
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel 10,11-methylenedioxy-camptothecin glycoside derivatives and investigation of their anti-tumor effects in vivo†
Guanzhao Wu‡
ab,
Xiaoyuan Mai‡ab,
Feng Liu‡ac,
Mingming Linc,
Xueyang Dongab,
Qingliang Xua,
Cui Haod,
Lijuan Zhangd,
Rilei Yu*ab and
Tao Jiang *ab
*ab
aKey Laboratory of Marine Drugs, Chinese Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, 5 Yushan Road, Qingdao 266003, China. E-mail: jiangtao@ouc.edu.cn; ryu@ouc.edu.cn; Tel: +86-532-820-32712
bLaboratory for Marine Drugs and Bioproducts of Qingdao National Laboratory for Marine Science and Technology, Qingdao 266003, China
cDepartment of Medical Imaging, Wei Fang Medical University, 7166 Baotongxi Street, Weifang 261000, China
dInstitute of Cerebrovascular Diseases, Affiliated Hospital of Qingdao University, Qingdao, 266003, China
First published on 9th April 2019
Abstract
10,11-Methylenedioxy-camptothecin (FL118) is a novel camptothecin analogue that possesses exceptional antitumor efficacy in human tumor xenograft models. The aim of the current study was to develop novel 20-substituted FL118 derivatives coupled with glycosyl-succinic acid esters with improved antitumor efficacy. These FL118 glycoside derivatives were designed, synthesized and their cytotoxicity evaluated in three tumor cell lines (A-549, MDA-MB-231 and RM-1). All of the derivatives showed superior in vitro cytotoxic activity and were more potent than irinotecan in A549 and MDA-MB-231 cells. In mouse prostate cancer cells RM-1, 10,11-methylenedioxy-camptothecin rhamnoside 11b displayed significant activities with IC50 of 48.27 nM. Western blot analysis demonstrated that 11b inhibited survivin expression and induced cancer cells apoptosis. Further cell cycle analyses clearly showed 11b induced G2/M phase cell cycle arrest. Molecule docking studies suggested that the binding mode of 11b was different from that of the crystal complex of ligand topotecan in Top1/DNA. Importantly, 11b showed high in vivo antitumor efficacy in the RM-1 mouse model with transplantation of prostate cancer (TGI = 44.9%) at dose of 9 mg kg−1 without apparent toxicity.
1. Introduction
20(S)-Camptothecin (CPT) is a cytotoxic quinoline alkaloid (Fig. 1) that was discovered by Wall et al. in 1966.1 Over the past five decades, many CPT derivatives were synthesized and tested, but most of the compounds designed so far are highly toxic to normal tissues or have other shortcomings making them unsuitable candidates for cancer treatment. To date, only two camptothecin analogues (i.e. irinotecan and topotecan, Fig. 1) have been commercially approved by the FDA for treatment of cancer in clinic.2,3 However, resistance to irinotecan and topotecan is often observed in practice, especially in patients using these drugs for an extended period of time.4–9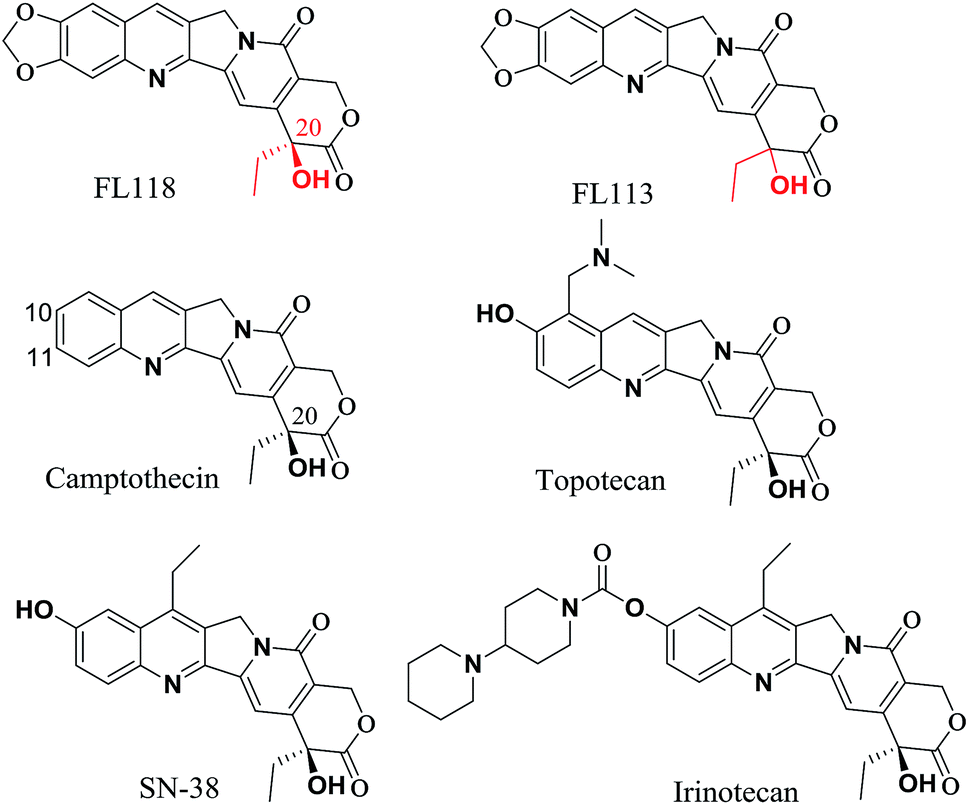 | ||
| Fig. 1 Comparison chemical structure of FL118 with FL113, camptothecin, irinotecan, SN-38 (active metabolite of irinotecan) and topotecan. | ||
The anticancer agent 10,11-methylenedioxy-camptothecin (FL118), a CPT analogue with a methylenedioxy group linked to positions 10 and 11 of the A-ring (Fig. 1), was recently identified through small molecule inhibitor screening and shows much higher anticancer activities in several different cancer types both in vitro and in vivo.10 It has been previously demonstrated that although FL118 is not a better Top1 inhibitor than the clinically used camptothecin analogues,10 FL118 is able to selectively inhibit multiple cancer survival and proliferation-associated antiapoptotic proteins (survivin,11–21 XIAP,22–26 and cIAP2 (ref. 27 and 28)) and the Bcl-2 family (Mcl-1 (ref. 29–35)), which contribute to FL118 function and antitumor activity.11,36
The superior antitumor efficacy of FL118 has inspired our interest in the development of antitumor drugs using the core structure of FL118 as a promising scaffold for the generation of novel FL118 analogs. Results from previous studies suggest that modification of the free hydroxyl group at 20-position in camptothecin via ester bonds could be a promising way to improve in vivo antitumor efficacy and reduce gastrointestinal toxicity.37–40 The idea of replacing the hydrogen atom in the hydroxyl group of FL118 with saccharide is to improve its water solubility and further decrease its normal tissue toxicity without affecting its antitumor activity.
