
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTiO2-coated LiCoO2 electrodes fabricated by a sputtering deposition method for lithium-ion batteries with enhanced electrochemical performance†
Sang-Hyun Moon,
Min-Cheol Kim,
Eun-Soo Kim,
Yeon-Kyung Shin,
Ji-Eun Lee,
Sojeong Choi and
Kyung-Won Park *
*
Department of Chemical Engineering, Soongsil University, Seoul 06978, Republic of Korea. E-mail: kwpark@ssu.ac.kr; Fax: +82-2-812-5378; Tel: +82-2-820-0613
First published on 11th March 2019
Abstract
We fabricated lithium cobalt oxide (LiCoO2, LCO) electrodes in the absence and presence of TiO2 layers as cathodes for lithium-ion batteries (LIBs) using a sputtering deposition method under an Ar atmosphere. In particular, TiO2 coating layers on sputtered LCO electrodes were directly deposited in a layer-by-layer form with varying TiO2 sputtering times from 60 to 120 s. These sputtered electrodes were heated at 600 °C in an air atmosphere for 3 h. The thicknesses of TiO2 layers in TiO2-coated LCO electrodes were controlled from ∼2 to ∼10 nm. These TiO2-coated LCO electrodes exhibited superior electrochemical performance, i.e. high capacities (93–107 mA h g−1@0.5C), improved retention of >60% after 100 cycles, and high-rate cycling properties (64 mA h g−1@1C after 100 cycles). Such an improved performance of TiO2-coated LCO electrodes was found to be attributed to relieved volumetric expansion of LCO and protection of LCO electrodes against HF generated during cycling.
1 Introduction
Li-ion batteries (LIBs) are attractive as electrochemical power sources for portable electronic devices and electric vehicles due to their high energy densities and long cycle life.1–5 Recently, thin-film technologies that can fabricate small-sized LIBs for drones and medical devices have been reported.6–8 In particular, lithium cobalt oxide (LiCoO2, LCO) can be utilized as a thin-film type cathode due to its high theoretical energy density of 274 mA h g−1, high working voltage of ∼3.9 V, and excellent electrical conductivity.9–13 Sputtering deposition, chemical vapor deposition, and e-beam evaporation methods have been used to fabricate LCO thin-film electrodes.14–19 Among them, sputtering deposition methods with high process efficiency and excellent availability have been intensively studied.Despite various merits of LCO, LCO cathode material has poor stability at high temperature with structural unstability during cycling at >4.2 V vs. Li/Li+.20–22 To overcome these disadvantages, LCO-based electrode structures partially exchanged with electrochemically active transition metals such Fe, Ti, and Ni instead of Co or doped with inactive materials such as Al and Mg have been proposed.23–26 Furthermore, LCO electrode coated with metal oxides (Al2O3, ZrO2, and TiO2) has exhibited significantly improved LIB performance.27–31 In particular, metal oxide coating on LCO electrodes can significantly enhance the stability of electrodes by protecting active materials due to HF generated by side reaction of LiPF6 + H2O → LiF + POF3 + 2HF.32,33 Among these oxides, TiO2 is a promising candidate for coating on LCO electrodes because of its low cost, non-toxicity, and stability.34–36 In this study, we fabricated TiO2-coated LCO electrodes using sputtering deposition method for LIBs. These TiO2-coated LCO electrodes were evaluated and compared with LCO electrode without TiO2.
