
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hyperpatulones A–F, polycyclic polyprenylated acylphloroglucinols from Hypericum patulum and their cytotoxic activities†
Zhong-Nan Wua,
Qian-Wen Niua,
Yu-Bo Zhangab,
Ding Luoa,
Qing-Guo Lic,
Ying-Ying Lia,
Guang-Kai Kuanga,
Li-Jun Hea,
Guo-Cai Wang *ab and
Yao-Lan Li*a
*ab and
Yao-Lan Li*a
aInstitute of Traditional Chinese Medicine & Natural Products, Guangdong Province Key Laboratory of Pharmacodynamic Constituents of TCM and New Drugs Research, College of Pharmacy, Jinan University, Guangzhou 510632, People's Republic of China. E-mail: twangguocai@jnu.edu.cn; tliyl@jnu.edu.cn
bIntegrated Chinese and Western Medicine Postdoctoral Research Station, Jinan University, Guangzhou 510632, People's Republic of China
cSchool of Pharmaceutical Sciences, Guangzhou University of Chinese Medicine, Guangzhou 510006, China
First published on 12th March 2019
Abstract
Six new compounds, hyperpatulones A–F (1–6), along with ten additional known related derivatives (7–16), were isolated from Hypericum patulum (Guttiferae). Their structures were elucidated by extensive analysis of spectroscopic data (IR, UV, HRESIMS, 1D and 2D NMR), X-ray crystallography, electronic circular dichroism (ECD) spectroscopy and Rh2(OCOCF3)4-induced ECD. All compounds were tested for their cytotoxic activities on human HepG-2, HeLa, MCF-7, and A549 cell lines via 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Compound 5 exhibited significant cytotoxicities against HepG-2, HeLa and A549 cell lines with IC50 values of 9.52 ± 0.27, 11.87 ± 0.22 and 12.63 ± 0.12 μM, respectively.
Introduction
Hypericum patulum (Guttiferae) is well known as “Jinsimei” in China, and is distributed mainly in southwest China, such as Guizhou, Sichuan and Yunnan Provinces.1 The herbs of H. patulum are used as a traditional medicine to clear heat, cool blood, relax tendons and activate collaterals, and to treat gonorrhea, hepatitis, colds, etc.2–6 Modern pharmacological investigations demonstrated that the plants of the genus