In the present study, different configurations of 10,11-methylenedioxy-camptothecin glycosyl substituted analogs were synthesized and their antitumor activity was evaluated. The two fragments, including saccharide moiety and succinic acid ester, were incorporated into the structure at the 20-position of 10,11-methylenedioxy-camptothecin to improve its water solubility.
2. Results and discussion
2.1 Chemistry
According to published procedures,41,42 the glycosyl donors 2,3,4,6-tetra-O-acetyl-D-glucopyranose 3a or 2,3,4-tri-O-acetyl-L-rhamnose 3b was prepared from D-glucose 1a or L-rhamnose 1b in 68% or 63% yield, respectively. The obtained compound 3a or 3b was then reacted with succinic anhydride to form β-O-(2,3,4,6-tetra-O-acetyl-β-D-glucosyl)-succinic acid monoester 4a by the yield of 89% or α-O-(2,3,4-tri-O-acetyl-α-L-rhamnosyl)-succinic acid monoester 4b by the yield of 87% (Scheme S1†).The synthetic routes to form target compounds (9a–11a, 9b, 11b) are outlined in Scheme 1. 2-Amino-4,5-methylenedioxybenzaldehyde 5 was prepared according to the published procedures.43,44 Different configurations of compounds 20(R)-10,11-methylenedioxy-camptothecin 9, 20(RS)-10,11-methylenedioxy-camptothecin 10 and 20(S)-10,11-methylenedioxy-camptothecin 11 was accomplished using Friedlander condensation with 80–85% yield between 6-amino piperonal 5 and the known tricyclic keto lactone 6, 7 or 8, respectively (Scheme S2†).45,46 Coupling of compound 9 with glycosyl donors 4a or 4b, catalyzed by EDCI and DMAP, resulted in pure R steric configuration 10,11-methylenedioxy-camptothecin glucoside 9a ([α]14D = 284.25°) or rhamnoside 9b ([α]14D = 98.75°) in 68% and 71% yield, respectively. Similarly, coupling of compound 10 with glycosyl donor 4a, resulted in racemic RS configuration 10,11-methylenedioxy-camptothecin glucoside 10a ([α]14D = 32.75°) with a 63% yield. The pure S steric configuration 10,11-methylenedioxy-camptothecin glycoside 11a ([α]14D = −27.75°) and 11b ([α]14D = −166.50°) were obtained by coupling of compound 11 with glycosyl donors 4a and 4b in 61% and 73% yield, respectively (Scheme 1).
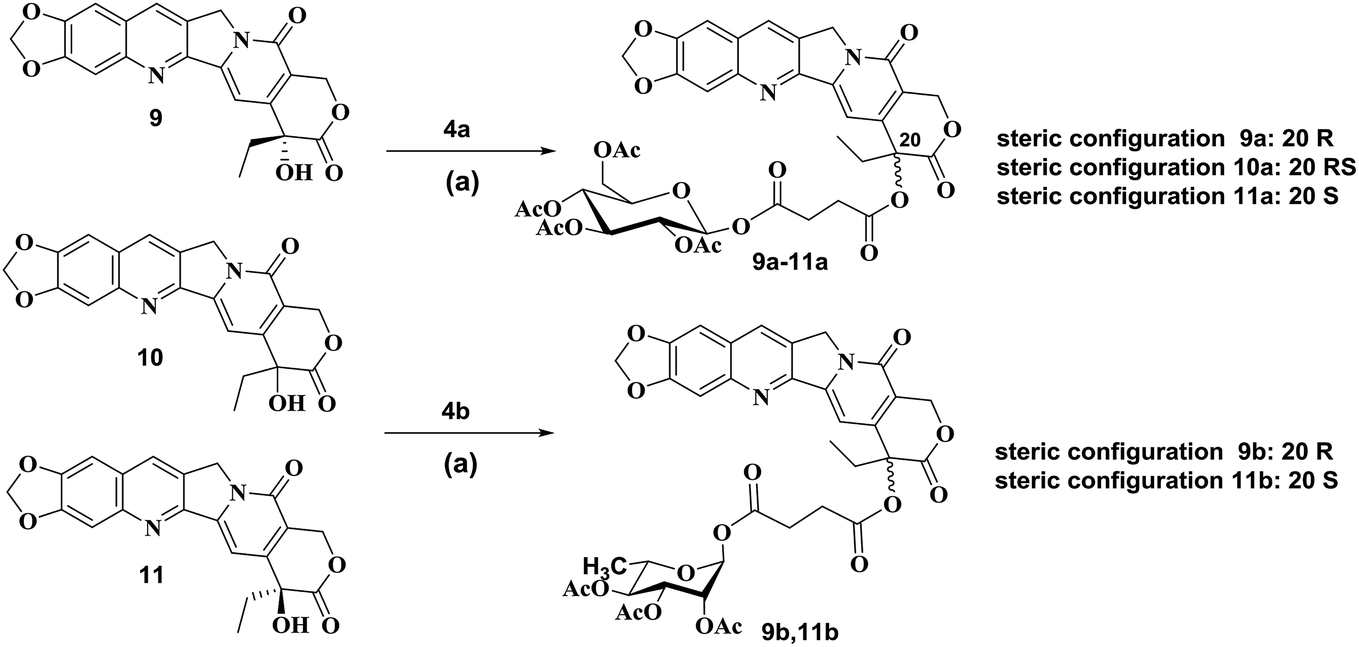 | ||
| Scheme 1 Synthesis of compounds 9a–11a, 9b, 11b. Reagents and conditions: (a) EDCI, DMAP, CH2Cl2, reflux. | ||
2.2 Cytotoxicity and IC50
The in vitro cytotoxicity of the newly synthesized compounds were determined in three tumor cell lines, A-549 (human lung carcinoma), MDA-MB-231 (human breast carcinoma) and RM-1 (mouse prostate carcinoma) while irinotecan and FL118 were used as positive controls. To obtain IC50 values for each compound, all three cancer cell lines were treated with series of concentrations of each compound. As shown in Table 1, all target compounds exhibited significant cytotoxic activities against the three tumor cell lines in vitro with IC50 values ranging from 2.32 nM to 4.53 μM. All of the new designed compounds were less potent than FL118, the core structure of these synthesized compounds, against A549 and MDA-MB-231 cell lines. In comparison, all the new compounds exhibited superior cytotoxicity to irinotecan in the two human cell lines. In addition, all of the compounds were more potent against A549 cells, moreover, 10,11-methylenedioxy-camptothecin rhamnoside 11b (the S-type enantiomer) showed the most significant cytotoxic effect with IC50 of 83 nM. The IC50 values in Table 1 indicated that the A-549 cell line was more sensitive to these compounds than the MDA-MB-231 cell lines.| Comp. | In vitro IC50 (nM)a | ||