2 Experimental
Fabrication of LCO–TiO2 electrodes
LCO–TiO2 electrodes as cathodes for lithium-ion batteries (LIBs) were fabricated using a radio-frequency (RF) magnetron sputtering deposition method on Al foil (Hohsen Corporation) as a substrate using LiCoO2 (99.9% LTS chemical) and TiO2 (99.9% LTS chemical) as sputtering targets. Prior to a main sputtering, a pre-sputtering was performed for 30 min to remove impurities on the target surface. The main sputtering process was carried out under a working pressure of 1.1 × 10−2 torr with an Ar gas flow rate of 30 standard cubic centimeter per min (sccm). First, LCO layers were deposited with an RF power of 80 W for 3 h. Then, TiO2 layers were formed on the LCO electrode layers with an RF power of 10 W for 0, 60, 90, and 120 s (denoted as LCO–TiO2(0), LCO–TiO2(60), LCO–TiO2(90), and LCO–TiO2(120), respectively). For a homogeneous deposition, a substrate was rotated with a rotating speed of 3 rpm. These as-deposited electrodes were heated at 600 °C under an air atmosphere for 3 h. The loading amount of each electrode was about 0.13–0.16 mg cm−2.Structural characterization
To confirm the crystal structure of the as-formed electrodes, an X-ray diffractometer (XRD, D2 PHASER, Bruker AXS) was operated at a working voltage of 30 kV with a Cu Kα (λ = 0.15418 nm) and an Ni filter. The morphology and elemental distribution of the electrodes were characterized using a field emission scanning electron microscope (FE-SEM, GeminiSEM 300, ZEISS) operating at 2 kV.Electrochemical measurement
To characterize the electrochemical properties of the electrodes, coin cells (size 2032, Hohsen corporation) were assembled using the sputtered samples as cathodes in an Ar-filled glove box (5 ppm, H2O and O2). Lithium foil (FMC Corporation) and porous polyethylene film were utilized as counter electrode and separator, respectively. Then, 1.1 M LiPF6 (Soulbrain Co., Ltd.) in a mixture solution of ethylene carbonate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dimethyl carbonate (1:1) was used as electrolyte. Electrochemical analysis of the assembled coin cells was perfromed using a multi-channel tester (WBCS300L, Wonatech Co.). Charge and discharge processes were carried out at 25 °C at a galvanostatic/potentiostatic and a galvanostatic modes, respectively, in the potential range of 3.01–4.30 V vs. Li/Li+ for 100 cycles. High-rate performance of each sample was characterized at varying current densities from 0.1 to 1C. To evaluate the stability of the electrodes at a high potential, the charge–discharge process was performed in the potential range of 3.01–4.50 V vs. Li/Li+ at a current density of 0.5C for 100 cycles. Cyclic voltammograms (CVs) of the electrodes were obtained by sweeping in the potential range of 3.01–4.30 V vs. Li/Li+ at a scan rate of 0.1 mV s−1.
:dimethyl carbonate (1:1) was used as electrolyte. Electrochemical analysis of the assembled coin cells was perfromed using a multi-channel tester (WBCS300L, Wonatech Co.). Charge and discharge processes were carried out at 25 °C at a galvanostatic/potentiostatic and a galvanostatic modes, respectively, in the potential range of 3.01–4.30 V vs. Li/Li+ for 100 cycles. High-rate performance of each sample was characterized at varying current densities from 0.1 to 1C. To evaluate the stability of the electrodes at a high potential, the charge–discharge process was performed in the potential range of 3.01–4.50 V vs. Li/Li+ at a current density of 0.5C for 100 cycles. Cyclic voltammograms (CVs) of the electrodes were obtained by sweeping in the potential range of 3.01–4.30 V vs. Li/Li+ at a scan rate of 0.1 mV s−1.
3 Results and discussion
Fig. 1(a) and (c) show the XRD patterns of the electrodes formed on Al foils using a sputtering deposition method. Prior to a heating process, the as-deposited electrodes exhibited amorphous LiCoO2 and TiO2 phases with the XRD peaks related to an Al foil used as a substrate. To obtain high-crystalline structure of a LiCoO2 cathode material, the electrodes were heated at 600 °C under an air atmosphere. Fig. 1(b) and (d) show the XRD patterns of the heated electrodes (LCO–TiO2(0), LCO–TiO2(60), LCO–TiO2(90), and LCO–TiO2(120)). The XRD peak at 2θ = 19° corresponds to the (003) plane of LiCoO2 layered structure.37–39 On the other hand, no XRD peak related to TiO2 phase appeared, demonstrating the formation of an amorphous TiO2 on the LiCoO2 crystalline structure. Fig. 2 shows SEM cross-sectional images of the as-prepared LCO–TiO2(0), LCO–TiO2(60), LCO–TiO2(90), and LCO–TiO2(120) electrodes. The thicknesses of LCO–TiO2(0), LCO–TiO2(60), LCO–TiO2(90), and LCO–TiO2(120) were 398, 402, 406, and 410 nm, respectively. With increasing deposition time of TiO2 from 60 to 120 s, the film thickness increased from 402 to 410 nm due to the additional deposition of TiO2 layer on the LCO electrode with a thickness of 398 nm (Fig. S1†). Thus, the thicknesses of the TiO2 layers in LCO–TiO2(60), LCO–TiO2(90), and LCO–TiO2(120) were 4, 8, and 12 nm, respectively.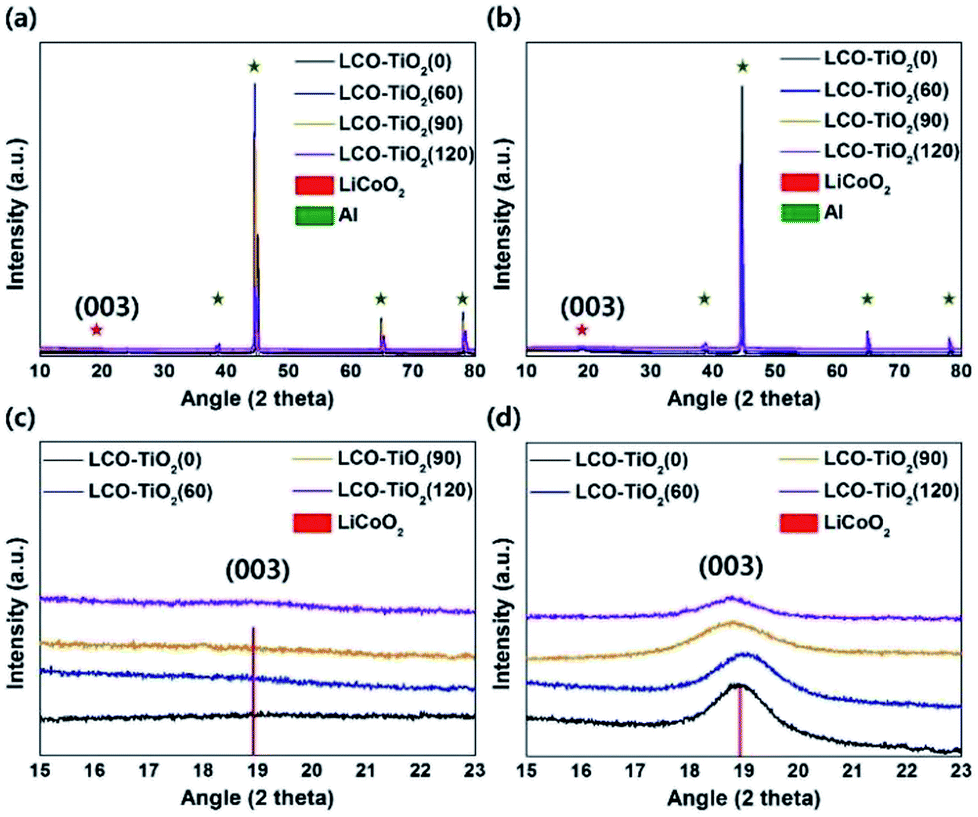 | ||
| Fig. 1 XRD patterns of (a) the as-prepared and (b) heated LCO–TiO2 electrodes. Enlarged XRD patterns to see the (003) plane of LiCoO2 of (c) the as-prepared and (d) heated LCO–TiO2 electrodes. | ||
 | ||
| Fig. 2 (a) Cross-section SEM images of LCO–TiO2(0), (b) LCO–TiO2(60), (c) LCO–TiO2(90), and (d) LCO–TiO2(120). | ||
Charge/discharge characteristic curves of LCO–TiO2(0), LCO–TiO2(60), LCO–TiO2(90), and LCO–TiO2(120) were obtained in the potential range of 0–4.3 V vs. Li/Li+ with 0.5 C-rate for 100 cycles (Fig. 3(a) and S2†). LCO–TiO2(0) exhibited an initial capacity of 80 mA h g−1 and a rapid capacity drop after 50 cycles. In contrast, LCO–TiO2(60), LCO–TiO2(90), and LCO–TiO2(120) with TiO2-coating layers showed relatively high capacities of 93, 103, and 107 mA h g−1, respectively without serious capacity drop up to 100 cycles. The capacity retentions of LCO–TiO2(0), LCO–TiO2(60), LCO–TiO2(90), and LCO–TiO2(120) were 2.5%, 26.6%, 60.2%, and 52.3%, respectively. Fig. 3(b) shows comparison of rate cycling performance of the electrodes measured with varying current densities from 0.1 to 1.0C. The average capacities of LCO–TiO2(0) measured at 0.1, 0.2, 0.3, 0.4, 0.5, 0.7, and 1.0C, were 80, 65, 51, 37, 21, 7, and 1 mA h g−1, respectively. To investigate recovery in capacity, the average capacities of LCO–TiO2(0) measured at 0.1 and 0.2C were 6 and 0.4 mA h g−1, respectively, demonstrating the damaged electrode due to fast cycling process. The average capacities of LCO–TiO2(60) measured at 0.1, 0.2, 0.3, 0.4, 0.5, 0.7, and 1.0C, were 75, 71, 65, 60, 53, 44, and 34 mA h g−1, respectively. The average capacities of LCO–TiO2(90) measured at 0.1, 0.2, 0.3, 0.4, 0.5, 0.7, and 1.0C, were 95, 82, 75, 69, 64, 58, and 51 mA h g−1, respectively. The average capacities of LCO–TiO2(120) measured at 0.1, 0.2, 0.3, 0.4, 0.5, 0.7, and 1.0C, were 78, 68, 60, 54, 48, 42, and 37 mA h g−1, respectively. Compared to LCO–TiO2(0), the TiO2-coated LCO electrodes showed high average capacities and improved recovery properties. Such enhanced electrochemical performance of the LCO electrodes containing amorphous TiO2 layers with nanometer scale thickness may be attributed to fast ionic conductivity and low charge transfer resistance.28,36,40
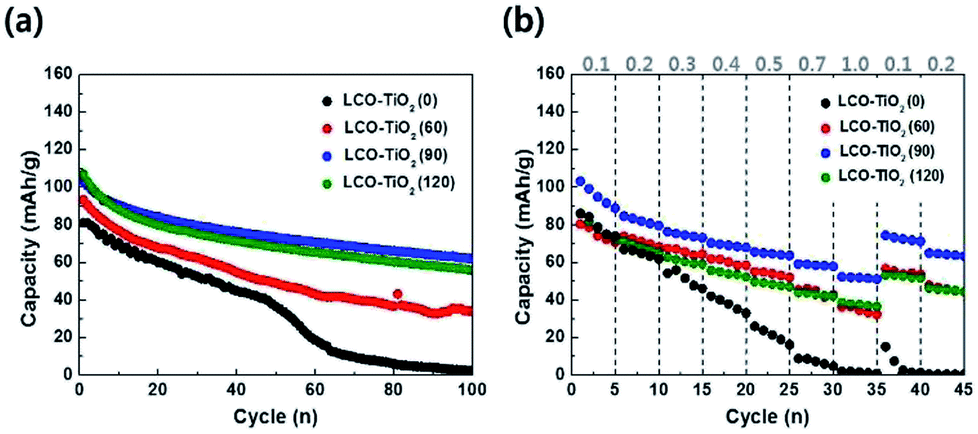 | ||
| Fig. 3 (a) Comparison of cycling performance for the electrodes measured at 0.5C for 100 cycles in the potential range of 3.01–4.30 V vs. Li/Li+. (b) High-rate performance of the electrodes measured with varying C-rates from 0.1 to 1.0 C. | ||
Fig. 4(a)–(d) shows cyclic voltammograms (CVs) measured with a scan rate of 0.1 mV s−1 in the potential range of 3.0–4.3 V for LCO–TiO2(0), LCO–TiO2(60), LCO–TiO2(90), and LCO–TiO2(120), respectively. All these electrodes exhibited anodic and cathodic peaks at 3.86 and 3.92 V, respectively, associated with deinsertion reaction and insertion of Li+ ions, respectively, in crystalline LiCoO2 electrodes. In general, LCO electrodes heated at relatively low temperatures showed anodic and cathodic peaks at 3.64 and 3.75 V, respectively. Interestingly, another cathodic peaks corresponding to insertion of Li+ ions in amorphous LiCoO2 electrode appeared at 3.82 V.39 Despite fairly high heating temperature of 600 °C, slight amorphous LiCoO2 phases were found to be remained in these electrodes.
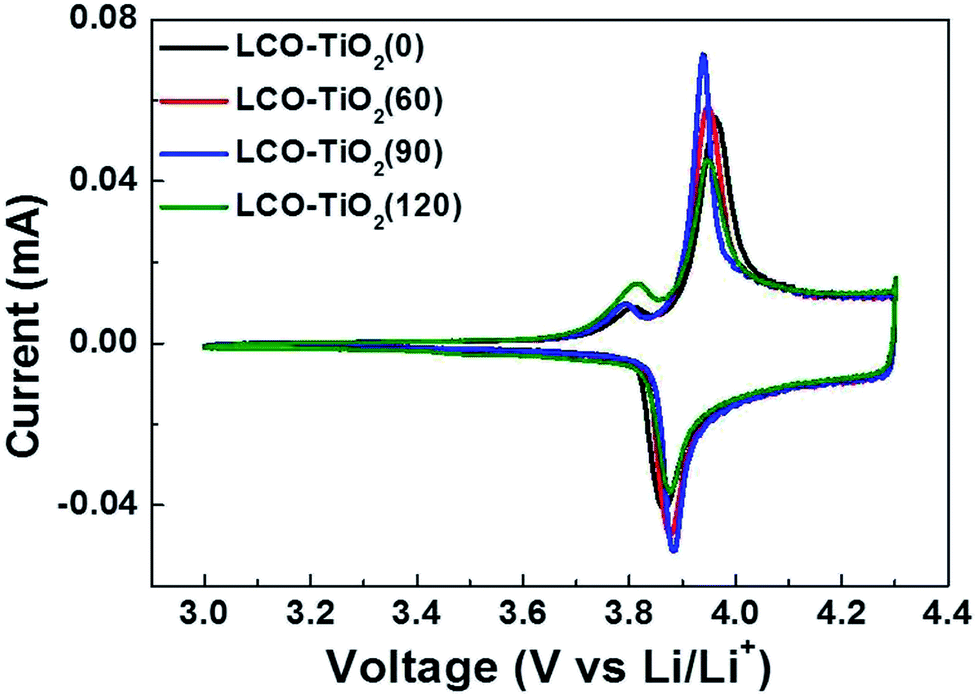 | ||
| Fig. 4 Cyclic volatmmograms (CVs) for the electrodes measured at a scan rate of 0.1 mV s−1 in the potential range of 3.01–4.30 V vs. Li/Li+. | ||
Fig. 5 shows SEM surface images of the electrodes before and after 100 cycles in the potential range of 0–4.3 V vs. Li/Li+ with 0.5 C. As shown in Fig. 5(a)–(d), the as-prepared LCO–TiO2(0), LCO–TiO2(60), LCO–TiO2(90), and LCO–TiO2(120) electrodes before cycling exhibited fairly homogeneous deposition of LCO and TiO2. In addition, from EDX mapping images, all electrodes showed the well-dispersion of elements related to LCO and TiO2 (Fig. S3†) and the increased TiO2 content with increasing sputtering time of TiO2 target (Table S1†). Fig. 5(e)–(h) and S4† show SEM surface and cross-sectional images of LCO–TiO2(0), LCO–TiO2(60), LCO–TiO2(90), and LCO–TiO2(120), respectively, after 100 cycles. LCO–TiO2(0) exhibited the surface crack and increased thickness (∼4.9 μm) of the film due to volume expansion of LCO during the cycling. LCO–TiO2(60) with 4 nm TiO2 layer also exhibited the surface crack and increased thickness (∼2.9 μm). On the other hand, LCO–TiO2(90) and LCO–TiO2(120) after 100 cycles showed significantly less surface damage and thickness (1.4 and 1.6 μm, respectively). The structural stability of LCO–TiO2(90) and LCO–TiO2(120) during cycling might be due to inhibition of volumetric expansion of LCO having TiO2 coating layer with proper thickness.
 | ||
| Fig. 5 SEM images of (a) and (e) LCO–TiO2(0), (b) and (f) LCO–TiO2(60), (c) and (g) LCO–TiO2(90), and (d) and (h) LCO–TiO2(120) before and after 100 cycles at a current density of 0.5C. | ||
To further investigate the effect of TiO2 layer on the LCO, as shown in Fig. 6, the charge/discharge characteristic curves of LCO–TiO2(0) and LCO–TiO2(90) were obtained in the potential range of 0–4.5 V with 0.5C-rate for 100 cycles. The initial capacities of LCO–TiO2(0) and LCO–TiO2(90) were 83 and 112 mA h g−1, respectively. However, LCO–TiO2(0) prepared without TiO2 layer exhibited a drastic capacity drop after 10 cycles and no capacity after 70 cycles whereas LCO–TiO2(90) with a TiO2 coating layer (8 nm in thickness) exhibited a discharge capacity of 45 mA h g−1 after 100 cycles with a retention rate of 40.2%. The surface images of LCO–TiO2(0) and LCO–TiO2(90) after 100 cycles in the potential range of 0–4.5 V were shown in Fig. S5.† LCO–TiO2(0) exhibited the serous surface damage such as the formation of holes (Fig. S5(a)†) whereas LCO–TiO2(90) exhibited a fairly clean surface state after cycling up to 4.5 V (Fig. S5(b)†). The surface states of LCO–TiO2(90) before and after cycling were characterized using XPS analysis (Fig. S6†). In the Ti2p spectra, the characteristic peaks corresponding to TiO2 were observed before and after cycling. Furthermore, the Li1s spectra contained the characteristic peaks at 24.1 and 54.4 eV before cycling with an additional peak at 55.2 eV associated with LiF after cycling. In the O1s spectra, the peaks at 529.6 eV corresponding to LiCoO2 lattice were maintained during cycling with additional peaks at 531–534 eV related to LiO2 and Li2CO3. In general, the formation reaction of HF (LiPF6 + H2O → LiF + POF3 + 2HF) during charge/discharge process can occur at a high electrode potential of >4.4 V.29 The formed HF can lead to the deteriorated electrochemical performance of electrodes due to the damage of electrochemically active materials by HF.41,42 However, the TiO2 coating layer in LCO–TiO2(90) can protect the LCO cathode from attacking by HF during the cycling, exhibiting improved electrochemical performance.
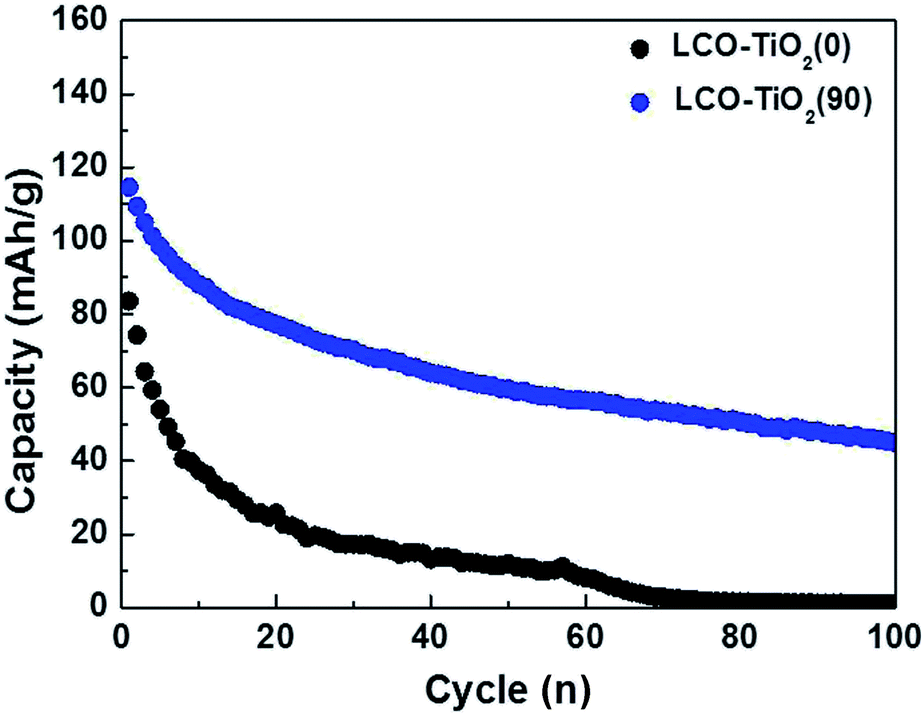 | ||
| Fig. 6 Comparison of cycling performance for LCO–TiO2(0) and LCO–TiO2(90) measured at 0.5C for 100 cycles in the potential range of 3.01–4.50 V vs. Li/Li+. | ||
4 Conclusions
In summary, LCO electrodes in the absence and presence of TiO2 layers as cathodes for LIBs were fabricated using sputtering deposition method. TiO2 coating layers on the LCO electrodes were formed with varying TiO2 sputtering time. Compared to LCO–TiO2(0) prepared without TiO2 coating layer, the TiO2-coated LCO electrodes with proper layer thicknesses exhibited enhanced electrochemical performance, i.e. high capacity, improved retention, and high-rate cycling properties. The superior performance of the TiO2-coated LCO electrodes was found to be attributed to the reduced volumetric expansion of LCO having TiO2 coating layer with a proper thickness and the protection of the LCO electrodes against HF generated during cycling. Consequently, such TiO2-coated LCO electrode can be used as a cathode candidate for high-performance thin-film LIBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Technology Development Program to Solve Climate Changes of the National Research Foundation (NRF) funded by the Ministry of Science, ICT (NRF-2017M1A2A2086648).Notes and references
- K. Kang, Y. S. Meng, J. Breger, C. P. Grey and G. Ceder, Science, 2014, 311, 977–980 CrossRef PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- S. Kim, M. Kim, S. Han, G. Lee, H. Choe, D. Kwak, S. Choi, B. Son, M. Shin and K. Park, Nano Energy, 2016, 27, 545–553 CrossRef CAS.
- Y. Wang, B. Liu, Q. Li, S. Cartmell, S. Ferrara, Z. D. Deng and J. Xiao, J. Power Sources, 2015, 286, 330–345 CrossRef CAS.
- L. Xue, S. V. Savilov, V. V. Lunin and H. Xia, Adv. Funct. Mater., 2018, 28, 1–10 Search PubMed.
- S. Pat, S. Özen, H. H. Yudar, Ş. Korkmaz and Z. Pat, J. Alloys Compd., 2017, 713, 64–68 CrossRef CAS.
- R. Robert, C. Villevieille and P. Novák, J. Mater. Chem. A, 2014, 2, 8589–8598 RSC.
- S. Schuld, R. Hausbrand, M. Fingerle, W. Jaegermann and K. M. Weitzel, Adv. Energy Mater., 2018, 8, 1703411 CrossRef.
- Z. Wang, Z. Wang, W. Peng, H. Guo, X. Li, J. Wang and A. Qi, Ionics, 2014, 20, 1525–1534 CrossRef CAS.
- Q. Yang, J. Huang, Y. Li, Y. Wang, J. Qiu, J. Zhang, H. Yu, X. Yu, H. Li and L. Chen, J. Power Sources, 2018, 388, 65–70 CrossRef CAS.
- G. Ting-Kuo Fey, C. Z. Lu, T. Prem Kumar and Y. C. Chang, Surf. Coat. Technol., 2005, 199, 22–31 CrossRef CAS.
- B. Wang, J. B. Bates, F. X. Hart, B. C. Sales, R. A. Zuhr and J. D. Robertson, J. Electrochem. Soc., 1996, 143, 3203–3213 CrossRef CAS.
- W. S. Yoon, S. H. Ban, K. K. Lee, K. B. Kim, M. G. Kim and J. M. Lee, J. Power Sources, 2001, 97–98, 282–286 CrossRef CAS.
- J. S. Wook and S.-M. Lee, J. Electrochem. Soc., 2007, 154, A22–A25 CrossRef CAS.
- H. HakanYudar, S. Pat, S. Özen, R. Mohammadigharehbagh, C. Musaoğlu, Ş. Korkmaz and Z. Pat, Vacuum, 2018, 152, 248–251 CrossRef CAS.
- J. B. Bates, N. J. Dudney, B. J. Neudecker, F. X. Hart, H. P. Jun and S. A. Hackney, J. Electrochem. Soc., 2000, 147, 59–70 CrossRef CAS.
- Z. Li, S. Yasui, S. Takeuchi, A. Creuziger, S. Maruyama, A. A. Herzing, I. Takeuchi and L. A. Bendersky, Thin Solid Films, 2016, 612, 472–482 CrossRef CAS.
- Q. Liu, X. Su, D. Lei, Y. Qin, J. Wen, F. Guo, Y. A. Wu, Y. Rong, R. Kou, X. Xiao, F. Aguesse, J. Bareño, Y. Ren, W. Lu and Y. Li, Nat. Energy, 2018, 1–8 Search PubMed.
- T. Zerrin, M. Ozkan and C. S. Ozkan, MRS Adv., 2018, 3, 3513–3518 CrossRef CAS.
- S. P. da Silva, L. E. Sita, C. S. dos Santos, F. H. Pavoni, H. de Santana, A. C. Andrello, P. R. C. da Silva and J. Scarminio, Mater. Chem. Phys., 2018, 213, 198–207 CrossRef.
- S. Hu, C. Wang, L. Zhou, X. Zeng, L. Shao, J. Zhou, C. Zhou, C. Huang, X. Xi and L. Yang, Ceram. Int., 2018, 44, 14995–15000 CrossRef CAS.
- K. Sivajee Ganesh, B. Purusottam reddy, P. Jeevan Kumar and O. M. Hussain, J. Electroanal. Chem., 2018, 828, 71–79 CrossRef CAS.
- B. J. Kwon, P. J. Phillips, B. Key, F. Dogan, J. W. Freeland, C. Kim, R. F. Klie and J. Cabana, Nanoscale, 2018, 10, 6954–6961 RSC.
- Y. Gao and J. H. Li, Adv. Mater. Res., 2012, 630, 93–98 Search PubMed.
- D. Zuo, G. Tian, D. Chen and K. Shu, Int. J. Electrochem. Sci., 2017, 12, 5044–5057 CrossRef CAS.
- S. Tian, L. L. Liu, Y. S. Zhu, Y. Y. Hou, C. L. Hu and Y. P. Wu, Funct. Mater. Lett., 2013, 6, 10–13 Search PubMed.
- J. L. Tebbe, A. M. Holder and C. B. Musgrave, ACS Appl. Mater. Interfaces, 2015, 7, 24265–24278 CrossRef CAS PubMed.
- J.-P. Noh, K.-T. Jung, M.-S. Jang, T.-H. Kwon, G.-B. Cho, K.-W. Kim and T.-H. Nam, J. Nanosci. Nanotechnol., 2013, 13, 7152–7154 CrossRef PubMed.
- X. Li, J. Liu, X. Meng, Y. Tang, M. N. Banis, J. Yang, Y. Hu, R. Li, M. Cai and X. Sun, J. Power Sources, 2014, 247, 57–69 CrossRef CAS.
- S. Yang, H. Qin, X. Li, H. Li and P. Yao, J. Nanomater., 2017, 2017, 1–10 Search PubMed.
- Y. Bi, T. Wang, M. Liu, R. Du, W. Yang, Z. Liu, Z. Peng, Y. Liu, D. Wang and X. Sun, RSC Adv., 2016, 6, 19233–19237 RSC.
- S.-J. Kim, M.-C. Kim, D.-H. Kwak, D.-M. Kim, G.-H. Lee, H.-S. Choe and K.-W. Park, J. Power Sources, 2016, 304, 119–127 CrossRef CAS.
- A. Zhou, Y. Lu, Q. Wang, J. Xu, W. Wang, X. Dai and J. Li, J. Power Sources, 2017, 346, 24–30 CrossRef CAS.
- S. S. Jayasree, S. Nair and D. Santhanagopalan, ChemistrySelect, 2018, 3, 2763–2766 CrossRef CAS.
- J.-P. Noh, G.-B. Cho, K.-T. Jung, W.-G. Kang, C.-W. Ha, H.-J. Ahn, J.-H. Ahn, T.-H. Nam and K.-W. Kim, Mater. Res. Bull., 2012, 47, 2823–2826 CrossRef CAS.
- H. Y. Park, S. C. Nam, Y. C. Lim, K. G. Choi, K. C. Lee, G. B. Park, H. P. Kim and S. B. Cho, Korean J. Chem. Eng., 2006, 23, 832–837 CrossRef CAS.
- J. P. Noh, K. T. Jung, G. B. Cho, S. H. Lee, K. W. Kim and T. H. Nam, J. Nanosci. Nanotechnol., 2012, 12, 5937–5941 CrossRef CAS PubMed.
- Y. Cho, J. Eom and J. Cho, J. Electrochem. Soc., 2010, 157, A617–A624 CrossRef CAS.
- Y. S. Jung, A. S. Cavanagh, A. C. Dillon, M. D. Groner, S. M. George and S. H. Lee, J. Electrochem. Soc., 2010, 157, A75–A81 CrossRef CAS.
- Y. Li, G. M. Veith, K. L. Browning, J. Chen, D. K. Hensley, M. P. Paranthaman, S. Dai and X. G. Sun, Nano Energy, 2017, 40, 9–19 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10451d |
| This journal is © The Royal Society of Chemistry 2019 |