Hypericum possessed anti-depression,7–11 anti-tumor,12–18 anti-bacterial,19,20 anti-viral,17,18,21–23 and liver protective activities.24 Previous phytochemical studies on these plants showed that derivatives of polycyclic polyprenylated acylphloroglucinols (PPAPs), which possessed a highly oxygenated bicyclo[3.3.1]nonane-2,4,9-trione or other related core decorated with C5H9 or C10H17 (prenyl or geranyl) side chains, were the main bioactive components.7,13–19,21,24,25In this paper, we report the isolation and structural elucidation of six new PPAPs (1–6) (Fig. 1), together with ten known ones (7–16). Their structures were elucidated using spectroscopic data, X-ray crystallography, ECD spectroscopy and Rh2(OCOCF3)4-induced ECD. Moreover, compounds 1–16 were evaluated for their cytotoxic activities on human HepG-2, HeLa, MCF-7, and A549 cell lines using the MTT assay. Among them, compound 5 shows significant cytotoxicities toward HepG-2, HeLa and A549 cell lines (IC50 = 9.52 ± 0.27, 11.87 ± 0.22 and 12.63 ± 0.12 μM).
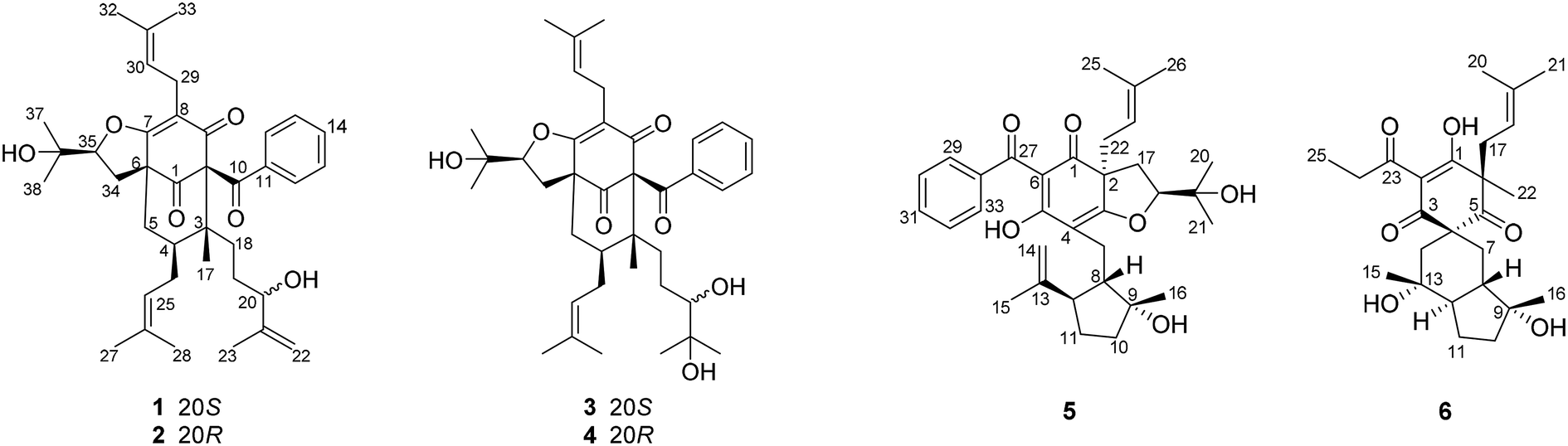 | ||
| Fig. 1 Chemical structures of 1–6. | ||
Results and discussion
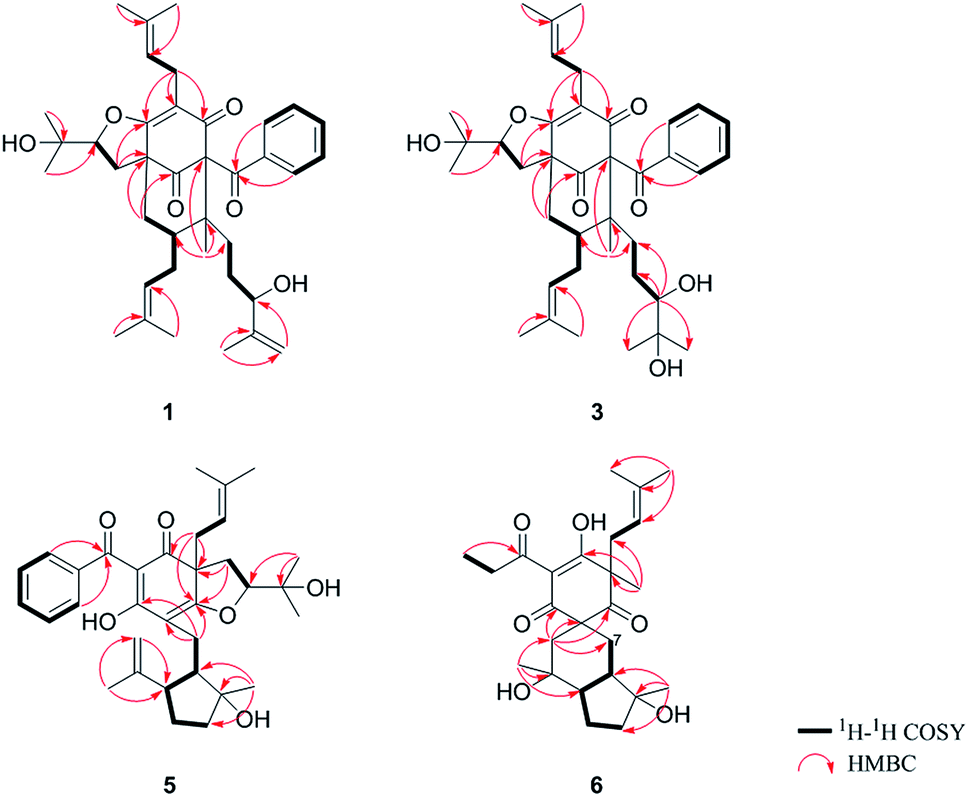
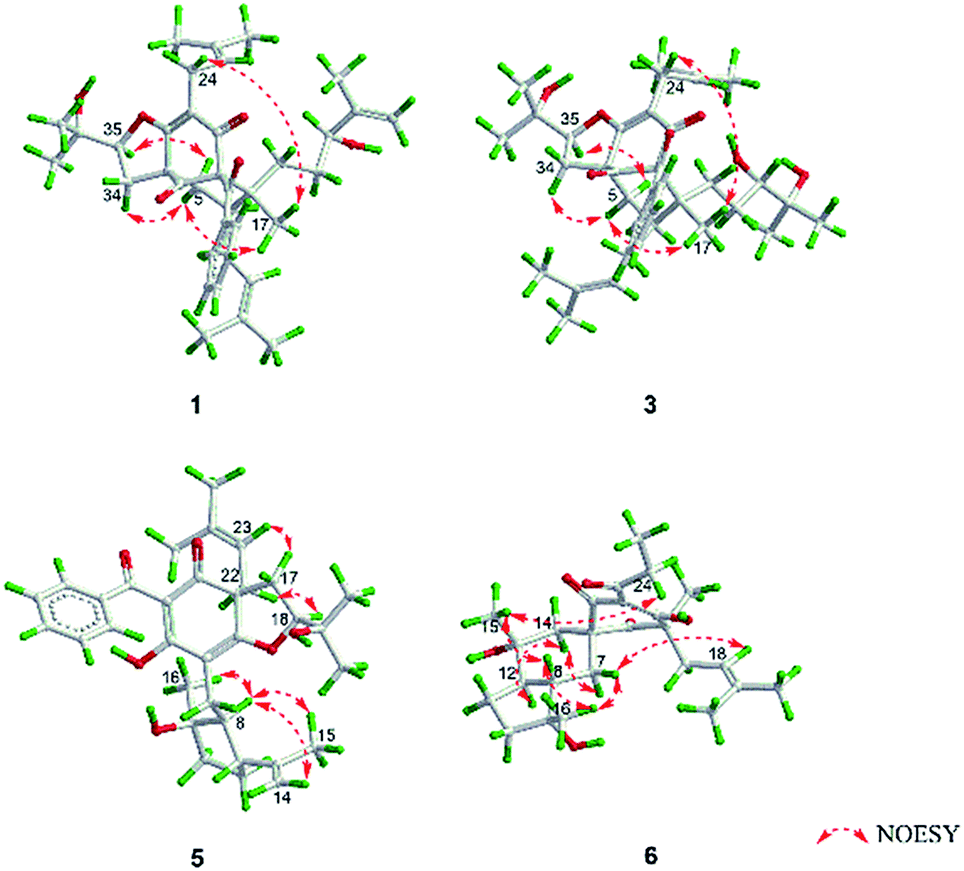
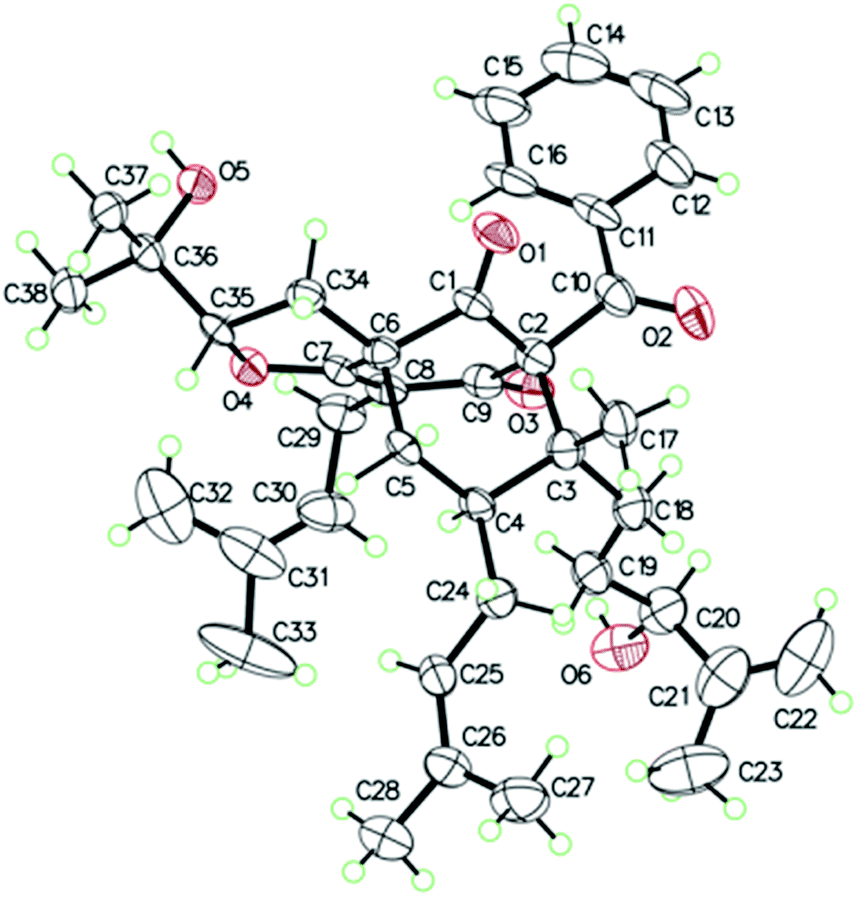
The 95% EtOH extract of Hypericum patulum was subjected to liquid–liquid fractionation to afford a petroleum ether (PE)-soluble fraction and an ethyl acetate (EtOAc)-soluble fraction. The PE fraction was separated by silica gel column chromatography, Sephadex LH-20 and preparative HPLC to obtain six new compounds (1–6) and ten known ones (7–16).Compound 1 was isolated from CH3OH as colorless crystals with [α]25D +39.6 (c 1.0, MeOH). Its molecular formula was deduced as C38H50O6 on the basis of 13C NMR and HRESIMS (m/z 625.3515 [M + Na]+, calcd for C38H50NaO6 625.3500) data. IR spectroscopy suggested the presence of hydroxyl (3456 cm−1), carbonyl (1716 cm−1) and aromatic double bond (1624, 1450 cm−1) groups. The NMR data of 1 (Table S1 and S2, ESI†) indicated the presence of an