|---|---|---|---|
| A549 | MDA-MB-231 | RM-1 | |
| a Each IC50 value was calculated from 3 independent experiments performed in triplicate. Data are shown as mean ± SD. | |||
| 9a | 1612.50 ± 276.48 | 4527.40 ± 174.66 | >1000 |
| 9b | 604.50 ± 208.17 | 1300.50 ± 276.48 | >1000 |
| 10a | 102.18 ± 33.41 | 213.00 ± 81.46 | >1000 |
| 11a | 135.80 ± 61.10 | 252.40 ± 70.75 | >1000 |
| 11b | 83.34 ± 11.04 | 154.50 ± 35.50 | 48.27 ± 6.25 |
| FL118 | 8.94 ± 1.54 | 24.73 ± 13.82 | 69.19 ± 8.34 |
| Irinotecan | 9140.30 ± 1054.87 | 7817.50 ± 2386.18 | >1000 |
For both human cancer cell lines, the compounds coupled with rhamnose (9b and 11b) were generally more potent than those coupled with glucose (9a and 11a), indicating that the rhamnose may be a better group in developing new antitumor agents. Furthermore, the configuration of the 20-position is crucial for its activity. Activity of the pure S-configuration derivatives (11a and 11b) was higher than the pure R-configuration derivatives (9a and 9b) in the two human cancer cell lines. In A549 cells, the mixture of R- and S- configuration derivative (10a) was the most active compound among the three compounds (9a, 10a, 11a). However, these compounds were less effective than FL118 in A549 and MDA-MB-231 cells. For RM-1 mouse prostate cancer cells, compounds 11b (IC50: 48.27 nM) showed the greatest cytotoxic effects, whilst the other glycoside compounds showed lower inhibitory activity at 1 μM (Table 1).
2.3 Effects of compound 11b on cell apoptosis
To study how 11b affected the cell apoptosis, human lung cancer cells A549 were treated with FL118, irinotecan or 11b for 48 h, respectively (Fig. 2). Specifically, the data revealed that treatment of cancer cells with 11b and FL118 resulted in the downregulation of survivin, while irinotecan treatment showed minimal effect on survivin. Furthermore, 11b and FL118 treatment increased the production of PARP cleavage in a concentration-dependent manner, which is the hallmark of apoptosis.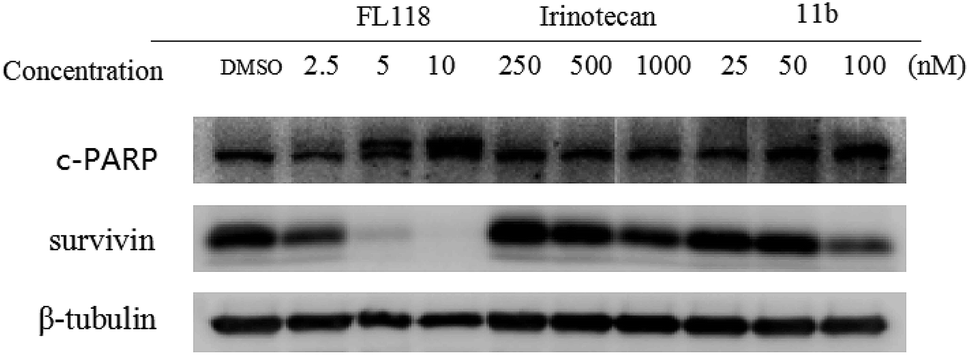 | ||
| Fig. 2 Effects of compounds irinotecan, FL118 and 11b on cell apoptosis related proteins c-parp and survivin in A549 human lung cancer cells. The cells were seeded in 15 cm dishes for 24 h, and then cells were treated with serial concentrations of three compounds, respectively. After 48 h of incubation, cells were harvested for western blotting analysis. β-Actin was served as an equal loading control. | ||
2.4 Effects of compound 11b on cell cycle
Since survivin is a central molecule for normal cell cycle progression in most of cancer cells, we next tested how the compound 11b affected the cell cycles in the A549 lung cancer cells by flow cytometry analysis. When A549 cells were treated with 11b from 25 nM to 100 nM for 48 h, the population of cells in G2/M phase dramatically increased as compared to that of vehicle (DMSO) group (Fig. 3A), along with concomitant losses in the G1 phase. As shown in Fig. 3B, compound FL118 at the concentrations of 2.5 nM, 5 nM, and 10 nM also increased G2/M cell population in a concentration-dependent manner. The cell cycle data clearly showed compound 11b arrested A549 cells mainly at the G2/M phase.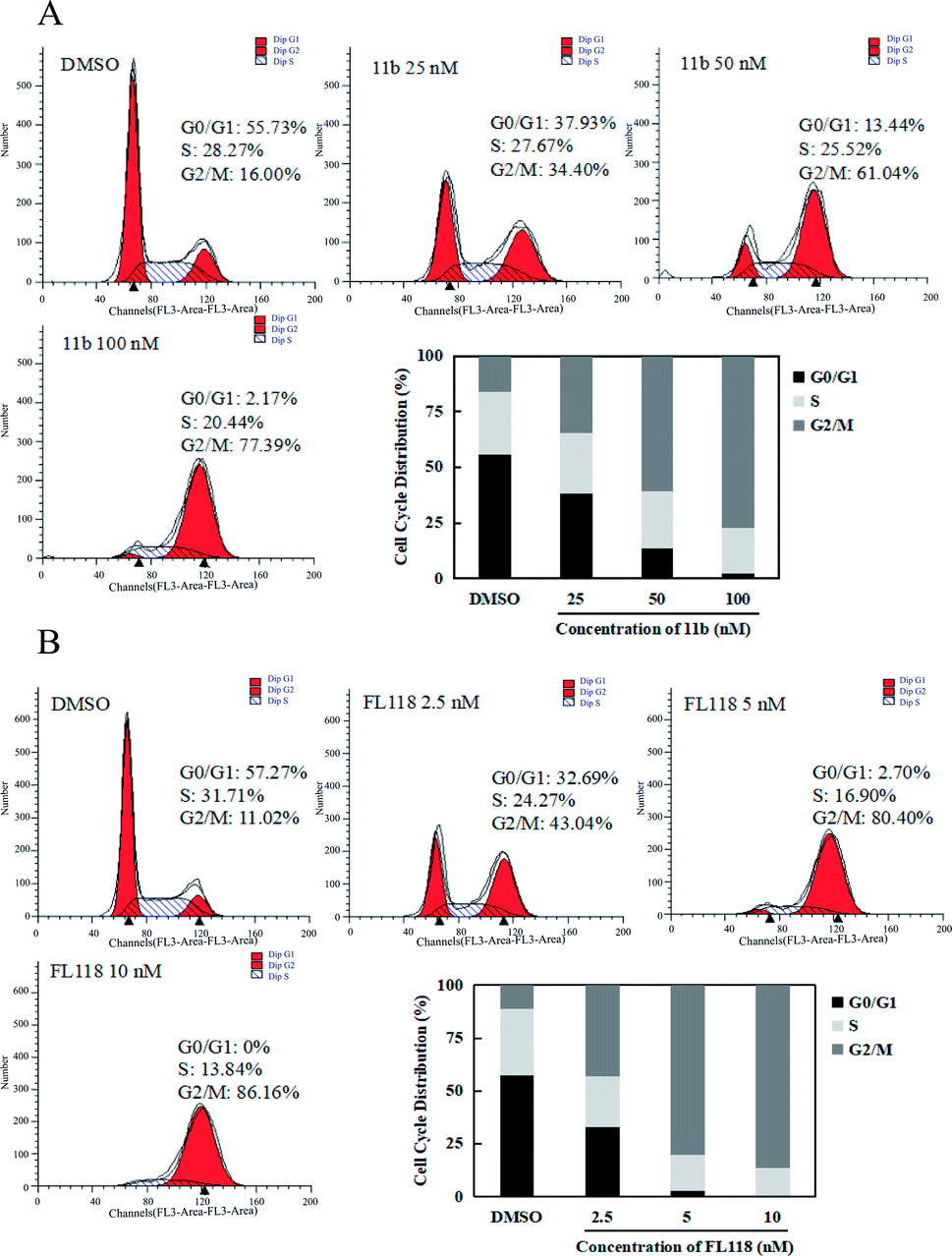 | ||
| Fig. 3 A cell cycle distribution of A549 human lung cancer cells after treatments with compound 11b (A) and FL118 (B). The cells were seeded in 6-well plates for 24 h, and then the cells were treated with compound 11b or FL118. After 48 h of treatments, cells were harvested and subject to cell cycle analyses as described in the Experimental section. Statistical analysis was conducted among control and treated groups in G0/G1, S, and G2/M phases, separately. | ||
2.5 In vivo growth inhibition
The in vivo antitumor efficacy of compound 11b was analyzed using RM-1 mouse model with transplantation of prostate cancer at doses of 3, 6 and 9 mg kg−1 per dose. Treatment was initiated 10 days after subcutaneous tumor implantation when the individual tumors had grown to about 200–300 mm3. Compound 11b was administered by intratumoral injection to the mice (three times, every other day) for 7 days. As shown in Fig. 4, compound 11b showed superior antitumor activity in vivo at a dose of 9 mg kg−1, whilst this concentration was much lower than the clinical dose of irinotecan (200 mg kg−1) and topotecan (12.5 mg kg−1). Tumor growth inhibition (TGI) was calculated at the end of the treatment. Compound 11b demonstrated siginificant antitumor activity (TGI = 44.9%) at a dose of 9 mg kg−1. It also can be seen from Fig. 4C that all the groups had nearly the same body weights, indicating the low toxicity of 11b toward mice.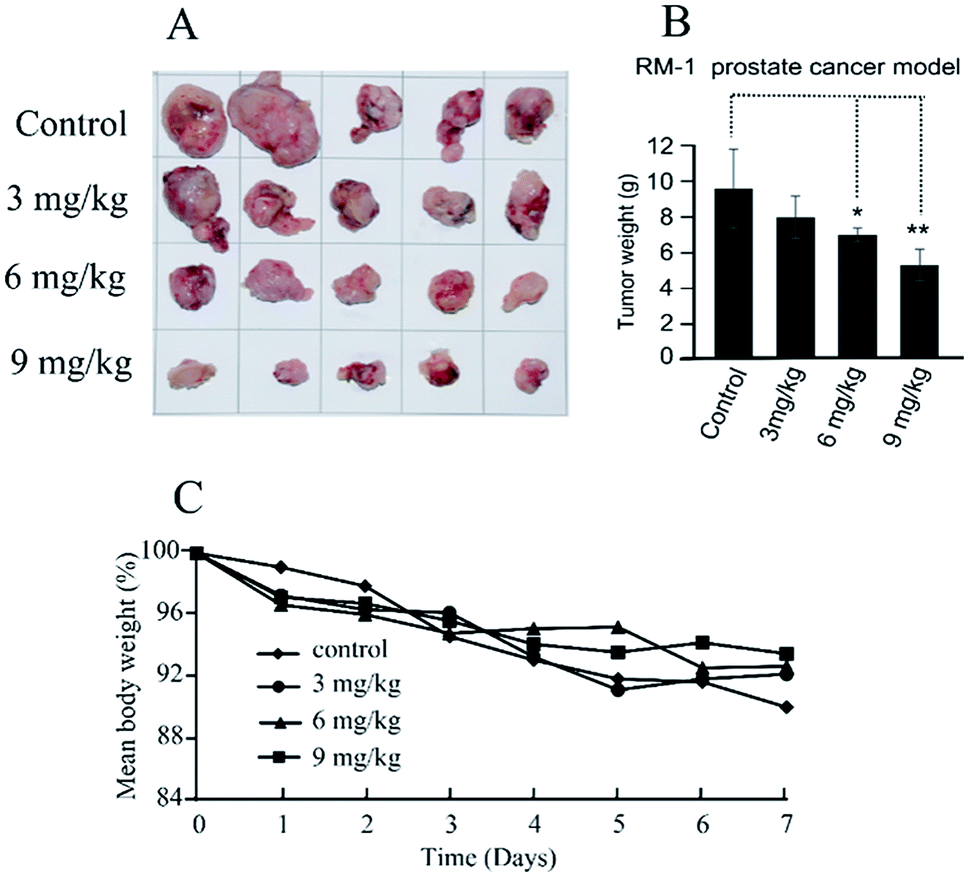 | ||
| Fig. 4 In vivo antitumor effects of compound 11b in the mouse prostate tumor of RM-1 cancer model. (A) Images of excised tumors in each group. (B) Weight of the excised tumors in each group (***P < 0.001, **P < 0.01, *P < 0.05; Student's t-test). Data are expressed as the mean ± standard deviation. (C) The mean mouse body weight curves derived from five mice treated with vehicle (control) or with one of the three doses of 11b. Of note, the standard error (SE) of body weight loss variation is within 10% among each group. | ||
2.6 Molecule docking studies
In order to understand the different molecular binding mechanism of compounds topotecan, FL118 and 11b in Top1/DNA complex, computational docking was performed using the Top1/DNA complex (PDB code: 1K4T).47 The docking study showed that the binding mode of topotecan was similar to that of the crystallographic topotecan in Top1/DNA complex (Fig. 5A), with a stacking interaction between 5-thio-2-deoxy-guanosine phosphonic acid (TGP)/cytosine on one side of the ligand and a thymine base pair on the opposite side of the ligand. The topotecan interacts with Asp533, Arg364, Lys532 and a water molecule through a hydrogen bonding interaction. As shown in Fig. 5B, the predicted binding pose of FL118 is shown to have a similar disposition with respect to the interacting residues. The MOE docking energy of FL118 is about 1.28 kcal mol−1 higher than the SN-38 in the crystal structure suggesting that FL118 is significantly less active than the SN-38 for targeting the DNA topoisomerase I (Table S1†).10 10,11-Methylenedioxy-camptothecin rhamnoside 11b displayed an energetically less favourable binding mode with the Top1/DNA complex (Fig. 5C), which indicates 11b may also show lower topoisomerase I inhibition activity than topotecan.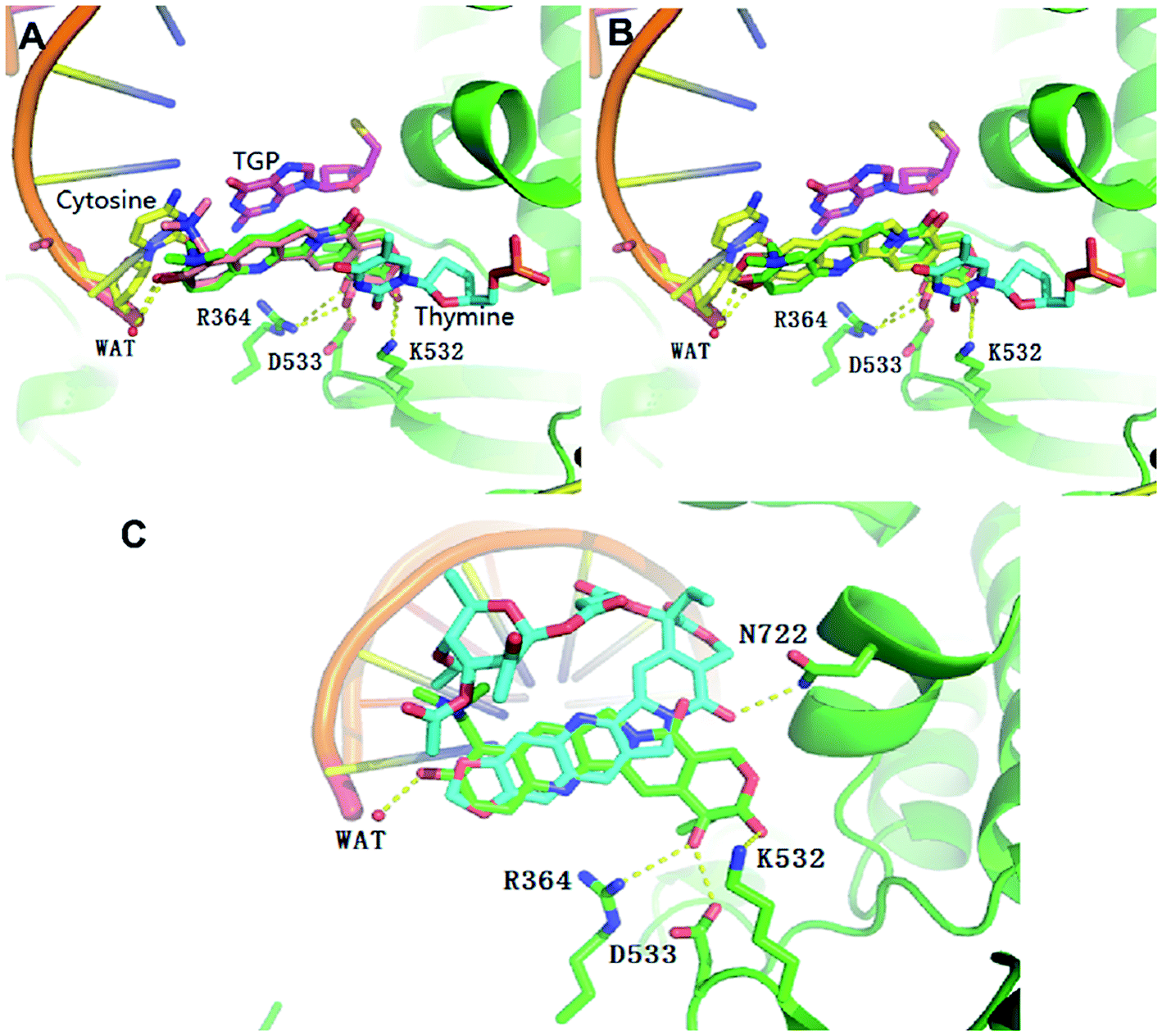 | ||
| Fig. 5 Overlap of the binding mode of compounds topotecan (pink) (A), FL118 (yellow) (B), 11b (blue) (C) with the crystallographic topotecan (green) in Top1/DNA complex (PDB code: 1K4T). The oxygen, nitrogen, and sulfur atoms of compounds are shown in red, blue, and yellow, respectively. The side chains of the binding site are colored according to the atom types (carbon, green; oxygen, red; nitrogen, blue) and the hydrogen bonds are shown as dashed lines. The docking poses were visualized using PyMOL1.8. | ||
2.7 In silico ADMET predictions
Nowadays, the computational prediction of descriptors representing absorption, distribution, metabolism, excretion and toxicity properties (ADMET) are considered useful in silico tools, decreasing the proportion of drug candidates that can fail in clinical trials for ADMET reasons. Owing to the excellent in vitro activity, we initiated in silico calculations for ADMET prediction of compounds. As shown in Table 2, the predicted central nervous system (CNS) activity was computed on a −2 (inactive) to +2 (active) scale and showed that all these four compounds (9a, 11a, 9b and 11b) could be CNS inactive due to low values. The blood/brain partition coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) B/B) were computed and also indicted all the four derivatives were difficult to access to the CNS. Human ether-a-go-go related gene (HERG) K+ channel blockers are potentially toxic. The recommended range for predicted logIC 50 values for blockage of HERG K+ channels is > −5. The logHERG values (−5.213 to −7.012) for compounds (9a, 11a, 9b and 11b) indicate that these compounds could show cardiac toxicity. Madin–Darby canine kidney (MDCK) monolayers are widely used to make oral absorption estimates. The values showed that compounds (9a, 11a, 9b and 11b) had poor MDCK cell permeability. However, all these four derivatives showed good Caco-2 permeability (PCaco).
B/B) were computed and also indicted all the four derivatives were difficult to access to the CNS. Human ether-a-go-go related gene (HERG) K+ channel blockers are potentially toxic. The recommended range for predicted logIC 50 values for blockage of HERG K+ channels is > −5. The logHERG values (−5.213 to −7.012) for compounds (9a, 11a, 9b and 11b) indicate that these compounds could show cardiac toxicity. Madin–Darby canine kidney (MDCK) monolayers are widely used to make oral absorption estimates. The values showed that compounds (9a, 11a, 9b and 11b) had poor MDCK cell permeability. However, all these four derivatives showed good Caco-2 permeability (PCaco).
| Comp. | CNS | logHERG |
PCaco | logB/B |
PMDCK | F% |
|---|---|---|---|---|---|---|
| a CNS: −2 (inactive),+2 (active); logHERG > −5; PCaCo: < 25 poor, > 500 great; logB/B (−3–1.2); PMDCK: < 25 poor, > 500 great. |
||||||
| 9a | −2 | −5.213 | 159.608 | −2.027 | 68.069 | 44.6 |
| 11a | −2 | −6.569 | 39.646 | −3.132 | 15.107 | 32.978 |
| 9b | −2 | −6.666 | 68.091 | −2.827 | 27.106 | 41.469 |
| 11b | −2 | −7.012 | 43.907 | −3.17 | 16.869 | 38.065 |
3. Conclusions
In summary, a series of 20-substituted-10,11-methylenedioxy-camptothecin glycoside derivatives were synthesized and their cytotoxic activities were evaluated against three tumor cell lines (A-549, MDA-MB-231, RM-1). All of the synthesized compounds showed superior cytotoxic activity to the irinotecan, but were less potent than FL118 in the two human cancer cell lines (A549 and MDA-MB-231). In the mouse prostate cancer cell model RM-1, 10,11-methylenedioxy-camptothecin rhamnoside 11b displayed excellent activity with IC50 of 48.27 nM, which is lower than that for FL118 and irinotecan. Western blot analysis indicated 11b inhibited survivin expression and increased the expression of apoptotic marker PARP cleavage. Cell cycle analyses showed that 11b induced A549 lung cancer cell cycle arrest in a concentration-dependent manner. Molecular docking studies suggested that the binding mode of 11b was different from that of the crystal ligand topotecan in Top1/DNA complex. In addition, compound 11b exhibited significant antitumor effects in the RM-1 prostate cancer model in vivo (TGI = 44.9%) at a dose of 9 mg kg−1. Overall, our findings provide strong evidence that FL118 is a promising scaffold for design of novel anti-tumor compounds with high anti-tumor efficacy.4. Experimental
4.1 Chemistry
Taking the 9 for example. A round bottom flask, 5 (1.06 g, 4 mmol), 6 (1.2 g, 7.2 mmol) and 4-methylbenzenesulfonic acid (0.16 g, 0.8 mmol) were dissolved in toluene (200 mL). Then the reaction mixture was refluxed under N2 atmosphere for 12 h at 110 °C. The solvent was evaporated under reduced pressure and the residue was purified by flash chromatography on silica gel (dichloromethane/acetone: 3/1) to afford pure product 9.
Taking the 9a for example. To a stirred solution of 9 (0.25 mmol) in dry DCM (40 mL) was added EDCI (2.2 mmol), DMAP (0.51 mmol) and 4a (1.0 mmol) at room temperature. Then the reaction mixture was refluxed under N2 atmosphere for 12 h at 40 °C. The mixture was cooled down to room temperature and DCM (30 mL) was poured into the mixture. The solution was washed with HCl (1 M), brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to provided colorless oily and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (dichloromethane/acetone: 5/1) to afford white powder 9a.
4.2 Bioactivity study
000 × g for 10 min at 4 °C, then supernatants were collected. Proteins were quantified by BCA Protein Assay Kit (Beyotime, Beijing, China). For immunoblot analysis, equal amount of proteins (25–100 μg, depending on the proteins of interest) were resolved over 10% or 12% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane. The membranes containing the transferred protein were blocked in blocking buffer (5% nonfat dry milk, 1% Tween-20 in 20 mM Tris-buffered saline, pH 7.6) for 2 h at room temperature, and then incubated with appropriate monoclonal primary antibody in blocking buffer overnight at 4 °C. After incubation with appropriate secondary antibodies, the membranes were washed three times with Tris buffer with Tween-20, and then visualized using Western Lightning (PerkinElmer, Waltham, MA, USA). Antibody for survivin was purchased from Santa Cruz. Antibody for PARP was purchased from Cell Signaling.4.3 Molecular modeling
Molecular docking was performed using MOE using AMBER10:EHT forcefield. Compounds topotecan, FL118 and 11b were drawn in Chem3D Pro saved the format as mol2 and minimized using 10000 steps of steepest minimization in MOE. The X-ray crystal structures of the Top1/DNA complex (PDB code: 1K4T) was downloaded from the protein data bank (http://www.rcsb.org). In consideration of the flexibility of the side chains of the residues at the binding site, the induced fit docking approach was applied in the docking studies. The produced conformation of with the best score was selected for the analysis.
4.4 ADMET predictions
The prediction of ADMET properties were performed using the Advanced Chemistry Development (ACD) Percepta platform (http://www.acdlabs.com). Any ADMET descriptor was evaluated by Percepta based on training libraries implemented in the software, which include a consistent pool of molecules whose pharmacokinetic and toxicity profiles are experimentally known.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful for financial support granted by National Science and Technology Major Project of China (2018ZX09735-004), NSFC-Shandong Joint Fund (U1706213, U1406403), the Fundamental Research Funds for the Central Universities (No. 201512007, 201762011 for R. Y.), Innovation Project from Qingdao National Laboratory for Marine Science and Technology (No. 2015ASKJ02), the Marine S&T Fund of Shandong Province (No. 2018SDKJ0403) and Taishan Scholar Project Fund of Shandong Province (TS201511011).Notes and references
- M. E. Wall, M. C. Wani, C. E. Cook, K. H. Palmer, A. T. Mcphail and G. A. Sim, J. Med. Chem., 1966, 88, 3888–3890 CAS.
- S. T. Liew and L. X. Yang, Curr. Pharm. Des., 2008, 14, 1078–1097 CrossRef CAS PubMed.
- Q. Y. Li, Y. G. Zu, R. Z. Shi and L. P. Yao, Curr. Med. Chem., 2006, 13, 2021–2039 CrossRef CAS PubMed.
- M. C. Chen, N. H. Lee, T. J. Ho, H. H. Hsu, C. H. Kuo, W. W. Kuo, Y. M. Lin, F. J. Tsai, C. H. Tsai and C. Y. Huang, Cancer Lett., 2014, 349, 51–60 CrossRef CAS PubMed.
- Y. Bessho, T. Oguri, H. Achiwa, H. Muramatsu, H. Maeda, T. Niimi, S. Sato and R. Ueda, Cancer Sci., 2006, 97, 192–198 CrossRef CAS PubMed.
- M. Leggas, M. Adachi, G. L. Scheffer, D. Sun, P. Wielinga, G. Du, K. E. Mercer, Y. Zhuang, J. C. Panetta, B. Johnston, R. J. Scheper, C. F. Stewart and J. D. Schuetz, Mol. Cell. Biol., 2004, 24, 7612–7621 CrossRef CAS PubMed.
- W. Cruz-Muñoz, T. Di. Desidero, S. Man, P. Xu, M. L. Jaramillo, K. Hashimoto, C. Collins, M. Banville, M. D. O'Connor-McCourt and R. S. Kerbel, Angiogenesis, 2014, 17, 661–673 Search PubMed.
- J. Nemunaitis, J. Cox, W. Meyer, A. Courtney and G. Mues, Am. J. Clin. Oncol., 1997, 20, 527–529 CrossRef CAS PubMed.
- B. Saraiya, M. Gounder, J. Dutta, A. Saleem, C. Collazo, L. Zimmerman, A. Nazar, M. Gharibo, D. Schaar, Y. Lin, W. Shih, J. Aisner, R. K. Strair and E. H. Rubin, Anticancer Drugs, 2008, 19, 411–420 CrossRef CAS PubMed.
- X. Ling, S. Cao, Q. Cheng, J. T. Keefe, Y. M. Rustum and F. Li, PLoS One, 2012, 7, e45571 CrossRef CAS PubMed.
- F. Li, E. J. Ackermann, C. F. Bennett, A. L. Rothermel, J. Plescia, S. Tognin, A. Villa, P. C. Marchisio and D. C. Altieri, Nat. Cell Biol., 1999, 1, 461–466 CrossRef CAS PubMed.
- M. J. Yoon, S. S. Park, Y. J. Kang, I. Y. Kim, J. A. Lee, J. S. Lee, E. G. Kim, C. W. Lee and K. S. Choi, Carcinogenesis, 2012, 33, 492–500 CrossRef CAS PubMed.
- S. Trabulo, A. M. Cardoso, T. Santos-Ferreira, A. L. Cardoso, S. Simões and M. C. Pedroso de Lima, Mol. Pharm., 2011, 8, 1120–1131 CrossRef CAS PubMed.
- K. M. Rahman, S. Banerjee and S. Ali, Cancer Res., 2009, 69, 4468–4475 CrossRef CAS PubMed.
- F. Li and X. Ling, J. Cell. Physiol., 2006, 208, 476–486 CrossRef CAS PubMed.
- F. Rödel, J. Hoffmann, L. Distel, M. Herrmann, T. Noisternig, T. Papadopoulos, R. Sauer and C. Rödel, Cancer Res., 2005, 65, 4881–4887 CrossRef PubMed.
- B. Z. Carter, D. H. Mak, W. D. Schober, M. Cabreira-Hansen, M. Beran, T. McQueen, W. Chen and M. Andreeff, Blood, 2006, 107, 1555–1563 CrossRef CAS PubMed.
- J. Wu, X. Ling, D. Pan, P. Apontes, L. Song, P. Liang, D. C. Altieri, T. Beerman and F. Li, J. Biol. Chem., 2005, 280, 9745–9751 CrossRef CAS PubMed.
- T. Saito, S. Hama, H. Izumi, F. Yamasaki, Y. Kajiwara, S. Matsuura, K. Morishima, T. Hidaka, P. Shrestha, K. Sugiyama and K. Kurisu, Br. J. Cancer, 2008, 98, 345–355 CrossRef CAS PubMed.
- M. Zhang, D. E. Latham, M. A. Delaney and A. Chakravarti, Oncogene, 2005, 24, 2474–2482 CrossRef CAS PubMed.
