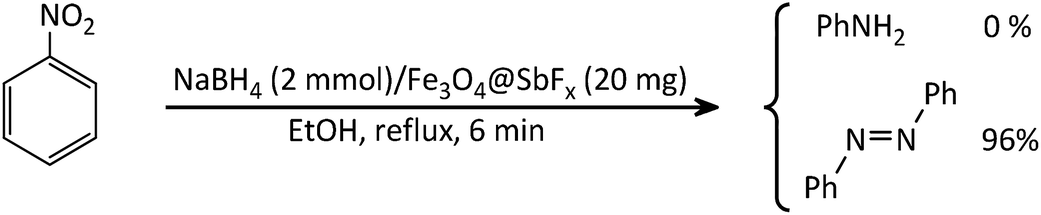DOI:
10.1039/C9RA01249D
(Paper)
RSC Adv., 2019,
9, 13112-13121
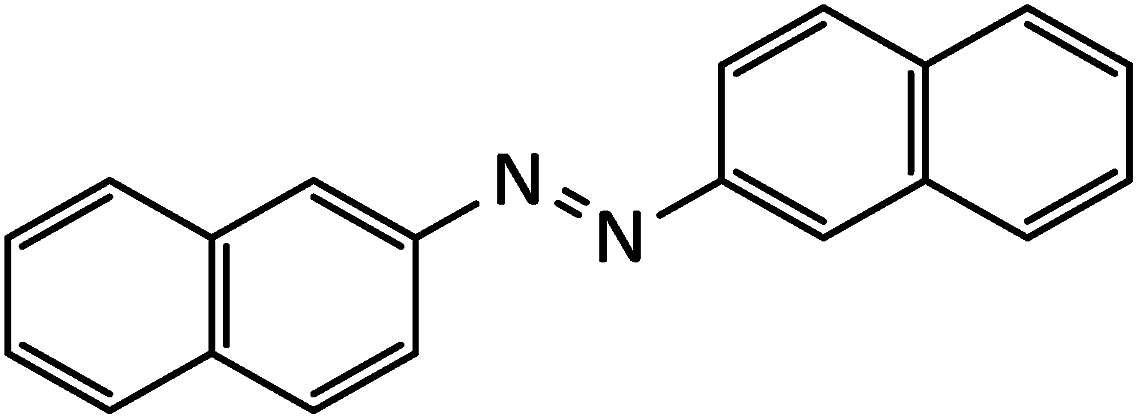
Immobilized antimony species on magnetite: a novel and highly efficient magnetically reusable nanocatalyst for direct and gram-scale reductive-coupling of nitroarenes to azoarenes†
Received
18th February 2019
, Accepted 23rd April 2019
First published on 29th April 2019
Abstract
In this study, magnetic nanoparticles of Fe3O4@SbFx from the immobilization of SbF3 on magnetite were synthesized. The prepared nanocomposite system was then characterized using scanning electron microscopy, Fourier transform infrared spectroscopy, X-ray diffraction, energy-dispersive X-ray spectroscopy, vibrating sample magnetometry and inductively coupled plasma optical emission spectroscopy. Next, the catalytic activity of Fe3O4@SbFx MNPs was highlighted by one-pot reductive-coupling of aromatic nitro compounds to the corresponding azoarene materials with NaBH4. The reactions were carried out in refluxing EtOH within 6–25 min to afford the products in high yields. The reusability of the Sb-magnetite system was also studied for 6 consecutive cycles without significant loss of catalytic activity. This synthetic protocol provided several advantages in terms of introducing a novel catalytic system based on antimony species for direct and gram-scale preparation of azoarenes from nitroarenes, low loading of the nanocatalyst, mild reaction conditions, using ethanol as a green and economic solvent and high yield of the products.
Introduction
Synthesis of azoarenes is one of the valuable topics in organic synthesis. These materials have widespread capabilities and are utilized in numerous fields of modern technologies such as synthesis of organic dyes and pigments,1–3 and as initiators of radical reactions,4 food additives, indicators5 and drug delivery systems.6 They possess excellent photoelectric and optic-memory properties.7 The recent studies also show that azobenzenes were successfully used in image storage and optical switches,8 molecular shuttles based on a rotaxane,9 nanotubes,10 electronic devices11 and production of filters as well as protection of eye glasses.12 Due to the cis–trans isomerization of the azo functionality by adequate radiation, azobenzenes were also used in photochemical molecular switches13 and photoregulation of DNA triplex formation.14 It was also reported that azoarenes can play a crucial role in the development of synthetic15 and mechanistic organic chemistry.16 Based on these great capabilities, much attention has been devoted to the synthesis of azobenzenes by various methods.
The literature review shows that the multistep synthesis of azoarene materials was primarily carried out through the coupling of diazonium ions17 and Anspon reaction.18 In due course, the multistep preparation of azoarenes has been extensively reviewed by Merino.19 Although most of the reported protocols provided the useful synthetic capabilities, however, they generally suffer from low to moderate yield of the products, the prolonged reaction times, cyclization, isomerization and rearrangement reactions.20,21 To overcome the mentioned shortcomings, several synthetic methods through the improvement of the catalyst activity have been reported.22,23 In contrast to the multistep strategy, the straightforward oxidative-coupling of aromatic amines to azoarenes24–26 and reductive-coupling of nitroarenes to azoxyarenes27–33 was also a subject of more interest. In this context, the application of HCOONH4/Pb,34 CH3COONH4/Pb,35 HCOONHEt3/Pb,36 HCOONHEt3/Mg,37 LiAlH4,38 NaH2Al(OCH2CH2OMe)2,39 NaBH4/DMSO,40 (MeCN)3Cr(CO)3,41 Co2(CO)8,42 HOCH2CH2ONa,43 Zn/NaOH,44 Bi/KOH,45 Mg/NH4Br,46 Mg/MeOH,47 Mg/MCln (MCln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TiCl4, VCl3, CrCl3, MoOCl3, WCl6 and FeCl3),48 Pd(acac)2/H2,49 InBr3/Et3SiH,50 In(OTf)3/Et3SiH/O2,51 FeCl2·4H2O/Li/DTBB,52 electroreduction,53 Au/meso CeO2/CO,54 Li/THF,55 Bi/ball mill56 and Cr57 has also been documented for direct conversion of nitroarenes to azoarene materials.
:TiCl4, VCl3, CrCl3, MoOCl3, WCl6 and FeCl3),48 Pd(acac)2/H2,49 InBr3/Et3SiH,50 In(OTf)3/Et3SiH/O2,51 FeCl2·4H2O/Li/DTBB,52 electroreduction,53 Au/meso CeO2/CO,54 Li/THF,55 Bi/ball mill56 and Cr57 has also been documented for direct conversion of nitroarenes to azoarene materials.
It is also known that in the promoted reactions by metal nano-particles, the electronic influence of metal species, nano-size dimension of particles and porosity of the surface play the synergic roles to improve the rate of examined reactions. However, metal nanoparticles generally due van der Waals, electrostatic or magnetic forces represent a strong tendency for agglomeration while it decreases the active surface area and catalytic activity of the applied catalyst system. In order to overcome this shortcoming and preserve the original characteristics of nanoparticles, the immobilization of metal nanoparticles on organic and inorganic stabilizers/supports has been frequently documented. Organic ligand,58 polymer,59 zeolites,60,61 reduced graphene oxide,62–65 CuO,66 gums67 and magnetic materials68 are the stabilizers which have been successfully served for the immobilization of metal nanoparticles. Among these, the magnetic nanoparticles (MNPs) of Fe3O4 due extreme specific surface area, high magnetic property, chemical solidity and ease of preparation and separation are more appropriate for anchoring the metal species.69
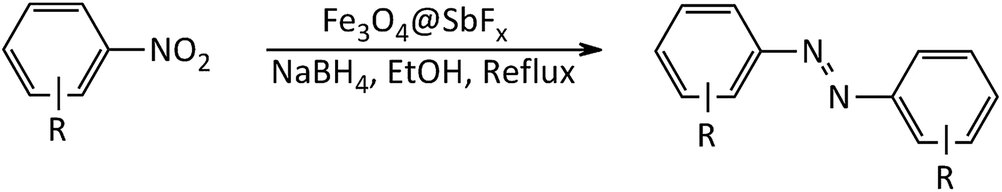
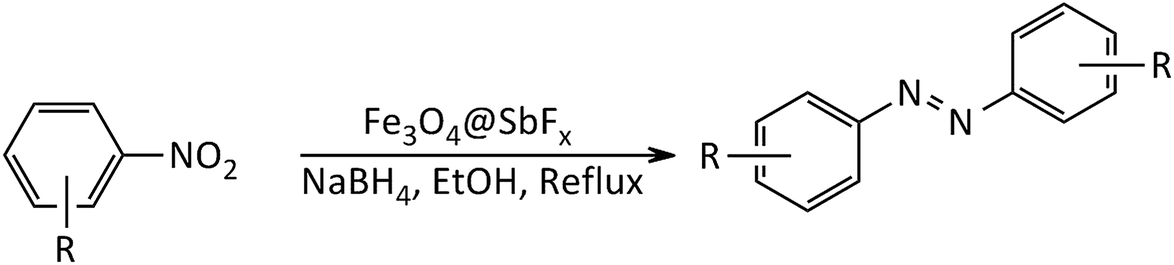
In line with the outlined strategies and continuation of our research program directed to synthetic usefulness of magnetic nanocatalysts,70–76 herein, we wish to report synthesis of the immobilized antimony species on magnetite, Fe3O4@SbFx, as novel, highly efficient and reusable magnetic nanocatalyst for straightforward reductive-coupling of nitroarenes to symmetrically substituted azoarene materials in refluxing EtOH (Scheme 1).
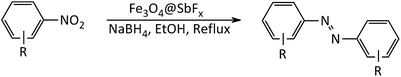 |
| | Scheme 1 Synthesis of azoarenes catalyzed by NaBH4/Fe3O4@SbFx MNPs system. | |
Results and discussion
Preparation and characterization of Fe3O4@SbFx MNPs
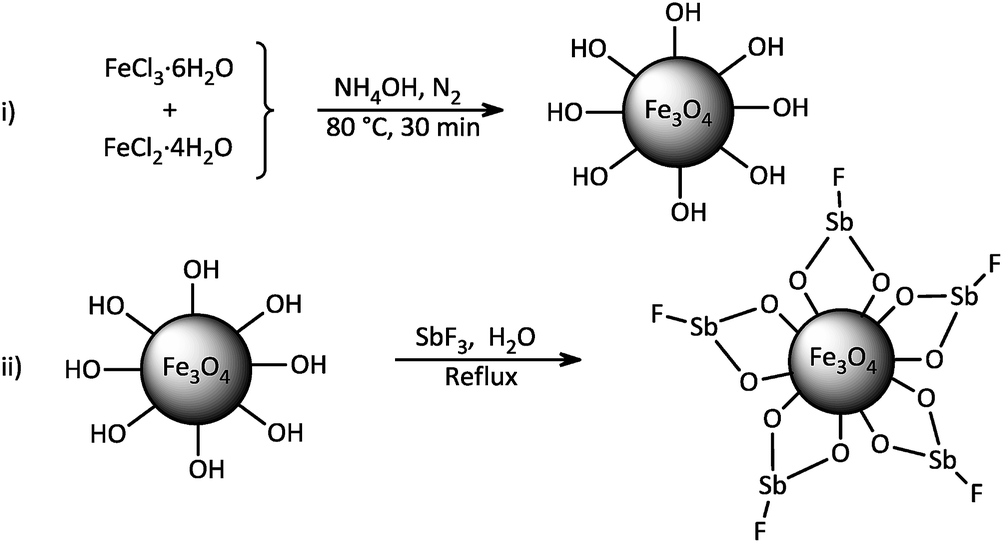
The study was started by preliminary preparation of magnetically nanoparticles of Fe3O4@SbFx with 15% Sb species via a two-step procedure: (i) preparation of Fe3O4 MNPs by chemical co-precipitation of FeCl3·6H2O and FeCl2·4H2O in aqueous ammonia and (ii) the immobilization of SbFx on magnetite by simply mixing of an aqueous solution of SbF3 with Fe3O4 MNPs under reflux conditions (Scheme 2).
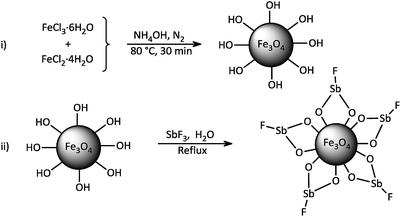 |
| | Scheme 2 Synthesis of magnetically nanoparticles of Fe3O4@SbFx. | |
Characterization of Fe3O4@SbFx MNPs
After the successful synthesis of Fe3O4@SbFx MNPs, the nano-catalyst was then characterized using Fourier transform infrared spectroscopy (FT-IR), scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDX), X-ray diffraction (XRD), vibrating sample magnetometer (VSM), N2 adsorption–desorption analysis and inductively coupled plasma-optical emission spectroscopy (ICP-OES).
FT-IR analysis
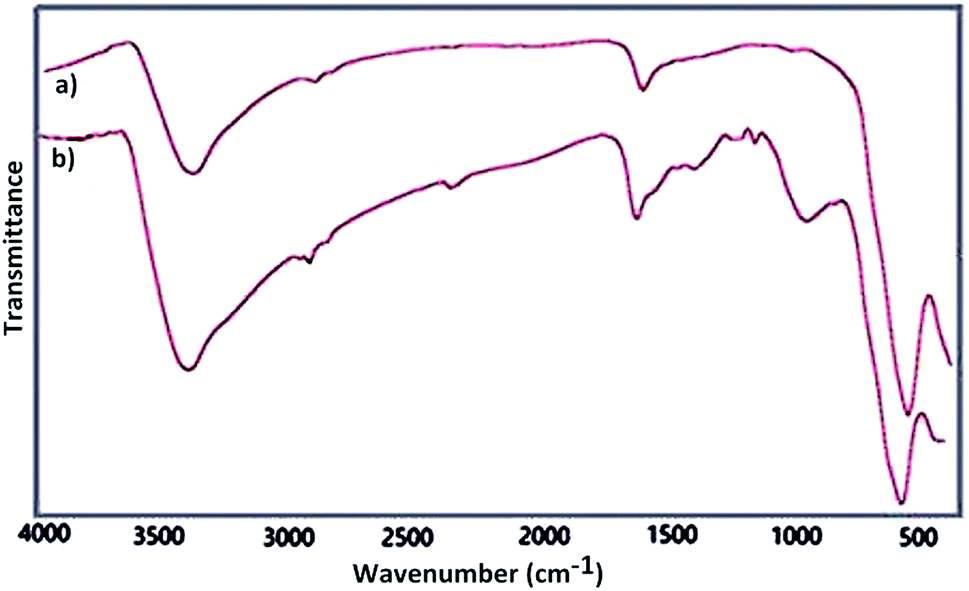
Fourier transform infrared spectroscopy as a primarily tool was used for structural elucidation of Fe3O4 and Fe3O4@SbFx MNPs. In this context, FT-IR spectrum of Fe3O4 (Fig. 1a) shows a strong absorption band at 575 cm−1 showing the stretching vibration of Fe–O bond. The absorption peaks at 1625 and 3400 cm−1 are also attributed to O–H deforming and stretching vibrations of adsorbed water, respectively. FT-IR spectrum of Fe3O4@SbFx MNPs (Fig. 1b) also shows the absorption peaks at 3421 and 1631 cm−1 related to the stretching and bending vibrations of OH groups on the surface of nanoparticles. The peaks around 1021 and 566 cm−1 are respectively attributed to stretching vibrations of Sb–O and Sb–F bonds. Therefore, the later absorption bands successfully verify the immobilization of SbFx species on the surface of magnetite.
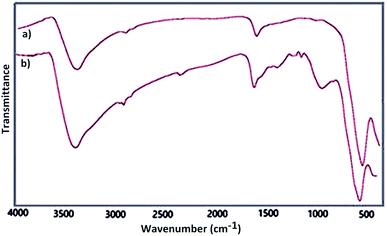 |
| | Fig. 1 FT-IR spectra of (a) Fe3O4 and (b) Fe3O4@SbFx MNPs. | |
SEM analysis
Morphology of the surface and size distribution of nanoparticles in Fe3O4 and Fe3O4@SbFx MNPs were also investigated using scanning electron microscopy technique (Fig. 2). The surface-microscopy image of Fe3O4 MNPs (Fig. 2a) represents that the magnetite was constituted from uniform and ultrafine granular particles distributed in the range of 10–21 nm. As well, SEM image of Fe3O4@SbFx MNPs (Fig. 2b) shows that through the reaction of magnetite with SbF3, the surface of Sb-magnetite was thoroughly modified and constituted from porous and uniform granular nanoparticles distributed in the range of 45–68 nm.
 |
| | Fig. 2 SEM images of (a) Fe3O4 and (b) Fe3O4@SbFx MNPs. | |
EDX analysis
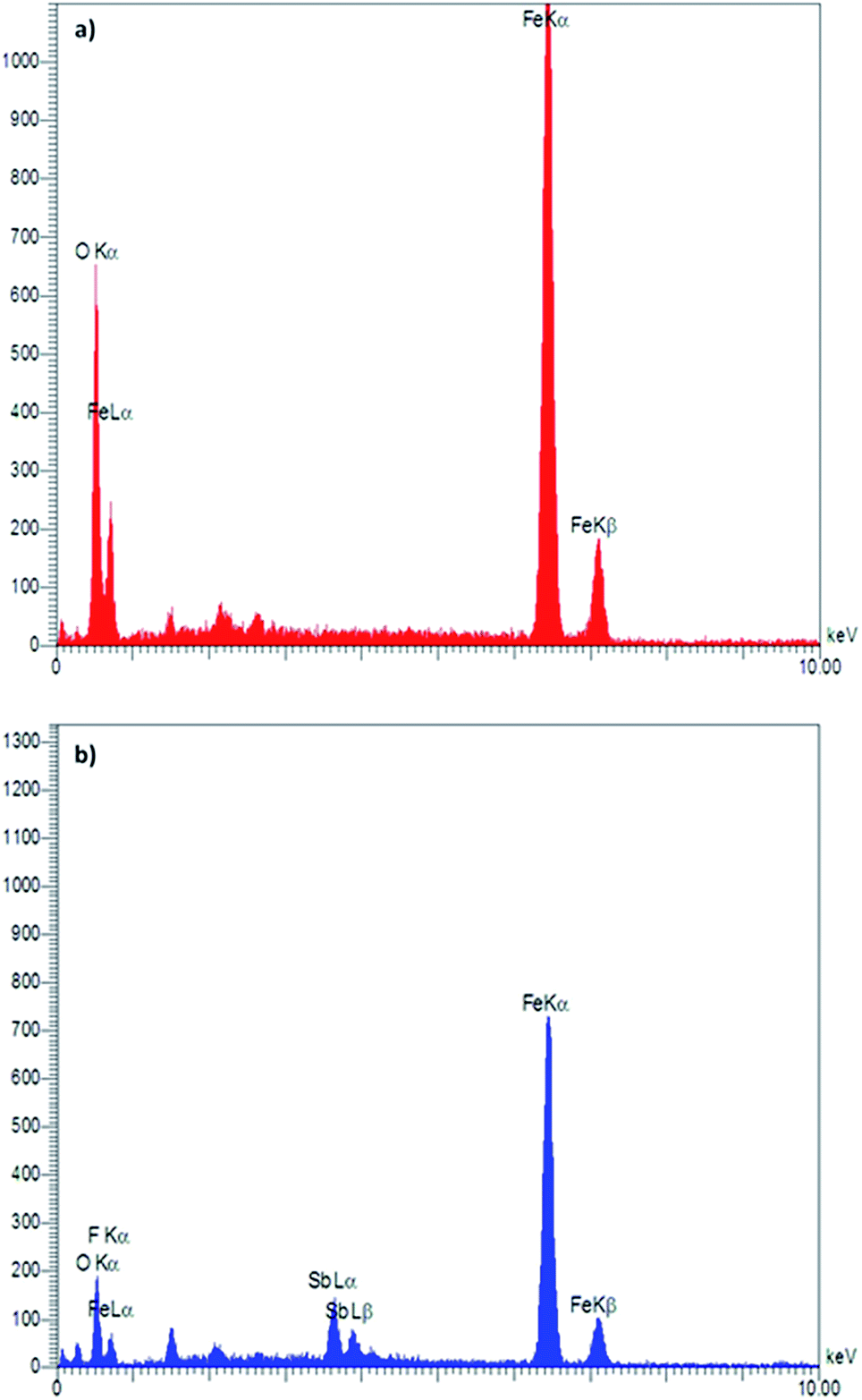
Energy-dispersive X-ray (EDX) spectroscopy as an effective technique was used to determine the presence of elements in a material. According to the depicted graphs for EDX analysis of Fe3O4 and Fe3O4@SbFx MNPs (Fig. 3), the presence of Fe and O in Fe3O4 as well Fe, O, Sb and F elements in Fe3O4@SbFx MNPs is proved. This analysis clearly shows the successful immobilization of SbFx species on the surface of Fe3O4.
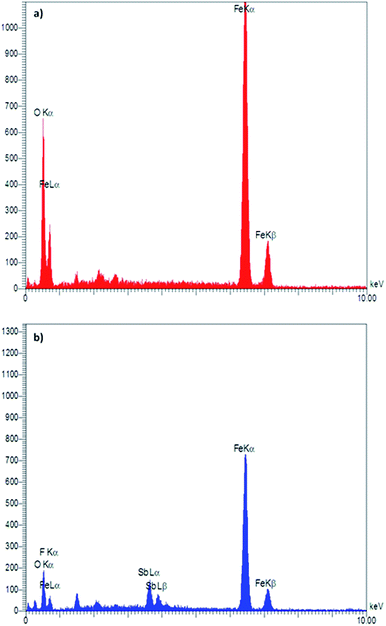 |
| | Fig. 3 EDX spectra of (a) Fe3O4 and (b) Fe3O4@SbFx MNPs. | |
ICP-OES analysis
In continuation to EDX analysis, the exact amounts of Fe and Sb in Fe3O4@SbFx MNPs were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES). Based on this analysis, the amounts of Sb and Fe in Fe3O4@SbFx MNPs were found to be 15.06% and 54.1%, respectively.
XRD analysis
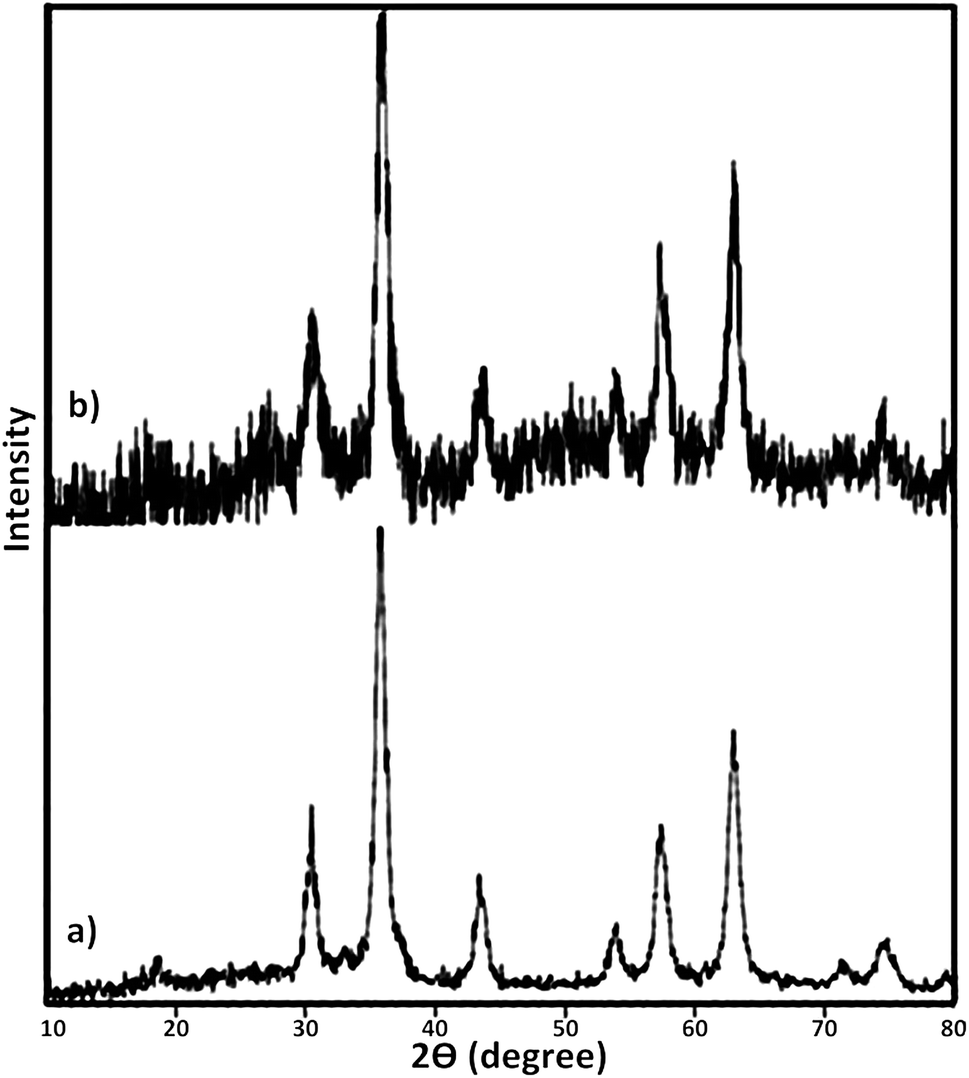
At the next, the phase purity and crystallinity structure of the prepared nanocatalysts was determined using X-ray diffraction (XRD) analysis. Fig. 4 represents XRD diffractograms of Fe3O4 and Fe3O4@SbFx MNPs. In XRD pattern of Fe3O4, the peaks at 2θ = 30.2°, 35.5°, 43.3°, 53.7°, 57.2° and 62.9° are attributed to (220), (311), (400), (422), (511) and (440) reflection planes of cubic spinal Fe3O4 (JCPDS 65-3107).77,78 In XRD pattern of Fe3O4@SbFx MNPs (Fig. 4b), the presence of characteristic peaks related to Fe3O4 is verifying that during the immobilization of Sb species, no structural change has been taken place in the spinal structure of magnetite. Moreover, the absorption peak at 2θ = 26° is exactly attributed to the bond of Sb–F showing the immobilization of Sb species on the surface of Fe3O4 MNPs.79
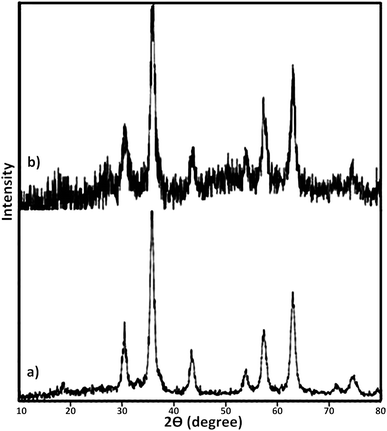 |
| | Fig. 4 XRD diffractograms of (a) Fe3O4 and (b) Fe3O4@SbFx MNPs. | |
BET analysis
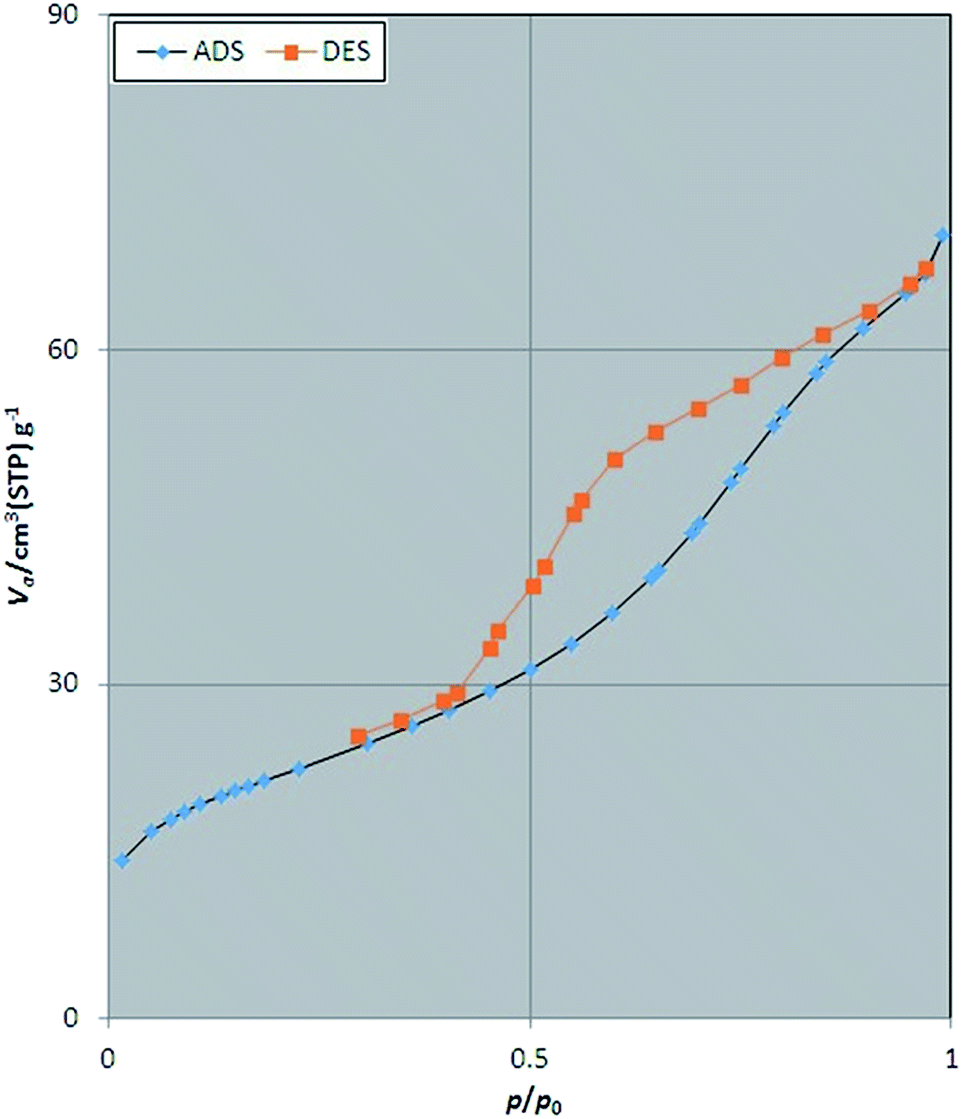
The porosity and surface characteristics of Fe3O4@SbFx MNPs was studied through the N2 adsorption–desorption analysis. The results of this investigation are illustrated in Fig. 5. According to categories of adsorption isotherms (BDDT), the shape of isotherm for Fe3O4@SbFx MNPs is belonged to type of IV with H3 hysteresis loop of desorption pathway. This type of isotherm is a characteristic of mesoporous materials. Table 1 represents total pore volume (Vtotal), pore diameter (DBJH) and BET surface area (SBET) of Fe3O4@SbFx MNPs. Investigation of the results shows that through modifying of the magnetite with SbFx species, porosity of the surface was increased. Subsequently, the specific surface area was raised to 75.7 m2 g−1 followed by decreasing the pores diameter to 5.74 nm.
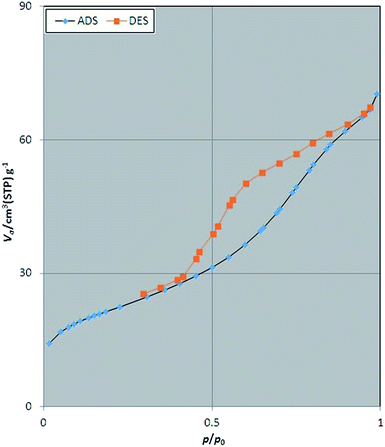 |
| | Fig. 5 N2 adsorption–desorption isotherm of Fe3O4@SbFx MNPs. | |
Table 1 The surface analysis of Fe3O4@SbFx MNPs
| Sample |
SBET (m2 g−1) |
Pore diameter (nm) |
Total pore volume (cm3 g−1) |
| Fe3O4@SbFx MNPs |
75.739 |
5.738 |
0.1086 |
VSM analysis
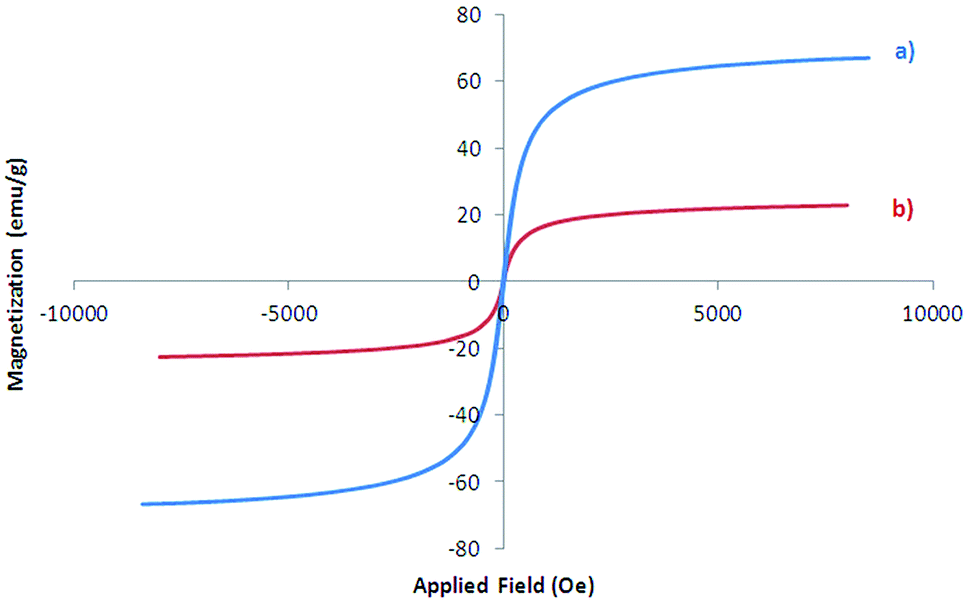
Magnetic property of Fe3O4 and Fe3O4@SbFx MNPs were also studied by vibrating sample magnetometer (VSM) analysis in the applied magnetic field up to 20 kOe (Fig. 6). The saturation magnetization (Ms) value of Fe3O4 (Fig. 6a) and Fe3O4@SbFx (Fig. 6b) were found to be 66.976 and 23.229 emu g−1, respectively. The graphs are clearly showing that through the immobilization of SbFx species on the surface of magnetite, Ms value of Fe3O4@SbFx was notably decreased. However, the magnetization value was still large enough for any magnetic separation.
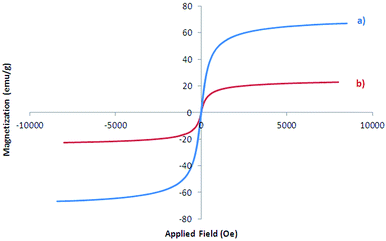 |
| | Fig. 6 Magnetization curves of (a) Fe3O4 and (b) Fe3O4@SbFx MNPs. | |
One-pot reductive-coupling of nitroarenes to azoarenes with NaBH4 catalyzed by Fe3O4@SbFx MNPs
After the successful synthesis and characterization of the antimony-coated magnetite, catalytic activity of the prepared nanocatalyst was further investigated towards one-pot reductive-coupling of aromatic nitro compounds to the corresponding azoarenes.
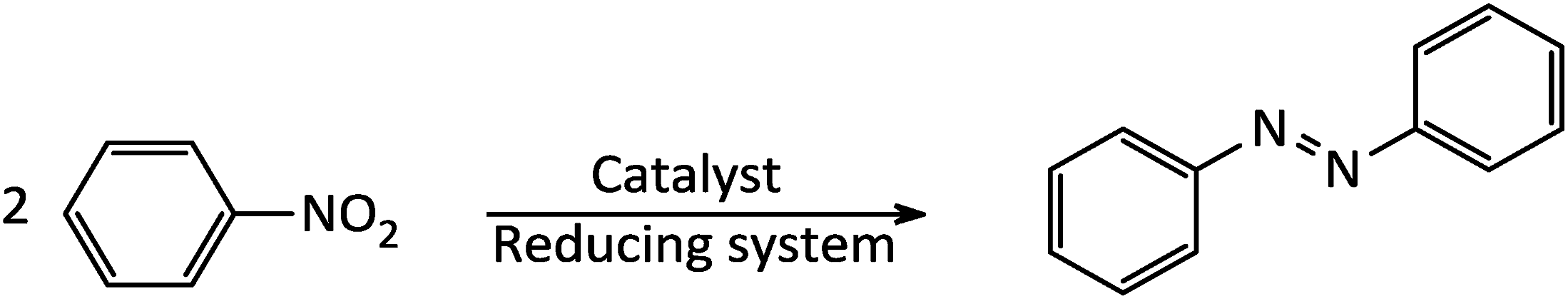
The study was started with the preliminary aim of using antimony-coated magnetite as a catalyst towards reduction of nitroarenes to the corresponding anilines with NaBH4. Therefore, reduction of nitrobenzene with NaBH4 (as a model reaction) in the presence and absence of antimony-coated magnetite was investigated with the change of reaction-solvent and amounts of nanocatalyst as well as verifying the influence of temperature (Table 2). The results of this study exhibited that progress of the reaction in the absence of Sb-nanocatalyst led to unsatisfactory yield of the product. However, the influence of small amounts of nanocatalyst on the rate of reaction was noteworthy. In addition, it was very amazing that the product of reaction was azobenzene while it was expected to obtain aniline as a product. This transformation was clearly identifiable through the color change of the reaction to orange-red solution. As well, instrumental analysis also verified the formation of azobenzene as a sole product. The examinations represented that using NaBH4 (2 mmol) and Fe3O4@SbFx MNPs (20 mg) in refluxing EtOH as a green and economic solvent was sufficient to perform one-pot reductive-coupling of nitrobenzene (1 mmol) to azobenzene within 6 min and 96% isolated yield (Scheme 3) (Table 2, entry 6).
Table 2 Optimization experiments for reductive-coupling of nitrobenzene to azobenzene with NaBH4/Fe3O4@SbFx MNPs systema
| Entry |
NaBH4 (mmol) |
Fe3O4@SbFx (mg) |
Solvent (2 mL) |
Condition |
Time (min) |
Conversion (%) |
| All reactions were carried out with 1 mmol of nitrobenzene. |
| 1 |
2 |
— |
H2O |
Reflux |
240 |
— |
| 2 |
1 |
10 |
H2O |
Reflux |
120 |
40 |
| 3 |
2 |
10 |
H2O |
Reflux |
120 |
60 |
| 4 |
2 |
20 |
H2O |
Reflux |
45 |
100 |
| 5 |
2 |
20 |
H2O–EtOH (1:1) |
Reflux |
300 |
100 |
| 6 |
2 |
20 |
EtOH |
Reflux |
6 |
100 |
| 7 |
2 |
20 |
CH3CN |
Reflux |
120 |
30 |
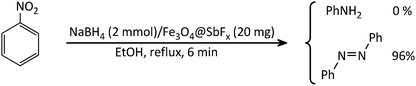 |
| | Scheme 3 Reductive-coupling of nitrobenzene to azobenzene with NaBH4/Fe3O4@SbFx MNPs. | |
At the next, the generality and usefulness of this synthetic protocol was further studied using structurally diverse aromatic nitro compounds at the optimized reaction conditions (Table 2, entry 6). The results of this investigation are summarized in Table 3. The table shows that all reductive-coupling of nitroarenes containing electron-withdrawing and releasing functionalities were carried out successfully using NaBH4 (2 mmol)/Fe3O4@SbFx (0.02 g) in refluxing ethanol within 6–25 min giving the corresponding azoarene materials in high yields. In addition, the capability of this protocol for gram scale synthesis of azoarene materials was also investigated by one-pot reductive-coupling of nitrobenzene (1.0 g, 8.1 mmol) with NaBH4 (0.61 g, 16 mmol)/Fe3O4@SbFx (160 mg) in refluxing EtOH (5 mL). The obtained result exhibited that the reaction was taken place successfully within 10 min to afford azobenzene in 94% yield (0.7 g).
Table 3 Reductive-coupling of nitroarenes with NaBH4/Fe3O4@SbFx systema
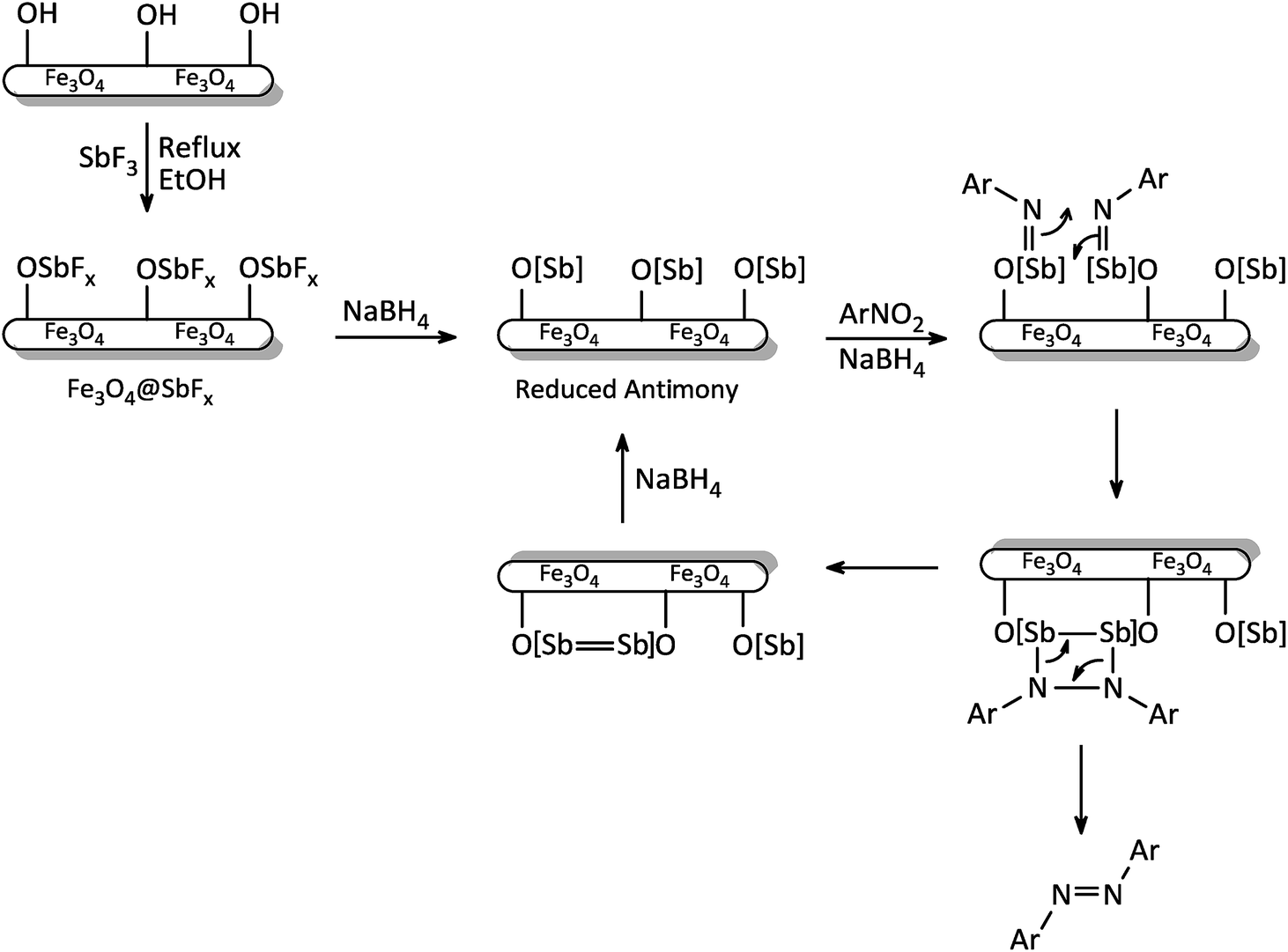
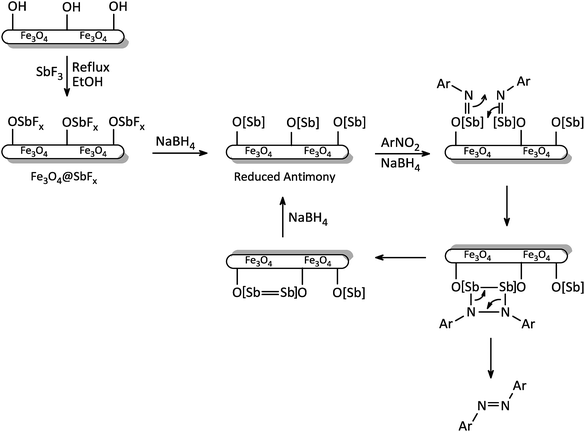
The exact mechanism of this synthetic protocol is not clear; however, a depicted mechanism (Scheme 4) shows a pathway for the influence of NaBH4/Fe3O4@SbFx system on one-pot reductive-coupling of nitroarenes to azoarene materials. The scheme shows that the Sb-magnetite nanocatalyst in the presence of NaBH4 was converted to the reduced-antimony species. Through the reaction of this activated nanocatalyst with nitroarene, the formation of imino-antimony intermediate was carried out. At the next, through the [2 + 2] cycloaddition reaction of two imino-antimony species followed by skeletal rearrangement, the formation of azoarenes product was taken place.
 |
| | Scheme 4 The proposed mechanism for reductive-coupling of nitroarenes to azoarenes with NaBH4/Fe3O4@SbFx system. | |
In order to show the suitability and usefulness of NaBH4/Fe3O4@SbFx system towards one-pot synthesis of azoarene materials, the result of reductive-coupling of nitrobenzene to azobenzene was compared with the previously reported procedures (Table 4). An investigation shows that in terms of the amount of nanocatalyst, mild reaction conditions, selective formation of azobenzene, short reaction time, high yield of the product and great reusability of the Sb-nanocatalyst, the current system represents the outstanding advantages.
Table 4 Reductive-coupling of nitrobenzene to azobenzene with NaBH4/Fe3O4@SbFx and other reported systems
|
| Entry |
Catalyst system |
Condition |
Time (min) |
Yield (%) |
Reusability |
Ref. |
| Present work. |
| 1 |
NaBH4/Fe3O4@SbFx |
EtOH/reflux |
6 |
95 |
6 |
a |
| 2 |
HCO2NH4/Pb |
MeOH/reflux |
60 |
92 |
— |
34 |
| 3 |
HCO2HNEt3/Pb |
MeOH/r.t. |
120 |
92 |
— |
36 |
| 4 |
LiAlH4 |
Et2O/−80 °C |
— |
84 |
— |
38 |
| 5 |
NaBH4 |
DMSO/85 °C |
90 |
75 |
— |
40 |
| 6 |
HOCH2CH2ONa |
Ethylene glycol |
3 |
88 |
— |
43 |
| 7 |
Bi/KOH |
MeOH/microwave |
8 |
85 |
— |
45 |
| 8 |
Mg/NH4Br |
MeOH/r.t. |
90 |
90 |
— |
46 |
| 9 |
Pd(acac)2/H2 (1 atm) |
KOH/70 °C/EtOH |
360 |
90 |
— |
49 |
| 10 |
In(OTf)3–Et3SiH |
O2/DMF/60 °C |
720 |
84 |
— |
51 |
| 11 |
FeCl2·4H2O/Li/DTBB |
THF/reflux |
150 |
97 |
— |
52 |
| 12 |
Au/meso-CeO2/CO (5 atm) |
Toluene/150 °C |
300 |
99 |
— |
54 |
Recyclability of Fe3O4@SbFx MNPs
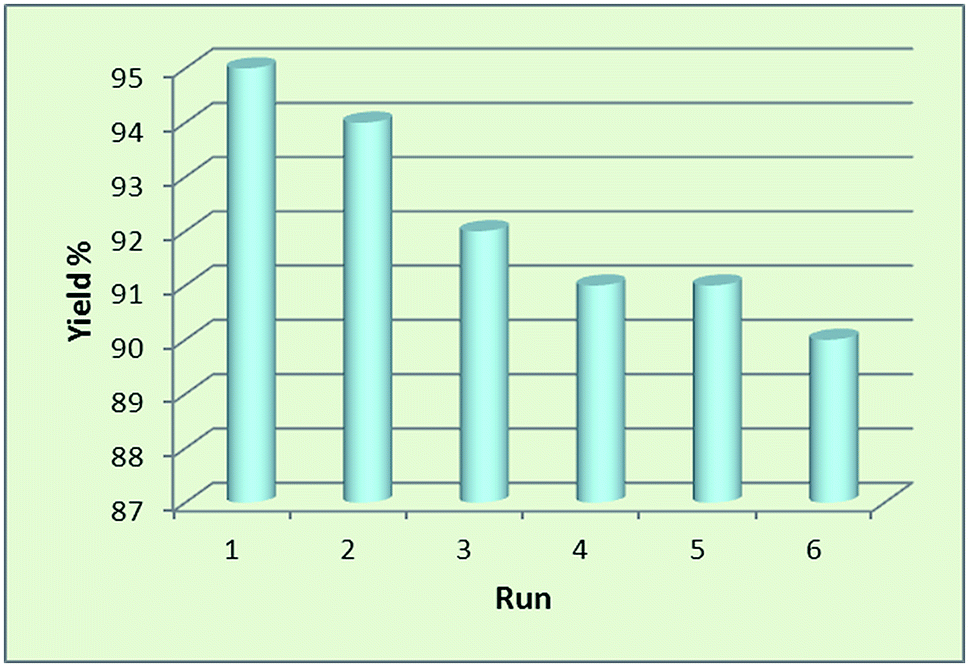
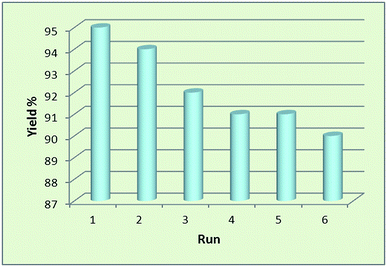
Nowadays, the recovery and reusability of the applied catalyst system has become a crucial subject in academic and industrial points of view. The green and economic aspect of this synthetic method was also studied by investigation of the reusability of Fe3O4@SbFx MNPs in reductive-coupling of nitrobenzene to azobenzene with NaBH4 at the optimized reaction conditions (Table 2, entry 6). After completion of the reaction, the Sb-magnetite nanocatalyst was magnetically recovered from the reaction mixture, washed with EtOAc and then dried for reusing at the next runs. The model reaction was again charged with the fresh NaBH4, nitrobenzene, ethanol and the recovered Sb-nanocatalyst. Fig. 7 shows that the nanocatalyst can be reused for six consecutive cycles without the significant loss of catalytic activity. It is also notable that due small decreasing in the activity of nanocatalyst and therefore remaining of the starting material for 1–5% over the six recycling of the nanocatalyst, isolated yield of the product was reduced for 1–5% in respect to the first run of model reaction (96%).
 |
| | Fig. 7 Reusability of Fe3O4@SbFx MNPs in reductive-coupling of PhNO2 with NaBH4. | |
Conclusions
In this study, magnetically nanoparticles of the novel antimony-coated magnetite, Fe3O4@SbFx, were prepared. The prepared nanocomposite was then characterized using FT-IR, SEM, EDX, XRD, BET, VSM and ICP analyses. The heterogeneous Sb-nanocatalyst showed an extraordinary catalytic activity toward rapid and efficient reductive-coupling of nitroarenes to azoarene materials with NaBH4 in refluxing ethanol as a green solvent. All reactions were carried out using NaBH4 (2 mmol)/Fe3O4@SbFx (20 mg) within 6–25 min to give the products in high yields. This synthetic protocol offers numerous advantages in terms of the novel and easy synthesis of Sb-magnetite nanocatalyst, mild reaction conditions in comparison to the previously reported methods, using EtOH as green solvent, short reaction times, high yield of the products, simple work-up procedure and the great reusability of the nanocatalyst.
Experimental
Chemicals and apparatus
All chemicals in analytical grade were purchased from chemical companies and were used without further purification. FT-IR and 1H, 13C NMR spectra were recorded on Thermo Nicolet Nexus 670 and Bruker Avance 300 MHz spectrometers, respectively. Melting points were measured on Electrothermal 9100 apparatus and were uncorrected. Mass spectra of the samples were obtained from Mass spectrometer (Agilent, 5975C, 20–70 eV). Morphology and size distribution of nanoparticles were determined by scanning electron microscopy (SEM) using FESEM-TESCAN followed by energy-dispersive X-ray analysis (EDX). Magnetic property of materials were measured by vibrating sample magnetometer (VSM, model MDKFT) under magnetic fields up to 20 kOe. Elemental composition of the samples was determined using inductively coupled plasma-optical emission spectrometer (ICP-OES). Specific surface area and pore size distribution of nanoparticles were determined on Belsorp-Max (Japan) instrument using N2 adsorption–desorption isotherm. X-ray diffraction (XRD) analysis was carried out by X'PertPro diffractometer. TLC was applied for monitoring of the reactions over silica gel 60 F254 aluminum sheet. All products were identified by physical and spectral data followed by comparison with authentic data.
Preparation of Fe3O4 MNPs
Magnetically nanoparticles of Fe3O4 were prepared via a chemical co-precipitation protocol.70 Due course, a solution of FeCl3·6H2O (0.0216 mol, 5.838 g) and FeCl2·4H2O (0.0108 mol, 2.147 g) in deionized water (100 mL) was prepared. The solution was stirred for 10 min at 85 °C under N2 atmosphere. Next, aqueous ammonia (25%, 10 mL) was quickly added and the black nanoparticles of Fe3O4 were instantly precipitated. The resulting mixture was again stirred for 30 min at 85 °C under N2 atmosphere. The mixture was cooled to the room temperature and Fe3O4 MNPs were magnetically separated from the reaction mixture. The nanoparticles were washed with distilled water, a solution of NaCl (0.02 M) and again with distilled water. Drying under air atmosphere afforded the nanoparticles of Fe3O4.
Synthesis of Fe3O4@SbFx MNPs
To a solution of SbF3 (0.15 g, 0.84 mmol) in deionized water (20 mL), magnetically nanoparticles of Fe3O4 (0.5 g, 2.15 mmol) was added. The resulting suspension was stirred for 20 h at 80 °C. After that, magnetically nanoparticles of Fe3O4@SbFx were separated by an external magnetic field, washed with deionized water and then dried at 60 °C.
A typical procedure for reductive-coupling of nitrobenzene to azobenzene with NaBH4/Fe3O4@SbFx MNPs
A mixture of nitrobenzene (0.123 g, 1 mmol) and Fe3O4@SbFx (0.02 g) in EtOH (3 mL) was well stirred for 2 min. NaBH4 (0.076 g, 2 mmol) was then added and the resulting mixture was stirred under reflux conditions for 6 min. During the progress of the reaction, the color of mixture was changed to orange-red showing the formation of azobenzene product. After completion of the reaction (monitored by TLC, n-hexane/EtOAc: 10/2), the mixture was cooled to the room temperature. The nanocatalyst was magnetically separated from the reaction mixture followed by the addition of H2O (4 mL). The mixture was stirred for additional 2 min and then extracted with EtOAc (2 × 5 mL). The combined color extracts were dried over anhydrous Na2SO4. Evaporation of the solvent under reduced pressure affords the pure crystals of azobenzene in 95% yield (0.077 g, Table 3, entry 1).
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
We gratefully acknowledge the financial support of this work by research councils of Urmia University.
References
- K. Hunger, Industrial Dyes: Chemistry, Properties, Applications, Wiley-VCH, Weinheim, 2003 Search PubMed.
- H. Zollinger, Colour Chemistry: Syntheses, Properties and Applications of Organic Dyes and Pigments, Wiley-VCH, Switzerland, 3rd edn, 2003 Search PubMed.
- P. F. Gordon and P. Gregory, Organic Chemistry in Colour, Springer, New York, 1983, pp. 95–162 Search PubMed.
- R. D. Athey Jr, Eur. Coat. J., 1998, 3, 146 Search PubMed.
- P. N. D. Ashutosh and J. K. Mehrotra, Colourage, 1979, 26, 25 Search PubMed.
- A. Jain, Y. Gupta and S. K. Jain, Crit. Rev. Ther. Drug Carrier Syst., 2006, 23, 349–400 CrossRef CAS PubMed.
- Z. F. Liu, K. Hashimoto and A. Fujishima, Nature, 1990, 347, 658–660 CrossRef CAS.
- T. Ikeda and O. Tsutsumi, Science, 1995, 268, 1873–1875 CrossRef CAS PubMed.
- H. Murakami, A. Kawabuchi, K. Kotoo, M. Kutinake and N. Nakashima, J. Am. Chem. Soc., 1997, 119, 7605–7606 CrossRef CAS.
- I. A. Banerjee, L. Yu and H. Matsui, J. Am. Chem. Soc., 2003, 125, 9542–9543 CrossRef CAS PubMed.
- F. Cisnetti, R. Ballardini, A. Credi, M. T. Gandolfi, S. Masiero, F. Negri, S. Pieraccini and G. P. Spada, Chem.–Eur. J., 2004, 10, 2011–2021 CrossRef CAS PubMed.
- J. C. Crano and R. J. Guglielmetti, Organic Photochromic and Thermochromic Compounds, Plenum Press, New York, 1999 Search PubMed.
- B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789–1816 CrossRef CAS PubMed.
- X. Liang, H. Asanuma and M. Komiyama, J. Am. Chem. Soc., 2002, 124, 1877–1883 CrossRef CAS PubMed.
- H. E. Zimmerman, R. J. Boettcher, N. E. Buehler, G. E. Keck and M. G. Steinmetz, J. Am. Chem. Soc., 1976, 98, 7680–7689 CrossRef CAS.
- B. M. Trost, R. M. Cory, P. H. Scudder and H. B. Neubold, J. Am. Chem. Soc., 1973, 95, 7813–7820 CrossRef CAS.
- K. Haghbeen and E. W. Tan, J. Org. Chem., 1998, 63, 4503–4505 CrossRef CAS.
- H. D. Anspon, Org. Synth. Coll., 1955, 3, 711 Search PubMed.
- E. Merino, Chem. Soc. Rev., 2011, 40, 3835–3853 RSC.
- M. Rahimizadeh, H. Eshghi, A. Shiri, Z. Ghadamyari, M. M. Matin, F. Oroojalian and P. Pordeli, J. Korean Chem. Soc., 2012, 56, 716–719 CrossRef CAS.
- A. Zarei, A. R. Hajipour, L. Khazdooz, B. F. Mirjalili and A. N. Chermahini, Dyes Pigm., 2009, 81, 240–244 CrossRef CAS.
- A. Mitsutani, Catal. Today, 2002, 73, 57–63 CrossRef CAS.
- J. Safari, S. H. Banitaba and S. D. Khalili, J. Mol. Catal. A: Chem., 2011, 335, 46–50 CrossRef CAS.
- A. Grirrane, A. Corma and H. Garcia, Science, 2008, 322, 1661–1664 CrossRef CAS PubMed.
- C. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2010, 49, 6174–6177 CrossRef CAS PubMed.
- X. Geng and C. Wang, Org. Biomol. Chem., 2015, 13, 7619–7623 RSC.
- F. Ahmed Khan and C. Sudheer, Tetrahedron Lett., 2009, 50, 3394–3396 CrossRef.
- Y. Lu, J. Liu, G. Diffee, D. Liu and B. Liu, Tetrahedron Lett., 2006, 47, 4597–4599 CrossRef CAS.
- J. R. Hwu, A. R. Das, C. W. Yang, J. J. Huang and M. H. Hsu, Org. Lett., 2005, 7, 3211–3214 CrossRef CAS PubMed.
- H. J. Shine and H. E. Mallory, J. Org. Chem., 1962, 27, 2390–2391 CrossRef CAS.
- H. W. Galbraith, E. F. Degering and E. F. Hitch, J. Am. Chem. Soc., 1951, 73, 1323–1324 CrossRef CAS.
- P. D. Ren, S. F. Pan, T. W. Dong and S. H. Wu, Synth. Commun., 1996, 26, 3903–3908 CrossRef CAS.
- T. F. Chung, Y. M. Wu and C. H. Cheng, J. Org. Chem., 1984, 49, 1215–1217 CrossRef CAS.
- S. Gowda and D. C. Gowda, Synthesis, 2002, 460–462 CrossRef CAS.
- G. R. Srinivasa, K. Abiraj and D. C. Gowda, Synth. Commun., 2003, 33, 4221–4227 CrossRef CAS.
- G. R. Srinivasa, K. Abiraj and D. C. Gowda, Tetrahedron Lett., 2003, 44, 5835–5837 CrossRef CAS.
- G. R. Srinivasa, K. Abiraj and D. C. Gowda, Aust. J. Chem., 2004, 57, 609–610 CrossRef CAS.
- R. F. Nystrom and W. G. Brown, J. Am. Chem. Soc., 1948, 70, 3738–3740 CrossRef CAS PubMed.
- J. F. Corbett, Chem. Commun., 1968, 1257b–1258 RSC.
- R. O. Hutchins, D. W. Lamson, L. Rua, C. Milewski and B. Maryanoff, J. Org. Chem., 1971, 36, 803–806 CrossRef CAS.
- A. C. Knipe, S. J. McGuinness and W. E. Watts, J. Organomet. Chem., 1979, 172, 463–466 CrossRef CAS.
- H. Alper and H. N. Paik, J. Organomet. Chem., 1978, 144, C18–C20 CrossRef CAS.
- W. Tadros, M. S. Ishak and E. Bassili, J. Chem. Soc., 1959, 627–629 RSC.
- A. Khan and S. Hecht, Chem.–Eur. J., 2006, 12, 4764–4774 CrossRef CAS PubMed.
- D. D. Laskar, D. Prajapati and J. S. Shandu, J. Chem. Soc., Perkin Trans. 1, 2000, 1, 67–69 RSC.
- M. B. Sridhara, R. Suhas and D. G. C. Gowda, Eur. J. Chem., 2013, 1, 61–63 CrossRef.
- J. M. Khurana and A. Ray, Bull. Chem. Soc. Jpn., 1996, 69, 407–410 CrossRef CAS.
- P. Sobota, T. Pluzinski and S. Rummel, Tetrahedron, 1981, 37, 939–942 CrossRef CAS.
- J. Wang, L. Hu, X. Cao, J. Lu, X. Li and H. Gu, RSC Adv., 2013, 3, 4899–4902 RSC.
- N. Sakai, K. Fujii, S. Nabeshima, R. Ikeda and T. Konakahara, Chem. Commun., 2010, 46, 3173–3175 RSC.
- N. Sakai, S. Asama, S. Anai and T. Konakahara, Tetrahedron, 2014, 70, 2027–2033 CrossRef CAS.
- Y. Moglie, C. Vitale and G. Radivoy, Tetrahedron Lett., 2008, 49, 1828–1831 CrossRef CAS.
- C. Luo, X. Ji, S. Hou, N. Eidson, X. Fan, Y. Liang, T. Deng, J. Jiang and C. Wang, Adv. Mater., 2018, 30, e1706498 CrossRef PubMed.
- H. Q. Li, X. Liu, Q. Zhang, S. S. Li, Y. M. Liu, H. Y. He and Y. Cao, Chem. Commun., 2015, 51, 11217–11220 RSC.
- V. Kalyanaraman and M. V. George, J. Org. Chem., 1973, 38, 507–514 CrossRef CAS.
- S. Wada, M. Urano and H. Suzuki, J. Org. Chem., 2002, 67, 8254–8257 CrossRef CAS PubMed.
- J. A. Gladysz, J. G. Fulcher and S. Togashi, Tetrahedron Lett., 1977, 18, 521–524 CrossRef.
- M. Tamura and H. Fujihara, J. Am. Chem. Soc., 2003, 125, 15742–15743 CrossRef CAS PubMed.
- V. K. Sharma, R. A. Yngard and Y. Lin, Adv. Colloid Interface Sci., 2009, 145, 83–96 CrossRef CAS PubMed.
- M. Nasrollahzadeh, S. M. Sajadi and A. Hatamifard, J. Colloid Interface Sci., 2015, 460, 146–153 CrossRef CAS PubMed.
- A. Hatamifard, M. Nasrollahzadeh and J. Lipkowski, RSC Adv., 2015, 5, 91372–91381 RSC.
- M. Atarod, M. Nasrollahzadeh and S. M. Sajadi, RSC Adv., 2015, 5, 91532–91543 RSC.
- M. Atarod, M. Nasrollahzadeh and S. M. Sajadi, J. Colloid Interface Sci., 2016, 465, 249–258 CrossRef CAS PubMed.
- M. Nasrollahzadeh, M. Maham, A. Rostami-Vartooni, M. Bagherzadeh and S. M. Sajadi, RSC Adv., 2015, 5, 64769–64780 RSC.
- M. Nasrollahzadeh, S. M. Sajadi, A. Rostami-Vartooni, M. Alizadeh and M. Bagherzadeh, J. Colloid Interface Sci., 2016, 466, 360–368 CrossRef CAS PubMed.
- M. Nasrollahzadeh, Tetrahedron Lett., 2016, 57, 337–339 CrossRef CAS.
- M. Nasrollahzadeh, New J. Chem., 2014, 38, 5544–5550 RSC.
- C. W. Lim and I. S. Lee, Nano Today, 2010, 5, 412–434 CrossRef CAS.
- M. Nasrollahzadeh and S. M. Sajadi, J. Colloid Interface Sci., 2016, 464, 147–152 CrossRef CAS PubMed.
- Z. Shokri, B. Zeynizadeh and S. A. Hosseini, J. Colloid Interface Sci., 2017, 485, 99–105 CrossRef CAS PubMed.
- B. Zeynizadeh, I. Mohammadzadeh, Z. Shokri and S. A. Hosseini, J. Colloid Interface Sci., 2017, 500, 285–293 CrossRef CAS PubMed.
- Z. Shokri, B. Zeynizadeh, S. A. Hosseini and B. Azizi, J. Iran. Chem. Soc., 2017, 14, 101–109 CrossRef CAS.
- B. Zeynizadeh and F. Sepehraddin, J. Iran. Chem. Soc., 2017, 14, 2649–2657 CrossRef CAS.
- S. Karami, B. Zeynizadeh and Z. Shokri, Cellulose, 2018, 25, 3295–3305 CrossRef CAS.
- B. Zeynizadeh and F. Sepehraddin, J. Organomet. Chem., 2018, 856, 70–77 CrossRef CAS.
- M. Gilanizadeh and B. Zeynizadeh, New J. Chem., 2018, 42, 8553–8566 RSC.
- G. Y. Li, Y. R. Jiang, K. L. Huang, P. Ding and L. L. Yao, Colloids Surf., A, 2008, 320, 11–18 CrossRef CAS.
- J. A. Lopez, F. Gonzalez, F. A. Bonilla, G. Zambrano and M. E. Gomez, Rev. LatinAm. Metal. Mat., 2010, 30, 60–66 Search PubMed.
- G. Zhang, T. Liu, T. Zhu, J. Qin, Y. Wu and C. Chen, Opt. Mater., 2008, 31, 110–113 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01249d |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Fariba Faraji
* and
Fariba Faraji