enolized 1,3-dicarbonyl ether group (δC 193.8, C-9; 116.3, C-8; 172.9, C-7), an unconjugated carbonyl carbon (δC 205.0, C-1), a methylene (δC 38.8, C-5), a methine (δC 43.2, C-4), and three quaternary carbons at δC 79.7 (C-2), 60.2 (C-6), and 49.7 (C-3), which suggested that 1 was a polycyclic polyprenylated acylphloroglucinol.7,26,27 Besides the above carbons, signals for eight methyls, six methylenes, nine methines and six quaternary carbons were observed. The NMR spectroscopic data of 1 resembled those of 32-epi-hyperforatin E.27 The main differences were that the absence of the 2-methylpropanoyl group [δH 2.00 (CH), 1.04 (CH3), 0.96 (CH3); δC 211.5 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 43.0 (CH), 21.8 (CH3), 20.8 (CH3)], and the presence of a benzoyl group [δH 7.41 (2CH), 7.36 (CH), 7.20 (2CH); δC 194.2 (CO), 137.1 (C), 132.3 (CH), 128.3 (2CH), 128.1 (2CH)] in 1 (Fig. 1), which implied that the 2-methylpropanoyl group in 32-epi-hyperforatin E was replaced by a benzoyl group in 1. This was confirmed by the 1H–1H COSY cross-peaks between H-13/15 (δH 7.20) and H-12/16 (δH 7.41)/H-14 (δH 7.36), as well as the HMBC cross-peaks from H-12/16 to C-10 (δC 194.2)/C-14 (δC 132.3) (Fig. 2). The relative stereochemistry of 1 resembled those of 32-epi-hyperforatin E, basing on the NOESY correlations of Me-17 (δH 1.17) with H-5b (δH 1.63)/H-24, H-5b with H-34 and of H-5a (δH 2.10) with H-35 (δH 4.61) (Fig. 3). The absolute configuration at C-20 was confirmed by the induced ECD of the in situ formed [Rh2(OCOCF3)4] complex.28,29 According to the bulkiness rule,28–30 the 20S configuration of 1 was confirmed by the Cotton effect (positive E band) of the Rh complex (Fig. S1, ESI†). Additionally, the absolute configuration of 1 was unequivocally confirmed by X-ray crystallography (Fig. 4, CCDC 1865373) and ECD calculations (Fig. S2, ESI†), allowing the assignment of the absolute configuration of 1 as 2R, 3R, 4S, 6S, 20S, 35S (Fig. 1). Based on the above analysis, the structure of 1 was elucidated and named hyperpatulone A.
O), 43.0 (CH), 21.8 (CH3), 20.8 (CH3)], and the presence of a benzoyl group [δH 7.41 (2CH), 7.36 (CH), 7.20 (2CH); δC 194.2 (CO), 137.1 (C), 132.3 (CH), 128.3 (2CH), 128.1 (2CH)] in 1 (Fig. 1), which implied that the 2-methylpropanoyl group in 32-epi-hyperforatin E was replaced by a benzoyl group in 1. This was confirmed by the 1H–1H COSY cross-peaks between H-13/15 (δH 7.20) and H-12/16 (δH 7.41)/H-14 (δH 7.36), as well as the HMBC cross-peaks from H-12/16 to C-10 (δC 194.2)/C-14 (δC 132.3) (Fig. 2). The relative stereochemistry of 1 resembled those of 32-epi-hyperforatin E, basing on the NOESY correlations of Me-17 (δH 1.17) with H-5b (δH 1.63)/H-24, H-5b with H-34 and of H-5a (δH 2.10) with H-35 (δH 4.61) (Fig. 3). The absolute configuration at C-20 was confirmed by the induced ECD of the in situ formed [Rh2(OCOCF3)4] complex.28,29 According to the bulkiness rule,28–30 the 20S configuration of 1 was confirmed by the Cotton effect (positive E band) of the Rh complex (Fig. S1, ESI†). Additionally, the absolute configuration of 1 was unequivocally confirmed by X-ray crystallography (Fig. 4, CCDC 1865373) and ECD calculations (Fig. S2, ESI†), allowing the assignment of the absolute configuration of 1 as 2R, 3R, 4S, 6S, 20S, 35S (Fig. 1). Based on the above analysis, the structure of 1 was elucidated and named hyperpatulone A.
 | ||
| Fig. 2 Key 1H–1H COSY and HMBC correlations of 1, 3, 5 and 6. | ||
 | ||
| Fig. 3 Key NOESY correlations of 1, 3, 5 and 6. | ||
 | ||
| Fig. 4 X-ray ORTEP drawing of 1. | ||
Compound 2 was isolated as a colorless oil with [α]25D +41.7 (c 1.0, MeOH). The HRESIMS of compound 2 showed an [M + Na]+ ion peak at m/z 625.3506 (calcd for C38H50NaO6, 625.3500), consistent with the molecular formula of C38H50O6. Compounds 2 and 1 were separated by using chiral HPLC over a CHIRALPAK IC column. And the NMR spectroscopic data of 2 (Table S1 and S2, ESI†) was almost identical to those of 1, which indicated that 2 possessed the same planar structure as that of 1. However, compound 2 showed a negative E band in the in situ [Rh2(OCOCF3)4] complex-induced ECD spectrum (Fig. S1, ESI†), which is different from that of 1, suggesting an 20R configuration in compound 2. Thus, structure 2 was established, and named hyperpatulone B.
Compound 3 had the molecular formula C38H52O7, which was assigned by HRESIMS (m/z 643.3640 [M + Na]+, calcd for C38H52O7Na, 643.3605). According to its 1D NMR spectra (Tables S1 and S2, ESI†), compound 3 has the same skeleton as that of 1 except for the C-18-C-23 side chain. The differences between them were the absence of a terminal double bond (δC 147.9, 111.2) between C-21 and C-22, but the presence of one additional oxygenated quaternary carbon (δC 73.1) and one additional methyl group (δC 26.4) in 3, and the chemical shifts of C-18, 19, 20, 23 shifted from δC 32.7, 32.0, 76.5, 17.8 in 1 to δC 34.2, 28.2, 79.5, 23.8 in 3, which indicated the olefinic carbons (C-21, C-22) in 1 were replaced by a tertiary alcohol hydroxy group and a methyl group in 3. This was confirmed by the HMBC cross-peaks from H-20 (δH 3.26)/H-22 (δH 1.18)/H-23 (δH 1.12) to C-21 (δC 73.1) (Fig. 2). NOESY correlations of Me-17 (δH 1.18) with H-5b (δH 1.63)/H-24, of H-5b with H-34 and of H-5a (δH 2.10) with H-35 (δH 4.62) indicated that the relative configuration of 3 was identical to that of 1 (Fig. 3). The in situ [Rh2(OCOCF3)4] complex-induced ECD spectrum of 3 exhibited a positive E band for a 20S configuration (Fig. S3, ESI†). Therefore, structure 3 was determined and named hyperpatulone C.
The molecular formula of 4 was established to be C33H42O6 by its HRESIMS m/z 643.3626 [M + Na]+ (calcd for C38H52O7Na, 643.3605). The NMR data (Tables S1 and S2, ESI†) of 4 showed lots of similarities to those of 3, suggesting that 4 and 3 possessed the same planar structure. The only difference between 4 and 3 was the orientation of H-20, which was determined by a negative E band for a 20R configuration in the in situ [Rh2(OCOCF3)4] complex-induced ECD spectrum of 4 (Fig. S3, ESI†). Accordingly, compound 4 was elucidated and named hyperpatulone D.
The molecular formula C33H42O6 of compound 5 was assigned by HRESIMS (m/z 557.2893 [M + Na]+, calcd for C33H42O6Na, 557.2874). The 1D NMR data (Tables S1 and S3, ESI†) of 5 showed lots of similarities to those of hyperascyrone G,18 with a 6/6/5 tricyclic spiro ring system. The natural occurring polyprenylated spirocyclic acylphloroglucinol derivatives (PSAPs), with a 6/6/5 tricyclic spiro ring system, were a special subgroup of PPAPs. Detailed comparison of the NMR spectra of 5 with those of hyperascyrone G indicated the absence of a 3-methylbutanoyl group [δH 3.04 and 2.88 (CH2), 2.30 (CH), 1.01 (CH3), 0.98 (CH3); δC 197.6 (CO), 45.9 (CH2), 27.1 (CH), 22.9 (CH3), 22.6 (CH3)] in hyperascyrone G, but the presence of a benzoyl group [δH 7.44 (2CH), 7.43 (CH), 7.37 (2CH); δC 191.5 (CO), 136.6 (C), 131.3 (CH), 128.1 (2CH), 127.9 (2CH)] in 5. Thus, it could be deduced that the 3-methylbutanoyl group in hyperascyrone G was replaced by a benzoyl group in 5. This was confirmed by the 1H–1H COSY cross-peaks between H-30/32 (δH 7.37) and H-29/33 (δH 7.44)/H-31 (δH 7.43), as well as the HMBC cross-peaks from H-29/33 to C-27 (δC 191.5)/C-31 (δC 131.3) (Fig. 2). The relative configurations of 5 and hyperascyrone G were very similar by analysis of the NOESY correlations between H-18 (δH 4.55) and H-22a (δH 2.66), between H-23 (δH 5.13) and H-17a (δH 2.15), between H-8 (δH 1.83) and Me-15 (δH 1.71)/Me-16 (δH 1.18)/H-14a (δH 4.78) (Fig. 3). The ECD data obtained for 5 showed positive Cotton effects at λmax 201 and 278 nm and a negative Cotton effect at λmax 242 and 311 nm (Fig. S4, ESI†) comparable to those of hyperascyrone G.18 Thus, structure 5 was established, and named hyperpatulone E.
Compound 6 was assigned the molecular formula C25H36O6 by HRESIMS (m/z 455.2412 [M + Na]+, calcd for C25H36O6Na, 455.2404). The 1D NMR data (Tables S1 and S3, ESI†) of 6 showed lots of similarities to chipericumin D (14).31 Detailed comparison of the NMR spectra of 6 with those of chipericumin D indicated the absence of a 2-methylbutanoyl group [δH 3.16 (CH), 1.78 and 1.44 (CH2), 1.22 (CH3), 0.83 (CH3); δC 205.1 (CO), 41.9 (CH), 25.3 (CH2), 19.5 (CH3), 12.3 (CH3)] in chipericumin D, but the presence of a propanoyl group [δH 2.98 (CH2), 0.97 (CH3); δC 201.0 (CO), 46.2 (CH2), 23.0 (CH3)] in 6. Thus, it could be deduced that the 2-methylbutanoyl group in chipericumin D was replaced by a propanoyl group in 6. The structure was supported by the 1H–1H COSY correlations between H-24 (δH 2.98) and Me-25 (δH 0.97) together with the HMBC correlations between Me-25 and C-23 (δC 201.0) (Fig. 2). The relative configuration of 6 was same as that of chipericumin D with the analysis of the NOESY correlations of H-7a (δH 1.91)/H-14a (δH 1.45), H-12 (δH 1.78)/H-14a, H-8 (δH 1.67)/Me-15 (δH 0.95), H-7b (δH 1.76)/Me-16, H-8/Me-16 (δH 1.40), Me-15/H-24 (δH 2.98) and H-7b/H-18(δH 4.55) (Fig. 3). In addition, compounds 6 and chipericumin D (14) gave closely correlated Cotton effects in the ECD spectrum (Fig. S4, ESI†). Thus, structure 6 was established, and named hyperpatulone F.
Ten known compounds were identified as uralodin A (7),32 uralodin B (8),13 attenuatumione H (9),26 uralione D (10),7 uralione I (11),7 tomoeone A (12),15 tomoeone B (13),15 chipericumin D (14),31 hyperascyrone F (15),18 hypercohone G (16),33 by comparison of their spectroscopic and physical data with those of related literature.
The isolates 1–16 were tested for their cytotoxic activities by MTT assay on human HepG-2, HeLa, MCF-7 and A549 cell lines. Cisplatin was used as the positive control. As shown in Table 1, PSAPs compounds (5–6, 12–16) exhibited more potent cytotoxic activities than other PPAPs compounds (1–4, 7–11), with IC50 values of 9.52 ± 0.27 to 42.33 ± 1.91 μM. Especially, compound 5 shows significant cytotoxicities toward HepG-2, HeLa and A549 cell lines (IC50 = 9.52 ± 0.27, 11.87 ± 0.22 and 12.63 ± 0.12 μM).
| Compounds | IC50a (μM) | A549 | ||
|---|---|---|---|---|
| HepG-2 | HeLa | MCF-7 | ||
| a IC50 values of 1–16 were detected by MTT assay after incubation for 48 h; data are expressed as mean ± SD.b Positive control. | ||||
| 1 | >50 | >50 | >50 | >50 |
| 2 | >50 | >50 | 46.83 ± 1.26 | >50 |
| 3 | >50 | >50 | >50 | >50 |
| 4 | >50 | 45.79 ± 1.21 | >50 | 44.35 ± 0.62 |
| 5 | 9.52 ± 0.27 | 11.87 ± 0.22 | 20.83 ± 0.52 | 12.63 ± 0.12 |
| 6 | 26.73 ± 0.23 | 39.67 ± 0.27 | 42.33 ± 1.91 | 36.89 ± 0.81 |
| 7 | >50 | >50 | >50 | 47.82 ± 1.17 |
| 8 | 41.03 ± 0.68 | 39.27 ± 1.23 | 35.72 ± 0.93 | 42.90 ± 1.04 |
| 9 | >50 | >50 | >50 | >50 |
| 10 | >50 | 42.67 ± 0.42 | 39.31 ± 0.67 | 41.32 ± 1.32 |
| 11 | >50 | >50 | 42.97 ± 1.21 | >50 |
| 12 | 30.91 ± 0.25 | 27.46 ± 0.37 | 35.29 ± 0.82 | 21.78 ± 0.57 |
| 13 | 35.67 ± 0.49 | 29.67 ± 0.21 | 31.44 ± 0.95 | 32.47 ± 0.31 |
| 14 | 22.83 ± 0.53 | 25.59 ± 0.32 | 26.92 ± 0.58 | 27.41 ± 0.71 |
| 15 | 29.38 ± 0.28 | 24.39 ± 0.28 | 27.37 ± 0.53 | 23.76 ± 0.17 |
| 16 | 19.28 ± 0.37 | 28.59 ± 0.35 | 22.91 ± 0.32 | 17.92 ± 0.23 |
| Cisplatinb | 5.9 ± 0.45 | 4.7 ± 0.17 | 6.7 ± 0.61 | 5.1 ± 0.21 |
Experimental
General experimental procedures
Optical rotations were obtained on a JASCO P-1020 polarimeter. UV spectra were recorded using a JASCO V-550 UV/VIS spectrophotometer. CD spectra were measured on a JASCO J-810 spectrometer. 1D and 2D NMR spectra were recorded on Bruker AV-500 NMR spectrometers with TMS as an internal standard. HRESIMS analyses were recorded on an Agilent 6210 ESI/TOF mass spectrometer. Column chromatography (CC) was performed with Silica gel (Qingdao Marine Chemical Plant, Qingdao, P. R. China), ODS (50 μm, YMC, Kyoto, Japan) and Sephadex LH-20 (Pharmacia Biotech, Uppsala, Sweden). Preparative HPLC was conducted on a Cosmosil C18 preparative column (5 μm, 20 × 250 mm) equipped with a G1311C pump and a G1315D photodiode array detector (Agilent Technologies, CA, USA). All chemical reagents were purchased from Tianjin Damao Chemical Company (Tianjin, P. R. China).Plant material
The whole plant of Hypericum patulum was collected in Guizhou Province of China, in August of 2016 and authenticated by Zhenqiu Mai, the senior engineer of Guangdong Province. A voucher specimen (no. 20160817) was deposited in the Institute of Traditional Chinese Medicine & Natural Products, Jinan University, Guangzhou, China.Extraction and isolation
The dried and powdered herbs of Hypericum patulum (12 kg) were extracted under reflux with 95% EtOH (30 L × 3) at room temperature. The combined ethanol extract was concentrated to afford a residue (654 g), which was suspended in water (4 L) and then extracted with petroleum ether (PE) (4 L × 3) and ethyl acetate (EtOAc) (4 L × 3). The PE extract (217 g) was subjected to silica gel column chromatography, eluting with PE-EtOAc (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 0:1, v/v) to yield seven fractions (Fr. A–F). Fr. C (18.7 g) was further applied to a silica gel CC with PE/EtOAc (10:1 to 1:1, v/v) to afford five subfractions (Fr. C1–C5). Fr. C2 (1.5 g) was purified by Sephadex LH-20 (CHCl3/MeOH, 2:1, v/v) and further separated by preparative HPLC (MeOH/H2O, 70:30, v/v) to yield compounds 1 (21.2 mg), 2 (18.5 mg), 7 (11.7 mg) and 8 (16.9 mg). Fr. C3 (7.5 g) was purified by ODS CC and Sephadex LH-20 to obtain compounds 3 (12.7 mg), 4 (13.9 mg), 9 (19.7 mg), 10 (15.7 mg) and 11 (13.2 mg). Fr. D (21.9 g) was applied to ODS CC using a MeOH/H2O gradient (40:60 to 100:0, v/v) to afford five subfractions (Fr. D.1–D.5). Fr. D.3 (3.6 g) was further purified by Sephadex LH-20 CC (CHCl3/MeOH, 1:1, v/v) and preparative HPLC (MeOH/H2O, 80:20, v/v) and to yield compounds 5 (9.5 mg), 12 (21.3 mg) and 13 (25.1 mg). Fr. D.4 (5.8 g) was separated by preparative HPLC (MeOH/H2O, 80:20, v/v) to yield compounds 6 (15.8 mg) and 14 (11.9 mg). Fr. D.5 (4.9 g) was purified by preparative HPLC (MeOH/H2O, 80:20, v/v) to achieve compounds 15 (9.2 mg) and 16 (15.2 mg).
:0 to 0:1, v/v) to yield seven fractions (Fr. A–F). Fr. C (18.7 g) was further applied to a silica gel CC with PE/EtOAc (10:1 to 1:1, v/v) to afford five subfractions (Fr. C1–C5). Fr. C2 (1.5 g) was purified by Sephadex LH-20 (CHCl3/MeOH, 2:1, v/v) and further separated by preparative HPLC (MeOH/H2O, 70:30, v/v) to yield compounds 1 (21.2 mg), 2 (18.5 mg), 7 (11.7 mg) and 8 (16.9 mg). Fr. C3 (7.5 g) was purified by ODS CC and Sephadex LH-20 to obtain compounds 3 (12.7 mg), 4 (13.9 mg), 9 (19.7 mg), 10 (15.7 mg) and 11 (13.2 mg). Fr. D (21.9 g) was applied to ODS CC using a MeOH/H2O gradient (40:60 to 100:0, v/v) to afford five subfractions (Fr. D.1–D.5). Fr. D.3 (3.6 g) was further purified by Sephadex LH-20 CC (CHCl3/MeOH, 1:1, v/v) and preparative HPLC (MeOH/H2O, 80:20, v/v) and to yield compounds 5 (9.5 mg), 12 (21.3 mg) and 13 (25.1 mg). Fr. D.4 (5.8 g) was separated by preparative HPLC (MeOH/H2O, 80:20, v/v) to yield compounds 6 (15.8 mg) and 14 (11.9 mg). Fr. D.5 (4.9 g) was purified by preparative HPLC (MeOH/H2O, 80:20, v/v) to achieve compounds 15 (9.2 mg) and 16 (15.2 mg).
X-ray crystallographic analysis of 1 (Table S4, ESI†). C38H50O6, M = 602.78, orthorhombic, space group P212121; a = 19.2963(4) Å, b = 16.3762(4) Å, c = 11.0039(2) Å, α = 90°, β = 90°, γ = 90°, V = 3477.23(13) Å3, T = 100.00(10) K, Z = 4, Dcalcd = 1.151 g m−3, F (000) = 1304.0. The final R values were R1 = 0.0809, wR2 = 0.2257, and the goodness of fit on F2 was equal to 1.156. Flack parameter = 0.0(2). The crystal data of compound 1 was deposited with the Cambridge Crystallographic Data Centre (CCDC 1865373, http://www.ccdc.cam.ac.uk/).†
Cell culture
Human HepG-2, HeLa, MCF-7, and A549 cells were obtained from the Human Virology Institute of Sun Yat-Sen University. Cells were maintained in RPMI-1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (Gibco, USA) and 1% penicillin/streptomycin at 37 °C with 5% CO2 for 24 h.Cytotoxic assay in vitro
Four selected human cancer cell lines at the logarithmic phase were seeded in 96-well plates at 5 × 103 cells per well, respectively. After incubating for 24 h, cells were treated with various concentrations of compounds 1–16 and incubated at 37 °C for 48 h. Then, the medium of each well was removed and 5 mg mL−1 MTT (30 μL) was added. After incubating for 4 h, the supernatant of each well was removed and DMSO (200 μL) was added to dissolve the formazan produced in the cells. The absorbance was recorded using an enzyme immunoassay reader (Thermo Labsystems Multiskan MK3) at 570 nm. The IC50 was calculated by the Bliss method: inhibitory rate = [(absorbance of the test group − absorbance of the blank control)/(absorbance of the control group-absorbance of the blank control)] × 100.Conclusions
In summary, six new PPAPs derivatives, hyperpatulone A–F (1–6), together with ten known analogs, were obtained from the dried herbs of Hypericum patulum. Their structures were determined by spectroscopic data, X-ray crystallography, ECD spectrum and Rh2(OCOCF3)4-induced ECD. Moreover, compounds 1–16 were evaluated for their cytotoxic activities on human HepG-2, HeLa, MCF-7, and A549 cell lines using the MTT assay. Compound 5 shows significant cytotoxicity toward HepG-2, HeLa and A549 cell lines (IC50 = 9.52 ± 0.27, 11.87 ± 0.22 and 12.63 ± 0.12 μM).Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81803376, 81673319, 81673670), the Science and Technology Planning Project of Guangdong Province (No. 2016A030303011, 2016B030301004), and China Postdoctoral Science Foundation (No. 2017M620405).Notes and references
- Q. L. Wu, S. P. Wang, L. W. Wang, J. S. Yang and P. G. Xiao, Nat. Prod. Res. Dev., 1997, 10, 15–18 Search PubMed.
- H. F. Lv, Q. G. Chu and H. Z. Hu, Chin. Tradit. Herb. Drugs, 2002, 33, 1135–1138 Search PubMed.
- Z. Y. Xiao and Q. Mu, Nat. Prod. Res. Dev., 2007, 19, 344–355 CAS.
- Z. Q. Yin, Y. Wang, D. M. Zhang, W. C. Ye and S. X. Zhao, Chinese Wild Plant Resources, 2004, 23, 6–11 Search PubMed.
- Y. H. Cui and J. Li, J. Northeast Agric. Univ., 2006, 37, 105–110 Search PubMed.
- L. S. Zhang, G. P. Dong and G. M. Liu, J. Chin. Med. Mater., 2009, 32, 224–226 CAS.
- Z. B. Zhou, Z. R. Li, X. B. Wang, J. G. Luo and L. Y. Kong, J. Nat. Prod., 2016, 79, 1231–1240 CrossRef CAS PubMed.
- E. Ernst, J. I. Rand, J. Barnes and C. Stevinson, Eur. J. Clin. Pharmacol., 1998, 54, 589–594 CrossRef CAS PubMed.
- A. Singer, M. Wonnemann and W. E. Müller, J. Pharmacol. Exp. Ther., 1999, 290, 1363–1368 CAS.
- V. Butterweck, F. Petereit, H. Winterhoff and A. Nahrstedt, Planta Med., 1998, 64, 291–294 CrossRef CAS PubMed.
- S. S. Chatterjee, S. K. Bhattacharya, M. Wonnemann, A. Singer and W. E. Muller, Life Sci., 1998, 63, 499–510 CrossRef CAS PubMed.
- D. Albert, I. Zundorf, T. Dingermann, W. E. Muller, D. Steinhilber and O. Werz, Biochem. Pharmacol., 2002, 64, 1767–1775 CrossRef CAS PubMed.
- X. Q. Chen, Y. Li, X. Cheng, K. Wang, J. He, Z. H. Pan, M. M. Li, L. Y. Peng, G. Xu and Q. S. Zhao, Chem. Biodiversity, 2010, 7, 196–204 CrossRef CAS PubMed.
- L. H. Hu and K. Y. Sim, Tetrahedron, 2000, 56, 1379–1386 CrossRef CAS.
- W. Hashida, N. Tanaka, Y. Kashiwada, M. Sekiya, Y. Ikeshiro and Y. Takaishi, Phytochemistry, 2008, 69, 2225–2230 CrossRef CAS PubMed.
- J. J. Zhang, X. W. Yang, J. Z. Ma, Y. Ye, X. L. Shen and G. Xu, Tetrahedron, 2015, 71, 8315–8319 CrossRef CAS.
- S. A. T. Fobofou, K. Franke, G. Sanna, A. Porzel, E. Bullita, P. L. Colla and L. A. Wessjohann, Bioorg. Med. Chem., 2015, 23, 6327–6334 CrossRef CAS PubMed.
- H. C. Zhu, C. M. Chen, J. J. Liu, B. Sun, G. Z. Wei, Y. Li, J. W. Zhang, G. M. Yao, Z. W. Luo, Y. B. Xue and Y. H. Zhang, Phytochemistry, 2015, 115, 222–230 CrossRef CAS PubMed.
- T. Naonobu, Y. Yuki, T. Yutaka and K. Yoshiki, Org. Lett., 2016, 18, 5360–5363 CrossRef PubMed.
- H. Jayasariya, A. M. Clark and C. J. D. Mc, J. Nat. Prod., 1991, 54, 1314–1320 CrossRef.
- S. A. T. Fobofou, C. R. Harmon, A. H. N. Lonfouo, K. Franke, S. M. Wright and L. A. Wessjohann, Phytochemistry, 2016, 124, 108–113 CrossRef CAS PubMed.
- A. K. George, P. Dan, T. John and C. Susan, Biochem. Biophys. Res. Commun., 1990, 172, 149–153 CrossRef.
- J. Zhao, Z. P. Zhang, H. S. Chen and X. H. Chen, Acta Pharmacol. Sin., 1998, 33, 67–72 CAS.
- W. Gao, W. Z. Hou, J. Zhao, F. Xu, L. Li, F. Xu, H. Sun, J. G. Xing, Y. Peng, X. L. Wang, T. F. Ji and Z. Y. Gu, J. Nat. Prod., 2016, 79, 1538–1547 CrossRef CAS PubMed.
- X. W. Yang, Y. Q. Ding, J. J. Zhang, X. Liu, L. X. Yang, X. N. Li, D. Ferreira, L. A. Walker and G. Xu, Org. Lett., 2014, 16, 2434–2437 CrossRef CAS PubMed.
- Z. B. Zhou, Y. M. Zhang, J. G. Luo and L. Y. Kong, Phytochem. Lett., 2016, 15, 215–219 CrossRef CAS.
- Y. Guo, N. Zhang, C. M. Chen, J. F. Huang, X. N. Li, J. J. Liu, H. C. Zhu, Q. Y. Tong, J. W. Zhang, Z. W. Luo, Y. B. Xue and Y. H. Zhang, J. Nat. Prod., 2017, 80, 1493–1504 CrossRef CAS PubMed.
- J. Frelek and W. J. Szczepek, Tetrahedron, 1999, 10, 1507–1520 CrossRef CAS.
- M. Gerards and G. Snatzke, Tetrahedron, 1990, 1, 221–236 CrossRef CAS.
- L. Liu, H. Gao, X. Chen, X. Cai, L. Yang, L. Guo, X. Yao and Y. Che, Eur. J. Org. Chem., 2010, 17, 3302–3306 CrossRef.
- S. Abe, N. Tanaka and J. Kobayashi, J. Nat. Prod., 2012, 75, 484–488 CrossRef CAS PubMed.
- N. Guo, X. Q. Chen and Q. S. Zhao, Acta Bot. Yunnanica, 2008, 30, 515–518 CrossRef CAS.
- J. J. Zhang, X. W. Yang, J. Z. Ma, X. Liu, L. X. Yang, S. C. Yang and G. Xu, Nat. Prod. Bioprospect., 2014, 4, 73–79 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ECD spectra of the [Rh2(OCOCF3)4] complexes of compounds 1–4 with the intrinsic ECD spectrum subtracted, calculated and experimental ECD spectra of 1–6, detailed HRESIMS, UV, IR, 1D, 2D NMR data of compounds 1–6. CCDC 1865373. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra00277d |
| This journal is © The Royal Society of Chemistry 2019 |