- R. Hernan, V. Zachary and K. J. Pienta, J. Biol. Chem., 2008, 283, 25057–25073 CrossRef PubMed.
- M. Vogler, H. Walczak, D. Stadel, T. L. Haas, F. Genze, M. Jovanovic, J. E. Gschwend, T. Simmet, K. M. Debatin and S. Fulda, Cancer Res., 2008, 68, 7956–7965 CrossRef CAS PubMed.
- X. Ding, A. B. Mohd, Z. Huang, T. Baba, M. Q. Bernardini, H. K. Lyerly, A. Berchuck, S. K. Murphy, A. B. Buermeyer and G. R. Devi, Br. J. Cancer, 2009, 101, 269–277 CrossRef CAS PubMed.
- K. Connolly, R. Mitter, M. Muir, D. Jodrell and S. Guichard, Cancer Chemother. Pharmacol., 2009, 64, 307–316 CrossRef CAS PubMed.
- X. He, A. Khurana, J. L. Maguire, J. Chien and V. Shridhar, Int. J. Cancer, 2012, 130, 1029–1035 CrossRef CAS PubMed.
- O. Ndozangue-Touriguine, M. Sebbagh, D. Mérino, O. Micheau, J. Bertoglio and J. A. Bréard, Oncogene, 2008, 27, 6012–6022 CrossRef CAS PubMed.
- X. Zhao, T. Laver, S. W. Hong, G. B. Jr Twitty, A. Devos, M. Devos, E. N. Benveniste and S. E. Nozell, J. Neuro-Oncol., 2011, 102, 367–381 CrossRef CAS PubMed.
- K. Miura, H. Karasawa and I. Sasaki, Expert Opin. Ther. Targets, 2009, 13, 1333–1345 CrossRef CAS PubMed.
- K. Shigemasa, O. Katoh, Y. Shiroyama, S. Mihara, K. Mukai, N. Nagai and K. Ohama, Jpn. J. Cancer Res., 2002, 93, 542–550 CrossRef CAS PubMed.
- H. Takahashi, M. C. Chen, H. Pham, E. Angst, J. C. King, J. Park, E. Y. Brovman, H. Ishiguro, D. M. Harris, H. A. Reber, O. J. Hines, A. S. Gukovskaya, V. L. Go and G. Eibl, Biochim. Biophys. Acta, 2011, 1813, 1465–1474 CrossRef CAS PubMed.
- K. Simonin, E. Brotin, S. Dufort, S. Dutoit, D. Goux, M. N'diaye, C. Denoyelle, P. Gauduchon and L. Poulain, Mol. Cancer Ther., 2009, 8, 3162–3170 CrossRef CAS PubMed.
- C. Mitchell, A. Yacoub, H. Hossein, A. P. Martin, M. D. Bareford, P. Eulitt, C. Yang, K. P. Nephew and P. Dent, Cancer Biol. Ther., 2010, 10, 903–917 CrossRef CAS PubMed.
- X. Guoan, W. Hanning, C. Kaiyun and L. Hao, Surgery, 2010, 147, 553–561 CrossRef PubMed.
- A. P. Martin, A. Miller, L. Emad, M. Rahmani, T. Walker, C. Mitchell, M. P. Hagan, M. A. Park, A. Yacoub, P. B. Fisher, S. Grant and P. Dent, Mol. Pharmacol., 2008, 74, 807–822 CrossRef CAS PubMed.
- M. Lee, A. Lapham, M. Brimmell, H. Wilkinson and G. Packham, Apoptosis, 2008, 13, 972–982 CrossRef CAS PubMed.
- J. Zhao, X. Ling, S. Cao, X. Liu, S. Wan, T. Jiang and F. Li, Mol. Pharm., 2014, 11, 457–467 CrossRef CAS PubMed.
- P. DiSaia, J. Sinkovics, F. Rutledge and J. Smith, Am. J. Obstet. Gynecol., 1972, 114, 979–989 CrossRef CAS PubMed.
- R. M. Wadkins, P. M. Potter, B. Vladu, J. Marty, G. Mangold, S. Weitman, G. Manikumar, M. C. Wani, M. E. Wall and D. D. Von Hoff, Cancer Res., 1999, 59, 3424–3428 CAS.
- H. G. Lerchen, J. Baumgarten, K. von. D. Bruch, T. E. Lehmann, M. Sperzel, G. Kempka and H. H. Fiebig, J. Med. Chem., 2001, 44, 4186–4195 CrossRef CAS PubMed.
- W. C. Rose, P. H. Marathe, G. R. Jang, T. M. Monticello, B. N. Balasubramanian, B. Long, C. R. Fairchild, M. E. Wall and M. C. Wani, Cancer Chemother. Pharmacol., 2006, 58, 73–85 CrossRef CAS PubMed.
- G. Gu, H. Lin, Y. Liu, L. Yang, Y. Zheng, B. Wang, C. Tang, H. Qian and W. Huang, Lett. Drug Des. Discovery, 2013, 10, 155–163 CrossRef CAS.
- S. M. Andersen, M. Heuckendorff and H. H. Jensen, Org. Lett., 2015, 17, 944–947 CrossRef CAS PubMed.
- I. Pendrak, R. Wittrock and W. D. Kingsbury, J. Org. Chem., 1995, 60, 2912–2915 CrossRef CAS.
- N. Mahindroo, Z. Ahmed, A. Bhagat, K. L. Bedi, R. K. Khajuria, V. K. Kapoor and K. L. Mara, Med. Chem. Res., 2005, 14, 347–368 CrossRef CAS.
- M. E. Wall, M. C. Wani, A. W. Nicholas, G. Manikumar, C. Tele, L. Moore, A. Truesdale, P. Leitner and J. M. Besterman, J. Med. Chem., 1993, 56, 2689–2700 CrossRef.
- Y. Bao, L. Zhang and F. Chen, Chin. J. Med. Chem., 2008, 4, 263–267 Search PubMed.
- B. L. Staker, K. Hjerrild, M. D. Feese, C. A. Behnke, A. B. Jr. Burgin and L. Stewart, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15387–15392 CrossRef CAS PubMed.
- J. Pan, Y. Qi, X. Zhou, H. Lu and L. Zhang, J. Biol. Chem., 2010, 285, 22966–22975 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00315k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |