
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning two-electron transfer in terpyridine-based platinum(II) pincer complexes†
Seher Kuyuldar ab,
Clemens Burda*b and
William B. Connick‡
a
ab,
Clemens Burda*b and
William B. Connick‡
a
aUniversity of Cincinnati, Department of Chemistry, 2600 Clifton Ave., Cincinnati, OH 45221, USA
bCase Western Reserve University, Department of Chemistry, 10900 Euclid Ave., Cleveland, OH 44106, USA. E-mail: burda@case.edu
First published on 5th July 2019
Abstract
An important factor in obtaining reversible multi-electron transfer is overcoming large changes in coordination geometry. One strategy is to use ligands that can support the geometries favored before and after the electron transfer. Pip2NCN− pincer and terpyridine ligands are used to support square planar Pt(II) and octahedral Pt(IV). For the Pt(II) complexes, [Pt(Z-pip2NCN)(R-tpy)]+ (Z = NO2, MeO, H; R = H, tertyl butyl, tolyl), 1H NMR spectroscopy shows that the Z-pip2NCN− ligand is monodentate whereas the R-terpyridyl ligand is tridentate. The availability of flanking piperidyl groups of the monodentate pincer ligand is essential for the stabilization of the metal center upon oxidation. Pt(Z-pip2NCN)(R-tpy)+ complexes undergo two-electron platinum centered oxidation near 0.4 V and two Pt(tpy) centered reductions near −1.0 V and −1.5 V. An estimate of nox/nred = 1.8 is consistent with an oxidation that involves two-electron transfer per Pt center. Variation in the pincer-(Z) and terpyridine-(R) substituents allows for tuning of the oxidation process over a 260 mV range and the two reduction processes over ranges of 230 mV (first reduction) and 290 mV (second reduction step).
Introduction
Many important reactions such as water splitting, activation of hydrocarbons and CO2 fixation require multiple redox equivalents.Several second- and third-row late transition metals have been shown to support multiple electron transfer reactivity leading to changes in their oxidation states along with the required geometrical changes. For example, Wilkinson's catalyst (RhCl(PPh3)3) for olefin hydrogenation,1 Shilov's catalyst (Pt(OH2)2Cl2) for alkane oxidation,2 and Heyduk's and Nocera's photocatalyst3 for the reduction of hydrohalic acids to hydrogen. The relative instability of the middle electron configuration, d7 in the above examples, compared to the initial and the two-electron-oxidized final configurations, d8 and d6 respectively, has been shown to be effective in generating cooperative two-electron transfer.4 Similar relative instability of the middle electron configuration leading to two-electron transfer has been reported for a few ruthenium complexes such as reversible oxidation of [RuIII(NH3)6]3+ to [RuV(NH3)6]5+ (ref. 5) and trans-[RuII(tpy)(CN-Me)(OH2)]2+ to [RuIV(tpy)(CN-Me)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)]4+.6
O)]4+.6
An important factor in obtaining reversible multi-electron transfer is overcoming large changes in coordination geometry. One strategy is to use ligands that can support the geometries favored by both oxidation states.7–16 Reversible outer-sphere two-electron transfer platinum and palladium complexes that utilize stabilization of Pt(II)/Pd(II) and Pt(IV)/Pd(IV) oxidation states with suitable ligands have been described recently.4,17 In the case of the platinum complex, Pt(pip2NCN)(tpy)+, the d8 square planar Pt(II) is coordinated to pip2NCN− (pip2NCNH = 1,3-bis(piperdylmethyl)benzene) in a monodentate fashion through the central binding site while tpy (2,2′:6′,2′′-terpyridine) is tridentate (Chart 1). Upon two-electron oxidation, the d6 octahedral Pt(IV) complex Pt(pip2NCN)(tpy)3+ forms. The oxidized complex binds both pip2NCN− and tpy ligands tridentate, occupying all six binding sites around the platinum cation.
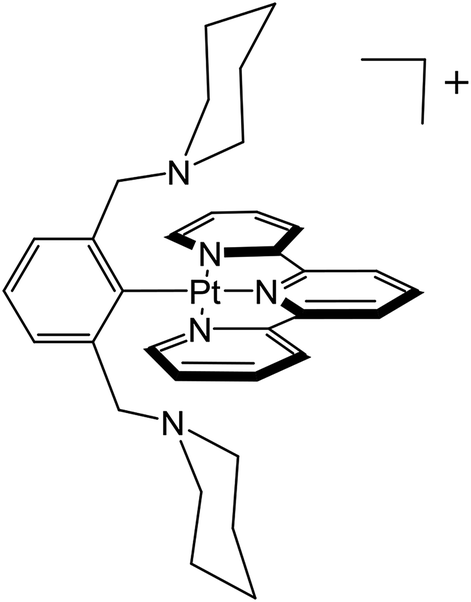 | ||
| Chart 1 The [Pt(pip2NCN)(tpy)]+ complex with tridentate tpy and monodentate pip2NCN ligand before two-electron oxidation. | ||
In contrast to well-studied platinum systems,18–26 which undergo irreversible metal-centered redox processes, the remarkable reversibility of the redox chemistry of Pt(pip2NCN)(tpy)+ is suggestive of a non-covalent interaction between the pip2NCN− amine groups and the Pt(II) metal center, which effectively preorganizes the square planar complex for electron transfer and formation of the Pt(VI) octahedral geometry. The notion of preorganization is supported by crystal structures of a series of two-electron reagents which reveal relatively short Pt⋯N(piperidyl) distances.4,27 In addition, UV-visible absorption spectra of these complexes exhibit long wavelength absorption features (>500 nm), which are absent from the spectra of complexes without Pt⋯N(piperidyl) interactions, such as [Pt(tpy)(phenyl)]+.27
In order to investigate the role of metal acidity on the redox potentials, we report the synthesis and redox chemistry of platinum complexes with electron-releasing and withdrawing groups (Z = MeO, NO2) at the para-positions of the pivoting pip2NCN− ligand and the 4′-substituted positions of the tridentate tpy ligand (R = t-butyl (C(CH3)3), tolyl (4-metylphenyl)) (Chart 2).
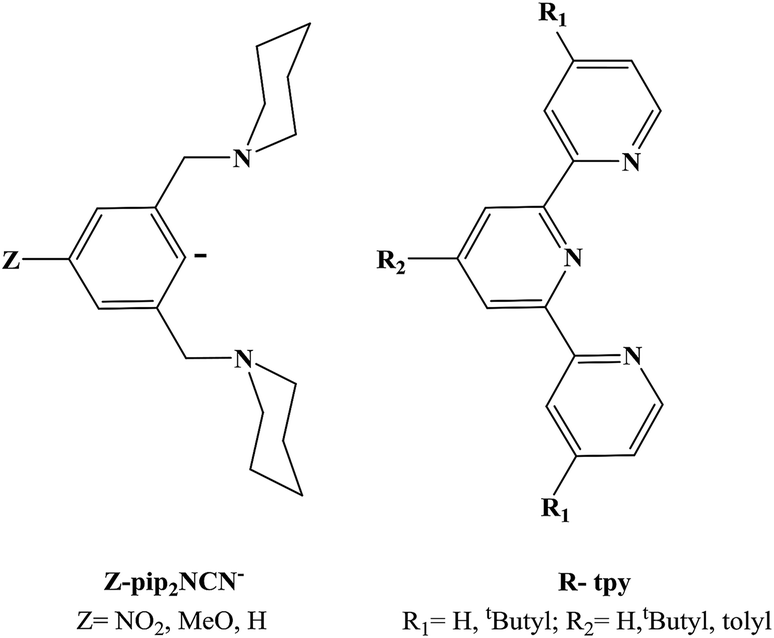 | ||
| Chart 2 Tridentate ligands prepared for this study. | ||
Results and discussion
Synthesis and 1H NMR Spectroscopy
Platinum(II) complexes with a para substituted NCN− pincer ligand and a terpyridyl ligand (tpy, tBu3tpy or toltpy) have been prepared (Chart 2). The synthesis of the Z-pip2NCNBr ligand precursors and their corresponding Pt(Z-pip2NCN)Br complexes along with their 1H NMR spectra are provided in the ESI (Scheme S1, Fig. S1 and S2†).The synthetic route to two-electron platinum reagents with NCN− and terpyridyl ligands involves treatment of an acetone solution of Pt(Z-pip2NCN)Br with a suitable silver salt (e.g., AgBF4), followed by filtration (Scheme 1). Within 15 minutes of adding one equivalent of tpy, tBu3tpy or toltpy to the filtrate, the pale yellow solution turns orange or red, indicating displacement of the piperidyl groups by the terpyridyl ligand. Full experimental details and characterization by 1H NMR spectroscopy and mass spectrometry are given in the ESI (Fig. S3†).
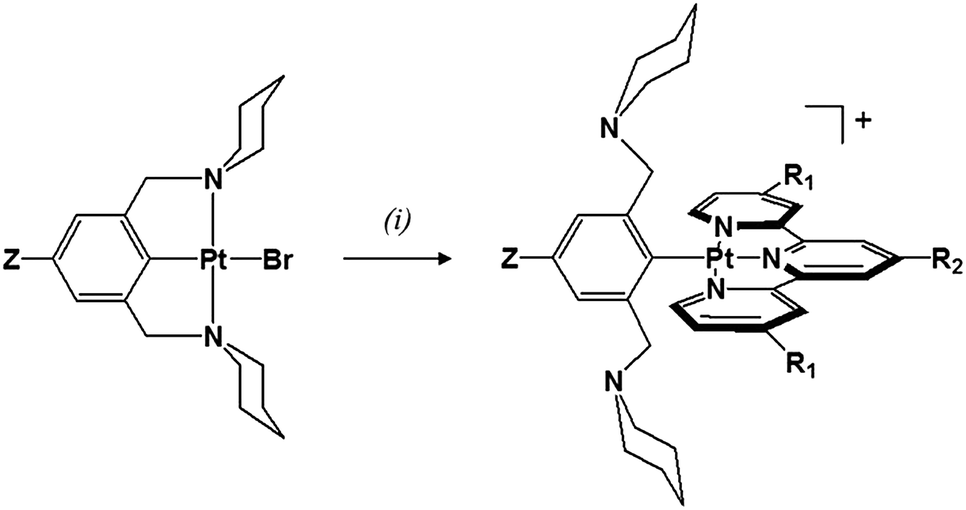 | ||
| Scheme 1 Synthesis of platinum(II) complexes (Z = H, NO2, MeO; R1 = H, t-butyl; R2 = H, t-butyl, tolyl) with two potentially meridional-coordinating tridentate ligands: (i) AgBF4 or AgOTf; tpy, tBu3tpy or toltpy; acetone. | ||
Interestingly, though solutions of the four complexes are red, their colors vary in the solid state from yellow [Pt(pip2NCN)(tBu3tpy)](BF4) and [Pt(NO2-pip2NCN)(tpy)](BF4) to orange-red [Pt(MeO-pip2NCN)(tpy)]OTf and [Pt(pip2NCN) (toltpy)](BF4).
The coordination geometry illustrated in Scheme 1, in which the tpy ligand is tridentate and the Z-pip2NCN− ligand is monodentate, is confirmed in the 1HNMR spectra of the complexes by (1) the presence of 195Pt satellites associated with the α-tpy proton (G) resonance, (2) the absence of 195Pt satellites on the benzylic proton (C) resonance, and (3) the appearance of a single α-piperidyl proton (D) resonance (Fig. 1). The substituents on pip2NCN− (Z) and tpy (R) ligands exert the greatest influence on nearest neighbor protons. For instance, substituting MeO for NO2 causes the meta-phenyl proton resonance to shift upfield by 1.1 ppm. Similarly, substitution of t-butyl groups on tpy, shifts the nearby terpyridyl J and K proton resonances upfield by 0.26 ppm. Changes in the Z and R substituents have little influence on the chemical shifts of the benzylic and piperidyl resonances.
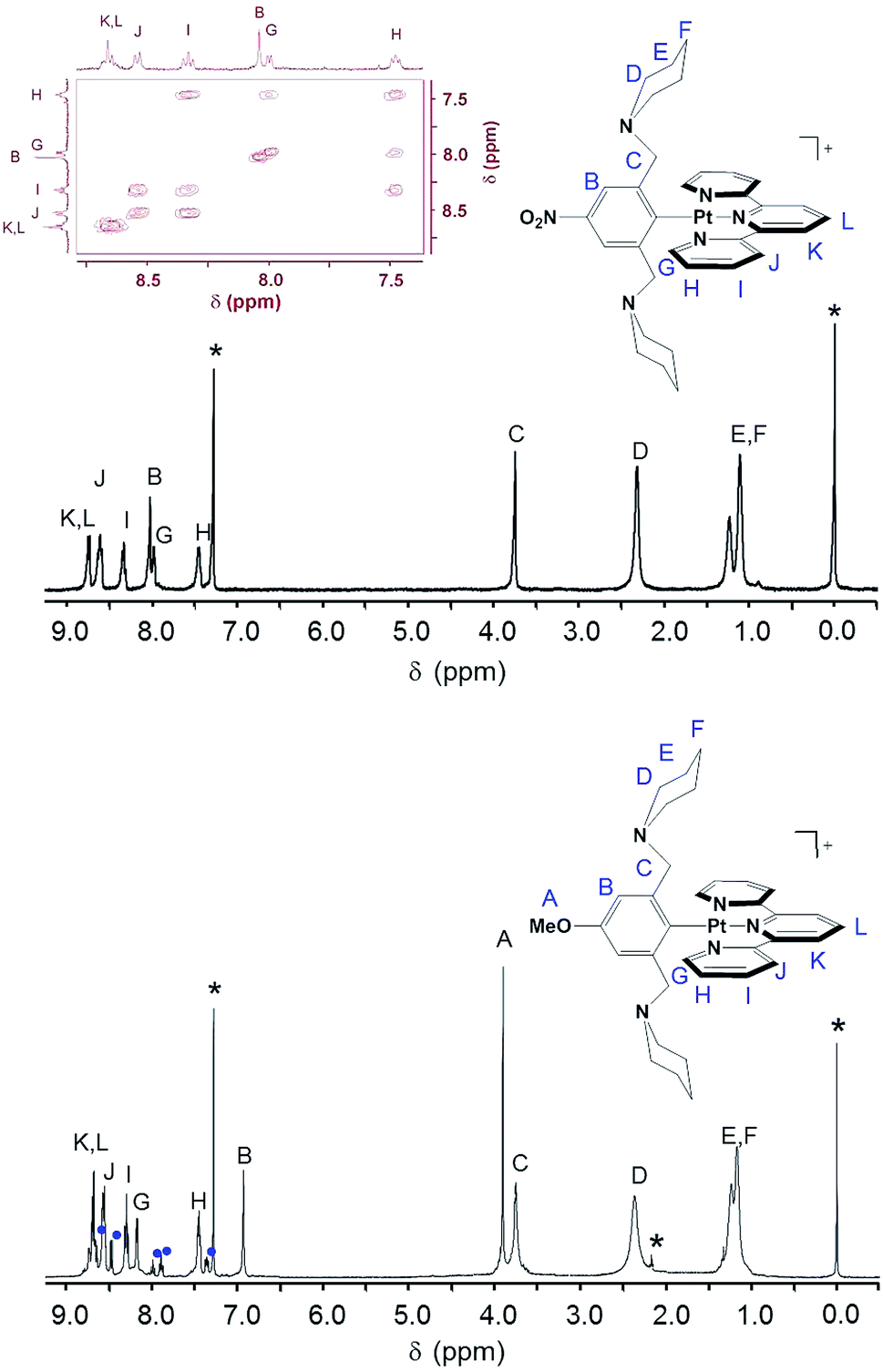 | ||
Fig. 1 1H NMR spectra of [Pt(NO2-pip2NCN)(tpy)](BF4) (top) and [Pt(MeO-pip2NCN)(tpy)]Otf (bottom) in CDCl3. Aromatic region of the COSY spectrum of [Pt(NO2-pip2NCN)(tpy)](BF4) in CDCl3 is shown embedded into the top spectrum. The asterisks * denote CHCl3 (7.26 ppm), acetone (2.17 ppm) and TMS (0.0 ppm) and  denote free tpy impurities (8.72–8.62 ppm, 8.46 ppm, 7.97 ppm, 7.88 ppm and 7.35 ppm). denote free tpy impurities (8.72–8.62 ppm, 8.46 ppm, 7.97 ppm, 7.88 ppm and 7.35 ppm). | ||
Interestingly, the pip2NCN− NO2 and terpyridyl t-butyl substituents shift the G proton resonances upfield by 0.17 ppm, whereas the pip2NCN− MeO shifts G downfield by 0.15 ppm and terpyridyl tolyl substituent does not shift it at all. The influence of the t-butyl group is consistent with its comparative electron-releasing properties. However, the origin of the effect of the NO2 and MeO substituents is more surprising since their electron withdrawing or donating properties are expected to have an opposite influence (decrease and increase, respectively) on the electron density of the platinum center and terpyridyl ligand through inductive effects. The MeO and NO2 substituents shift the remaining terpyridyl resonances upfield and downfield, respectively, by ≤0.07 ppm.
Mediation of electron donor properties through a metal has been investigated by different researchers. For a series of Ru(X-tpy)(Y-tpy)2+ complexes (X,Y = substituents on the para position of the central pyridine, i.e., NO2, NH2, Cl) in acetone, Constable et al. reported that the chemical shifts of each tpy ligand are independent of the substituent on the other ligand.28 However, Fallahpour et al. observed that the substituent on one of the tpy ligands influences certain resonances on the other tpy for a series of similar Ru(II) and Fe(II) complexes in acetonitrile.29
Interestingly, they report that the resonance that is most sensitive to substituent effects changes depending on the metal. For instance, replacing one of the NH2-tpy ligands with NO2-tpy in Ru(NH2-tpy)22+ shifts the α-pyridyl resonance (G) of the other NH2-tpy ligand downfield by 0.2 ppm, but the effect of the same replacement in Fe(NH2-tpy)22+ is a downfield shift of only 0.02 ppm. From these observations, we suggest that the mediation of electron donor properties of a substituent on a ligand to another ligand is influenced by the metal, solvent, type of ligand and coordination geometry.
Pt(pip2NCN)(tpy)+ is reported to be unstable in acetonitrile solution with an half-life of ∼2 hours. The tpy ligand is dissociated to form a Pt(pip2NCN)(solvent)+ adduct.4 Pt(pip2NCN)(tBu3tpy)+ is considerably more stable in acetonitrile. The loss in the intensity of the 420 nm electronic absorption band is consistent with 3% decomposition in one hour. The increased stability of the tBu3tpy complex is attributable to the comparatively greater electron-donor properties of the tBu3tpy ligand, which are anticipated to decrease the electrophilicity of the metal center and thereby increase the reaction barrier of an associative mechanism. This observation implicates modulation of ligand electron-donor properties as an effective strategy for tuning the relative stabilities of two-electron platinum reagents.
Each piperidyl group of Pt(pip2NCN)(tBu3tpy)+ was readily protonated by dropwise addition of 1 M HNO3 to the deep red acetone solution of the complex until the solution turned bright yellow. The 1H NMR spectrum of the protonated complex is given in the ESI (Fig. S3†). However, attempts to protonate the other complexes with substituted pincer ligands did not yield similar diprotonated compounds. In the case of Pt(MeO-pip2NCN)(tBu3tpy)+, the product obtained from treatment with acetic acid gave a platinum(IV) salt, [Pt(tpy)(CH3COO)3](PF6) with a rare tridentate terpyridine and three monodentate acetate groups.30 In the case of [Pt(NO2-pip2NCN)(tpy)]+, treatment with acid caused dissociation of the terpyridine ligand, as determined by 1H NMR spectroscopy.
Electronic spectroscopy
To assess the influence of the pincer piperidyl groups on the electronic structures of these complexes, UV-visible absorption spectra were recorded of dichloromethane solutions of [Pt(Ph)(tBu3tpy)](BF4), [Pt(pip2NCN)(tBu3tpy)](BF4), [Pt(pip2NCN)(toltpy)](BF4) and [Pt(NO2-pip2NCN)(tpy)] (BF4) (Fig. 2 and Table S1 in the ESI†). [Pt(Ph)(tBu3tpy)](BF4) and [Pt(pip2NCN)(tBu3tpy)](BF4) are yellow solids whereas [Pt(pip2NCN)(toltpy)](BF4) and [Pt(NO2-pip2NCN)(tpy)](BF4) are orange solids. [Pt(Ph)(tBu3tpy)](BF4) dissolves to give yellow solutions, whereas the two-electron reagents dissolve to give red solutions. The UV-visible absorption spectra of the terpyridyl complexes are qualitatively similar for wavelengths <500 nm, consistent with previous studies.31–37 For example, each complex exhibits intense absorption bands between 200 nm and 300 nm (>30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1). Platinum(II) terpyridyl complexes, such as Pt(tBu3tpy)Cl+ (215 nm, 46000 M−1 cm−1; 256 nm, 45100 M−1 cm−1; 282 nm, 33700 M−1 cm−1; in acetonitrile)38 and Pt pincer complexes such as Pt(pip2NCN)Cl (275 nm, 9050 M−1 cm−1; in dichloromethane)39 give rise to intense bands in this region. The moderately intense bands observed between 300–365 nm and the low intensity band observed near 425 nm are similar to those observed for other Pt(II) pip2NCN− or phenyl complexes with terpyridyl ligands. These features have been assigned to π → π* transitions associated with the tpy ligand and spin allowed 5d(Pt) → π* (tpy) MLCT transitions, respectively.27 The red shift in the transitions near 425 nm from where similar absorption features occur in Pt(II) terpyridine complexes, such as Pt(tBu3tpy)Cl (373 nm, 3990 M−1 cm−1; 386 nm, 3530 M−1 cm−1in acetonitrile)38 and Pt(tpy)Cl (372 nm, 1300 M−1 cm−1; 398 nm, 1800 M−1 cm−1 in acetonitrile),34 is consistent an aryl group. For all complexes, a weak shoulder is observed near 500 nm in the tail of the 1MLCT transition. As expected from a corresponding 3MLCT transition, this feature is shifted 2000 to 3500 cm−1 to the red of the 1MLCT maximum.38,40,41 The apparent singlet–triplet MLCT splitting is in agreement with that observed for Pt(tBu3tpy)Cl (4800 cm−1)38 and Pt(6-phenyl-2,2′-bipyridine)(4-aminopyridine)+ (2000 cm−1).40
000 M−1 cm−1). Platinum(II) terpyridyl complexes, such as Pt(tBu3tpy)Cl+ (215 nm, 46000 M−1 cm−1; 256 nm, 45100 M−1 cm−1; 282 nm, 33700 M−1 cm−1; in acetonitrile)38 and Pt pincer complexes such as Pt(pip2NCN)Cl (275 nm, 9050 M−1 cm−1; in dichloromethane)39 give rise to intense bands in this region. The moderately intense bands observed between 300–365 nm and the low intensity band observed near 425 nm are similar to those observed for other Pt(II) pip2NCN− or phenyl complexes with terpyridyl ligands. These features have been assigned to π → π* transitions associated with the tpy ligand and spin allowed 5d(Pt) → π* (tpy) MLCT transitions, respectively.27 The red shift in the transitions near 425 nm from where similar absorption features occur in Pt(II) terpyridine complexes, such as Pt(tBu3tpy)Cl (373 nm, 3990 M−1 cm−1; 386 nm, 3530 M−1 cm−1in acetonitrile)38 and Pt(tpy)Cl (372 nm, 1300 M−1 cm−1; 398 nm, 1800 M−1 cm−1 in acetonitrile),34 is consistent an aryl group. For all complexes, a weak shoulder is observed near 500 nm in the tail of the 1MLCT transition. As expected from a corresponding 3MLCT transition, this feature is shifted 2000 to 3500 cm−1 to the red of the 1MLCT maximum.38,40,41 The apparent singlet–triplet MLCT splitting is in agreement with that observed for Pt(tBu3tpy)Cl (4800 cm−1)38 and Pt(6-phenyl-2,2′-bipyridine)(4-aminopyridine)+ (2000 cm−1).40
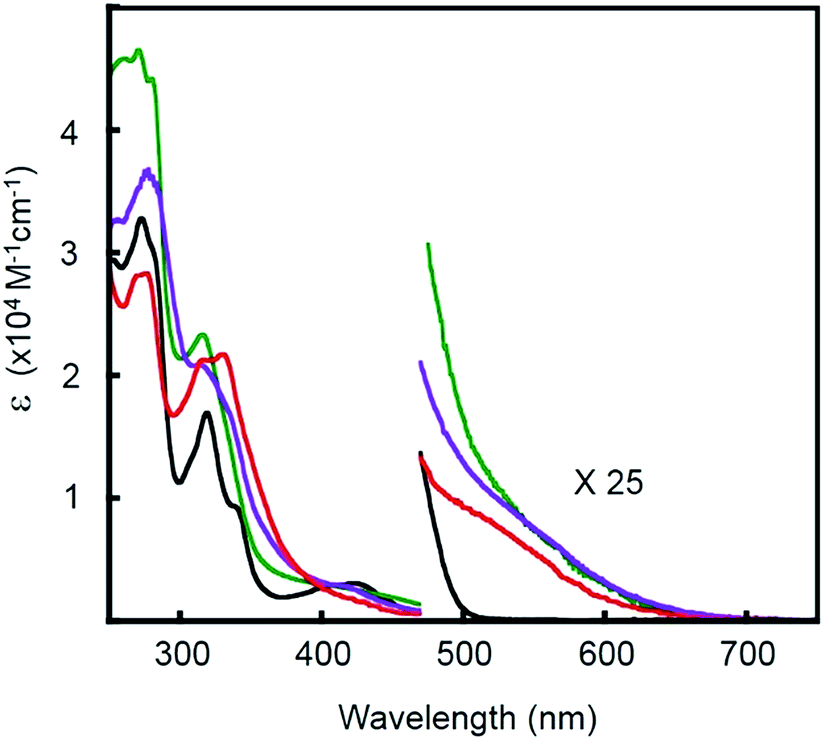 | ||
Fig. 2 UV-visible absorption spectra of 0.01 mM [Pt(ph)(tBu3tpy)](BF4) (—), [Pt(pip2NCN)(tBu3tpy)](BF4) ( ), [Pt(pip2NCN)(toltpy)](BF4) ( ), [Pt(pip2NCN)(toltpy)](BF4) ( ) and [Pt(NO2-pip2NCN)(tpy)](BF4) ( ) and [Pt(NO2-pip2NCN)(tpy)](BF4) ( ) in dichloromethane. ) in dichloromethane. | ||
In the spectra of Pt(pip2NCN)(tBu3tpy)+, Pt(pip2NCN)(toltpy)+ and Pt(NO2-pip2NCN)(tpy)+, there is an additional long wavelength absorption feature appearing near 550 nm (∼300 M−1 cm−1). This weak band is absent from the spectra of model complexes such as Pt(Ph)(tBu3tpy)+. In fact, this band has been observed for only the [Pt(pip2NCN)(R-tpy)]+ complexes and is proposed to be the result of a weak interaction between the N(piperidyl) groups and the platinum center. This hypothesis is supported by the fact that this feature is not observed if the piperidyl groups are placed further away from the platinum center, as in [Pt(3,5-pip2NCN)(tpy)]+,27 or when the piperidyl groups are protected by protonation, as in Pt(pip2NCNH2)(tBu3tpy)3+. The sp3 hybridized N(piperidyl) lone-pair orbital is expected to combine with the 6pz(Pt) orbital to give rise to a filled σ(Pt–N) orbital and an empty σ(Pt–N)* orbital. These axial interactions also are expected to destabilize the metal d orbitals, especially the dz2(Pt) level, resulting in a red-shift in MLCT transitions, as previously noted for other platinum(II) complexes with dangling nucleophiles.42 The presence of a new long-wavelength absorption band is suggestive of a σ(Pt–N) → π* (tpy) charge-transfer transition. The influence of piperidyl and terpyridyl substituents on the energy of this transition is consistent with this assignment. Substitution of NO2 for H on the pip2NCN− ligand and substitution of t-butyl groups for H on the terpydiyl ligand cause the band to blue shift 7600 cm−1 and 5100 cm−1, respectively. By contrast, substitution of a tolyl group for H on the terpyridyl ligand causes the band to red shift by 3900 cm−1. The influence of the Z substituents is consistent with modulation of the energy of the σ(Pt–N) level, whereas the influence of the R substituents is largely through modulations of the π*(tpy) level. These results also give deeper insight into the relation between the colors of these two-electron reagents and the stability of the charge-transfer states since the t-butyl substituents are anticipated to raise the π*(tpy) level, which further destabilizes the charge-transfer transition.
In keeping with this description, we note that [Pt(pip2NCNH2)(tBu3tpy)](PF6)3 is a yellow solid that dissolves to give yellow solutions (Fig. S3, and Table S1†). The spectrum is identical to that obtained when two equivalents of TFA (triflouroacidic acid) are added to an acetonitrile solution of [Pt(pip2NCN)(tBu3tpy)](BF4). During the addition of acid, the intensities of the low-energy absorption bands (>400 nm) decrease, whereas a band at 382 nm emerges. Additionally, small shifts observed at wavelengths <350 nm, and the overall spectrum becomes qualitatively similar to that of [Pt(ph)(tBu3tpy)](BF4). This observation is consistent with protonation of the N(piperidyl) groups, which prevents interaction with the metal center. The intensity increase near 380 nm is likely a consequence of the long-wavelength MLCT band shifting slightly to shorter wavelengths due to the reduced donor properties of the phenyl group.
The room-temperature solution emission spectra of [Pt(pip2NCN)(tBu3tpy)](BF4) and [Pt(pip2NCNH2)(tBu3tpy)](PF6)3 are shown in Fig. 3. [Pt(pip2NCN)(tBu3tpy)]+ is non-emissive, whereas [Pt(pip2NCNH2)(tBu3tpy)]3+ is weakly emissive or non-emissive. The emission from the protonated adduct is characteristically structured (λmax = 470, 502, 535, 585 nm). The significantly diminished intensity of emission from fluid solution samples of [Pt(pip2NCN)(tBu3tpy)](BF4) is consistent with the notion that the lowest excited states of platinum(II) terpyridyl complexes are susceptible to quenching by interactions of the metal center with nucleophiles.43 The deprotonated piperidyl groups are expected to be strong nucleophiles, and previously studied [Pt(pip2NCN)(tpy)]+ and [Pt(pip2NCN)(phtpy)]+ are also non-emissive. The structured emission profile of [Pt(pip2NCNH2)(tBu3tpy)]3+ is similar to those observed for previously reported platinum(II) terpyridyl complexes.34–38
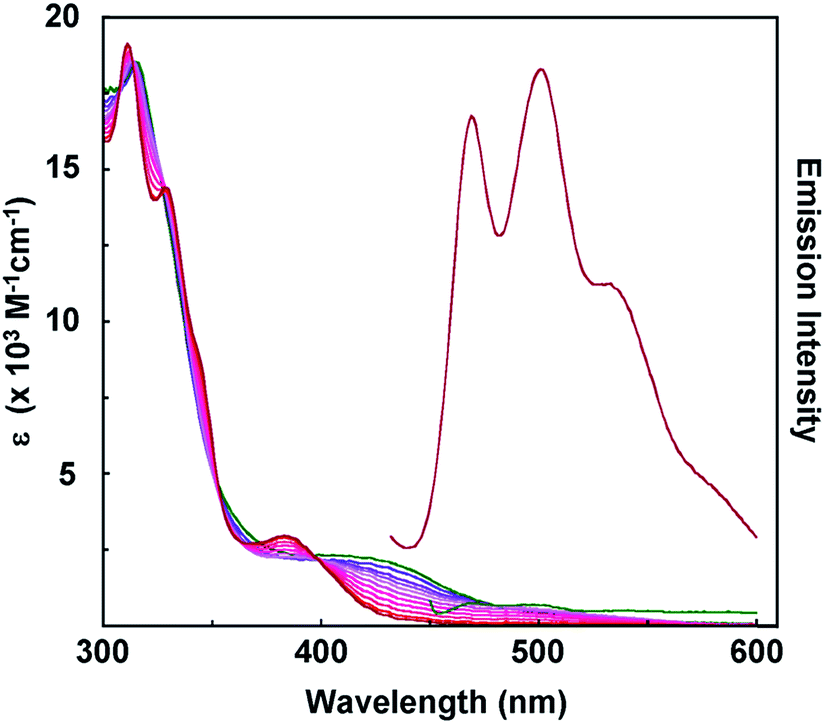 | ||
Fig. 3 UV-visible absorption spectra recorded during titration of a 0.12 mM acetonitrile solution of [Pt(pip2NCN)(tBu3tpy)](BF4) ( ) with 2 equivalents of TFA (triflouroacetic acid, 0.3 M in acetonitrile) in 0.2 equivalents increments. Relative emission spectra of [Pt(pip2NCN)(tBu3tpy)](BF4) ( ) with 2 equivalents of TFA (triflouroacetic acid, 0.3 M in acetonitrile) in 0.2 equivalents increments. Relative emission spectra of [Pt(pip2NCN)(tBu3tpy)](BF4) ( ) and [Pt(pip2NCNH2)(tBu3tpy)] (PF6)3 ( ) and [Pt(pip2NCNH2)(tBu3tpy)] (PF6)3 ( ) in acetonitrile. ) in acetonitrile. | ||
The emissions are assigned to predominantly spin-forbidden 3π–π* terpyridyl ligand-centered lowest excited states. The vibronic spacings (1200–1500 cm−1) are excited state.31 The origin of the emission observed for [Pt(pip2NCNH2)(tBu3tpy)](PF6)3 is only shifted by approximately 2000 cm−1 from the phosphorescence observed for free terpyridine.44 In addition, the bandshapes and Franck–Condon factors, as indicated by the Huang–Rhys ratios45 (I1,0/I0,0 ∼ 0.85), are similar to that of the free ligand (I1,0/I0,0 ∼ 0.85). Both facts are consistent with the 3π–π* assignment.
Cyclic voltammetry
Cyclic voltammograms (CVs) of [Pt(ph)(tBu3tpy)](BF4), [Pt(pip2NCN)(tBu3tpy)](BF4), [Pt(pip2NCN)(toltpy)](BF4), [Pt(NO2-pip2NCN)(tpy)] (BF4), and [Pt(MeO-pip2NCN)(tpy)](BF4) in 0.1 M TBAPF6 acetonitrile are shown in Fig. 4 and 5. The CV of [Pt(pip2NCN)(tpy)](BF4) in 0.1 M TBAPF6 acetonitrile is shown in Fig. 5 for comparison. Cyclic voltammetry data are summarized in Table 1.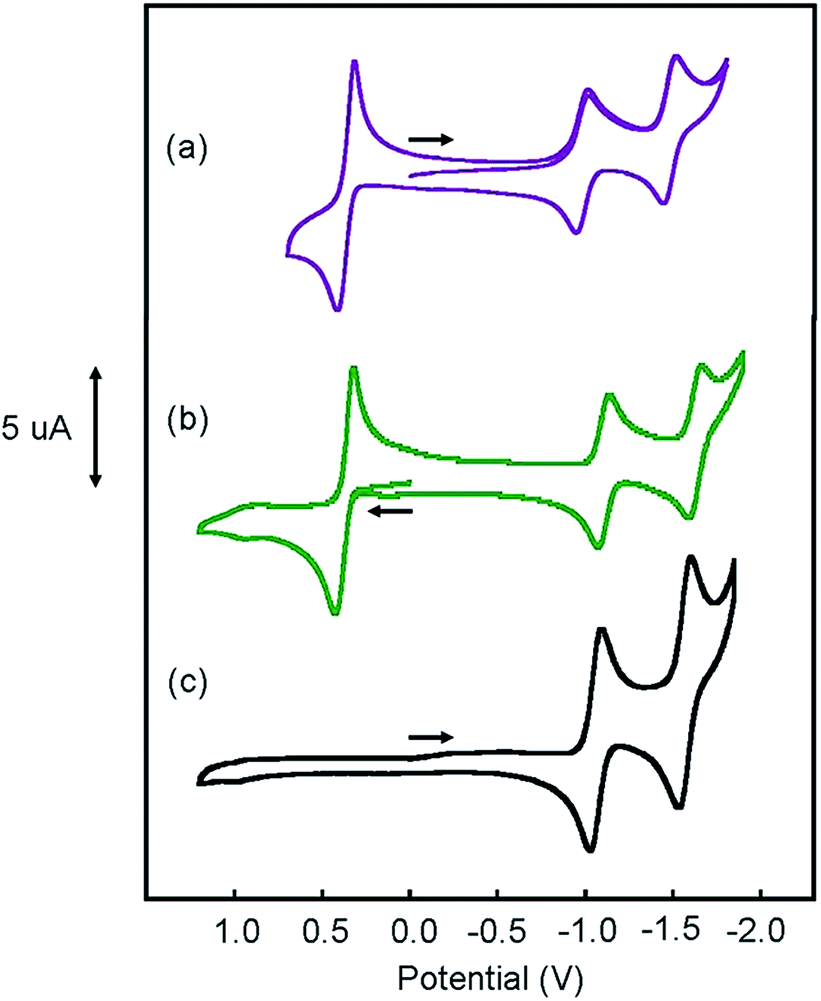 | ||
Fig. 4 Cyclic voltammograms of 1 mM (a) [Pt(pip2NCN)(toltpy)](BF4) ( ); (b) [Pt(pip2NCN)(tBu3tpy)](BF4) ( ); (b) [Pt(pip2NCN)(tBu3tpy)](BF4) ( ) and (c) 2 mM [Pt(ph)(tBu3tpy)](BF4) ( ) and (c) 2 mM [Pt(ph)(tBu3tpy)](BF4) ( ) in acetonitrile (0.1 M TBAPF6, gold working electrode, 0.25 V s−1). ) in acetonitrile (0.1 M TBAPF6, gold working electrode, 0.25 V s−1). | ||
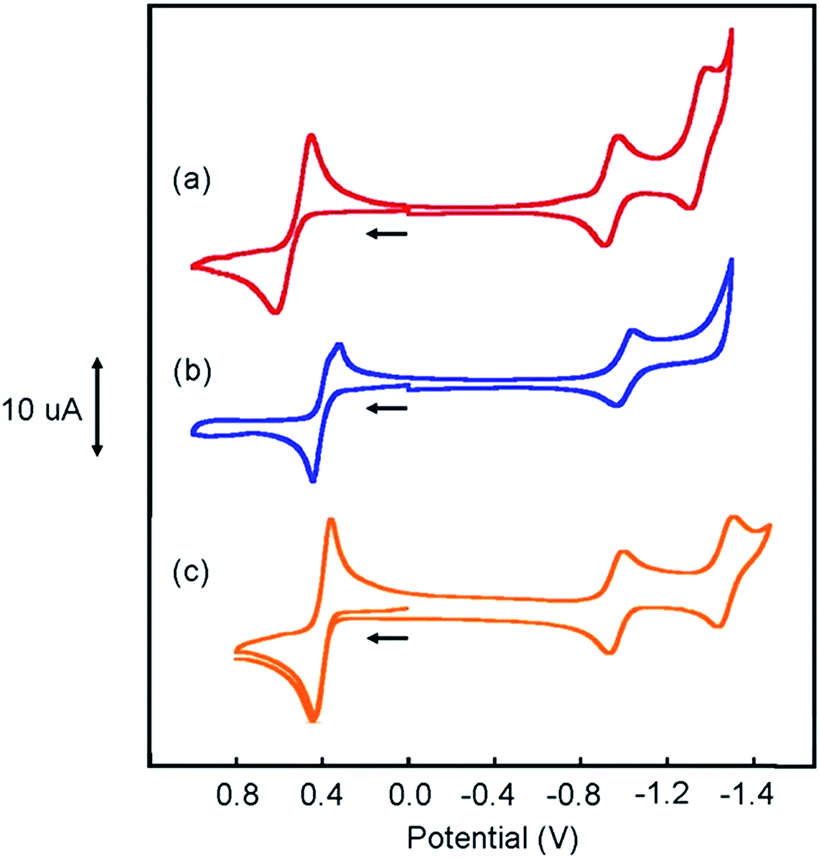 | ||
Fig. 5 Cyclic voltammograms of (a) 2 mM [Pt(NO2-pip2NCN)(tpy)](BF4) ( ), (b) 2 mM [Pt(MeO-pip2NCN)(tpy)]OTf ( ), (b) 2 mM [Pt(MeO-pip2NCN)(tpy)]OTf ( ) and (c) [Pt(pip2NCN)(tpy)](BF4) ( ) and (c) [Pt(pip2NCN)(tpy)](BF4) ( ) in 0.1 M TBAPF6 CH3CN (gold working electrode, 0.1 V s−1). ) in 0.1 M TBAPF6 CH3CN (gold working electrode, 0.1 V s−1). | ||
| Compound |
|
|
|---|---|---|
| [Pt(Ph)(tBu3tpy)]+ | — | −1.06(62), −1.57(69) |
| [Pt(pip2NCN)(tBu3tpy)]+ | 0.36(68) | −1.11(54), −1.62(58) |
| [Pt(pip2NCN)(toltpy)]+ | 0.37(94) | −0.98(67), −1.48(67) |
| [Pt(NO2-pip2NCN)(tpy)]+ | 0.62(89) | −0.88(61), −1.33(53) |
| [Pt(MeO-pip2NCN)(tpy)]+ | 0.37(102) | −1.00(70), −1.52(67) |
| [Pt(pip2NCN)(tpy)]+ | 0.40(54) | −0.98(65), −1.50(61) (ref. 4) |
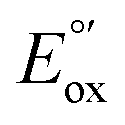
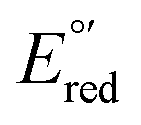
The CVs of each complex in acetonitrile solution (0.1 M TBAPF6, 0.1 V s−1) exhibit two reversible one-electron reduction waves near −1.0 V (E°′) and −1.5 V (E°′), with peak-to-peak separations (ΔEp) of 62 ± 8 and 61 ± 8 mV, respectively. Assignment of the observed redox processes can be inferred from comparison to the electrochemical behavior of a series of related compounds. Under identical conditions, neither free tpy, pip2NCNBr nor Pt(pip2NCN)Cl is reduced at potentials larger than −2.10 V, suggesting that the one-electron reduction processes are associated with the Pt(tpy) unit. Pt(tpy)Cl+ has been reported to undergo reversible one-electron reductions in DMF (0.1 M TBAPF6) at E°′ = −0.74 and E°′ = −1.30 V vs. Ag/AgCl whereas Zn(tpy)Cl2 undergoes reversible one-electron reduction at E°′ = −1.36 V.46 The cathodic shift of the ligand-centered couples in platinum(II) complexes is attributed to stabilization of the reduced tpy ligand as a result of coupling between the empty 6pz(Pt) and the π*(tpy) orbitals.46 As observed for [Pt(Z-pip2NCN)(R-tpy)]+complexes, the fact that [Pt(ph)(tpy)]+ and similar complexes without the piperidyl groups such as [Pt(2,6-dimethylphenyl)(tpy)]+ (E°′ = −0.96 V, ΔEp = 60 mV, ipc/ipa = 0.91; E°′ = −1.49 V, ΔEp = 64 mV, ipc/ipa = 0.90)27 in acetonitrile solutions also undergo two reversible one-electron reductions support this assignment.
The Z and R substituents influence the apparent potentials of both cathodic processes. The NO2 substituent that anodically shifts the reductions, waves by 0.1 V and 0.23 V, respectively (Fig. 5). The tolyl terpyridyl substituent shifts only the second reduction anodically by 0.02 V (Fig. 4). On the other hand, the pip2NCN− MeO and t-butyl terpyridyl substituents act as electron donating groups and shift both reductions cathodically by 0.02 V and 0.13 V from those of Pt(pip2NCN)(tpy)+, respectively (Fig. 4 and 5). Similar shifts (−0.11 V and −0.14 V, respectively) have been observed for the introduction of t-butyl substituent in Pt(R-tpy)–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–phenyl–CC–Re(N^N)(CO)3 (tpy, Ered1/2: −0.90 V, −1.37 V; tBu3tpy, Ered1/2: −1.01 V, −1.51 V, in 0.1 M TBAPF6 acetonitrile solution, vs. SCE, glassy carbon working electrode).47 However, for Pt(tpy)(CC–C6H5-Z)+ complexes, introduction of either NO2 or MeO substituents shift the first reduction process to more negative potentials (by −0.21 V and −0.08 V, respectively) while not influencing the second reduction process (Pt(tpy)(CC–C6H5)+, Ered1/2 = −0.97 V, −1.46 V, in 0.1 M TBAH acetonitrile, vs. SCE, glassy carbon working electrode).48 Apparently, the nature of the fourth ligand carrying the substituent influences the way the substituent effect Pt(tpy) reductions.
C–phenyl–CC–Re(N^N)(CO)3 (tpy, Ered1/2: −0.90 V, −1.37 V; tBu3tpy, Ered1/2: −1.01 V, −1.51 V, in 0.1 M TBAPF6 acetonitrile solution, vs. SCE, glassy carbon working electrode).47 However, for Pt(tpy)(CC–C6H5-Z)+ complexes, introduction of either NO2 or MeO substituents shift the first reduction process to more negative potentials (by −0.21 V and −0.08 V, respectively) while not influencing the second reduction process (Pt(tpy)(CC–C6H5)+, Ered1/2 = −0.97 V, −1.46 V, in 0.1 M TBAH acetonitrile, vs. SCE, glassy carbon working electrode).48 Apparently, the nature of the fourth ligand carrying the substituent influences the way the substituent effect Pt(tpy) reductions.
Pt(ph)(tBu3tpy)+ is not oxidized at <1.2 V vs. Ag/AgCl. By contrast, each of the platinum(II) complexes with both a Z-pip2NCN− and a terpyridyl ligand undergo a two-electron oxidation process in the 0.4–0.6 V range (E°′), as previously noted for [Pt(pip2NCN)(tpy)]+ (E°′ = 0.40 V, ΔEp = 74 mV, 0.25 V s−1),4 [Pt(pip2NCN)(tBu3tpy)]+ and [Pt(pip2NCN)(toltpy)]+ undergo a chemically reversible and nearly electrochemically reversible two-electron oxidation processes at E°′ = 0.36 V (ipc/ipa = 1.02, ΔEp = 68 mV, 0.1 V s−1) and E°′ = 0.37 V (ipc/ipa = 1.1, ΔEp = 94 mV, 0.25 V s−1), respectively. For [Pt(NO2-pip2NCN)(tpy)]+ and [Pt(MeO-pip2NCN)(tpy)]+, the oxidation process occurs at E°′ = 0.62 V (ipc/ipa = 1.33, ΔEp = 89 mV, 0.1 V s−1) and E°′ = 0.37 V (ipc/ipa = 0.97, ΔEp = 102 mV, 0.1 V s−1), respectively. No splitting of any of the waves was observed over the range of investigated scan rates. As assessed by the anodic–cathodic peak-to-peak separation (ΔEp) at a given sweep rate, the electrochemical reversibility varies among the five two-electron reagents (ΔEp, 50–100 mV, 0.1 V s−1). The reversibility does not vary with electron-donor properties of the substituents in an obvious manner; however it appears that the presence of the Z substituents on the pip2NCN− ligand tends to slow the electron-transfer kinetics. The reversibility of the Ru(II)/Ru(III) process for a library of ruthenium terpyridyl complexes also varies somewhat irratically.28,49 Nazeeruddin et al. have reported diminished reversibility for ruthenium bipyridyl complexes with multiple strong electron donor substituents such as NMe2.50 In the present case, it appears that ΔEp tracks with the severity of electrode passivation problems as indicated by the shift in Epa and loss of current with consecutive sweeps. For all five complexes, the ratios of the peak anodic current of the oxidation process to the peak cathodic current of the first reduction wave are between 2.0 and 2.3.
Although the ipa/ipc are less than the predicted value for a Nernstian two-electron process (2.8 (=23/2)), the ratios clearly exceed the expected value of 1.0 for a one-electron process. The ΔEp for a diffusion controlled two-electron Nernstian process is expected to be 29.5 mV.51 In fact, at 0.1 V s−1 the couples are clearly not electrochemically reversible as indicated by the relatively large values of ΔEp, and therefore the values of ipa/ipc cannot be expected to approach the Nernstian limits. Since the oxidations clearly involve transfer of considerably more charge than observed for the reduction, the processes are attributed to a net two-electron oxidation of the complexes.
The apparent two-electron oxidation waves observed for [Pt(Z-pip2NCN)(R-tpy)]+ complexes are absent in cyclic voltammograms of related compounds. For example, neither Pt(tpy)(dmph)+, pip2NCNBr nor pip2NCNBrH22+ is oxidized at potentials <1.2 V,4 and Pt(pip2NCN)Cl undergoes irreversible metal-centered oxidation near 0.8 V.39 Taken together, these data indicate that both the pip2NCN− and terpyridyl ligands play important roles in the unusual redox chemistry of Pt(Z-pip2NCN)(R-tpy)+complexes. The availability of the amine lone electron pairs is critical to facilitating reversible two-electron oxidation and stabilizing the resulting Pt(IV) center. For example, protonation of the piperidyl groups (e.g., Pt(pip2NCNH2)(tpy)3+),4 results in irreversible oxidation near 0.4 V accompanied by electrode fouling.
In order to further characterize the electrochemical behavior of these systems, CVs of Pt(pip2NCN)(tBu3tpy)+ were recorded for the first reduction process (−0.8 to −1.3 V) and the oxidation process (0.2 to 0.6 V) over a range of scan rates from 0.01 to 25 V s−1 (Fig. 6). ΔEp of the first reduction (E°′ = −1.11 V) is essentially invariant (59 ± 6 mV) for scan rates ranging from 0.01 to 25 V s−1. The cathodic peak current (ipc) exhibits an approximately linear dependence on the square root of the scan rate (ν1/2), as predicted by the Randles–Ševčik equation for Nernstian conditions.52–54
| ip = 2.69 × 105n3/2AD1/2Cν1/2 | (1) |
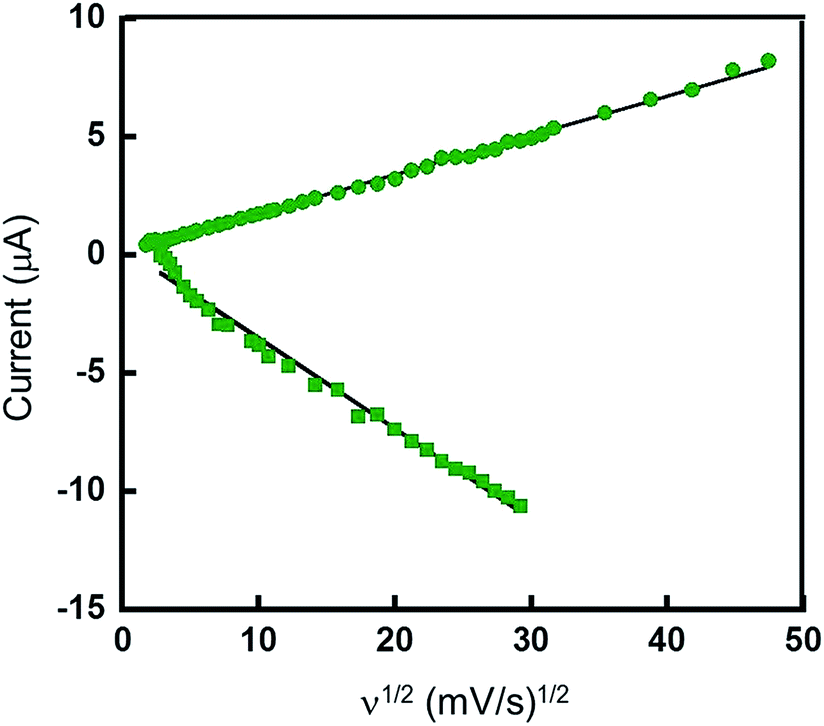 | ||
Fig. 6 Dependence of anodic peak current (ipa) for 0.36 V oxidation process ( ) and cathodic peak current (ipc) for −1.11 V reduction process ( ) and cathodic peak current (ipc) for −1.11 V reduction process ( ) on the square root of the scan rate (ν1/2) for 1 mM [Pt(pip2NCN)(tBu3tpy)](BF4) in acetonitrile (0.1 M TBAPF6). Scans recorded from 0.2 to 0.6 V and from −1.2 to −1.7 V. The lines represent linear fits of the anodic and cathodic currents. ) on the square root of the scan rate (ν1/2) for 1 mM [Pt(pip2NCN)(tBu3tpy)](BF4) in acetonitrile (0.1 M TBAPF6). Scans recorded from 0.2 to 0.6 V and from −1.2 to −1.7 V. The lines represent linear fits of the anodic and cathodic currents. | ||
In order to verify the electron stoichiometry of the oxidation process for [Pt(pip2NCN)(tBu3tpy)](BF4), the anodic peak current (ipa) is plotted against ν1/2 in Fig. 6. The data are remarkably linear over the entire range of scan rates (0.01 to 30 V s−1) as predicted by the Randles–Ševčik eqn (1). The ratio (2.40) of the slope of the best fit line to that obtained for the first reduction process is used to derive an estimate of nox/nred (=1.8). Since the reduction is known to be a one electron process, nox = 1.8 is consistent with an oxidation that involves two-electron transfer per Pt center. See more detailed explanation in the ESI.†
The substituents on the pip2NCN− and tpy ligands significantly influence the apparent Pt(IV/II) redox couple, E°′, the influence of the Rn substituents is comparatively small. For example, E°′ is cathodically shifted by 0.04 and 0.03 volts in [Pt(pip2NCN)(tBu3tpy)]+ and [Pt(pip2NCN)(toltpy)]+, respectively. A comparable cathodic shift (0.05 V) has been observed for the irreversible Pt(II)/Pt(III) process when tpy is replaced by tBu3tpy in Pt(R-tpy)–CC–phenyl–CC–Re(N^N)(CO)3 complexes (N^N = 4,4′-bis-t-butylbipyridine; R = H (tpy), Epa = 1.41 V; R = t-butyl (tBu3tpy), Epa = 1.36 V; in 0.1 M TBAPF6 acetonitrile solution, vs. SCE, glassy carbon working electrode).47 Similarly, replacing tpy ligands with phenyl-tpy in Ru(tpy)22+ causes a cathodic shift of 0.02 volts (Ru(tpy)22+, E°′ = 0.92 V; Ru(ph-tpy)22+, E°′ = 0.90 V; in acetonitrile, vs. Fc/Fc+).49
The Z substituents have a more substantial influence on the apparent redox potential. For Pt(NO2-pip2NCN)(tpy)+, E°′ (0.62 V) is anodically shifted by 0.22 V from the two-electron oxidation process observed for Pt(pip2NCN)(tpy)+ (E°′ = 0.40 V), as expected for a more electron-poor metal center. The involvement of the metal-center is confirmed by comparison to the redox chemistry of ruthenium(III/II) polypyridyl complexes. Previously, Nazeeruddin et al. have shown that there exists an approximate linear correlation between the ruthenium(III/II) redox potentials of Ru(Z,Z′-bpy)x(bpy)3−x2+ complexes and the Hammett parameters of the bipyridyl substituents.50 Fig. 7 shows the dependence of E°′ on the effective Hammett parameter ∑σp+, which derives from summing the σp+ values for each of the substituents of the six pyridyl groups. The use of σp+ is rationalized on grounds that it is a more suitable descriptor for a reaction that involves increasing positive charge on the metal center. We have applied this analysis to the five Pt(Z-pip2NCN)(tpy)+ two-electron reagents, where σp+ values are summed for the three substituents of the terpyridyl ligand and the single substituent of the pip2NCN− ligand. The analysis reveals a similar approximate linear relationship, albeit over a more narrow range of ∑σp+ (Fig. 7).
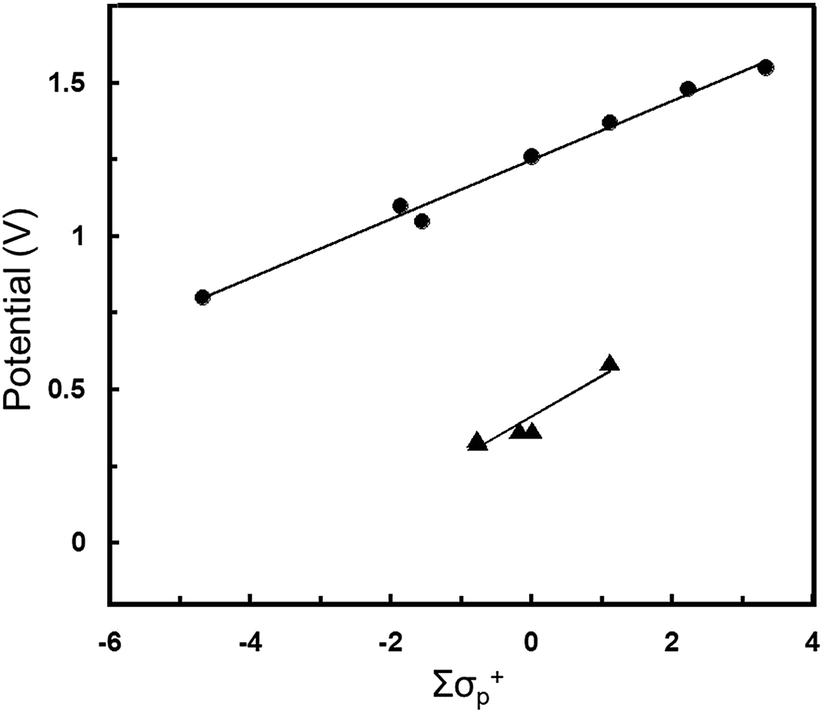 | ||
| Fig. 7 Correlation of oxidation potential (E°′ vs. SCE) to Hammett parameter ∑σ+. ▲ represents Pt(Z-pip2NCN)(R-tpy)+ complexes where Z = NO2, MeO, H and R = H, t-butyl, phenyl, E°′ = 0.13∑σp+ + 0.41 (R = 0.952). ● represents Ru(Z,Z′-bpy)x(bpy)3−x2+ complexes50 where Z = NO2, H, Me, MeO, (x = 0, 1, 2, 3), E°′ = 0.10∑σp+ + 1.25 (R = 0.995). ∑σp+ is calculated by summing the σp+ values for each substituent; a 6 coordinate Ru(Z,Z′-bpy)x(bpy)3−x2+ complex has six different substituents, whereas a Pt(Z-pip2NCN)(R-tpy)+ complex has four substituents. | ||
For both Pt(II) and Ru(II) complexes, the observed E°′ reveal, as expected, that the oxidation of the metal center becomes more difficult as the electron withdrawing character of the substituents increases. The relative slopes of best-fit lines E°′ versus ∑σp+ (Ru, 0.13 V; Pt, 0.11 V) reflect the fact that replacement of a MeO group with NO2 causes a slightly greater anodic shift in E°′ for the two-electron platinum reagents (0.25 V) than for the Ru(4,4′-R2bpy)(bpy)22+ (0.18 V per MeO/NO2 substitution). Potentials for Ru(R-tpy)22+ also would seem to suggest a more shallow dependence on EtO/NO2 substitution (0.19 V),29,49 however, for this limited data set, we regard the slopes of the best-fit lines in Fig. 7 as essentially identical within the scatter of the data. The general agreement between these data sets confirms that the two-electron process is metal-centered and suggests that, over this narrow range of potentials, the d6/d7-electron and d7/d8-electron couples are comparably affected by changes in ligand substituents.
Interestingly, in the cases of Ru(R-tpy)22+ and Ru(4,4′-R2bpy)(bpy)22+ complexes, the nitro and ethoxy substituents each strongly shift the redox couple from that of the unsubstituted complex (Ru(NO2-tpy)22+, 1.114 V; Ru(tpy)22+, 0.92 V; Ru(OEt-tpy)22+, 0.74 V, in acetonitrile vs. Fc/Fc+;43,49 Ru(4,4′-(NO2)2bpy)(bpy)22+, 1.48 V; Ru(bpy)32+, 1.26 V; Ru(4,4′-(MeO)2bpy)(bpy)22+, 1.05 V, in acetonitrile vs. SCE).50 The potential shifts per substituent upon by replacing H with MeO or with NO2 are comparable (Ru(R-tpy)22+, R = NO2, 0.97 V; R = EtO, −0.09 V;29,49 Ru(4,4′-R2bpy)(bpy)22+, R = NO2, 0.098 V; R = MeO, −0.075 V).50,56 By contrast in the [Pt(Z-pip2NCN)(tpy)]+ series, the nitro group has a much stronger influence (+0.22 V) on the apparent redox couple than the methoxy substituent (−0.03 V).
A similar effect is observed for Pt(tpy)(CC–C6H5-Z)+ (Z = NO2, MeO, H) complexes where the NO2 substituent is proposed to cause the irreversible one electron Pt(II)/Pt(III) oxidation potential to shift anodically by 0.24 volts from that of Pt(tpy)(CC–C6H5)+ (Epa = 1.22 V, in 0.1 M TBAH acetonitrile solution, vs. SCE, glassy carbon working electrode); however, in that case the influence of the OMe substituent also is substantial (−0.22 V), and it is not clear that these shifts represent thermodynamic potentials.48 In the case of the Ru(III/II) systems, the redox process involves a dπ orbital, whereas the oxidation of platinum(II) formally involves a dz2 orbital. On the other hand, the Hammett inductive (σI) and mesomeric (σm) constants for nitro (0.7, 0) and methoxy (0.31, −0.41) groups suggest significant differences in the coupling of substituent electron-donor properties. Therefore, the comparative insensitivity of the platinum system to the methoxy substituent is consistent with the influence of resonance on the dz2 level being significantly less compared to its influence on the dπ levels of ruthenium(III/II) polypyridyl complexes. Similarly, the increased sensitivity to nitro substituent suggests that the influence of induction on the dz2 level is significantly increased.
Conclusions
Substituted pip2NCN− ligands, their Pt(II) halide complexes, and novel [Pt(Z-pip2NCN)(R-tpy)]+ complexes with Z = NO2, MeO, H and R = H, tertyl butyl, tolyl substituents have been prepared. For [Pt(Z-pip2NCN)(R-tpy)]+, 1H NMR spectroscopy shows that the Z-pip2NCN− ligand is bonded monodentate whereas the R-terpyridyl ligand is bonded tridentate. The approximate square planar coordination geometry around the metal center is consistent with structures of Pt(II) complexes. The piperidyl groups are located above and below the platinum center suggesting that the amine groups are available to stabilize the metal center upon oxidation. As in the case of Pt(pip2NCN)(tpy)+, the electronic absorption spectra of [Pt(Z-pip2NCN)(R-tpy)]+ exhibit a band at ∼550 nm assigned to a σ(Pt–N) → π*(tpy) charge-transfer transition, resulting from the weak interaction of a piperidyl group with the metal center. Pt(Z-pip2NCN)(R-tpy)+ complexes undergo two-electron platinum centered oxidation near 0.4 V and two Pt(tpy) centered reductions near −1.0 V and −1.5 V. The scan rate dependence of the peak currents of the oxidation and the first reduction processes for Pt(pip2NCN)(tBu3tpy)+ gives nox/nred = 1.8.Variation in the ligand substituents (Z and R) allows for tuning of the two-electron-oxidation process over a 260 mV range. Whereby the Z substituents on the pip2NCN− ligand have a more substantial influence (220 mV) on the oxidation potential than the R substituents (40 mV) on the terpyridine ligand. In addition, the two reduction processes are tunable over a range of 230 mV (first reduction step) and 290 mV (second reduction step). The electron-withdrawing NO2 substituent shifts the two-electron-oxidation potential anodically, whereas electron-donating MeO-shifts the redox potential cathodically. The electrochemical sensitivity to the nitro-substituent suggests that the redox tuning occurs predominantly through electronic induction at the dz2 orbital.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express their gratitude to Dr Janette Krause for help with characterization. We thank the National Science Foundation (Grant CHE0134975) and the Arnold and Mabel Beckman Foundation for support.References
- J. F. Young, J. A. Osborn, F. H. Jardine and G. Wilkinson, Chem. Commun., 1965, 131 RSC.
- A. E. Shilov and G. B. Shul'pin, Russ. Chem. Rev., 1987, 56, 442 CrossRef.
- A. F. Heyduk and D. G. Nocera, Science, 2001, 293, 1639 CrossRef CAS.
- H. Jude, J. A. K. Bauer and W. B. Connick, J. Am. Chem. Soc., 2003, 125, 3446 CrossRef CAS.
- R. Ramasamy, K. Masao and K. Akira, Bull. Chem. Soc. Jpn., 1991, 64, 1028 CrossRef.
- M. Dakkach, X. Fontrodona, T. Parella, A. Atlamsani, I. Romeo and M. Rodrigues, Adv. Synth. Catal., 2011, 353, 231 CrossRef CAS.
- R. G. Finke, R. H. Voegeli, E. D. Laganis and V. Boekelheide, Organometallics, 1983, 2, 347 CrossRef CAS.
- W. J. Bowyer, J. W. Merkert, W. E. Geiger and A. L. Rheingold, Organometallics, 1989, 8, 191 CrossRef CAS.
- W. J. Bowyer and W. E. Geiger, J. Am. Chem. Soc., 1985, 107, 5657 CrossRef CAS.
- K. D. Plitzko, B. Rapko, B. Gollas, G. Wehrle, T. Weakley, D. T. Pierce, W. E. Geiger, R. C. Haddon and V. Boekelheide, J. Am. Chem. Soc., 1990, 112, 6545 CrossRef CAS.
- K. D. Plitzko, G. Wehrle, B. Gollas, B. Rapko, J. Dannheim and V. J. Boekelheide, J. Am. Chem. Soc., 1990, 112, 6556 CrossRef CAS.
- J. Merkert, R. M. Nielson, M. J. Weaver and W. E. Geiger, J. Am. Chem. Soc., 1989, 111, 7084 CrossRef CAS.
- J. Edwin and W. E. Geiger, J. Am. Chem. Soc., 1990, 112, 7104 CrossRef CAS.
- K. R. Mann, N. S. Lewis, V. M. Miskowski, D. K. Erwin, G. S. Hammond and H. B. Gray, J. Am. Chem. Soc., 1977, 99, 5525 CrossRef CAS.
- K. R. Mann, M. J. DiPierro and T. P. Gill, J. Am. Chem. Soc., 1980, 102, 3965 CrossRef CAS.
- W. J. Bowyer and W. E. Geiger, J. Electroanal. Chem. Interfacial Electrochem., 1988, 239, 253 CrossRef CAS.
- S. Chatterjee, PhD thesis, University of Cincinnati, 2009.
- F. G. Baddour, M. I. Kahn, J. A. Golen, A. L. Rheingold and L. H. Doerrer, Chem. Commun., 2010, 46, 4968 RSC.
- A. J. Blake, R. D. Crofts and M. Schroder, J. Chem. Soc., Dalton Trans., 1993, 2259 RSC.
- A. J. Blake, R. O. Gould, A. J. Holder, T. I. Hyde, A. J. Lavery, M. O. Odulate and M. Schroder, J. Chem. Soc., Chem. Commun., 1987, 118 RSC.
- J. Fornies, B. Menjon, R. M. Sanz-Carrillo, M. Tomas, N. G. Connelly, J. G. Crossley and A. G. Orpen, J. Am. Chem. Soc., 1995, 117, 4295 CrossRef CAS.
- G. J. Grant, N. J. Spangler, W. N. Setzer, D. G. VanDerveer and L. F. Mehne, Inorg. Chim. Acta, 1996, 246, 31 CrossRef CAS.
- R. I. Haines, D. R. Hutchings and T. M. McCormack, J. Inorg. Biochem., 2001, 85, 1 CrossRef CAS.
- M. Matsumoto, M. Itoh, S. Funahashi and H. D. Takagi, Can. J. Chem., 1999, 77, 1638 CAS.
- R. Uson, J. Fornies, M. Tomas, B. Menjon, R. Bau, K. Suenkel and E. Kuwabara, Organometallics, 1986, 5, 1576 CrossRef CAS.
- M. A. Watzky, D. Waknine, M. J. Heeg, J. F. Endicott and L. A. Ochrymowyzc, Inorg. Chem., 1993, 32, 4882 CrossRef CAS.
- H. Jude, PhD thesis, University of Cincinnati, 2004.
- E. C. Constable, A. M. W. C. Thompson, D. A. Tocher and M. A. M. Daniels, New J. Chem., 1992, 16, 855 CAS.
- R. A. Fallahpour, Eur. J. Inorg. Chem., 1998, 9, 1205 CrossRef.
- S. Kuyuldar, PhD thesis, University of Cincinnati, 2009.
- J. A. Bailey, M. G. Hill, R. E. Marsh, V. M. Miskowski, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1995, 34, 4591 CrossRef CAS.
- R. Romeo, L. M. Scolaro, M. R. Plutino and A. J. Albinati, Organomet. Chem., 2000, 593–594, 403 CrossRef CAS.
- T. K. Aldridge, E. M. Stacy and D. R. McMillin, Inorg. Chem., 1994, 33, 722 CrossRef CAS.
- H. K. Yip, L. K. Cheng, K. K. Cheung and C. M. Che, J. Chem. Soc., Dalton Trans., 1993, 2933 RSC.
- J. S. Field, R. J. Haines, D. R. McMillin and G. C. Summerton, J. Chem. Soc., Dalton Trans., 2002, 1369 RSC.
- G. Arena, G. Calogero, S. Campagna, L. M. Scolaro, V. Ricevuto and R. Romeo, Inorg. Chem., 1998, 2763 CrossRef CAS.
- S. E. Hobert, J. T. Carney and S. D. Cummings, Inorg. Chim. Acta, 2001, 318, 89 CrossRef CAS.
- S. W. Lai, M. C. W. Chan, K. K. Cheung and C. M. Che, Inorg. Chem., 1999, 38, 4262 CrossRef CAS.
- H. Jude, J. A. Krause-Bauer and W. B. Connick, Inorg. Chem., 2002, 41, 2275 CrossRef CAS.
- J. H. K. Yip and J. J. Vittal, Inorg. Chem., 2000, 39, 3537 CrossRef CAS.
- W. B. Connick, V. M. Miskowski, V. H. Houlding and H. B. Gray, Inorg. Chem., 2000, 39, 2585 CrossRef CAS.
- T. W. Green, R. Lieberman, N. Mitchell, J. A. Krause Bauer and W. B. Connick, Inorg. Chem., 2005, 44, 1955 CrossRef CAS.
- D. K. Crites-Tears and D. R. McMillin, Coord. Chem. Rev., 2001, 211, 195 CrossRef CAS.
- A. Sarkar and S. J. Chakravorti, J. Lumin., 1995, 63, 143 CrossRef CAS.
- K. Huang and A. Rhys, Proc. - R. Soc. Edinburgh, Sect. A: Math. Phys. Sci., 1950, 204, 406 CAS.
- M. G. Hill, J. A. Bailey, V. M. Miskowsk and H. B. Gray, Inorg. Chem., 1996, 35, 4585 CrossRef CAS.
- S. C. F. Lam, V. W. W. Yam, K. M. C. Wong, E. C. C. Cheng and N. Zhu, Organometallics, 2005, 24, 4298 CrossRef CAS.
- V. W. W. Yam, R. P. L. Tang, K. M. C. Wong and K. K. Cheung, Organometallics, 2001, 20, 4476 CrossRef CAS.
- M. Maestri, N. Armaroli, V. Balzani, E. C. Constable and A. M. W. C. Thompson, Inorg. Chem., 1995, 34, 2759 CrossRef CAS.
- M. K. Nazeeruddin, S. M. Zakeeruddin and K. Kalyanasundaram, J. Phys. Chem., 1993, 97, 9607 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, Wiley, New York, 1980 Search PubMed.
- J. E. B. Randles, Trans. Faraday Soc., 1948, 44, 327 RSC.
- A. Sevcik, Collect. Czech. Chem. Commun., 1948, 13, 349–377 CrossRef CAS.
- J. J. van Benschoten, J. Y. Lewis, W. R. Heineman, D. A. Roston and P. T. Kissinger, J. Chem. Educ., 1983, 60, 772 CrossRef CAS.
- R. L. Myers and I. Shain, Anal. Chem., 1969, 41, 980 CrossRef CAS.
- V. Balzani, A. Juris, F. Barigelletti, P. Belser and A. Von Zelewsky, Sci. Pap. Inst. Phys. Chem. Res., 1984, 78, 78 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03939b |
| ‡ William B. Connick passed away in 2018. |
| This journal is © The Royal Society of Chemistry 2019 |