
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: MiR-132 enhances proliferation and migration of HaCaT cells by targeting TIMP3
Lina Jianga,
Yizhou Jiangb,
Xiaohui Jic,
Jiangtao Lid and
Ximei Zhai *a
*a
aDepartment of Plastic Surgery, The First Affiliated Hospital of Zhengzhou University, No. 1, East Jianshe Rd, Zhengzhou, 450052, China. E-mail: qinhzhouygwcshan@163.com; Tel: +86-371-66913114
bDepartment of Breast Surgery, Shanghai Cancer Center, Fudan University, Shanghai, 200032, China
cDepartment of Pathology, China
dDepartment of Breast Surgery, The People's Hospital of Zhengzhou, Zhengzhou, 450003, China
First published on 5th July 2019
Abstract
MicroRNAs (miRNAs) are involved in multiple skin pathologies, including wound healing. Here, we explored the detailed role and molecular mechanism of miR-132 on HaCaT cells proliferation and migration. qRT-PCR assay was used to assess miR-132 expression and Western blot analysis was performed to detect inhibitor of matrix metalloproteinase-3 (TIMP3) level in HaCaT cells and normal human epidermal keratinocytes (NHEK) under transforming growth factor β1 (TGF-β1) treatment. Dual-luciferase reporter assay and RNA immunoprecipitation (RIP) assay were employed to confirm the endogenous interaction between miR-132 and TIMP3. Cell proliferation ability was determined by MTT assay and the migration capacity was evaluated by transwell assay. TGF-β1 treatment resulted in a increase of miR-132 expression and a decrease of TIMP3 level in HaCaT cells and NHEK cells. The proliferation and migration abilities of TGF-β1-treated HaCaT cells were promoted by miR-132 upregulation, while them were inhibited by TIMP3 overexpression. Moreover, TIMP3 was a direct target of miR-132. MiR-132-mediated pro-proliferation and pro-migration effects were antagonized by TIMP3 in HaCaT cells under TGF-β1 treatment. Our data supported that miR-132 promoted the proliferation and migration of HaCaT cells at least partly by targeting TIMP3, highlighting miR-132 as a potential therapeutic strategy of wound healing.
Introduction
Wound healing, a natural response to skin injury, is a highly orchestrated biological phenomenon that consists of hemostasis, inflammatory response, re-epithelialization and remodeling.1 The re-epithelialization process is an essential component of wound healing and mainly relies on the migration and proliferation of keratinocytes.2 The migration of keratinocyte is initiated by various growth factors and cytokines, including transforming growth factor β1 (TGF-β1).3 However, TGF-β1 was manifested to suppress keratinocyte proliferation, indicating its paradoxical role on keratinocyte migration and proliferation.4 Interestingly, TGF-β1 is upregulated during wound healing, while non-healing wounds often show a loss of TGF-β1 signalling.5 Accumulating evidence has suggested that TGF-β1 plays a crucial role in enhancing or repressing wound healing by its downstream molecules or receptors.6,7 Therefore, a better understanding of the molecular mechanism of the cellular response to TGF-β1 signaling is vital in wound healing.MicroRNAs (miRNAs) are small, evolutionarily conserved, non-coding RNAs that are about 20–22 nucleotides in length and direct posttranscriptional suppressor of gene expression by binding to 3′-untranslated region (UTR) of target mRNAs.8 Dysregulation of miRNAs plays critical roles in the development and progression of mutiple human diseases.9 It has been reported that miRNAs are involved in skin development and associated with multiple skin pathologies, including wound healing.10,11 MiR-132 has been identified as a top upregulated miRNA in epidermal keratinocytes during the wound healing, and miR-132 overexpression promoted the proliferation of keratinocytes.12 In mouse in vivo and human ex vivo wound models, miR-132 knockdown impaired wound healing.12 High expression of miR-132 was found in dermal fibroblasts from human skin wounds and miR-132 enhanced the migration of fibroblast by targeting RAS p21 protein activator 1 in wound healing.13 Additionally, miR-132 expression was induced by TGF-β1 in keratinocytes and dermal fibroblasts.12,13
In this study, our data supported that miR-132 was upregulated by TGF-β1 and miR-132 promoted the proliferation in HaCaT cells. We firstly verified that miR-132 enhanced HaCaT cells migration under TGF-β1 treatment. Furthermore, miR-132 promoted the proliferation and migration of HaCaT cells at least partly by targeting inhibitor of matrix metalloproteinase-3 (TIMP3), highlighting miR-132 as a potential therapeutic marker of wound healing.
Materials and methods
Cell isolation, culture and treatment
Normal human epidermal keratinocytes (NHEK) were isolated as previously described.14 Briefly, foreskin tissues from 5 cases patients (20–25 years old, with informed consent) at the First Affiliated Hospital of Zhengzhou University were digested by Dispase II (Roche, Mannhein, Germany) for removing the dermis from the epidermis, and then were incubated with trypsinization (Roche). Our study was approved by the Ethic Committee of the First Affiliated Hospital of Zhengzhou University in accordance with the Declaration of Helsinki Principles. Informed consents were obtained from all participants.Human immortalized keratinocytes HaCaT cells were purchased from China Center for Type Culture Collection (CCTCC, Wuhan, China). NHEK cells were cultured in EpiLife serum-free medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with HKGS (Thermo Fisher Scientific), and HaCaT cells were maintained in DMEM medium (Hyclone, Logan, UT), supplemented with 10% fetal calf serum (FCS, Life Technologies, Carlsbad, CA, USA), 1% streptomycin/penicillin (Thermo Fisher Scientific). All cells were maintained at 37 °C in the presence of 5% CO2.
HaCaT and NHEK cells were treated with different concentration (0, 2, 5 and 10 ng ml−1) of TGF-β1 (Abbkine, Redlands, CA, USA) for 24 h, or exposed with TGF-β1 (5 ng ml−1) at different time period (0, 6, 12 and 24 h) followed by the detection of miR-132 and TIMP3 expression levels. For proliferation and migration assays, HaCaT cells were treated with TGF-β1 (5 ng ml−1) for the indicated time.
Cell transfection
The modified mature miRNA mimics (miR-132 mimics or miR-NC mimics), modified antisense oligonucleotides (anti-miR-NC or anti-miR-132), silencer select predesigned siRNA for TIMP3 or siRNA control (si-TIMP3 or si-NC), TIMP3 overexpression plasmid or negative control (pcDNA-TIMP3 or pcDNA-NC) were designed and constructed by Thermo Fisher Scientific. 20 nM of the indicated oligonucleotides or/and 10 ng of plasmid were transfected into HaCaT cells (1 × 105) by using Lipofectamine 2000 (Invitrogen, Waltham, MA, USA) according to the direction of manufacturers. After 6 h transfection, fresh medium containing 10% FBS was changed every three days.RNA extraction, reverse transcription PCR and quantitative real-time PCR (qRT-PCR)
For miR-132 quantification, total RNA from cells was extracted by using mirVana miRNA Isolation Kit (Ambion, Austin, TX, USA), and then was reversely transcripted into cDNA with TaqMan MiRNA Reverse Transcription Kit (Thermo Fisher Scientific). qRT-PCR was performed using TaqMan MiRNA Assay Kit (Thermo Fisher Scientific) on the ABI 7000 system (Applied Biosystems, Grand Island, NY, USA), and U6 was used as endogenous control. The fold changes were calculated by 2−ΔΔCt method. For TIMP3 mRNA analysis, total RNA extraction from cells was carried out with RNAeasy Plus (Qiagen, Hilden, Germany) according to the standard instruction. cDNA was synthesized from RNA extract with M-MVL reverse transcriptase (Promega, Madison, WI, USA) and qRT-PCR was performed by using One-Step SYBR Green Kit (Invitrogen). The relative expression of TIMP3 mRNA was quantified compared to that of GAPDH.MTT assay of cell proliferation
Cell proliferation ability was evaluated by MTT assay with MTT Cell Proliferation Reagent Kit (Roche) according to the protocol of manufacturers. To be brief, HaCaT cells were seeded into 96-well plates and then transfected or/and treated with TGF-β1. At the indicated time, 10 μl of MTT solution (5 mg ml−1) was added into each well and incubated for 4 h at 37 °C in 5% CO2 incubator, followed by the incubation with 100 μl of crystal dissolving solution. The absorbance at 540 nm was measured by a Multiscan Ex microplate reader (Thermo Fisher Scientific).Transwell assay of cell migration
The 24-well transwell chamber with 8 μm polycarbonate nucleopore filter (Corning, NY, USA) was used to assess cell migration capacity. About 5.0 × 104 cells was seeded into the upper chamber for 6–8 h to attach. 200 μl of serum-free medium was changed in the upper chamber and 500 μl of growth medium containing 10% FCS was added to the lower chamber. After incubation for 24 h, migrated cells were fixed with 4% paraformaldehyde (Sangon Biotech, Shanghai, China), and stained with 0.1% crystal violet (Sangon Biotech). The number of migrated cells was analyzed under a microscope (Leica, Wetzlar, Germany).Western blot of TIMP3 level
Total cellular protein was isolated by lysis buffer (150 mM NaCl, 50 mM Tris–HCl pH = 7.4, 0.1% SDS, 1% Triton X-100, 1 mM EDTA, 0.5% deoxycholate) supplemented with protease inhibitor cocktail (Roche). Equal amounts of protein lysates (100 μg) was subjected to 10% SDS-PAGE and then electrotransferred to PVDF membrances (Bio-Rad, Hercules, CA, USA). Blocked by a buffer containing 5% non-fat milk in TBS with 0.1% Tween-20, the membrances were probed overnight with specific primary antibodies against TIMP3 (sc-373839, Santa Cruz Biotechnology, Santa Cruz, CA, USA; dilution 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500) or β-actin (sc-517582, Santa Cruz Biotechnology; dilution 1:1000) at 4 °C, followed by the incubation with HRP-conjugated secondary antibodies (sc-516102, Santa Cruz Biotechnology; dilution 1:5000). The protein bands were visualized by the ECL assay (GE Healthcare, Princeton, NJ, USA).
:500) or β-actin (sc-517582, Santa Cruz Biotechnology; dilution 1:1000) at 4 °C, followed by the incubation with HRP-conjugated secondary antibodies (sc-516102, Santa Cruz Biotechnology; dilution 1:5000). The protein bands were visualized by the ECL assay (GE Healthcare, Princeton, NJ, USA).
Dual-luciferase reporter assay
The modified TIMP3 overexpression vector (TIMP3-WT) containing the complementary sites of miR-132 and its mutant-type vector (TIMP3-MUT) were designed and constructed by Sangon Biotech. HaCaT cells were cotransfected with 10 ng of TIMP3-WT or TIMP3-MUT and 20 nM of miR-132 mimics or miR-NC mimics using Lipofectamine 2000. The relative luciferase activity was determined by using a dual-luciferase assay system (Promega) according to Renilla luciferase activity.RNA immunoprecipitation (RIP) assay
RIP assay was performed using the EZ Magna RIP Kit (Millipore, Billerica, MA, USA) according to the manufacturer's direction. HaCaT cells were transfected with miR-132 mimics or miR-NC mimics, and then were lysed in lysis buffer. Whole cell lysates were incubated with RIP buffer containing magnetic beads conjugated with anti-Argonaute2 (anti-Ago2, Abcam, Cambridge, UK) or negative control anti-IgG (Abcam) antibody. Samples were purified by Dnase I (Sigma-Aldrich, St. Louis, MO, USA) and Proteinase K (Sigma-Aldrich), and then the coprecipitated RNAs were isolated. The relative enrichment of TIMP3 mRNA was measured by qRT-PCR assay in the coprecipitated RNAs.Statistical analysis
All data were presented as the mean ± SD based on at least three replicates. The Student's t-test was used to compare the differences between two groups, and one-way ANOVA was performed to analyze the differences of multiple groups. P < 0.05 was considered statistically significant.Results
MiR-132 expression was induced by TGF-β1 in HaCaT and NHEK cells
TGF-β1 was manifested to enhance the expression of miR-132 in epidermal keratinocytes and human dermal fibroblasts.12,13 Here, we validated whether TGF-β1 regulated miR-132 expression in HaCaT cells. HaCaT cells were treated with different concentration (0, 2, 5 and 10 ng ml−1) of TGF-β1 for 24 h, and then miR-132 expression was detected by qRT-PCR. As expect, miR-132 levels in HaCaT cells were highly elevated by TGF-β1 at the concentrations of 5 and 10 ng ml−1 (Fig. 1A). Then, NHEK cells were obtained and treated with different concentration (0, 2, 5 and 10 ng ml−1) of TGF-β1. qRT-PCR results revealed that TGF-β1 upregulated miR-132 expression in NHEK cells in a dose-dependent manner (Fig. 1B). Subsequently, HaCaT and NHEK cells were exposed with TGF-β1 (5 ng ml−1) at different time period (0, 6, 12 and 24 h). Results demonstrated that TGF-β1 treatment in HaCaT and NHEK cells resulted in a significant increase of miR-132 expression in a time-dependent manner (Fig. 1C and D).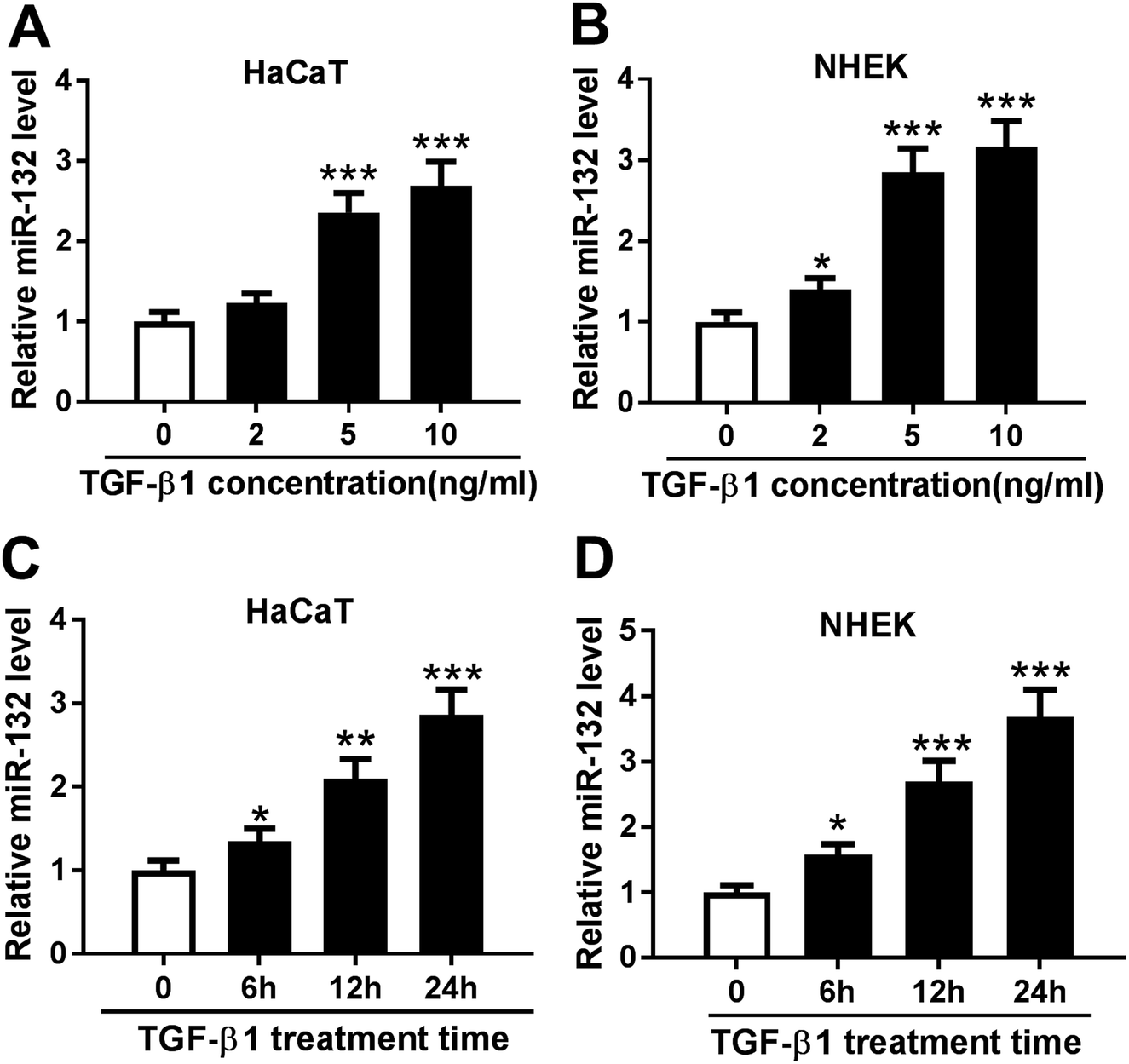 | ||
| Fig. 1 TGF-β1 upregulated miR-132 expression in HaCaT and NHEK cells. HaCaT (A) and NHEK (B) cells were treated with different concentration (0, 2, 5 and 10 ng ml−1) of TGF-β1 for 24 h, and miR-132 expression was detected by qRT-PCR assay. HaCaT (C) and NHEK (D) cells were exposed with TGF-β1 (5 ng ml−1) at different time period (0, 6, 12 and 24 h), followed by the measurement of miR-132 expression. *P < 0.05 or **P < 0.01 or ***P < 0.001 vs. control. | ||
MiR-132 promoted the proliferation and migration of HaCaT cells under TGF-β1 treatment
Given our data on TGF-β1-induced miR-132 upregulation (Fig. 2A and B), we further observed whether TGF-β1 treatment affected the proliferation and migration of HaCaT cells. MTT assay revealed that TGF-β1 treatment strikingly repressed cell proliferation compared with negative control (Fig. 2C and D). Whereas, transwell assay showed that TGF-β1 treatment resulted in increased migration ability of HaCaT cells (Fig. 2E and F).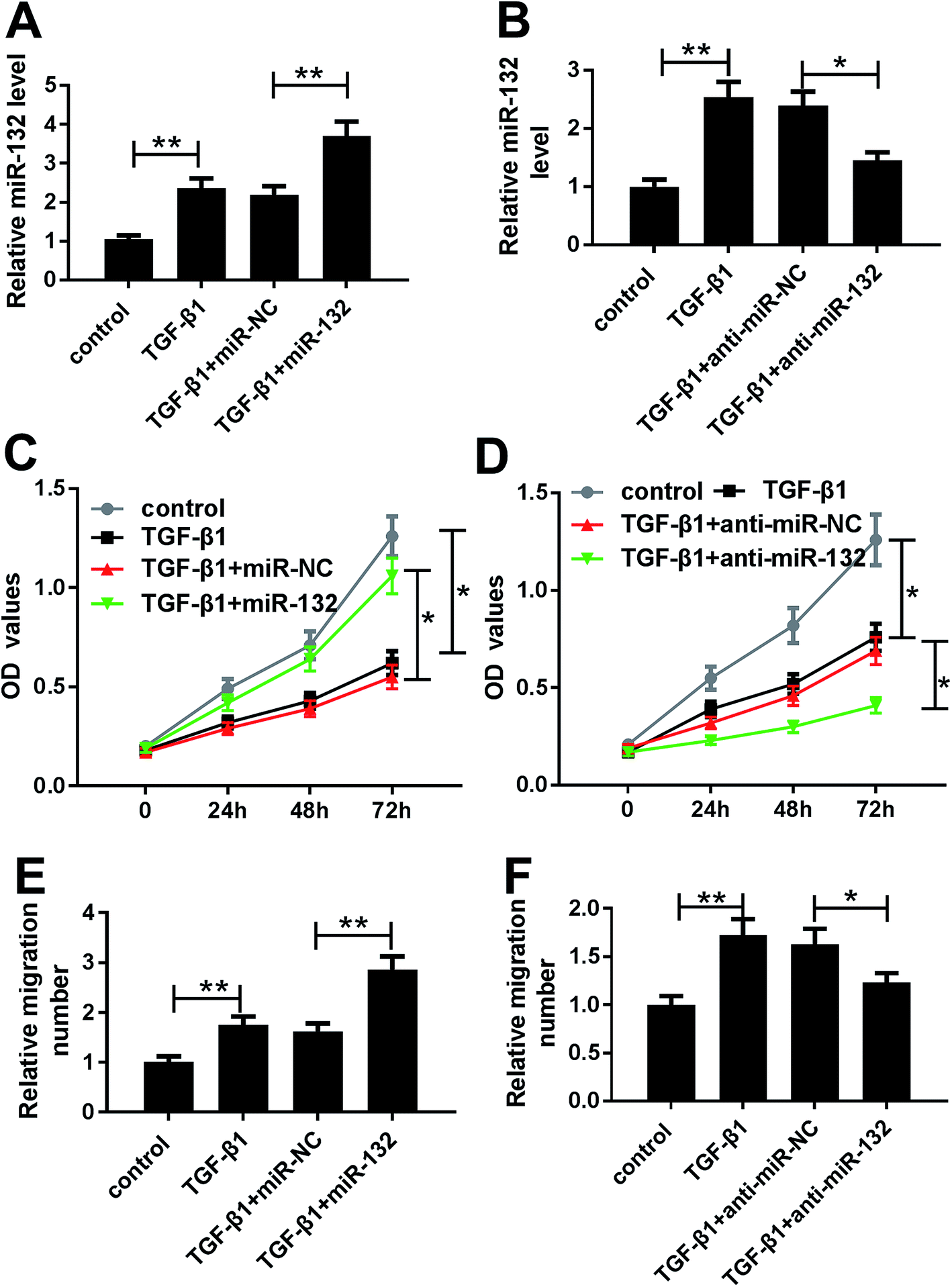 | ||
| Fig. 2 MiR-132 promoted the proliferation and migration of HaCaT cells under TGF-β1 treatment. HaCaT cells were treated with TGF-β1 (5 ng ml−1) or transfected with miR-132 mimics or anti-miR-132 prior to TGF-β1 treatment, followed by the determination of miR-132 expression by qRT-PCR assay (A) and (B), cell proliferation ability by MTT assay (C) and (D), cell migration capacity by transwell assay (E) and (F). *P < 0.05 or **P < 0.01 vs. control or TGF-β1 + miR-NC. | ||
To determine the role of miR-132 in HaCaT cells, we manipulated miR-132 expression by transfecting with miR-132 mimics or anti-miR-132 into HaCaT cells prior to TGF-β1 treatment. These data presented that in comparison to respective control, miR-132 mimics transfection led to a more distinct increase of miR-132 expression, while anti-miR-132 introduction markedly abrogated TGF-β1-induced miR-132 overexpression (Fig. 2A and B). Subsequently, functional experiments showed that cell proliferation and migration abilities in miR-132 mimics group were higher than those in control group (Fig. 2C and E). Conversely, cell proliferation and migration abilities in anti-miR-132 group were lower than those in anti-miR-NC group (Fig. 2D and F).
TIMP3 was a direct target of miR-132
To further explore the molecular mechanism involved in the promotional effects of miR-132 on HaCaT cells proliferation and migration, online software Starbase was used to predict the targets of miR-132. Intriguingly, we found that the 3′-UTR of TIMP3 mRNA contained the putative complementary sites for miR-132 (Fig. 3A). To confirm whether miR-132 could target TIMP3, luciferase-based reporter constructs containing the wild-type or mutant-type miR-132 targeted-sequences for TIMP3 3′-UTR (TIMP3-WT or TIMP3-MUT) were cotransfected into HaCaT cells with miR-132 mimics. Results demonstrated that compared to corresponding control, the relative luciferase activity of TIMP3-WT was markedly repressed by miR-132 overexpression, while little change was observed in TIMP3-MUT in response to the upregulation of miR-132 (Fig. 3B). Moreover, RIP assay showed an abundant enrichment of TIMP3 mRNA in the presence of miR-132 mimics, indicating the endogenous interaction between miR-132 and TIMP3 mRNA (Fig. 3C). Further, we examined the effect of miR-132 on TIMP3 expression by Western blot. These data revealed that TIMP3 expression in HaCaT cells was highly decreased by miR-132 overexpression, while it was remarkably increased following miR-132 downregulation, in comparison to their counterparts (Fig. 3D).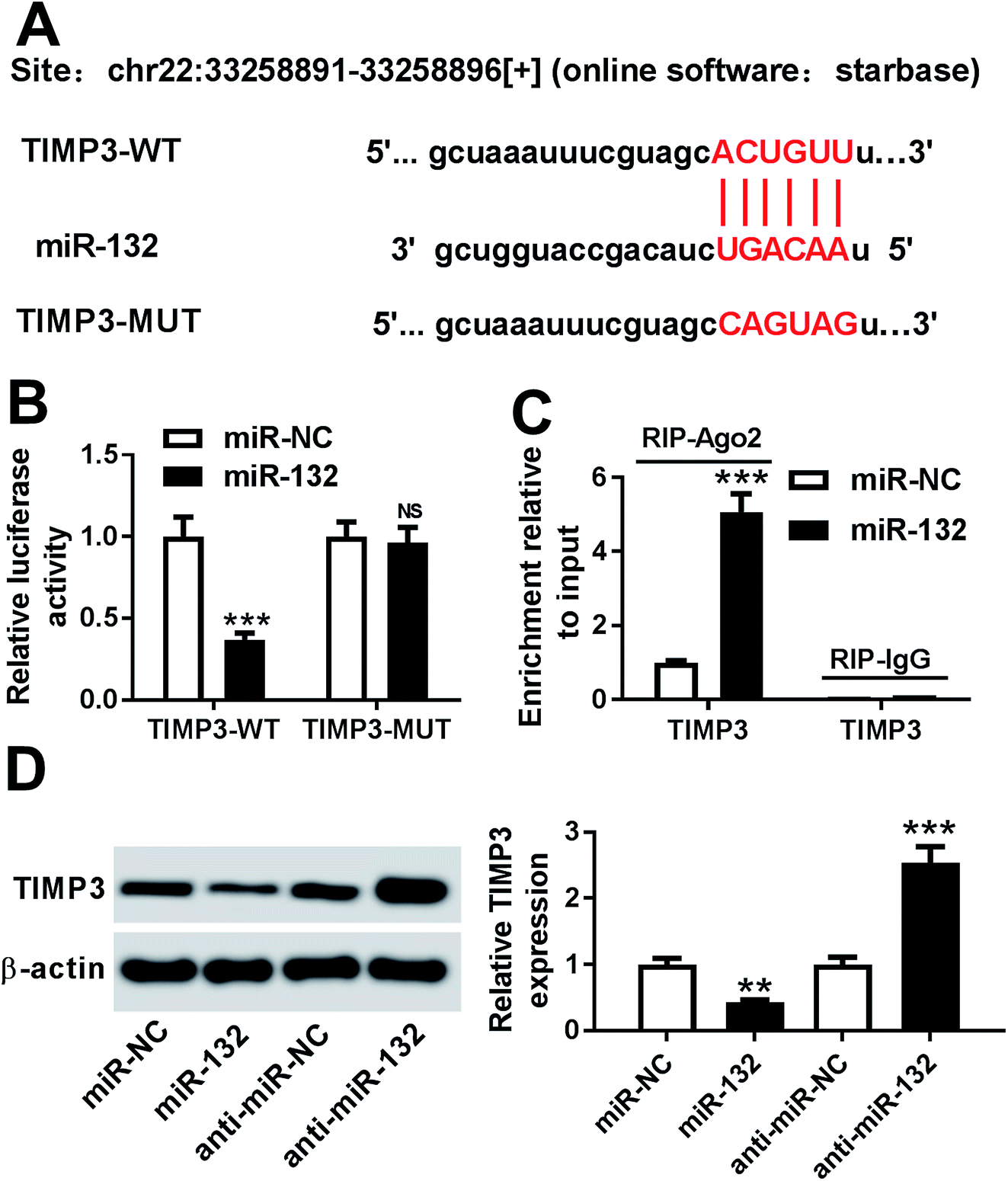 | ||
| Fig. 3 TIMP3 was a direct target of miR-132 in HaCaT cells. (A) The miR-132 binding sites predicted in the 3′-UTR of TIMP3 mRNA and the mutant in seed sites. (B) The relative luciferase activity was detected in HaCaT cells cotransfected with TIMP3-WT or TIMP3-MUT constructs and miR-NC mimics or miR-132 mimics. (C) HaCaT cells were transfected with miR-NC mimics or miR-132 mimics, and the enrichment of TIMP3 mRNA was measured with anti-Ago2 or anti-IgG by qRT-PCR assay. (D) HaCaT cells were transfected with miR-NC mimics, miR-132 mimics, anti-miR-NC or anti-miR-132, followed by the detection of TIMP3 level by Western blot. **P < 0.01 or ***P < 0.001 vs. respective control. | ||
TIMP3 expression was inhibited by TGF-β1 in HaCaT and NHEK cells
Then, we evaluated the effect of TGF-β1 on TIMP3 expression in HaCaT and NHEK cells. HaCaT and NHEK cells were treated with different concentration (0, 2, 5 and 10 ng ml−1) of TGF-β1 or TGF-β1 (5 ng ml−1) at different time period (0, 6, 12 and 24 h), followed by the measurement of TIMP3 expression by Western blot. Results revealed that TGF-β1 repressed the expression of TIMP3 in a dose- and time-dependent manner in HaCaT and NHEK cells (Fig. 4A–D).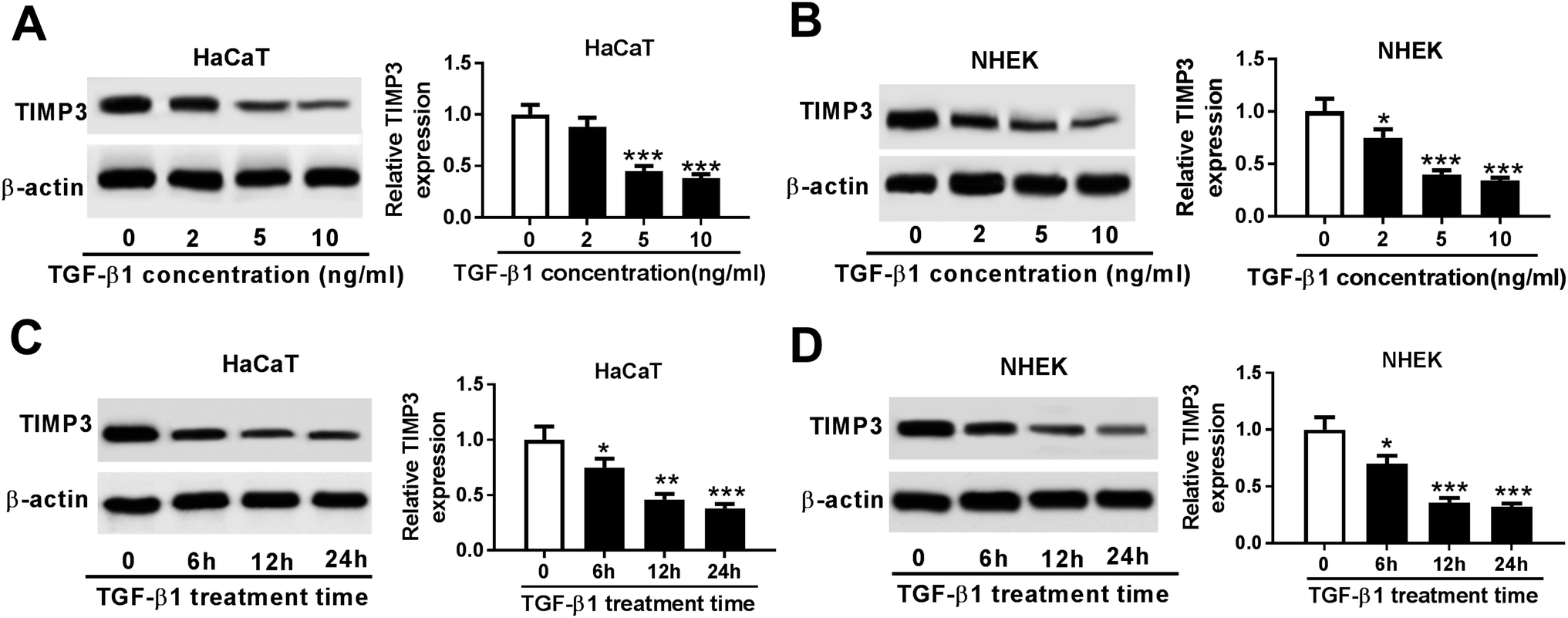 | ||
| Fig. 4 TGF-β1 repressed the expression of TIMP3. HaCaT (A) and NHEK (B) cells were treated with different concentration (0, 2, 5 and 10 ng ml−1) of TGF-β1 for 24 h, and then TIMP3 expression was detected by Western blot. HaCaT (C) and NHEK (D) cells were treated with 5 ng ml−1 of TGF-β1 at different time period (0, 6, 12 and 24 h), followed by the measurement of TIMP3 expression by Western blot. *P < 0.05 or **P < 0.01 or ***P < 0.001 vs. control. | ||
TIMP3 inhibited the proliferation and migration of TGF-β1-treated HaCaT cells
To investigate the role of TIMP3 on HaCaT cells proliferation and migration, gain-of-function and loss-of-function experiments were performed by transfecting with pcDNA-TIMP3 and si-TIMP3 into HaCaT cells prior to TGF-β1 treatment, respectively. Western blot results presented that compared with homologous control, TIMP3 expression in HaCaT cells was significantly increased by pcDNA-TIMP3 transfection under TGF-β1 treatment, while it was dramatically decreased after si-TIMP3 introduction (Fig. 5A and B). Further, functional experiments demonstrated that in comparison to their counterparts, cell proliferation and migration abilities were highly repressed by TIMP3 upregulation, while them were evidently enhanced following TIMP3 knockdown in TGF-β1-treated HaCaT cells (Fig. 5C–D).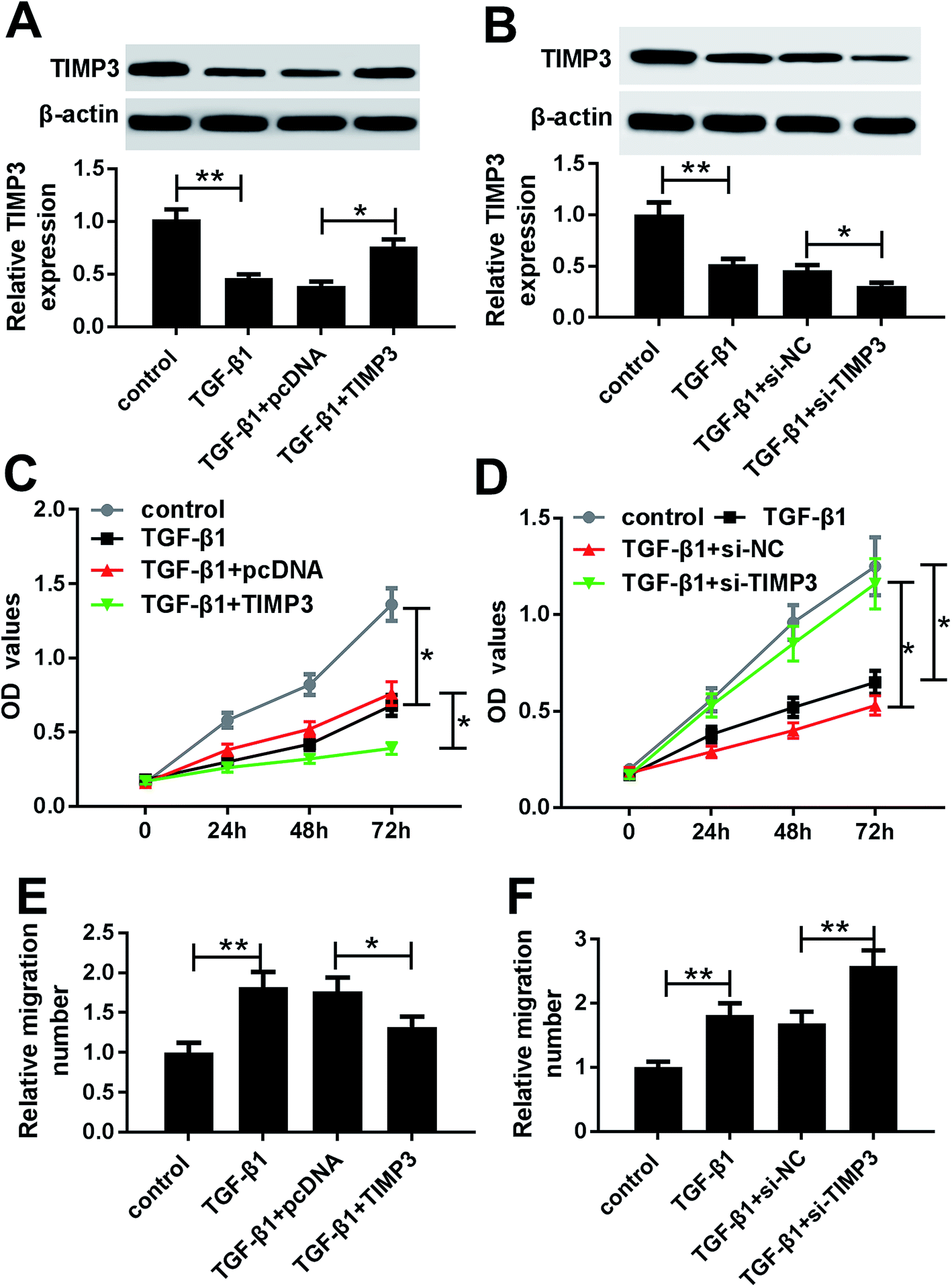 | ||
| Fig. 5 TIMP3 inhibited the proliferation and migration of HaCaT cells under TGF-β1 treatment. HaCaT cells were treated with TGF-β1 (5 ng ml−1) or transfected with pcDNA or pcDNA-TIMP3 prior to TGF-β1 treatment. (A) and (B) qRT-PCR for miR-132 expression in treated cells. (C) and (D) MTT assay for the proliferation ability in treated cells. (E) and (F) Transwell assay for the migration capacity in treated cells. *P < 0.05 or **P < 0.01 vs. control or TGF-β1 + pcDNA. | ||
MiR-132-mediated pro-proliferation and pro-migration effects were antagonized by TIMP3 in TGF-β1-treated HaCaT cells
To provide further mechanistic insight into the link between miR-132 and TIMP3 on HaCaT cells proliferation and migration, HaCaT cells were cotransfected with miR-132 mimics and pcDNA-TIMP3 or anti-miR-132 and si-TIMP3 prior to TGF-β1 treatment. MTT assays showed that miR-132-mediated pro-proliferation effect was significantly abrogated by pcDNA-TIMP3 cotransfection, and cotransfection of si-TIMP3 markedly antagonized the effect of anti-miR-132-mediated anti-proliferation in TGF-β1-treated HaCaT cells (Fig. 6A and B). In parallel, cell migration capacity in miR-132 mimics + pcDNA-TIMP3 group was lower than that in control group, and it in anti-miR-132 + si-TIMP3 group was higher than that of control (Fig. 6C and D).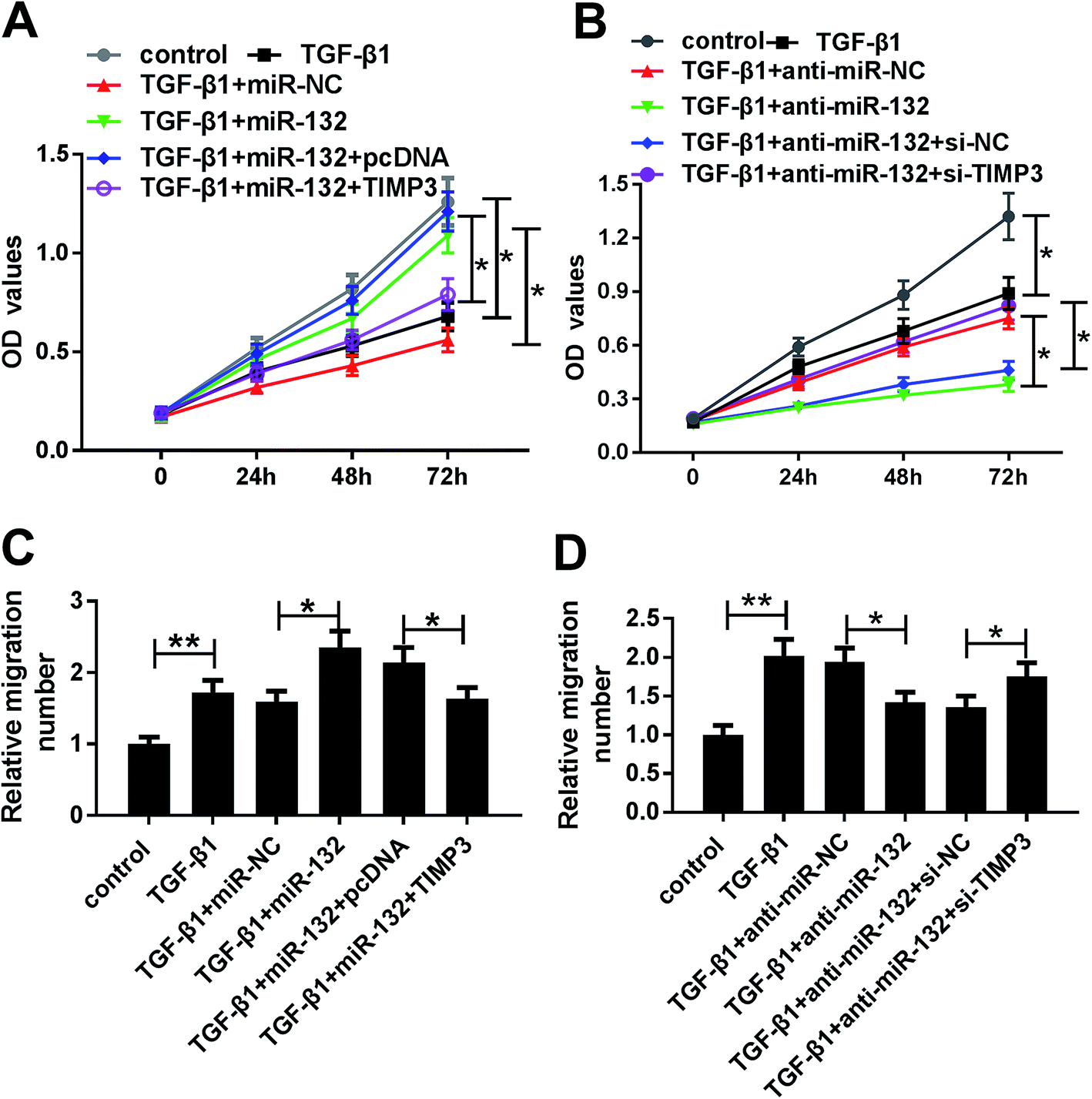 | ||
| Fig. 6 MiR-132-mediated pro-proliferation and pro-migration effects were antagonized by TIMP3 in TGF-β1-treated HaCaT cells. HaCaT cells were transfected with miR-132 mimics + pcDNA, miR-132 mimics + pcDNA-TIMP3, anti-miR-132 + si-NC or anti-miR-132 + si-TIMP3 prior to TGF-β1 (5 ng ml−1) treatment, followed by the detection of cell proliferation ability by MTT assay (A) and (B), cell migration capacity by transwell assay (C) and (D). *P < 0.05 or **P < 0.01 vs. control or TGF-β1 + miR-NC or TGF-β1 + miR-132 + pcDNA. | ||
Discussion
The abnormal expression of miRNAs is discovered at a specific phase of wound healing process and aberrant regulation of these miRNAs plays important roles during wound healing.15 For example, miR-21 was reported to promote keratinocyte migration and enhance re-epithelialization through regulating TIMP3 and TIAM1 expression in skin wound healing.16 High expression of miR-155 accelerated wound healing and re-epithelization in vivo, and it promoted keratinocyte migration through modulating MMP-2 in vitro.17 Additionally, miR-31 was identified to enhance keratinocyte proliferation and migration via targeting EMP-1.11 Conversely, miR-26a repressed the proliferation and migration of HaCaT cells by modulating PTEN expression.18 MiR-205 knockdown was demonstrated to drive keratinocyte migration and accelerate re-epithelialization by targeting ITGA5, providing a potential therapeutic target for chronic wounds.19In the present study, we verified that miR-132 expression was induced by TGF-β1 in HaCaT and NHEK cells, consistent with previous results.12 MiR-132 has been considered to play a pivotal role in various biological and pathological events, such as ocular dominance plasticity, visual cortex plasticity and cancers.20–23 MiR-132 also was manifested to accelerate wound healing by promoting keratinocyte proliferation and fibroblast migration.12,13 Additionally, miR-132 was downregulated in human diabetic ulcers, and miR-132 replenishment effectively facilitated wound closure by promoting keratinocyte proliferation in the wounds of db/db mice.24 In the study, our data also demonstrated that miR-132 promoted the proliferation of HaCaT cells under TGF-β1 treatment, in accordance with former works.12 Moreover, we firstly confirmed that miR-132 enhanced the migration of TGF-β1-treated HaCaT cells. Besides, we found that TGF-β1 treatment resulted in a inhibition of cell proliferation and a promotion of migration in HaCaT cells, which were consistent with a recent document,4 indicating its paradoxical role on keratinocyte migration and proliferation. All these results implied that miR-132 might accelerate wound healing through promoting the proliferation and migration of HaCaT cells.
MiRNAs play critical roles in various biological process by negatively regulating target genes expression in a posttranscriptional manner.25 Thus, online software Starbase was used to predict the targets of miR-132. Among the predicted candidates, TIMP3 was of interest in the study, considering the pivotal role of TIMP3 in wound re-epithelialization.26 Subsequently, we firstly confirmed that TIMP3 was a direct target of miR-132 in HaCaT cells. TIMP3 acts as an inhibitor of matrix metalloproteinase, in regulating extracellular matrix deposition and degradation that is essential for wound healing.26 TIMP3 expression was validated to be decreased in cutaneous wound repair and psoriatic skin.27,28 TIMP3 implicated in the promotion effect of miR-21 on keratinocyte migration.16 Moreover, previous studies reported that TIMP3 was capacity of repressing cell proliferation and migration in hepatocellular carcinoma cells and melanomas cells.29,30 Additionally, TIMP3 was reported to play a vital role in kidney injury,31 traumatic brain injury32 and sciatic nerve injury.33
Further, our data revealed a significant inhibition of TIMP3 expression by TGF-β1 treatment in a dose- and time-independent manner in HaCaT and NHEK cells. To investigate the role of TIMP3 on cell proliferation and migration, we performed gain-of-function and loss-of-function experiments. These results indicated that TIMP3 replenishment inhibited the proliferation and migration of TGF-β1-treated HaCaT cells, partly consistent with a previous study.16 Lastly, we firstly elucidated that miR-132 enhanced the proliferation and migration of HaCaT cells that could be reversed by TIMP3. Similar with our findings, miR-21 promoted keratinocyte migration by targeting TIMP3 and TIAM1.16 MiR-31 promoted the proliferation and migration of human primary keratinocytes through modulating epithelial membrance protein 1 (EMP-1).11 Moreover, miR-132 enhanced fibroblast migration by regulating RAS p21 protein activator 1 (RASA1).13
In conclusion, our study indicated that miR-132 promoted proliferation and migration of HaCaT cells by targeting TIMP3, highlighting miR-132 as a potential therapeutic strategy of wound healing.
Ethics approval and consent to participate
Foreskin tissues from 5 cases patients (20–25 years old, with informed consent) at the First Affiliated Hospital of Zhengzhou University were digested by Dispase II (Roche, Mannhein, Germany) for removing the dermis from the epidermis, and then were incubated with trypsinization (Roche). The study was approved by Ethics and Committees of the First Affiliated Hospital of Zhengzhou University.Conflicts of interest
No financial and non-financial conflicts are declared by all authors.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant no: 81502278).References
- G. Han and R. Ceilley, Adv. Ther., 2017, 34, 599–610 CrossRef.
- I. Pastar, O. Stojadinovic, N. C. Yin, H. Ramirez, A. G. Nusbaum, A. Sawaya, S. B. Patel, L. Khalid, R. R. Isseroff and M. Tomic-Canic, Adv. Wound Care, 2014, 3, 445–464 CrossRef.
- H. J. Nam, Y. Y. Park, G. Yoon, H. Cho and J. H. Lee, Exp. Mol. Med., 2010, 42, 270–279 CrossRef CAS.
- K. Rasanen and A. Vaheri, J. Dermatol. Sci., 2010, 58, 97–104 CrossRef CAS.
- J. W. Penn, A. O. Grobbelaar and K. J. Rolfe, Int. J. Burns Trauma, 2012, 2, 18–28 CAS.
- B. R. Klass, A. O. Grobbelaar and K. J. Rolfe, Postgrad. Med. J., 2009, 85, 9–14 CrossRef CAS.
- C. Margadant and A. Sonnenberg, EMBO Rep., 2010, 11, 97–105 CrossRef CAS.
- D. P. Bartel, Cell, 2018, 173, 20–51 CrossRef CAS.
- J. Hayes, P. P. Peruzzi and S. Lawler, Trends Mol. Med., 2014, 20, 460–469 CrossRef CAS.
- J. Wang, Y. Qiu, N. W. Shi, J. N. Zhao, Y. C. Wang, H. Jiang and H. B. Qian, Eur. Rev. Med. Pharmacol. Sci., 2016, 20, 3748–3759 CAS.
- D. Li, X. I. Li, A. Wang, F. Meisgen, A. Pivarcsi, E. Sonkoly, M. Stahle and N. X. Landen, J. Invest. Dermatol., 2015, 135, 1676–1685 CrossRef CAS.
- D. Li, A. Wang, X. Liu, F. Meisgen, J. Grunler, I. R. Botusan, S. Narayanan, E. Erikci, X. Li, L. Blomqvist, L. Du, A. Pivarcsi, E. Sonkoly, K. Chowdhury and S. B. Catrina, et al., J. Clin. Invest., 2015, 125, 3008–3026 CrossRef.
- X. Li, D. Li, J. D. Wikstrom, A. Pivarcsi, E. Sonkoly, M. Stahle and N. X. Landen, Sci. Rep., 2017, 7, 7797 CrossRef.
- D. Zhang, J. Wang, Z. Wang, T. Zhang, P. Shi, X. Wang, F. Zhao, X. Liu, X. Lin and X. Pang, BioMed Res. Int., 2015, 2015, 453518 Search PubMed.
- J. Banerjee, Y. C. Chan and C. K. Sen, Physiol. Genom., 2010, 43, 543–556 CrossRef.
- X. Yang, J. Wang, S. L. Guo, K. J. Fan, J. Li, Y. L. Wang, Y. Teng and X. Yang, Int. J. Biol. Sci., 2011, 7, 685–690 CrossRef CAS.
- L. Yang, Z. Zheng, Q. Zhou, X. Bai, L. Fan, C. Yang, L. Su and D. Hu, J. Mol. Histol., 2017, 48, 147–155 CrossRef CAS.
- N. Yu, Y. Yang, X. Li, M. Zhang, J. Huang, X. Wang and X. Long, Gene, 2016, 594, 117–124 CrossRef CAS.
- T. Wang, N. Zhao, S. Long, L. Ge, A. Wang, H. Sun, X. Ran, Z. Zou, J. Wang and Y. Su, Biochim. Biophys. Acta, 2016, 1862, 1443–1452 CrossRef CAS.
- P. Tognini, E. Putignano, A. Coatti and T. Pizzorusso, Nat. Neurosci., 2011, 14, 1237–1239 CrossRef CAS.
- N. Mellios, H. Sugihara, J. Castro, A. Banerjee, C. Le, A. Kumar, B. Crawford, J. Strathmann, D. Tropea, S. S. Levine, D. Edbauer and M. Sur, Nat. Neurosci., 2011, 14, 1240–1242 CrossRef CAS.
- A. Formosa, A. M. Lena, E. K. Markert, S. Cortelli, R. Miano, A. Mauriello, N. Croce, J. Vandesompele, P. Mestdagh, E. Finazzi-Agro, A. J. Levine, G. Melino, S. Bernardini and E. Candi, Oncogene, 2013, 32, 127–134 CrossRef CAS.
- W. Renjie and L. Haiqian, Cancer Lett., 2015, 356, 568–578 CrossRef.
- X. Li, D. Li, A. Wang, T. Chu, W. Lohcharoenkal, X. Zheng, J. Grunler, S. Narayanan, S. Eliasson, E. K. Herter, Y. Wang, Y. Ma, M. Ehrstrom, L. Eidsmo and M. Kasper, et al., J. Invest. Dermatol., 2017, 137, 2630–2638 CrossRef CAS.
- S. M. Hammond, Adv. Drug Deliv. Rev., 2015, 87, 3–14 CrossRef CAS.
- M. P. Caley, V. L. Martins and E. A. O'Toole, Adv. Wound Care, 2015, 4, 225–234 CrossRef.
- J. R. Zibert, M. B. Lovendorf, T. Litman, J. Olsen, B. Kaczkowski and L. Skov, J. Dermatol. Sci., 2010, 58, 177–185 CrossRef CAS.
- C. Soo, W. W. Shaw, X. Zhang, M. T. Longaker, E. W. Howard and K. Ting, Plast. Reconstr. Surg., 2000, 105, 638–647 CrossRef CAS.
- B. Wang, S. H. Hsu, S. Majumder, H. Kutay, W. Huang, S. T. Jacob and K. Ghoshal, Oncogene, 2010, 29, 1787–1797 CrossRef CAS.
- A. M. Das, A. L. Seynhaeve, J. A. Rens, C. E. Vermeulen, G. A. Koning, A. M. Eggermont and T. L. Ten Hagen, Angiogenesis, 2014, 17, 163–177 CrossRef CAS.
- C. Schrimpf, C. Xin, G. Campanholle, S. E. Gill, W. Stallcup, S.-L. Lin, G. E. Davis, S. A. Gharib, B. D. Humphreys and J. S. Duffield, J. Am. Soc. Nephrol., 2012, 23(5), 868–883 CrossRef CAS.
- T. Menge, Y. Zhao, J. Zhao, K. Wataha, M. Gerber, J. Zhang, P. Letourneau, J. Redell, L. Shen and J. Wang, Sci. Transl. Med., 2012, 4, 161ra150 CrossRef.
- S. Zhou, S. Zhang, Y. Wang, S. Yi, L. Zhao, X. Tang, B. Yu, X. Gu and F. Ding, Neurosci. Lett., 2015, 586, 43–49 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |