentration of 100 μg mL−1 attenuated the up-regulation of NF-κB and promoted IκB-α (Fig. 4D and E; ##p < 0.01).
3.5. ACP2 regulated MAPKs signal pathways
Next, we evaluated the protein levels of p38, p-p38, JNK, p-JNK, ERK and p-ERK. As shown in Fig. 4F–H, ACP2 at the concentration of 25 μg mL−1 attenuated the levels of p-p38/p38, which were up-regulated by LPS (##p < 0.01). At a concentration of 100 μg mL−1, ACP2 down-regulated p-JNK/JNK (#p < 0.05) but had no significant regulatory effect on the expression of p-ERK/ERK. These results confirm that ACP2 exhibited an anti-inflammation effect in L02 cells, mainly through its effect on the p38/NF-κB signaling pathway.3.6. ACP2 down-regulated LPS-induced inflammation-related proteins
The NF-κB and p38 signaling pathways were involved in the regulation of LPS-induced IL-1β and TNF-α.33 Previous studies indicated that NF-κB activation and MAPK phosphorylation are pre-conditions for the production of inflammation-related factors, including IL-1β, COX-2, TNF-α and IL-6.35,36 Notably, TNF-α and IL-1β were released by some immune cells and contributed to the expression of cytokines, particularly IL-6, which was produced via macrophages and then induced an inflammatory response when exposed to LPS in the liver. Furthermore, COX-2, which is one inducible isoform of cyclooxygenase, is considered as response to miscellaneous inflammatory stimuli such as LPS.37 Therefore, to determine the protective effect of ACP2 against LPS-induced damage to L02 cells, we measured the expressions of a series of mediators, including IL-1β, COX-2, TNF-α, and IL-6. Compared to the control group, the expressions of these factors were significantly increased after treatment with LPS (Fig. 4I–M), and ACP2 attenuated these increases, in agreement with a previous study.38 Therefore, ACP2 exhibited a protective effect by down-regulating inflammation-related proteins.4. Conclusion
In the present study, ACP2, a galactoglucan from the mycelium of cultured A. camphorata with 6-deoxyglucose in the sugar chain, was reported for the first time. Purified, homogeneous ACP2 was water soluble and had a molecular weight of 1.93 × 104 Da. Based on by monosaccharide composition analysis, ACP2 consisted mainly of glucose, galactose and 6-deoxyglucose in a molar ratio of 5:2:1. The backbone of ACP2 had the following composition: →6)-linked-β-D-Galp-(1→, →6)-linked-α-D-Gclp-(1→, →3)-linked-α-D-Glcp-(1→, and →2,4)-linked-β-D-Glcp-(1→, with terminal 6-deoxy-α-D-Glcp and α-D-Glcp. In tests with L02 cells, ACP2 exhibited protective effects by attenuating the expressions of IL-1β, COX-2, TNF-α and IL-6. ACP2 achieved these anti-inflammatory effects through the regulation of the TLR4-MyD88 signaling pathway by down-regulating the expression of proteins associated with the MAPK and NF-κB signaling pathways. In summary, the results indicate that ACP2 can attenuate liver inflammation by down-regulating the expressions of inflammation-related factors via the TLR4 receptor and p38/NF-κB signaling pathways. The relationship between the anti-inflammatory activity and monosaccharide composition or glycosidic linkage (e.g., the existence of 6-deoxyglucose) should be discussed in a further study.
Conflicts of interest
The authors confirm that there are no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81773985), “Double-First-Class” University project (CPU2018GF/GY16), and the 2018 Fundamental Research Funds for the Central Universities (No. 2632018ZD06). The project was funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).References
- O. Krenkel and F. Tacke, Nat. Rev. Immunol., 2017, 17, 306–321 CrossRef CAS PubMed.
- W. Yu, Z. Pan, Y. Zhu, F. An and Y. Lu, Eur. J. Pharmacol., 2017, 812, 234–242 CrossRef CAS PubMed.
- K. Hamesch, E. Borkham-Kamphorst, P. Strnad and R. Weiskirchen, Lab. Anim., 2015, 49, 37–46 CrossRef CAS PubMed.
- W. Lee, M. H. Hwang, Y. Lee and J. S. Bae, Chem.-Biol. Interact., 2018, 281, 106–110 CrossRef CAS.
- S. T. Anderson, S. Commins, P. N. Moynagh and A. N. Coogan, Brain, Behav., Immun., 2015, 43, 98–109 CrossRef CAS PubMed.
- X. Xia, J. Fu, X. Song, Q. Shi, C. Su, E. Song and Y. Song, Free Radicals Biol. Med., 2015, 89, 522–532 CrossRef CAS PubMed.
- L. A. Orci, S. Lacotte, V. Delaune, F. Slits, G. Oldani, V. Lazarevic, C. Rossetti, L. Rubbia-Brandt, P. Morel and C. Toso, J. Hepatol., 2018, 68, 978–985 CrossRef CAS PubMed.
- S. Y. Park, M. L. Jin, Y. H. Kim, Y. Kim and S. J. Lee, Int. Immunopharmacol., 2012, 14, 13–20 CrossRef CAS PubMed.
- Z. Rasheed and T. M. Haqqi, Biochim. Biophys. Acta, 2012, 1823, 2179–2189 CrossRef CAS PubMed.
- D. Cao, J. Luo, D. Chen, H. Xu, H. Shi, X. Jing and W. Zang, Sci. Rep., 2016, 6, 23132 CrossRef CAS PubMed.
- Y. Li, L. Chen and C. Wang, et al., Am. J. Transl. Res., 2016, 8, 5619–5627 CAS.
- J. Xue, F. Chen, J. Wang, S. Wu, M. Zheng, H. Zhu, Y. Liu, J. He and Z. Chen, Cell. Physiol. Biochem., 2015, 35, 1557–1570 CrossRef CAS PubMed.
- J. J. Cheng, C. H. Chao, P. C. Chang and M. K. Lu, Food Hydrocolloids, 2016, 53, 37–45 CrossRef CAS.
- G. J. Huang, J. S. Deng, S. S. Huang, Y. Y. Shao, C. C. Chen and Y. H. Kuo, Food Chem., 2012, 132, 709–716 CrossRef CAS.
- Z. M. Lu, W. Y. Tao, H. Y. Xu, Z. H. Ao, X. M. Zhang and Z. H. Xu, Nat. Prod. Res., 2011, 25, 684–695 CrossRef CAS PubMed.
- H. W. Chiu and K. F. Hua, PLoS One, 2016, 11, e0153087 CrossRef PubMed.
- L. M. Meng, M. H. Pai, J. J. Liu and S. L. Yeh, Nutrition, 2012, 28, 942–949 CrossRef CAS PubMed.
- Q. Chen, H. Tang, Z. Zha, H. Yin, Y. Wang, Y. Wang, H. Li and L. Yue, Int. J. Biol. Macromol., 2017, 104, 768–777 CrossRef CAS PubMed.
- M. Wang, X. B. Yang, J. W. Zhao, C. J. Lu and W. Zhu, Carbohydr. Polym., 2017, 156, 390–402 CrossRef CAS PubMed.
- H. Tang, W. Wei, W. Wang, Z. Zha, T. Li, Z. Zhang, C. Luo, H. Yin, F. Huang and Y. Wang, Carbohydr. Polym., 2017, 163, 43–53 CrossRef CAS PubMed.
- K. Fukuda, T. Uematsu, A. Hamada and S. Akiya, Chem. Pharm. Bull., 1975, 23, 1955–1959 CrossRef CAS.
- I. Ciucanu and F. Kerek, Carbohydr. Res., 1984, 131, 209–217 CrossRef CAS.
- X. Xu, L. Yang, X. Xu, X. Wang, X. Chen, Q. Liang, J. Zeng and X. Jing, J. Controlled Release, 2005, 108, 33–42 CrossRef CAS.
- J. Chokboribal, W. Tachaboonyakiat, P. Sangvanich, V. Ruangpornvisuti, S. Jettanacheawchankit and P. Thunyakitpisal, Carbohydr. Polym., 2015, 133, 556–566 CrossRef CAS PubMed.
- W. Liu, Y. Liu, R. Zhu, J. Yu, W. Lu, C. Pan, W. Yao and X. Gao, Carbohydr. Polym., 2016, 147, 114–124 CrossRef CAS PubMed.
- M. X. Yan, W. J. Mao, X. Liu, S. Y. Wang, Z. Xia, S. J. Cao, J. Li, L. Qin and H. L. Xian, Carbohydr. Polym., 2016, 147, 272–281 CrossRef CAS.
- P. K. Agrawal, Phytochemistry, 1992, 31, 3307–3330 CrossRef CAS PubMed.
- L. A. Possamai, M. R. Thursz, J. A. Wendon and C. G. Antoniades, J. Hepatol., 2014, 61, 439–445 CrossRef CAS PubMed.
- D. H. Kim and T. R. Billiar, J. Korean Surg. Soc., 2011, 80, 194–203 CrossRef.
- L. Sen, Y. Qing and Z. Qi, et al., J. Neuroinflammation, 2012, 9, 2–14 Search PubMed.
- S. K. Ha, E. Moon, M. S. Ju, D. H. Kim, J. H. Ryu, M. S. Oh and S. Y. Kim, Neuropharmacology, 2012, 63, 211–223 CrossRef CAS.
- S. W. A. Himaya, B. Ryu, Z.-J. Qian and S.-K. Kim, Toxicol. In Vitro, 2012, 26, 878–887 CrossRef CAS.
- P. C. Liao, L. K. Chao, J. C. Chou, W. C. Dong, C. N. Lin, C. Y. Lin, A. Chen, S. M. Ka, C. L. Ho and K. F. Hua, Inflammation Res., 2012, 62, 89–96 CrossRef PubMed.
- W. B. Song, Y. Y. Wang, F. S. Meng, Q. H. Zhang, J. Y. Zeng, L. P. Xiao, X. P. Yu, D. D. Peng, L. Su, B. Xiao and Z. S. Zhang, PLoS One, 2010, 5, e12969 CrossRef PubMed.
- Y. Q. Chen, L. Rong and J. O. Qiao, Mol. Med. Rep., 2014, 10, 1400–1408 CrossRef CAS PubMed.
- M. S. Yoo, J. S. Shin, H. E. Choi, Y. W. Cho, M. H. Bang, N. I. Baek and K. T. Lee, Food Chem., 2012, 135, 967–975 CrossRef CAS PubMed.
- C. K. Khatri, K. S. Indalkar, C. R. Patil, S. N. Goyal and G. U. Chaturbhuj, Bioorg. Med. Chem. Lett., 2017, 27, 1721–1726 CrossRef CAS.
- D. K. Park and H. J. Park, BMC Complementary Altern. Med., 2013, 2013, 914524 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10347j |
| ‡ Both the authors worked equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |