
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental and theoretical study for CO2 activation and chemical fixation with epoxides†
Jinwei Gao ab,
Liuyi Lib,
Caiyan Cuib,
Muhammad Asad Ziaeeb,
Yaqiong Gonga,
Rongjian Sa*c and
Hong Zhong*ab
ab,
Liuyi Lib,
Caiyan Cuib,
Muhammad Asad Ziaeeb,
Yaqiong Gonga,
Rongjian Sa*c and
Hong Zhong*ab
aCollege of Science, North University of China, Taiyuan, Shanxi 030051, P. R. China
bState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China. E-mail: zhonghong@fjirsm.ac.cn
cInstitute of Oceanography, Ocean College, Fujian Provincial Key Laboratory of Information Processing and Intelligent Control, Minjiang University, Fuzhou, Fujian 350108, China. E-mail: rjsa@mju.edu.cn
First published on 29th April 2019
Abstract
The synthesis of five-membered cyclic carbonates via catalytic cycloaddition reaction of CO2 with epoxides is considered to be an effective technology for alleviation of the energy crisis and global warming. Various commercial organic bases and ionic salts were used as catalysts, while the relationship of catalytic activity and compound structure has been seldom explored. Herein, a facilely obtained binary catalytic system based on triethylamine/NBu4Br was developed for CO2 activation and chemical fixation. The highly efficient catalytic system showed outstanding conversion and above 99% selectivity under metal-free mild reaction conditions (100 °C, 1 atm) in one hour. The detailed process of CO2 activation and chemical fixation was investigated at the molecular level by a series of experiments and theoretical calculation, which provided a mode for the design and synthesis of a highly efficient catalytic system for conversion of CO2 under mild conditions.
Introduction
Carbon dioxide (CO2) excessive emission is a main component of greenhouse gases in the atmosphere.1–3 Its conversion to value-added products has offered one promising way to alleviate the energy crisis and global warming.4–7 As an economical and abundant C1 source, CO2 is renewable, inexpensive and non-toxic,8–11 but its chemical inertness is still the bottleneck for its applicability as a raw material in industry. The catalytic synthesis of cyclic carbonate from epoxides and CO2 is considered to be one of the most promising pathways to CO2 utilization.12–14 Cyclic carbonates have wide applications as green polar aprotic solvents, fuel additives, and chemical intermediates.15–18 The chemical fixations of CO2 into cyclic carbonates are relatively simple from a chemical reactivity point of view, while in fact, the high temperature, high pressure, high catalyst loading, or a combination of these are required to make the reaction effective, which is not economically suitable and poses safety concerns as well.19–22 To optimize the reaction conditions, a great variety of catalysts for the synthesis of cyclic carbonates have been developed so far, including alkali-metal halides, metal complexes, metal oxides, cellulose, MOFs, zeolites, ionic liquids, carbon nitride and so on.23–32 Though most catalysts were used to activate epoxide by a metal ion or hydrogen bond center, the effect of CO2 activity on the reaction efficiency has rarely been studied deeply.CO2 is the highest oxidation state of carbon, and it is thermodynamically stable and kinetically inert, which will consequently hinder the development of efficient catalysts that achieve CO2 activation and subsequently its functionalization.23,33,34 Thus the activation of CO2 is pivotal for its effective conversion. The introduce/use of Lewis basic species and transition metal system have been highly considered, while the detailed CO2 activation process was unclear,35–38 so developing a catalytic system that provides molecular level insight for CO2 activation process is still highly desired. Taking the aforementioned concerns into account, we propose to explore a new catalytic system that can activate and convert CO2 with epoxides under mild conditions, and serve as an ideal mold for providing a detailed mechanistic understanding of CO2 activation and fixation process.
Herein, a simple and efficient binary catalytic system based on organobase/NBu4Br was developed for CO2 cycloaddition reaction with epoxides under metal-free mild conditions. It was found that the synergistic effect between two components in this new catalytic system promote the cycloaddition reaction occur under atmospheric pressure during a short time period of 1 hour. Moreover, the relationship of catalytic activity and catalyst structure was investigated at molecular level by a series of experiments and theoretical calculation, which could not only offer in-depth understanding of the reaction mechanism but also provide a theoretical basis for the effect of triethylamine (NEt3) in activating CO2 and promoting the reaction process.
Results and discussion
To understand the effects of the counter anions on the catalytic activity, the catalytic cycloaddition reaction of CO2 with epoxides were initially investigated in the presence of 0.5 mL NEt3. As shown in Table 1, NBu4Br afforded a full conversion of 2-(chloromethyl)oxirane to 4-(chloromethyl)-1,3-dioxolan-2-one under 1.0 atm. CO2 at 100 °C for 1.0 h (entry 1), and the catalytic conversion showed unconspicuous change when the NBu4Br was replaced by NBu4Cl, NBu4I, respectively (entries 2–3). Notably, the yield of 2-(chloromethyl)oxirane to 4-(chloromethyl)-1,3-dioxolan-2-one decreased significantly when NBu4PF6 was used under the same condition, which is probably due to the weakest nucleophilicity of PF6− in selected Cl−, Br−, I−, and PF6−, suggesting the counter anions play a dominant role in cycloaddition reaction of CO2 with epoxides.39 When the common organic base NEt3 was used alone under the same condition, 36% yield of 2-(chloromethyl)oxirane to 4-(chloromethyl)-1,3-dioxolan-2-one was obtained (entry 6), indicating that NEt3 can activate CO2 in this reaction.40 Based on the above results, the high efficiency of the binary system is probably attributed to a synergistic effect between NBu4Br and NEt3 during the catalytic conversion of CO2 with epoxides to cyclic carbonates.| Entry | Catalyst | Time (h) | Yieldb (%) | Select.b (%) |
|---|---|---|---|---|
| a Reaction conditions: 2-(chloromethyl)oxirane (12.8 mmol), NBu4X (X = Br−, Cl−, I−, PF6−) (0.06 mmol), NEt3 (0.5 mL), CO2 (1 atm), 100 °C.b Yield was determined by GC and 1H NMR. The possibility of by-product was 3-chloro-1,2-propanediol. | ||||
| 1 | NBu4Br + NEt3 | 1 | 99 | >99 |
| 2 | NBu4Cl + NEt3 | 1 | 97 | >99 |
| 3 | NBu4I + NEt3 | 1 | 96 | >99 |
| 4 | NBu4PF6 + NEt3 | 1 | 43 | >99 |
| 5 | NBu4Br | 1 | 34 | >99 |
| 6 | NEt3 | 1 | 36 | >99 |
Additionally, the influence of the basicity on the conversion of 2-(chloromethyl)oxirane was investigated in the presence of NBu4Br with various organic bases at 100 °C for 1 h (Fig. 1a and b). As shown in Fig. 1b, the yield of 4-(chloromethyl)-1,3-dioxolan-2-one greatly related to the basicity of organic bases, and NEt3 gave rise to the highest conversion of 2-(chloromethyl)oxirane to corresponding product due to its strongest alkalinity and minimum steric hindrance. The activities of organic bases decreased sharply from 99% to 92 and 63% when the pKa decrease from 18.8 to 10.2, respectively. As reported previously, the base was weaker, the ΔG of this reaction was lower,41 so the basicity order might be DIPEA > NEt3 > TBA > TMEDA > MIm > Py > DMBA. However, the catalytic activity order of the organic bases is not in strict accordance with the established pKa, that was NEt3 > TBA > Py > TMEDA > MIm > DIPEA > DMBA, which indicates that the basicity of the organic bases is one important factor for promoting catalytic activity, but the steric-hindrance also play a role in this catalytic reaction.23
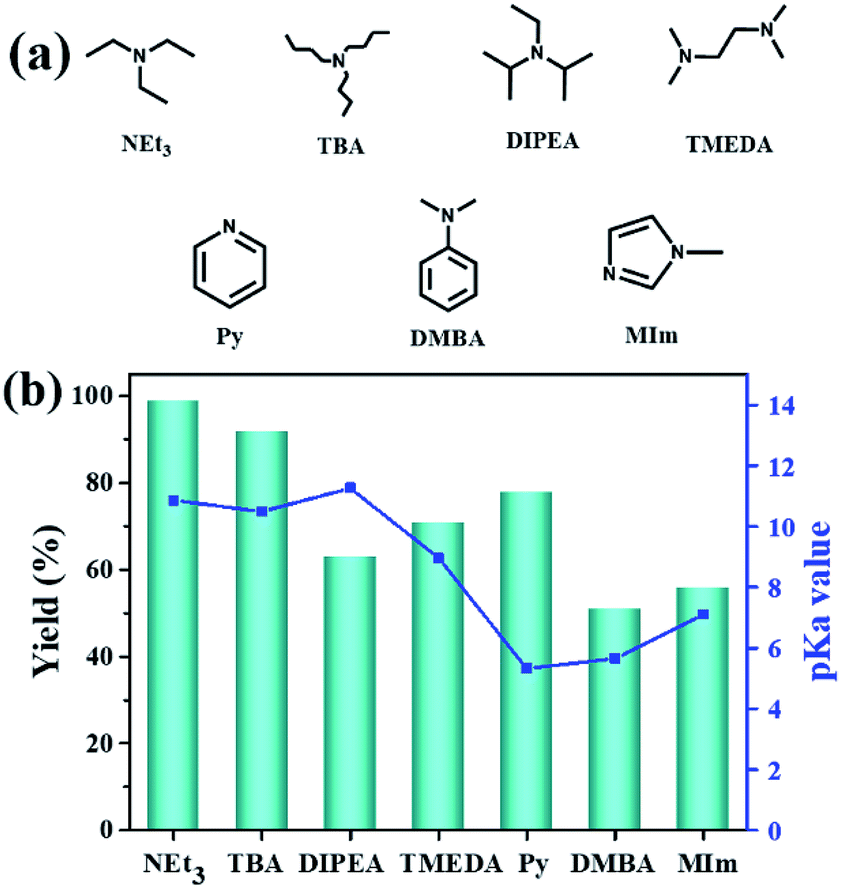 | ||
| Fig. 1 Organic bases structures (a); the effect of organic bases on the yield of cyclic carbonate (b). pKa value of the conjugated acid in water which was obtained from the U.S. National Library of Medicine database.42 Reaction conditions: 2-(chloromethyl)oxirane (12.8 mmol), NBu4Br (0.06 mmol), organic base (0.5 mL), CO2 (1 atm), 100 °C, 1 h. The possibility of by-product was 3-chloro-1,2-propanediol. | ||
The dependence of the cycloaddition reaction of CO2 and 2-(chloromethyl)oxirane on temperature is shown in Fig. 2a. The results indicated that the activity of this catalytic system is highly dependent on the reaction temperature. In the lower temperature region (25 to 50 °C), the yield of 4-(chloromethyl)-1,3-dioxolan-2-one increases slowly with increasing temperature. A further increase in temperature from 50 to 100 °C has significant effects on the 2-(chloromethyl)oxirane conversion, and gave the target product in 99% GC yield at 100 °C for 1 h.
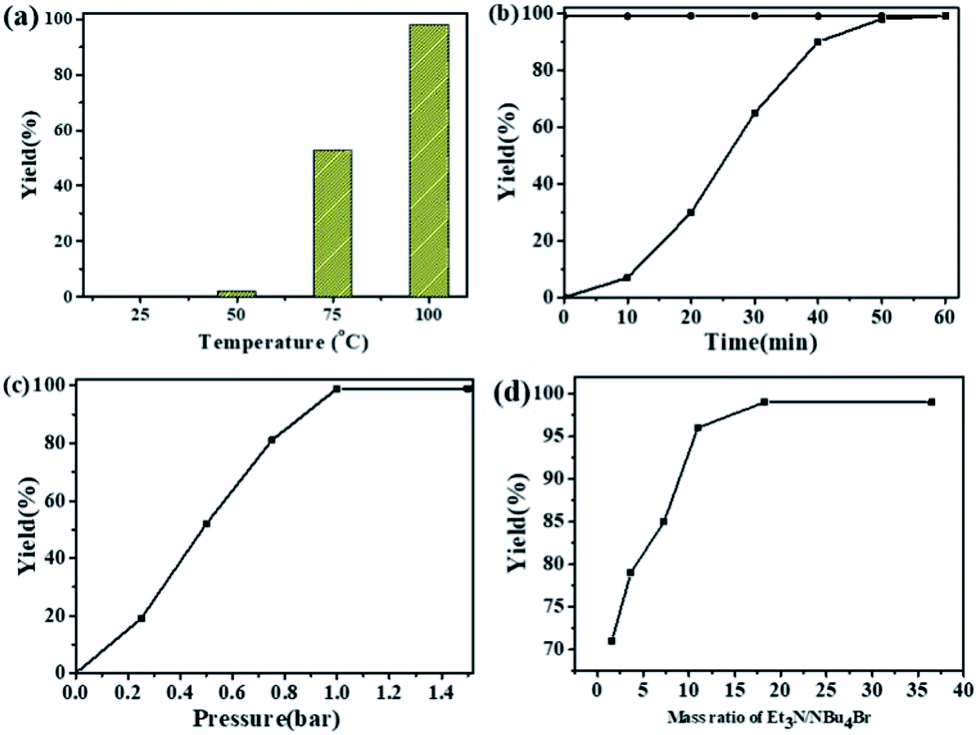 | ||
| Fig. 2 Effects of different reaction parameters. (a) Effects of reaction temperature, conditions: 2-(chloromethyl)oxirane (12.8 mmol), NBu4Br (0.06 mmol), NEt3 (0.5 mL), CO2 (1 atm), 1 h. (b) Effects of reaction time, conditions: 2-(chloromethyl)oxirane (12.8 mmol), NBu4Br (0.06 mmol), NEt3 (0.5 mL), CO2 (1 atm), 100 °C. (c) Effects of CO2 pressure, conditions: 2-(chloromethyl)oxirane (12.8 mmol), NBu4Br (0.06 mmol), NEt3 (0.5 mL), 100 °C, 1 h. (d) Effects of mass ratio of NEt3/NBu4Br, conditions: 2-(chloromethyl)oxirane (12.8 mmol), NBu4Br (0.06 mmol), CO2 (1 atm), 100 °C, 1 h. The possibility of by-product was 3-chloro-1,2-propanediol. | ||
The kinetic curve for catalytic conversion of CO2 into cyclic carbonates was also investigated in the presence of NEt3/NBu4Br at 100 °C. As shown in Fig. 2b, the yield of 4-(chloromethyl)-1,3-dioxolan-2-one increased rapidly in the first 40 min and then went up slowly. The complete consumption of 2-(chloromethyl)oxirane and synchronous formation of the desired product was achieved in 1 h. It should mentioned that the selectivity remains above 99% in the entire catalytic process. It could be obviously seen that the reaction pressure showed a great effect on the cycloaddition reaction (Fig. 2c). With the increase of CO2 from 0.25 to 1 atm, the 4-(chloromethyl)-1,3-dioxolan-2-one yield increases from 20 to 99%. A further increase in the CO2 pressure from 1 to 1.5 atm results in a same level in 2-(chloromethyl)oxirane conversion. A similar effect of CO2 pressure on catalytic activity was observed in other related catalytic systems.43,44
The influence of NEt3 to NBu4Br ratio on the yield of 4-(chloromethyl)-1,3-dioxolan-2-one was also investigated at 100 °C for 1 h with fixed NBu4Br (0.06 mmol). As shown in Fig. 2d, when the ratio of NEt3 to NBu4Br increases from 1.6 to 18.2, the 4-(chloromethyl)-1,3-dioxolan-2-one yield increased rapidly. Then the 2-(chloromethyl)oxirane conversion stayed almost constant when ratio of NEt3 to NBu4Br increased further.
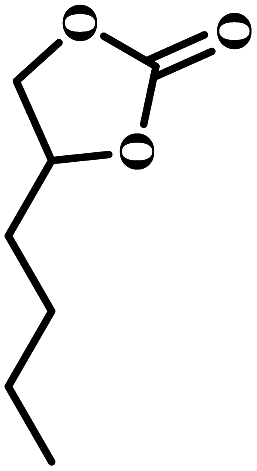
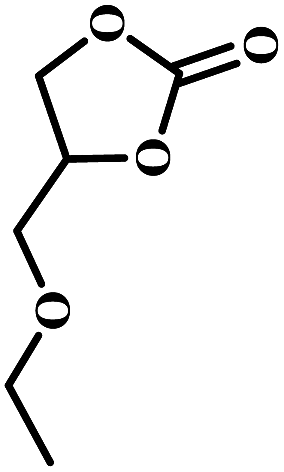
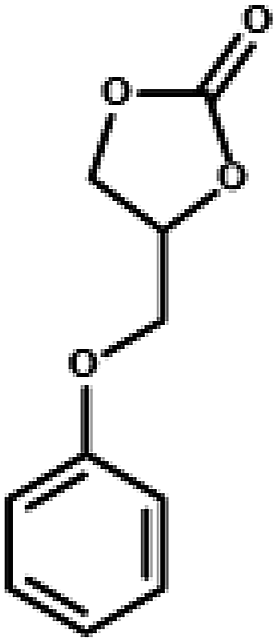
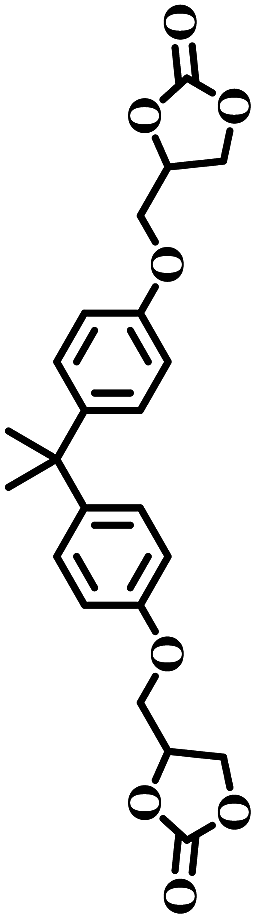
The excellent catalytic activity encouraged us to further explore the generality of the catalytic system, and several expoxides with different steric and electronic characters were tested. The results show that the various diverse epoxides were converted to the corresponding cyclic carbonates in high yields and excellent selective by this effective catalytic system (Table 2). The complete conversion of 2-(chloromethyl)oxirane is attributed to the presence of electron-withdrawing –CH2Cl group (entry 1), which facilitates nucleophilic attack of halide anions during the ring opening of the epoxide.45,46 The catalytic system is still effective for the epoxides with long alkyl carbon chain, such as 2-butyloxirane, 2-(ethoxymethyl)oxirane and 2-((allyloxy)methyl)oxirane (entries 2–4). The reactivity of epoxides with aromatic substituent is similar to that of long alkyl carbon chain (entries 5, 6). Interestingly, the complete conversion of 2,2′-(((propane-2,2-diylbis(4,1-phenylene))bis(oxy))bis(methylene))bis(oxirane) into 4,4′-(((propane-2,2-diylbis(4,1-phenylene))bis(oxy))bis(methylene))bis(1,3-dioxolan-2-one) could be achieved in 18 h (entry 7).
| Entry | Epoxide | Product | Time (h) | Yieldb (%) | Sel. (%) |
|---|---|---|---|---|---|
| a Reaction conditions: epoxide (12.8 mmol), NBu4Br (0.06 mmol), NEt3 (0.5 mL), CO2 (1 atm), 100 °C.b Yield was determined by GC and 1H NMR. The possibility of by-products were corresponding diols. | |||||
| 1 | 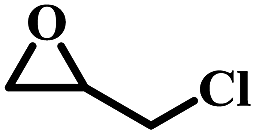 |
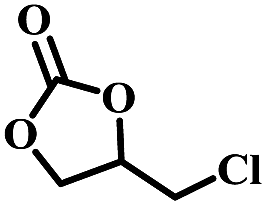 |
1 | 98 | >99 |
| 2 | 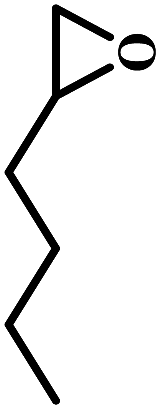 |
 |
1 | 7 | >99 |
| 10 | 59 | ||||
| 20 | 98 | ||||
| 3 | 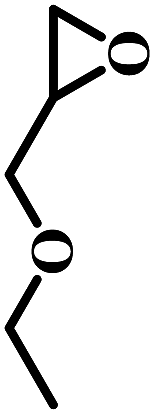 |
 |
1 | 17 | >99 |
| 8 | 82 | ||||
| 12 | 98 | ||||
| 4 | 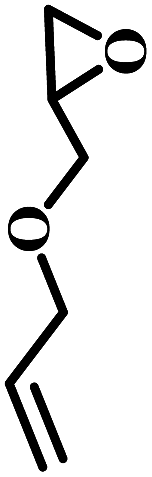 |
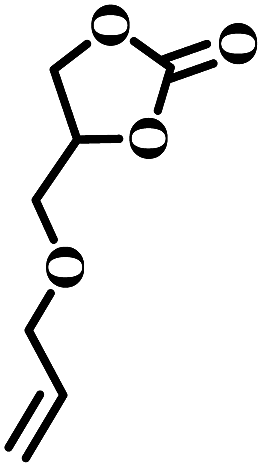 |
1 | 16 | >99 |
| 4 | 85 | ||||
| 8 | 99 | ||||
| 5 | 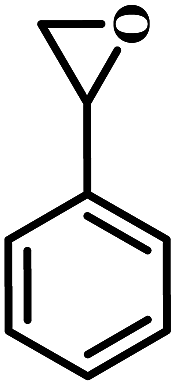 |
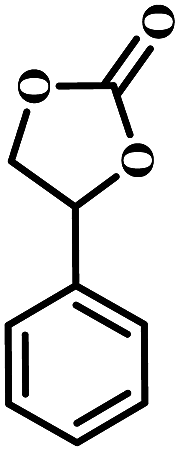 |
1 | 11 | >99 |
| 10 | 66 | ||||
| 20 | 99 | ||||
| 6 | 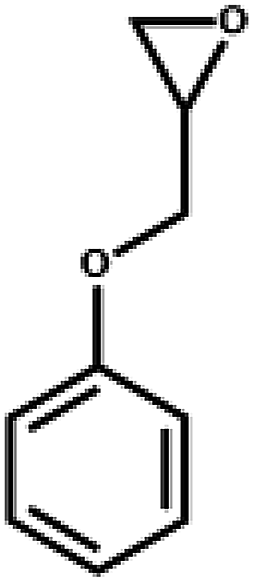 |
 |
1 | 17 | >99 |
| 4 | 95 | ||||
| 6 | 99 | ||||
| 7 | 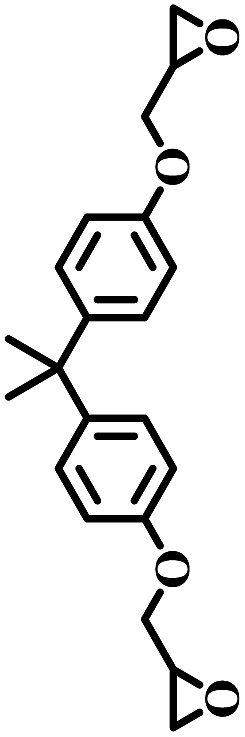 |
 |
1 | 21 | >99 |
| 8 | 79 | ||||
| 18 | 99 | ||||
To further understand the underlying principles of CO2 activation, the computation by the DFT (M06-2X) calculations was studied. Preliminary calculations indicated no involvement of NBu4+ cation (Fig. S1 in ESI†), which was consequently neglected from the elaborate calculations reported herein.47 With bromide as catalyst (Fig. 3), the ring opening through the attack of a nucleophile on epoxide and CO2 addition takes place simultaneously in aTS1, which is considered to be the rate-determining step with a free energy of 22.3 kcal mol−1. Subsequently, this reaction tended to undergo the carbonate ring-closure step (aTS2) has the energy of 10.8 kcal mol−1 compared to the reactants. This is consistent with the inefficient reaction under the mild experimental conditions.
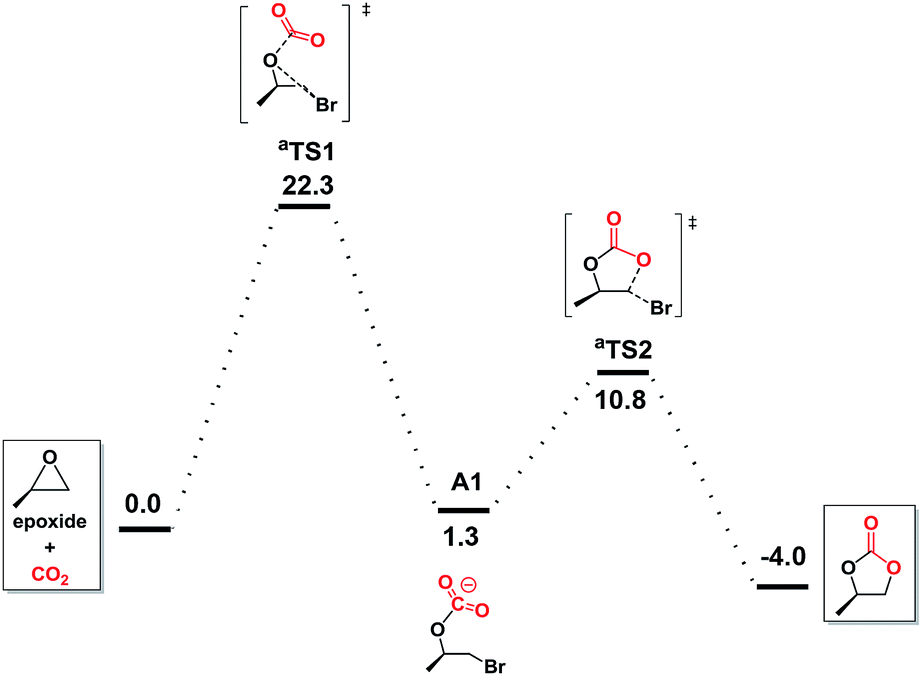 | ||
| Fig. 3 Free energy profiles for carbonate formation with bromine as catalyst in gas-phase. | ||
The efficiency of the reaction is promoted significantly when NEt3 is introduced as the catalyst in addition to the system. To gain further insight into the synergetic catalytic role played by NEt3 and NBu4Br, more detailed calculations were carried out. When the reaction was calculated in NEt3 solution, there different mechanisms were revealed (Fig. 4). The bromide-catalyzed process (black energy profile shown in Fig. 4) in NEt3 solution shows the same transition states and intermediates as in gas-phase. But the energy barrier of rate-determining step is 31.1 kcal mol−1, which is 8.8 kcal mol−1 larger this procedure without NEt3 solvent. Also the carbonate ring-closure step (aTS2) in NEt3 solution has an energy of 18.2 kcal mol−1, which is 7.4 kcal mol−1 larger than bromide-catalyzed process in gas. For comparison, we also calculated the reaction with NEt3 as catalyst in the absence of bromide (red profile). The NEt3-catalyzed epoxide ring-opening procedure has a high energy barrier (bTS1, 42.4 kcal mol−1, red profile in Fig. 4), which is 11.3 kcal mol−1 larger than bromide-catalyzed transition state in NEt3 solution. Afterwards, the CO2 addition step to form bTS2 has an energy of 46.0 kcal mol−1 relative to the energy of the reactants. Finally, this mechanism of ring-closure process may undergo a bTS3 transition states, which has an energy of 44.7 kcal mol−1 above the reactants. However, the energy barrier of 42.4 kcal mol−1 is quite high and consistent with a sluggish reaction and unable to take place under mild experimental conditions,48 indicating that NEt3 does not merely act as a solvent but also influences the course of the reaction. Thus, the mechanism when NEt3 participation tends to a bromine and NEt3 jointly catalyzed process (blue profile shown in Fig. 4), a new intermediate C1 appears, resulting from the CO2 addition upon interaction with NEt3 and bromine. This rate-determining step presented no transition states and has an energy of 17.5 kcal mol−1 the above reactants, 4.8 kcal mol−1 energy favorable than pure bromine-catalyzed process in gas-phase, 13.8 kcal mol−1 decrease than bromine-catalyzed procedure in NEt3 solvent, which allows the reaction to be performed under mild conditions. A potential energy surfaces scan calculation based on C1 intermediate validates transition states non-existent (Fig. S3 in ESI†). It is conceivable that the ring-opening of epoxide and CO2 addition might undergo synchronously with two catalysts. Afterwards, ring-closure takes place through cTS1. NEt3 then dissociates from the reacting system, which reverts to A1 and evolves to product through aTS2 with a free energy of 27.7 kcal mol−1 above the reactants. The calculated rate constant is 1.850 × 10−3 s−1 at 373 K. Then the corresponding half-life is ten minute, which accord with the experimental data. The intermediate of A1 with C1 were selected to compare the structure changes in reaction, the C–O bond of CO2 change slightly as shown in Fig. S4,† while the C–O bond between C atom of CO2 moiety and O atom of epoxide moiety decreases from 1.46 Å to 1.43 Å, then the interaction between CO2 and epoxide moieties enhanced with NEt3 as solution and catalyst. Moreover, the Mulliken charge of carbon atom of CO2 moiety decreases from 0.3 to 0.07 when compared C1 with A1. Therefore, the bond length and charge analysis suggest that the lone pair electron of nitrogen in NEt3 stabilized the formation of C1 and cTS1.
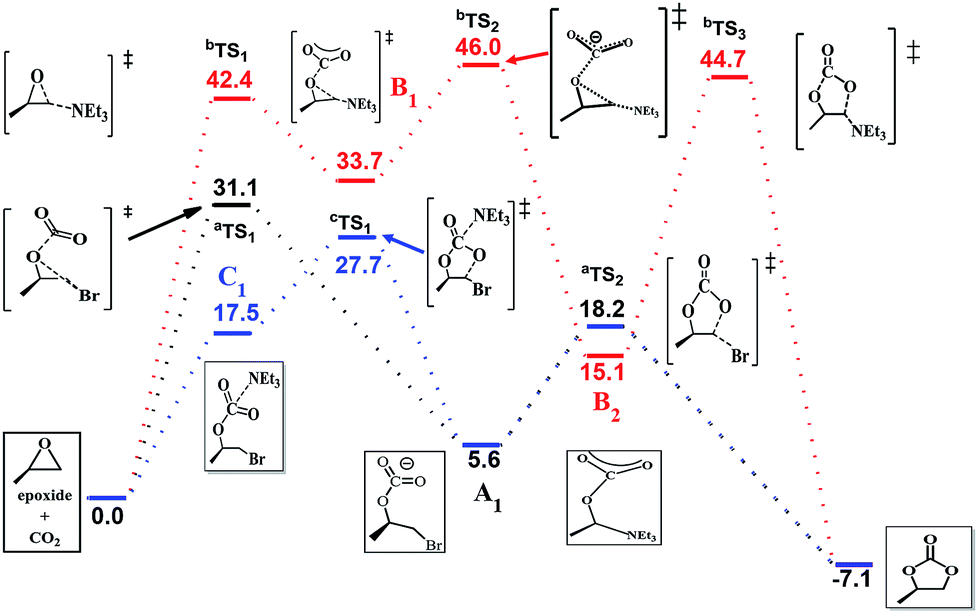 | ||
| Fig. 4 Free energy profiles of three different mechanisms in NEt3. | ||
Overall, a plausible mechanism for triethylamine-promoted catalytic conversion of CO2 into cyclic carbonates has been proposed based on the aforementioned results (Fig. 5). First, the epoxide ring opens through nucleophilic attack on the less sterically hindered β-carbon atom by Br− to produce an alkoxide ion, and simultaneously CO2 is activated by NEt3 via electrostatic interaction to form the carbamate salt. Then a new intermediate C1 is produced resulting from the nucleophilic attack on carbamate salt by alkoxide ion. Finally, cyclic carbonate is produced by following intramolecular ring-closure reaction of C1.
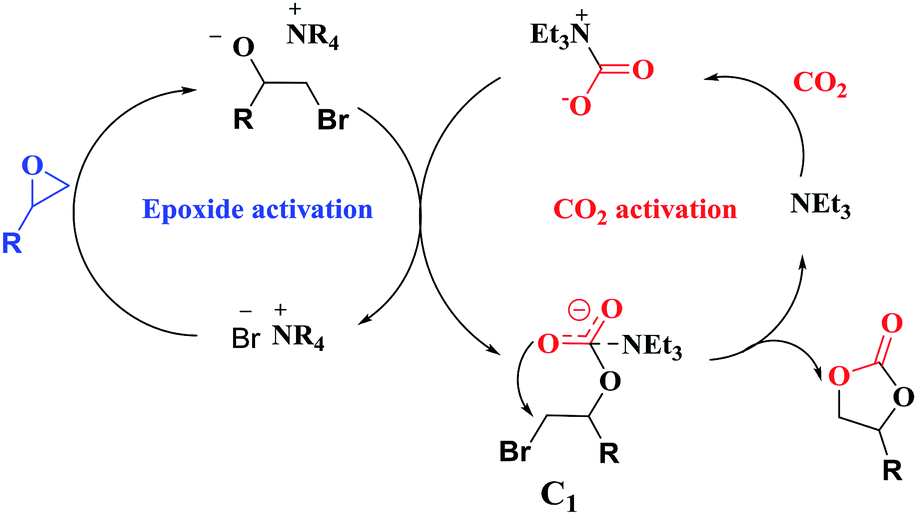 | ||
| Fig. 5 Proposed reaction pathway for cyclic carbonate formation catalyzed by NEt3 and NBu4Br. | ||
Conclusions
An efficient binary catalytic system containing triethylamine and NBu4Br was screened for catalytic conversion of CO2 and epoxides into cyclic carbonates under metal-free mild conditions. Especially, NEt3 could activate CO2 via electrostatic interaction and remarkably reduce the reaction energy to promote the reaction in the catalytic system. This work not only presents a simple and useful route for CO2 chemical fixation into high-value chemicals, but provides a detailed mechanistic understanding of CO2 activation and fixation process.Experimental
General
Chemicals including NEt3, NBu4Br, NBu4Cl, NBu4I, NBu4PF6, epoxides and CO2 are commercially available and used directly without further purification. Gas chromatography (GC) was performed on a Shimadzu GC-2014 equipped with a capillary column (RTX-5, 30 m × 0.25 μm) using a flame ionization detector.Typical catalytic reaction
The cycloaddition reaction was carried out by magnetic stirring, trimethylamine (0.5 ml), NBu4Br (0.06 mmol) and 2-(chloromethyl)oxirane (12.8 mmol) were added into a reactor at room temperature. Then, the reactor was sealed and purged with CO2 to remove air. CO2 was introduced into the reactor and the pressure was adjusted to 1 atm at room temperature. The reactor was placed into pre-heated oil bath and temperature was maintained at 100 °C. After the reaction was completed, the reactor was cooled to 0 °C in ice-water bath, and then the excess of CO2 was carefully vented. The mixture was diluted with ethyl acetate. The conversion of epoxide and yield of cyclic carbonate were determined by gas chromatography (Shimadzu GC-2014, a flame ionization detector) and 1H NMR.Computational details
The M06-2X functional49 was employed in this article to perform all the calculations. Our structure optimizations were as follows. In NBu4Br involved reaction, only gas-phase calculations were performed. For carbonate formation with bromine as catalyst, the structures were firstly optimized base on the level of 6-31+G(d,p) in gas-phase, then the solvent structure optimizations were carried out based on gas-phase results. As for the rest of other structure optimizations, the calculations were performed in solvent. Vibrational frequency analyses at the same basis sets were used on all optimized structures in order to characterize stationary points as local minima or transition states. Furthermore, the intrinsic reaction coordinate (IRC) calculations at the same level have been applied to validate that transition states connect appropriate reactants and products. The Gibbs free energy were further calculated by single-point energy calculations using M02-2X/6-311+G(d,p) method on previously 6-31+G(d,p) structures and thermal corrections at 298.15 K and 1 atm. No conformational sampling calculations were performed in this work. The continuum SMD model50 was applied. The Gaussian 09 package51 was used for all of our calculations in NEt3 solvent.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21603228 and 21671039) and Natural Science Foundation of Fujian Province (2015J01038), State Key Laboratory of Structural Chemistry and Program for Innovative Research Team in Science and Technology in Fujian Province University (IRTSTFJ) and the Open Project Program of the State Key Laboratory of Photocatalysis on Energy and Environment (SKLPEE-KF201813).Notes and references
- F. D. Bobbink, D. Vasilyev, M. Hulla, S. Chamam, F. Menoud, G. Laurenczy, S. Katsyuba and P. J. Dyson, ACS Catal., 2018, 8, 2589–2594 CrossRef CAS.
- B. B. Lu, W. Jiang, J. Yang, Y. Y. Liu and J. F. Ma, ACS Appl. Mater. Interfaces, 2017, 9, 39441–39449 CrossRef CAS PubMed.
- A. Buonerba, A. D. Nisi, A. Grassi, S. Milione, C. Capacchione, S. Vagin and B. Rieger, Catal. Sci. Technol., 2015, 5, 118–123 RSC.
- Q. L. Qian, J. J. Zhang, M. Cui and B. X. Han, Nat. Commun., 2016, 7, 11481 CrossRef PubMed.
- L. G. Ding, B. J. Yao, W. L. Jiang, J. T. Li, Q. J. Fu, Y. A. Li, Z. H. Liu, J. P. Ma and Y. B. Dong, Inorg. Chem., 2017, 56, 2337–2344 CrossRef CAS PubMed.
- C. Feng, X. L. Cao, L. G. Zhang, C. Y. Guo, N. Akram and J. D. Wang, RSC Adv., 2018, 8, 9192–9201 RSC.
- E. Fazekas, G. S. Nichol, M. P. Shaver and J. A. Garden, Dalton Trans., 2018, 47, 13106–13112 RSC.
- D. H. Lan, H. T. Wang, L. Chen, C. T. Au and S. F. Yin, Carbon, 2016, 100, 81–89 CrossRef CAS.
- L. Wang, R. L. Zhang, Q. X. Han, C. Xu, W. M. Chen, H. Yang, G. S. Gao, W. W. Qin and W. S. Liu, Green Chem., 2018, 20, 5311–5317 RSC.
- M. H. Anthofer, M. E. Wilhelm, M. Cokoja, M. Drees, W. A. Herrmann and F. E. Kühn, ChemCatChem, 2015, 7, 94–98 CrossRef CAS.
- L. Martinez-Rodriguez, J. Otalora Garmilla and A. W. Kleij, ChemSusChem, 2016, 9, 749–755 CrossRef CAS PubMed.
- J. K. Qiu, Y. L. Zhao, Z. Y. Li, H. Y. Wang, M. H. Fan and J. J. Wang, ChemSusChem, 2017, 10, 1120–1127 CrossRef CAS PubMed.
- Z. J. Guo, Q. W. Jiang, Y. M. Shi, J. Li, X. N. Yang, W. Hou, Y. Zhou and J. Wang, ACS Catal., 2017, 7, 6770–6780 CrossRef CAS.
- M. R. Li, M. C. Zhang, T. J. Yue, X. B. Lu and W. M. Ren, RSC Adv., 2018, 8, 39182–39186 RSC.
- L. Wang, G. Y. Zhang, K. Kodama and T. Hirose, Green Chem., 2016, 18, 1229–1233 RSC.
- C. Maeda, T. Taniguchi, K. Ogawa and T. Ema, Angew. Chem., Int. Ed., 2015, 54, 134–138 CrossRef CAS PubMed.
- H. L. Parker, J. Sherwood, A. J. Hunt and J. H. Clark, ACS Sustainable Chem. Eng., 2014, 2, 1739–1742 CrossRef CAS.
- A. H. Tamboli, A. A. Chaugule and H. Kim, Fuel, 2016, 184, 233–241 CrossRef CAS.
- Z. Z. Yang, L. N. He, Y. N. Zhao, B. Li and B. Yu, Energy Environ. Sci., 2011, 4, 3971–3975 RSC.
- J. Q. Wang and Y. G. Zhang, ACS Catal., 2016, 6, 4871–4876 CrossRef CAS.
- L. Guo, L. L. Deng, X. C. Jin, Y. R. Wang and H. Z. Wang, RSC Adv., 2018, 8, 26554–26562 RSC.
- C. K. Johannes Steinbauer, R. Ludwig and T. Werner, ACS Sustainable Chem. Eng., 2018, 6, 10778–10788 CrossRef.
- J. Sun, W. G. Cheng, Z. F. Yang, J. Q. Wang, T. T. Xu, J. Y. Xin and S. J. Zhang, Green Chem., 2014, 16, 3071–3078 RSC.
- R. C. Luo, Z. Yang, W. Y. Zhang, X. T. Zhou and H. B. Ji, Sci. China: Chem., 2017, 60, 979–989 CrossRef CAS.
- T. Berestok, P. Guardia, R. F. Du, J. B. Portals, M. Colombo, S. Estrade, F. Peiro, S. L. Brock and A. Cabot, ACS Appl. Mater. Interfaces, 2018, 10, 16041–16048 CrossRef CAS PubMed.
- J. Zhu, P. M. Usov, W. Q. Xu, P. J. Celis-Salazar, S. Y. Lin, M. C. Kessinger, C. Landaverde-Alvarado, M. Cai, A. M. May, C. Slebodnick, D. Zhu, S. D. Senanayake and A. J. Morris, J. Am. Chem. Soc., 2018, 140, 993–1003 CrossRef CAS PubMed.
- K. Zhou, B. Mousavi, Z. X. Luo, S. Phatanasri, S. Chaemchuen and F. Verpoort, J. Mater. Chem. A, 2017, 5, 952–957 RSC.
- J. Y. Hu, J. Ma, H. Z. Liu, Q. L. Qian, C. Xie and B. X. Han, Green Chem., 2018, 20, 2990–2994 RSC.
- X. H. Song, Y. F. Wu, D. H. Pan, F. F. Cai and G. M. Xiao, Mol. Catal., 2017, 436, 228–236 CrossRef CAS.
- J. Liang, Y. Q. Xie, Q. Wu, X. Y. Wang, T. T. Liu, H. F. Li, Y. B. Huang and R. Cao, Inorg. Chem., 2018, 57, 2584–2593 CrossRef CAS PubMed.
- J. J. Zhu, T. T. Diao, W. Y. Wang, X. L. Xu, X. Y. Sun, S. A. C. Carabineiro and Z. Zhao, Appl. Catal., B, 2017, 219, 92–100 CrossRef CAS.
- A.-L. Girard, N. Simon, M. Zanatta, S. Marmitt, P. Gonçalves and J. Dupont, Green Chem., 2014, 16, 2815–2825 RSC.
- Q. Liu, L. P. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- S. Marmitt and P. F. Goncalves, J. Comput. Chem., 2015, 36, 1322–1333 CrossRef CAS PubMed.
- S. Hayashi, S. Yamazoe, K. Koyasu and T. Tsukuda, Chem.–Asian J., 2017, 12, 1635–1640 CrossRef CAS PubMed.
- X. D. Lang and L. N. He, Chem. Rec., 2016, 16, 1337–1352 CrossRef CAS PubMed.
- X. F. Liu, Q. W. Song, S. Zhang and L. N. He, Catal. Today, 2016, 263, 69–74 CrossRef CAS.
- B. Sarmah and R. Srivastava, Ind. Eng. Chem. Res., 2017, 56, 8202–8215 CrossRef CAS.
- H. Zhong, Y. Q. Su, X. W. Chen, X. J. Li and R. H. Wang, ChemSusChem, 2017, 10, 4855–4863 CrossRef CAS PubMed.
- L. Wang, G. Zhang, K. Kodama and T. Hirose, Green Chem., 2016, 18, 1229–1233 RSC.
- D. J. Heldebrant, C. R. Yonker, P. G. Jessopc and L. Phan, Energy Environ. Sci., 2008, 1, 487–493 RSC.
- L. Gong and J. S. McCullagh, Rapid Commun. Mass Spectrom., 2014, 28, 339–350 CrossRef CAS PubMed.
- J. Q. Wang, W. G. Cheng, J. Sun, T. Y. Shi, X. P. Zhang and S. J. Zhang, RSC Adv., 2014, 4, 2360–2367 RSC.
- M. S. Liu, L. Liang, X. Li, X. X. Gao and J. M. Sun, Green Chem., 2016, 18, 2851–2863 RSC.
- Z. Z. Yang, Y. F. Zhao, G. P. Ji, H. Y. Zhang, B. Yu, X. Gao and Z. M. Liu, Green Chem., 2014, 16, 3724–3728 RSC.
- J. Liang, R. P. Chen, X. Y. Wang, T. T. Liu, X. S. Wang, Y. B. Huang and R. Cao, Chem. Sci., 2017, 8, 1570–1575 RSC.
- C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, ChemSusChem, 2012, 5, 2032–2038 CrossRef CAS PubMed.
- W. Cho, M. S. Shin, S. Hwang, H. Kim, M. Kim, J. G. Kim and Y. Kim, J. Ind. Eng. Chem., 2016, 44, 210–215 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10475a |
| This journal is © The Royal Society of Chemistry 2019 |