
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHepatitis B virus S gene therapy with 10-23 DNAzyme delivered by chitosan-g-stearic acid micelles
Yun Honga,
Dongsen Maob,
Rui Wuc,
Zhe Gaoa,
Tingting Mengc,
Rongrong Wanga,
Lin Liua and
Jing Miao *a
*a
aDepartment of Pharmacy, the First Affiliated Hospital, College of Medicine, Zhejiang University, 79 Qingchun Road, Hangzhou 310003, P. R. China. E-mail: miaojing@zju.edu.cn; Fax: +86-571-87236675; Tel: +86-571-87236675
bZhejiang Center of Laboratory Animals, Zhejiang Academy of Medical Sciences, 182 Tianmushan Road, Hangzhou 310007, P. R. China
cCollege of Pharmaceutical Sciences, Zhejiang University, 866 Yuhangtang Road, Hangzhou 310058, P. R. China
First published on 15th May 2019
Abstract
DNAzymes have the potential to suppress gene expression through sequence-specific mRNA cleavage and can therefore play an important role in various gene therapies. Hepatitis B virus (HBV) is still one of the most serious liver infections in people around the world and is difficult to treat. We previously designed a 10-23 DNAzyme called DrzBS, which targets HBV S gene expression, but this enzyme depends on exogenous delivery, and so its application has been limited. To overcome this limitation, we have now developed a chitosan-based nanocarrier (chitosan-g-stearic acid, CSO–SA) for intracellular delivery of DrzBS, then compared the inhibition effect of our CSO–SA/DrzBS complex to a common transfection reagent, Lipofectamine™ 2000/DrzBS, on hepatitis B surface antigen expression. The synthesized CSO–SA assembles into micelles in an aqueous solution and exhibits excellent cytoplasmic targeting, and could protect DrzBS from degradation by ribonuclease. CSO–SA/DrzBS showed a higher inhibition rate (IR) than Lipofectamine™ 2000/DrzBS. Moreover, at the same DrzBS concentration (1.2 μmol L−1), the maximum IR of CSO–SA/DrzBS micelles was 2.4-fold that of the Lipofectamine™ 2000/DrzBS complex, and held on for 96 hours. Compared with Lipofectamine™ 2000/DrzBS, CSO–SA/DrzBS achieved a higher HBV inhibition effect. This study demonstrates that CSO–SA micelles can serve as a potential vector for DrzBS and that CSO–SA/DrzBS micelles are a promising application for anti-HBV gene therapy.
Introduction
Hepatitis B, resulting from the infection of the hepatitis B virus (HBV), is one of the world's most common and serious infectious diseases. About two billion people globally have been exposed to the infection1 and more than 250 million people are suffering from chronic HBV infection.2–4 Twenty-five percent of all chronic HBV carriers develop serious liver diseases, such as chronic hepatitis, cirrhosis and primary hepatocellular carcinoma.5,6 HBV-related liver diseases still cause an estimated 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to one million deaths per year worldwide.7–9 Viral mutations10 and patient immune tolerances to HBV antigens are the main causes of persistent chronic HBV infection. Therefore, reactivation of HBV-specific immune responses is essential to reduce morbidity and mortality from chronic HBV infection.
000 to one million deaths per year worldwide.7–9 Viral mutations10 and patient immune tolerances to HBV antigens are the main causes of persistent chronic HBV infection. Therefore, reactivation of HBV-specific immune responses is essential to reduce morbidity and mortality from chronic HBV infection.
HBV is an enveloped virus and can only infect humans and primates. The S gene codes for the viral envelope proteins known as the hepatitis B surface antigen (HBsAg). Recent studies have shown that HBsAg accumulation was associated with specific viral mutations related to disease activities.11 In addition, when sufficient HBV envelope molecules are present, HBsAg is synthetized much more in excess than required for viral enveloping purposes, and most likely overwhelms any benefit of anti-HBs responses, causing immune tolerance. Persistence of HBsAg production is associated with an increased risk of liver cancer, even in the absence of productive HBV replication.12 Inhibition of HBsAg expression is a critical step in the functional control of HBV infection. It will unveil the unmaking of the existing anti-HBs response, which would favor clearance of virions, both infectious and noninfectious.13,14 In other words, the ability to clear HBsAg is the next frontier in hepatitis B therapeutics. The achievement of a ‘functional cure’ for chronic HBV infection with sustained HBsAg clearance, represents the next step in the pace towards HBV elimination.15
The discovery of DNAzymes breaks the traditional concept of protease-based biocatalysts. A DNAzyme is a structured DNA sequence with catalytic RNA-cleaving activity. The 10-23 DNAzyme consists of a catalytic domain formed by 15 deoxynucleotides in the center with recognition arms formed by seven to ten nucleotides on both sides.16 The enzyme specifically combines with the target mRNA in a highly sequence-specific manner through its recognition arms, while the central catalytic domain cleaves the mRNA target at purine–pyrimidine junctions (A–U sites), thereby blocking the expression of the corresponding mRNA and inhibiting the target virus gene expression.17–21
HBV is an attractive target for the 10-23 DNAzyme, and so we designed the HBV-specific 10-23 DNAzyme named DrzBS. Our previous research showed that DrzBS inhibited the expression of HBV S genes by suppressing the corresponding antigen proteins, thus suggesting that DrzBS could be a powerful, specific, and effective form of anti-HBV gene therapy. In addition, DrzBS has a flexible design, independent of cellular mechanisms and does not require expensive chemical modifications when be used in vivo. And it does not affect the RNA of the host cell. Thus, DrzBS has a potential application value in anti-HBV gene therapy research. However, a major challenge for the application of the 10-23 DNAzyme is that it cannot replicate endogenously and, consequently, relies on exogenous delivery. Delivery of the 10-23 DNAzyme is limited by poor cellular uptake, rapid degradation by nucleases, and poor endosomal release. Therefore, development of a safe and efficient gene carrier is a prerequisite for successful gene therapy.22,23
Several non-viral vectors have been developed for DNA delivery that use a wide range of materials such as cationic liposomes,24,25 and natural or synthetic polymers.26–28 Among these, chitosan has been extensively studied because it is inherently biocompatible, biodegradable, and has low cytotoxicity in comparison to other cationic polymers.23,29–31 Moreover, the positive surface charge and presence of easily modifiable primary amines and hydroxyl groups on the chitosan backbone make it an attractive tool for DNAzyme delivery. However, without laborious chemical modifications to its structure, it is of limited use as a gene delivery vehicle due to its low ability to efficiently transfect under physiological conditions.32,33 To address this problem, our recent studies34–36 have demonstrated that chitosan-g-stearic acid (CSO–SA), which is synthesized via a reaction between the carboxyl group of stearic acid and the amine group of chitosan oligosaccharide in the presence of 1-(3-Dimethylamino-propyl)-3-ethyl-carbodiimide (EDC), can greatly improve the cellular uptake and in vitro gene transfection efficacy of chitosan.37,38 This means that CSO–SA micelles could be an efficient approach for intracellular delivery of the 10-23 DNAzyme.
In this research, we designed and synthesized a kind of 10-23 DNAzyme, DrzBS, specific to the HBV S gene open reading frame (ORF) A157UG. The CSO–SA was achieved and bound with DrzBS by electrostatic interaction to form CSO–SA/DrzBS micelles, which could be an efficient approach for intracellular delivery of DrzBS. The characteristics and stability of CSO–SA and CSO–SA/DrzBS micelles were studied, as well as the inhibition rate (IR) on HBsAg expression. Thus, we investigated CSO–SA as a gene vector and its therapeutic effect with DrzBS in HepG2.2.15 cells.
Results and discussion
Synthesis and characterization of CSO–SA micelles
In this study, the schematic diagram of CSO–SA/DrzBS targeting of cell uptake and intracellular release were shown in Fig. 1.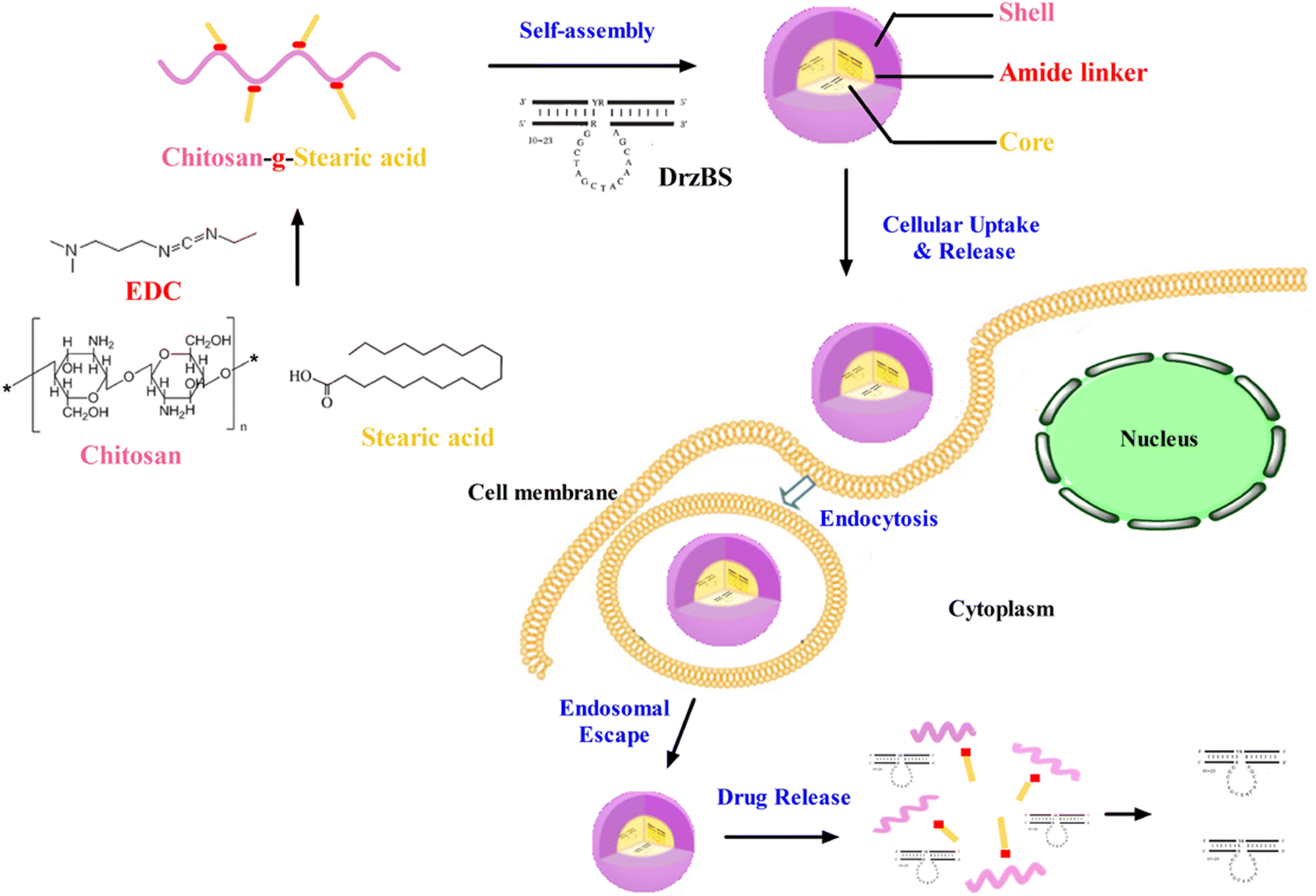 | ||
| Fig. 1 The schematic diagram of passive targeting of cell uptake and intracellular release. | ||
The synthesis route of the CSO–SA was shown in Fig. 2a, which was obtained by a coupling reaction between the amino groups of CSO and the carboxyl groups of SA in the presence of EDC. The final product, CSO–SA, was a white soluble powder after lyophilization. The chemical structure of CSO–SA was confirmed by 1H NMR (Fig. 2b). The peaks at about 1.2 ppm and 0.8 ppm were attributed to the –CH2 and –CH3 groups respectively. These results proved the successful synthesis of the CSO–SA micelles. The synthesized CSO–SA could be self-assembly in aqueous solution.
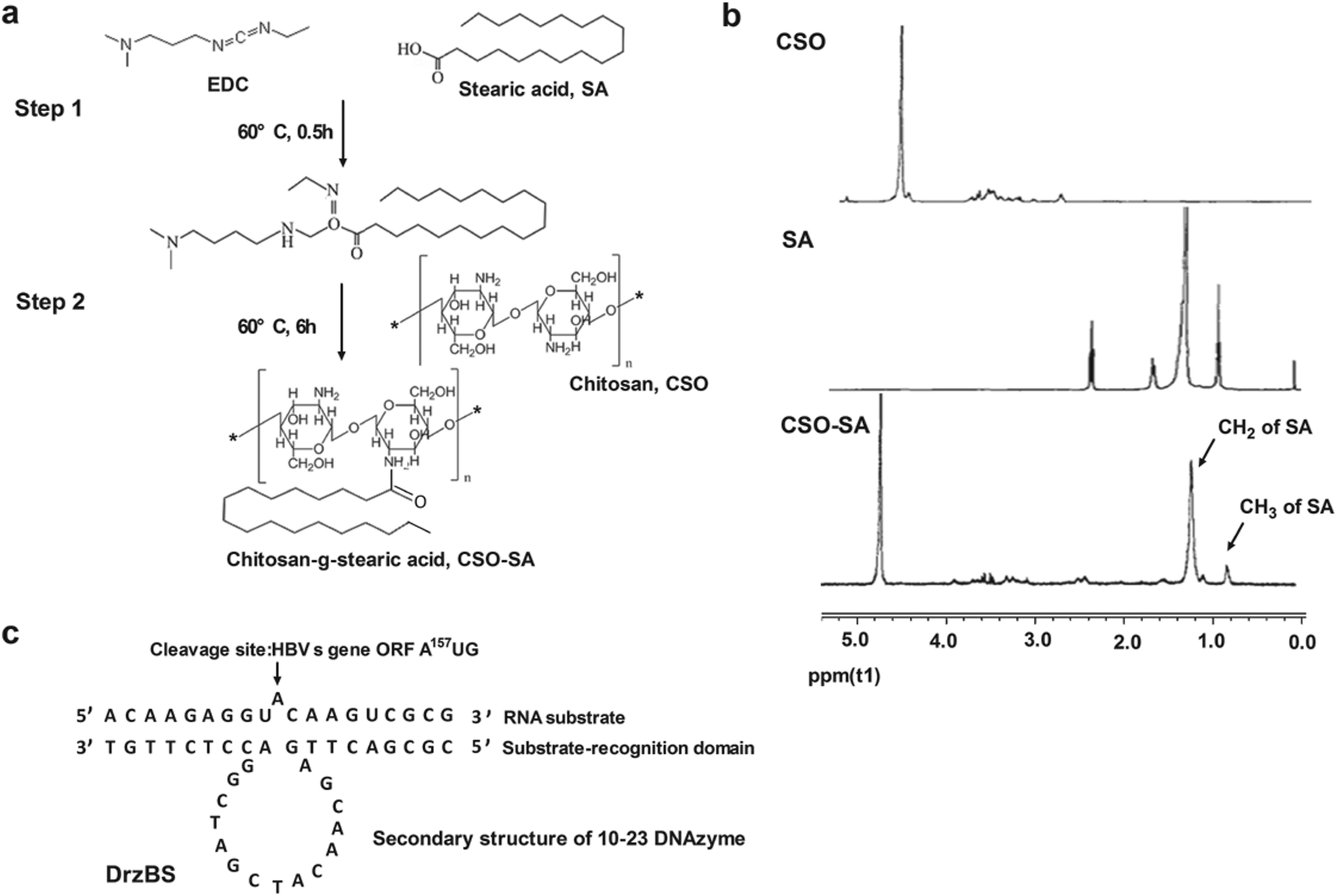 | ||
| Fig. 2 The schematic diagram of chitosan-g-stearic acid (CSO–SA). (a) Sequence of DrzBS and RNA substrates (arrow indicating cleavage site). (b) Synthesis route of CSO–SA. (c) 1H NMR spectra, from top to bottom: chitosan, stearic acid, and CSO–SA. The signals assigned to the –CH2-(1.2 ppm) and –CH3-(0.8 ppm) SA were detectable on the CSO–SA spectra. | ||
The average size and zeta potential of CSO–SA were 67.3 ± 3.4 nm and 48.3 ± 2.9 mV respectively (Table 1). However, the values of PI were slightly large, which might due to the micelles dispersed in the deionized water where the pH and ionic strength were unstable. The spherical-like shape of the micelles were revealed with TEM images (Fig. 3). The CMC of CSO–SA was 75 μg mL−1.
| Polymers | Volume average diameter (nm) | Zeta potential (mV) | PI |
|---|---|---|---|
| CS-SA | 67.3 ± 3.4 | 48.3 ± 2.9 | 0.57 ± 0.06 |
| CS-SA/DrzBS | 158.0 ± 2.5 | 39.1 ± 3.3 |
 | ||
| Fig. 3 TEM images of the CSO–SA and CSO–SA/DrzBS micelles. Scale bar, 100 nm. | ||
Characterization of CSO–SA/DrzBS micelles
Based on the 10-23 DNAzyme molecular structure reported by Santoro and Joyce,16 we designed and synthesized our 10-23 DNAzyme for the HBV S gene, named DrzBS (Fig. 2c). DrzBS is a DNA structure that can be combined with CSO–SA by electrostatic interaction to form CSO–SA/DrzBS micelles. The particle size and zeta potential of CSO–SA/DrzBS micelles are presented in Table 1. The average size was 158.0 ± 2.5 nm and the zeta potential was 39.1 ± 3.3 mV in deionized water. The value of the zeta potential in the CSO–SA and the CSO–SA/DrzBS did not change much, which shows that the composition of the complex is hardly affected by the surface charge of the particles. The TEM photos (Fig. 3) indicate that CSO–SA/DrzBS micelles had a well-formed shape and compact structure.Tightlock experiment and DNase I protection assay
Protection of DrzBS from nucleases as well as its polymer binding capability are both crucial factors for its efficient delivery to inhibit HBV gene expression. As the complexes pass through the cytoplasm, this protection effect is important.39,40 This is because the enzymes and nucleic acids within the cytoplasm make the DrzBS complexes susceptible to hydrolytic and enzymatic degradation. CSO–SA exert the proper protection relied on appropriate N/P ratio. In our previous studies,41 we have confirmed that CSO–SA micelles became to compact with 10-23 DNAzyme when N/P > 1.2, and further presented a good loading potency at the optimal N/P ratio of 10. In addition, the migration of DrzBS within the CSO–SA/DrzBS complexes was entirely retarded in the loading slot when the N/P ratio was 10. The DNase I protection assay also shown the same results. The naked DrzBS was significantly degraded, whereas DrzBS combined with the CSO–SA micelles were protected from the attack of DNase I at the N/P ratio of 10 as shown in Fig. 4.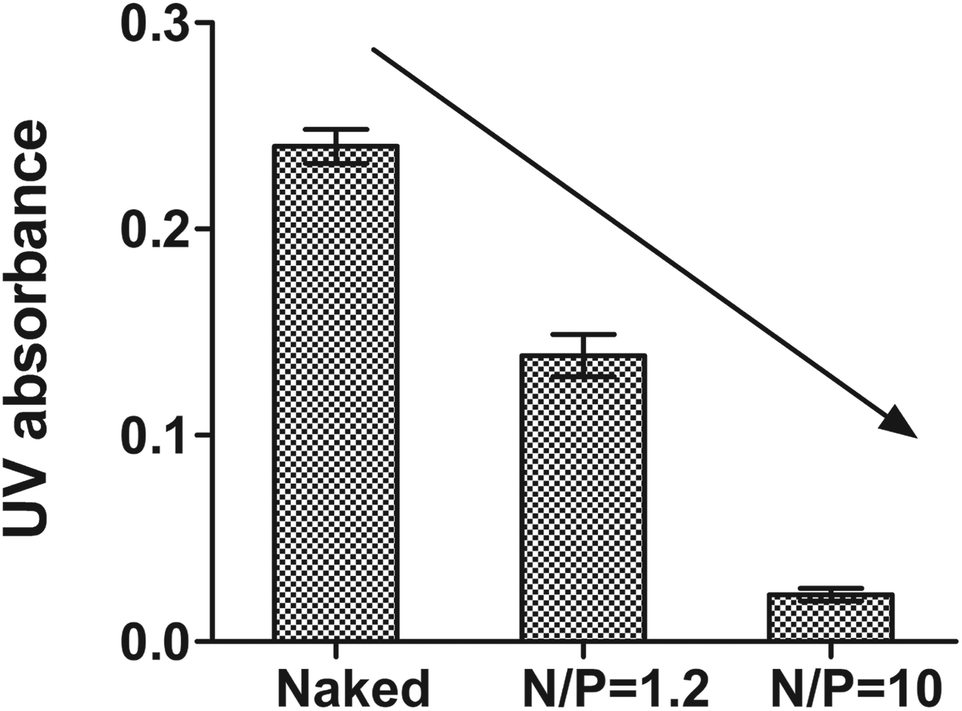 | ||
| Fig. 4 UV absorbance at 260 nm in DNase I protection assay with or without CSO–SA micelles (reaction time was 15 min, mean ± SD, n = 3). | ||
Cellular uptake
The SD% of CSO–SA, when concentrated in the nucleus and cytoplasm, were measured as 25.31% and 37.92% respectively. Since the inhibitory effect of DrzBS needs to be exerted in the cytoplasm, we chose the CSO–SA with an SD% of 37.92 ± 0.62%.In vitro cellular uptake of FITC-labeled micelles on HepG2.2.15 cells were visualized by fluorescence microscope at 2 h and 4 h. Fig. 5a shows that the cellular uptake of FITC-labeled CSO–SA micelles in cell lines was time dependent. The polymer showed favorable cellular internalization, which mainly depended on the hydrophobic core composed of SA and moderate surface charge. The CLSM results were also shown that the CSO–SA with an SD% of 37.92 ± 0.62% has the ability to target cytoplasm, and the cellular uptake of the CSO–SA micelles is time dependent (Fig. 5b). These results suggested that CSO–SA micelles were efficient intracellular delivery systems and could improve the cellular uptake into HepG2.2.15 cells.
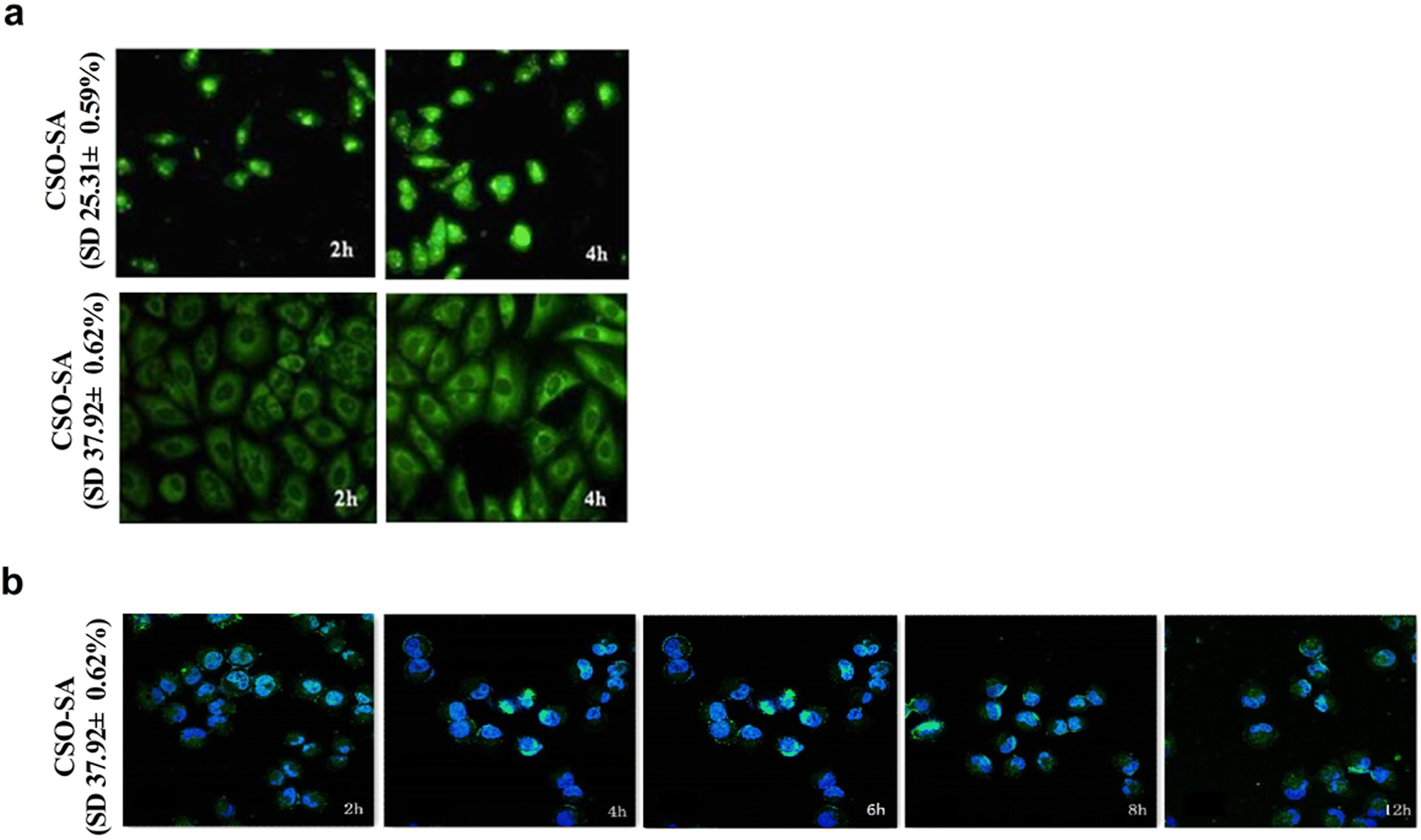 | ||
| Fig. 5 Representative confocal fluorescent images of the cellular uptake of FITC-CSO–SA/DrzBS on HepG2.2.15 cells. The CSO–SA concentration was 100 μg mL−1, SD% is the degrees of amino-substitution. | ||
In vitro cytotoxicity
A good quality vector should be biocompatible and not damage the cell structure or its normal function. To examine the cytotoxicity of the CSO–SA and Lipofectamine™ 2000, cell viability was determined by MTT assay. Fig. 6a shows that CSO–SA micelles exhibit excellent cell compatibility in HepG2.2.15 and L02 (non-tumour hepatocytes) cells, whereas the cytotoxicity of Lipofectamine™ 2000 was found to be much higher, as previously reported.42 The half maximal inhibitory concentration (IC50) value in HepG2.2.15 cells were 6.8 μg mL−1 for Lipofectamine™ 2000 and 412.5 μg mL−1 for CSO–SA. Similar to that, the IC50 value in L02 cells were 4.2 μg mL−1 and 254.0 μg mL−1, respectively. Its safety provided more extensive dosage range for DNAzymes transfection. The survival rates of the CSO–SA/DrzBS micelles were all approximately 100% in L02 cells and above 75% in HepG2.2.15 cells at various concentrations of DrzBS (DrzBS concentration = 0.1, 0.6, 1.2, 1.8, 2.4, 3.6, 4.8, 9.6 μmol L−1). As for Lipofectamine™ 2000, the cell viability drastically decreased (Fig. 6b).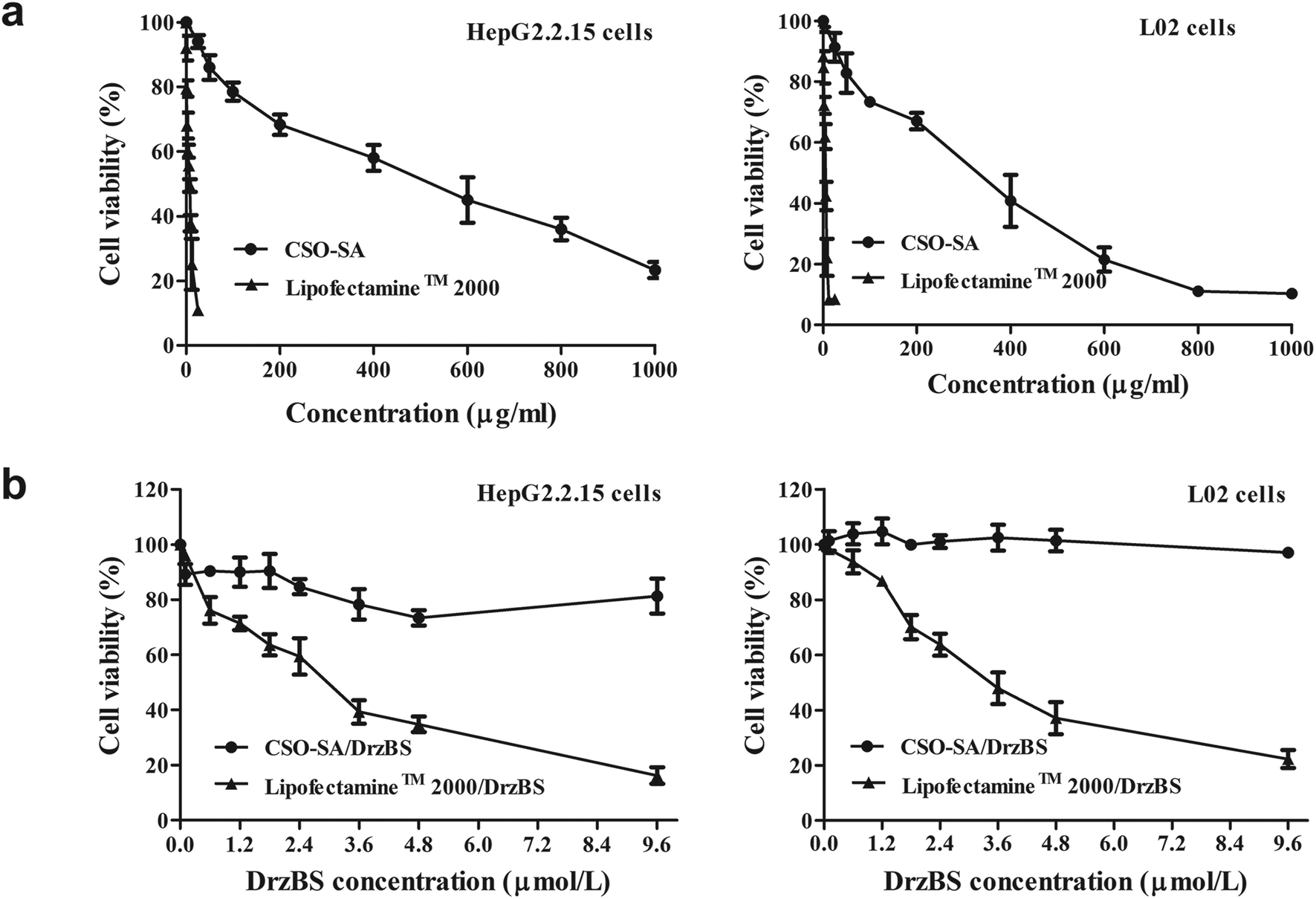 | ||
| Fig. 6 (a) Cell viabilities of Lipofectamine™ 2000 and CSO–SA on HepG2.2.15 cells and L02 cells. (b) Cell viabilities of Lipofectamine™ 2000/DrzBS and CSO–SA/DrzBS on HepG2.2.15 cells and L02 cells with different DrzBS concentration. Error bars represent the standard deviation (mean ± SD, n = 3). | ||
The cytotoxicity of Lipofectamine™ 2000 may be a result of its high positive surface charges and its aggregation on cell surfaces, thus impairing important membrane functions.28,43,44 In contrast, the sources for CSO–SA synthesis, CSO and SA, were either biodegradable, low toxic or biocompatible. Compared with Lipofectamine™ 2000, CSO–SA micelles had relatively low cytotoxicity. Together, these internalization and cytotoxicity results verified that the CSO–SA is safe and has potential as an anti-HBV gene therapy.
Transfection and inhibitory effects of DrzBS on HBsAg expression in HepG2.2.15 cells
Our experiments showed that the inhibition percentages of HBsAg expression in the HepG2.2.15 cells by our CSO–SA complexes were 16.7 ± 3.2% (for the N/P ratio = 5:1) and 51.3 ± 4.6% (for the N/P ratio = 10:1) (p < 0.001, Fig. 7a). This confirms that the N/P ratio of 10:1 achieves the best intracellular efficacy. It is also worth noticing that the inhibition percentage in the CSO–SA group was higher than that in Lipofectamine™ 2000 (35.8 ± 2.5%) at the similar N/P ratio of 10:1 (51.3 ± 4.6%, p < 0.01). These results indicate that, at the N/P ratio of 10:1, CSO–SA/DrzBS micelles can efficiently transfect into cells and release DrzBS into the cytoplasm to effectively inhibit HBsAg levels.
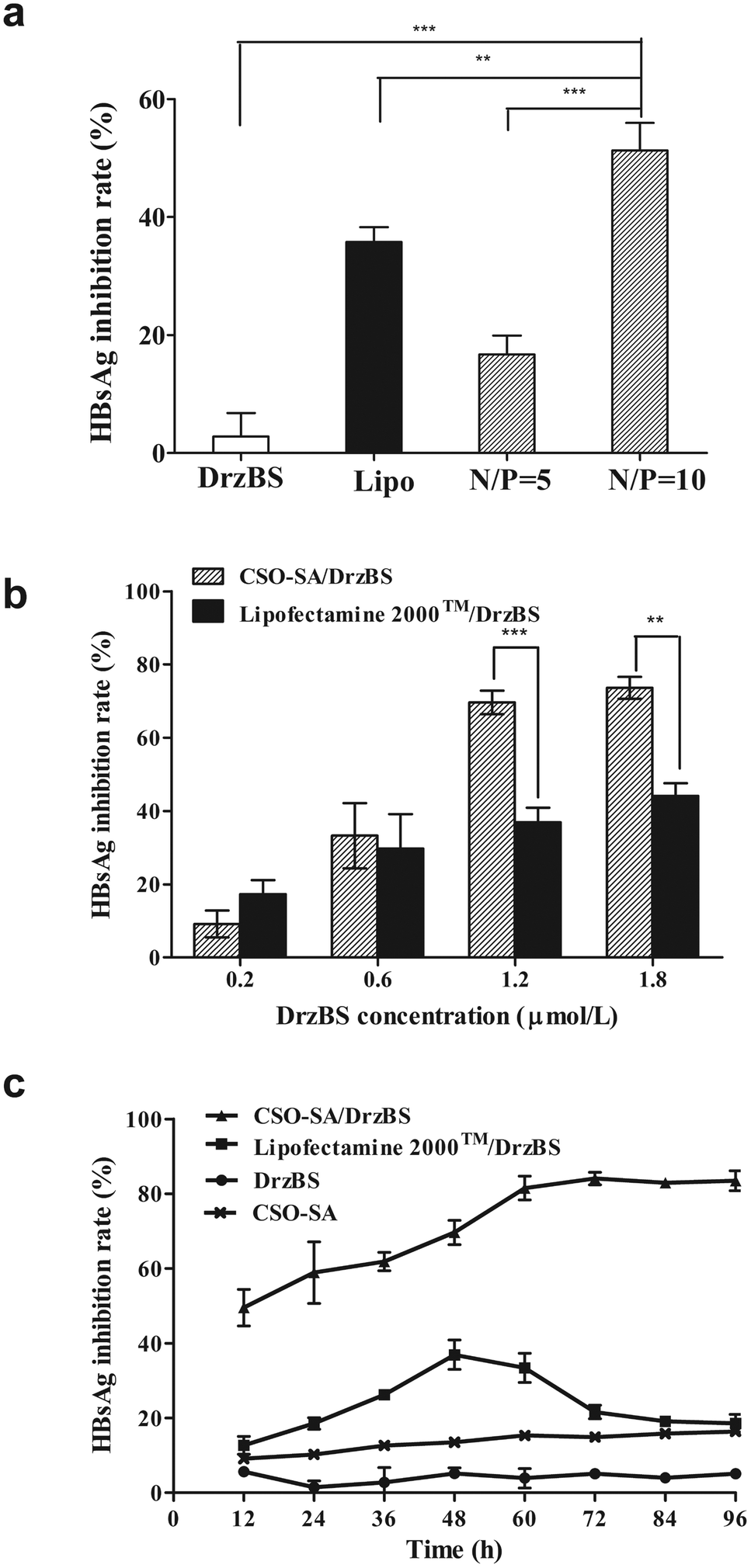 | ||
| Fig. 7 HBsAg inhibition effect of DrzBS transfected by the polymer complexes. (a) Inhibitory effect of CSO–SA/DrzBS complex on HepG2.2.15 cells for 48 h (mean ± SD, n = 3). Lipo was the abbreviation for Lipofectamine™ 2000. (b) For the same incubation time of 48 h with different DrzBS concentration (mean ± SD, n = 3, **p < 0.01, ***p < 0.001). (c) In the same DrzBS concentration of 1.2 μmol L−1 for different incubation time. The error is the standard deviation from the mean (mean ± SD). | ||
On the basis of the above experiment, different concentrations of DrzBS were selected to evaluate the IR on HBsAg for the same incubation time of 48 h. Within a concentration range of 0.2 to 1.8 μmol L−1, the IR on HBsAg expression of DrzBS delivered by Lipofectamine™ 2000 increased from 17.3 ± 3.8% to 44.1 ± 3.4%, while the IR on HBsAg expression of DrzBS delivered by CSO–SA micelles increased from 9.2 ± 3.6% to 73.5 ± 3.0%, with both complexes showing a distinct dose-dependent effectiveness (Fig. 7b). At low concentrations (0.2 μmol L−1), the IR of CSO–SA/DrzBS (9.2 ± 3.6%) was lower than that of Lipofectamine™ 2000/DrzBS (17.3 ± 3.8%), probably because the release of the drug from the CSO–SA micelles did not reach its intracellular peak, whereas the Lipofectamine™ 2000 complex had reached its peak. But, when the concentration of DrzBS reached 0.6 μmol L−1, the DrzBS delivered by the CSO–SA micelles exhibited much higher efficiency for inhibiting HBsAg expression than that delivered by Lipofectamine™ 2000 (33.3 ± 8.9% vs. 29.7 ± 9.4%). And when the concentration of DrzBS reached 1.2 μmol L−1, the IR of the CSO–SA/DrzBS micelles with the value of 69.6 ± 3.2% which was significantly higher than that of Lipofectamine™ 2000/DrzBS complexes (36.9 ± 3.9%, p < 0.001). Remarkably, at further increases of DrzBS dose concentrations, IR growth became more slowly, and platform-like effectiveness was observed. These results confirmed the capacity of the CSO–SA/DrzBS micelles selective release DrzBS in the cytoplasm.
To further understand the potential mechanism of the decreased cytotoxicity and intracellular effects observed with CSO–SA/DrzBS micelles, we examined their inhibitory effect on S gene expression over time and at the same concentration (1.2 μmol L−1). Fig. 7c showed the HBsAg inhibition effect after transfection. It was obvious that DrzBS cannot inhibit HBsAg itself, and the CSO–SA has a slight inhibition of HBsAg but much lower than the CSO–SA/DrzBS micelles. CSO–SA/DrzBS micelles displayed a significant HBsAg inhibition compared with that of Lipofectamine™ 2000/DrzBS complexes. The IR of Lipofectamine™ 2000/DrzBS on HBsAg expression increased during the first 48 h and reached a maximum IR of 34.8 ± 0.6% at 48 h, due to the relatively high cell transduction efficiency of Lipofectamine™ 2000 in HepG2.2.15 cells and the peak expression of DrzBS in the cells. However, after 48 h, the IR began to descend, and at 60 h, the IR started to rapidly decrease, possibly due to the system toxicity of Lipofectamine™ 2000. On the other hand, the IR of HBsAg expression treated by CSO–SA/DrzBS micelles continued to increase with the incubation time for 72 h (84.1 ± 2.6%) and remained stable. When viewed with CSO–SA/DrzBS micelles for 96 h, the IR value was 83.5 ± 2.7%, which was significantly higher than that of Lipofectamine™ 2000/DrzBS (18.6 ± 2.4%, p < 0.001). These results demonstrate that CSO–SA possesses a higher efficiency and a lower cytotoxicity for inhibiting HBsAg expression than Lipofectamine™ 2000. Furthermore, the drug release in intracellular compartments made the free DrzBS produce a better pharmacological action.
Experimental
Materials
Chitosan oligosaccharide (CSO) of low molecular weight (MW = 18.4 kDa) was obtained by enzymatic degradation of chitosan (95% deacetylated, MW = 450 kDa), and supplied by Yuhuan Marine Biochemistry Co., Ltd. (Zhejiang, China). Stearic acid (SA) was purchased from Shanghai Chemical Reagent Co., Ltd. (Shanghai, China). EDC hydrochloride and pyrene were provided by Aladdin Reagent Co., Ltd. (Shanghai, China). Methylthiazol-tetrazolium (MTT), 2,4,6-trinitrobenzenesulfonic acid (TNBS), and 5-fluorescein isothiocyanate (FITC) were provided by Sigma Chemical Co. Lipofectamine™ 2000 and Opti-MEMI medium were supplied by Invitrogen Corporation (Carlsbad, CA, USA). Trypsin and Dulbecco's minimum essential medium (DMEM) were purchased from Gibco (Merelbeke, Belgium). HepG2.2.15 cells were provided by the State Key Laboratory for Diagnosis and Treatment of Infectious Diseases of Zhejiang Province (Hangzhou, China). The HBsAg quantitative radioimmunoassay kit was purchased from Weifang 3V diagnostic technology company (Shandong, China).Design and synthesis of 10-23 DNAzyme
Based on the molecular structure of 10-23 DNAzyme, we designed a 10-23 DNAzyme, named DrzBS, using Watson–Crick base pairing, and specific to the HBV S gene (Genbank gi:59429) ORF A157UG. We consigned its synthesis to Sangon Biological Engineering Technology & Service Co., Ltd. (Shanghai, China). DrzBS contains the 15-nucleotide catalytic sequence GGCTAGCTACAACGA and has nine oligonucleotides on either side. Named DrzBS-9, this DNAzyme can specifically recognize the HBV S gene ORF A157UG sequence from the 148th to 166th nucleotide.Synthesis of chitosan-g-stearic acid (CSO–SA)
Chitosan oligosaccharide was prepared according to the previous study.45 CSO–SA was then synthesized via a reaction of the carboxyl group of stearic acid with the amine group of the chitosan oligosaccharide in the presence of EDC.46 Briefly, SA and EDC were dissolved in acetone and then added into an ethanol–acetone mixed solvent (ethanol:acetone ratio = 3:7, v/v). The mixture was stirred for 0.5 h at 60 °C. CSO (MW 18.4 kDa) was then dissolved in hot water, after which the SA/EDC mixture was added dropwise. The reaction solution was stirred for another 6 h and was then dialyzed against deionized water using a dialysis membrane (molecular weight cut-off (MWCO): 7 kDa, Spectrum Laboratories, Laguna Hills, CA) for 48 h. Finally, the product was collected by lyophilization followed by a further wash with ethanol to remove products. The washed product was then collected by lyophilization again.
Preparation of Lipofectamine™ 2000/DrzBS and CSO–SA/DrzBS micelles
The Lipofectamine™ 2000/DrzBS complex was prepared by gently mixing DrzBS with Lipofectamine™ 2000 in 50 μL OPTI-MEMI (total volume: 100 μL), followed by incubation for 20 min at room temperature.CSO–SA solutions with different concentrations (dissolved in 25 mM sodium acetate buffer, pH 5.5) were first purified with a 0.22 m Millipore filter. CSO–SA/DrzBS micelles with various N/P ratios (The N/P ratio is the number of unreacted free primary amines of CSO–SA to the number of phosphate groups of DrzBS) were prepared by mixing appropriate volumes of CSO–SA micelle solutions with DrzBS solutions (diluted with water, 500 μg mL−1). After mixing in a vortex for 30 s, the mixtures were incubated for 30 min at 37 °C. Electrophoresis was then carried out with a current of 100 V for 20 min in a TAE buffer solution (40 mM Tris–HCl, 1% (v/v) acetic acid, 1 mM EDTA).
Characterization of CSO–SA and CSO–SA/DrzBS micelles
The chemical structure of CSO–SA was determined with 1H nuclear magnetic resonance (NMR) spectroscopy, and this chemical was dispersed in D2O at pH 7 and 25 °C. The substitution degree (SD%) of amino groups of CSO–SA, defined as the molar ratio of stearate to anhydroglucosidic units in chitosan oligosaccharide, was detected with TNBS, and the ultraviolet (UV) absorbance of the final reaction mixture at 344 nm was measured by UV spectroscopy (TU-1800PC, Beijing Purkinje General Instrument Co., Ltd., China). The critical micelle concentration (CMC) of CSO–SA was measured by pyrene fluorescence using a fluorometer (F-2500, Hitachi Co., Japan). The intensity ratio (I1:I3) of the first peak (I1, 374 nm) to the third peak (I3, 385 nm) in the pyrene emission spectra was analyzed to calculate the CMC.
The size and zeta potential of CSO–SA micelles and CSO–SA/DrzBS micelles were measured by dynamic light scattering (Zetasizer 3000HS, Malvern Instruments Ltd., UK) in deionized water. Their morphology was examined by transmission electronic microscopy (TEM, Stereoscan, Leica, England).
DNase I protection assay
The DNase I protection assay was performed to investigate the efficiency of the CSO–SA micelles protecting the DrzBS from enzymatic degradation by DNase I. The CSO–SA/DrzBS micelles with 1.2 and 10 of N/P ratios were incubated in a buffer solution (10 mM Tris–HCl, 150 mM NaCl, 1 mM MgCl2, pH 7.4) with DNase I (25 units), then shaken at 100 rpm for 30 min at 37 °C. Electrophoresis was conducted in 1% agarose gel with a voltage of 100 V. UV absorbance was measured by UV spectrophotometer at 260 nm (UV-2450, Shimadzu, Japan).Cell culture
Human normal liver cells (L02 cell) were obtained from American Type Culture Collection (ATCC, Rockville, MD, USA). HBV-transfected human hepatoma cells (HepG2.2.15 cells) were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in a biological safety protection third-level laboratory (P3 laboratory, The First Affiliated Hospital, Zhejiang University). All cells lines were cultured at 37 °C in DMEM supplemented with 10% fetal bovine serum in a humidified atmosphere containing 5% CO2.Cellular uptake
To investigate the cell internalization, FITC-labeled CSO–SA was prepared. First, a FITC (C21H11NO5S) ethanol solution was added dropwise into a 1.0 mg mL−1 CSO–SA aqueous solution (CSO–SA:FITC ratio = 1:1, mol:mol), then stirred overnight at 400 rpm in an aphotic environment and subsequently dialyzed (MWCO = 3.5 kDa) against pure water for 6 h to remove ethanol and unreacted FITC. The HepG2.2.15 cells were then incubated with these FITC-labeled CSO–SA micelles (N/P ratio = 10), at a CSO–SA concentration of 100 μg mL−1, in a growth medium for 2 h, 4 h and then washed three times with a phosphate buffered solution (PBS) (pH 7.4). We then used a fluorescence microscope (OLYMPUS America, Melville, NY) to observe the internalization ability of CSO–SA and the cellular uptake of the CSO–SA/DrzBS micelles.
In vitro cytotoxicity assay
In vitro cytotoxicity of Lipofectamine™ 2000, CSO–SA and their complexes were evaluated by MTT assay on HepG2.2.15 cells and L02 cells. Briefly, 1.0 × 104 cells were plated in 96-well plates and incubated for 48 h to allow the cells to attach. Various concentrations of Lipofectamine™ 2000, CSO–SA and the controls were added. After 48 h, 50 μg MTT solution (5 mg mL−1) was added to each well, and the cells were incubated for a further 4 h. After the unreduced MTT and medium were removed, each well was washed with 100 μL of PBS, and then 200 μL of dimethyl sulfoxide (DMSO) was added to dissolve the formazan. Finally, the absorbance of each well was detected with a microplate reader (BioRad, Model 680, USA) at 570 nm. The survival percentage was calculated as compared to mock-treated cells (100% survival). The reported data represent the means of the triplicate measurements and the standard errors of the means were less than 15%. The transfection cytotoxicity of Lipofectamine™ 2000/DrzBS and CSO–SA/DrzBS (N/P = 10) complexes containing various DrzBS concentration were also measured as above.Transfection and inhibitory effects of DrzBS on HBsAg expression in HepG2.2.15 cells
HepG2.2.15 cells were seeded in 24-well plates at a density of 5 × 104 cells per well. After transfection with CSO–SA/DrzBS micelles at N/P ratios of 5 and 10 for 48 h, HepG2.2.15 cell culture supernatant was harvested. Naked DrzBS was used as the negative control. The Lipofectamine™ 2000/DrzBS complex served as the positive control. The level of HBsAg in the cell culture supernatant was determined by radioimmunoassay (under the same DrzBS concentration of 1.0 μmol L−1) to quantify the cell transfection and intracellular efficacy.To evaluate the therapeutic efficiency of the CSO–SA/DrzBS delivery system, HepG2.2.15 cells were seeded at 1.0 × 105 cells per well in a 24-well plate and grown for 48 h to allow the cells to attach. After the medium was replaced with 0.5 mL OPTI-MEMI, different amounts of CSO–SA/DrzBS micelles (N/P ratio = 10) and Lipofectamine™ 2000/DrzBS (the positive control) were added to each well and incubated with the cells for 6 h. The medium was then replaced with 1 mL of fresh complete medium, and the incubation was continued for a further 12 to 96 h at 37 °C. All the experiments were performed in triplicate. The culture media from the wells were collected for virological assessment. HBsAg production was determined by commercial enzyme immunoassay kits (AXSYM System, Abbott, Wiesbaden, Germany).
Conclusions
In this study, the CSO–SA was synthesized and evaluated as a novel non-viral DNA carrier by binding with a kind of 10-23 DNAzyme named DrzBS, which specifically targeted the expression of HBV S genes. The CSO–SA micelles demonstrated superior ability to form nanoscale complexes with DrzBS and had suitable physicochemical properties for DNAzyme delivery, thus finally leading to enhanced gene transfer efficiency, protected DrzBS from degradation, and effectively released DrzBS after cellular uptake into HepG2.2.15 cells. The CSO–SA/DrzBS micelles significantly inhibited HBsAg expression in HepG2.2.15 cells compared to Lipofectamine™ 2000/DrzBS micelles (p < 0.001). Most importantly, the inhibitory effect of the CSO–SA/DrzBS micelles on HBV S gene expression could be prolonged to 96 h, which is crucial for anti-HBV gene therapy. In summary, DrzBS delivered by the CSO–SA showed obvious inhibitory effects of HBsAg expression and therapeutic effects, indicating that the CSO–SA was a potential vector for gene therapy and might be used for the successful treatment of chronic HBV carriers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Zhejiang Provincial Natural Science Foundation of China (Qing Shan Hu Science&Technology City Union Foundation, LQY19H300001), the National Nature Science Foundation of China (81402862 and 81503256), the Hospital Pharmacy Special Research Project of Zhejiang Pharmaceutical Association (2018ZYY02, 2016ZYY01), and the Medical and Health Science and Technology Project of Zhejiang Province (No. 2018247580).References
- World Health Organization, Guidelines for the Prevention, Care and Treatment of Persons with Chronic Hepatitis B Infection, Geneva, 2015 Search PubMed.
- R. Orlando, M. Foggia, A. E. Maraolo, S. Mascolo, G. Palmiero, O. Tambaro and G. Tosone, Eur. J. Clin. Microbiol. Infect. Dis., 2015, 34, 1059–1070 CrossRef CAS PubMed.
- S. Mittal and H. B. El-Serag, J. Clin. Gastroenterol., 2013, 47(suppl.), S2–S6 CrossRef PubMed.
- P. Karayiannis, Hepatol. Int., 2017, 11, 500–508 CrossRef CAS PubMed.
- Y. F. Liaw, M. R. Brunetto and S. Hadziyannis, Antiviral Ther., 2010, 15, 25–33 CrossRef PubMed.
- The Korean Association for the Study of the Liver (KASL), KASL clinical practice guidelines: management of chronic hepatitis B, Clin. Mol. Hepatol., 2016, 22, 18–75 CrossRef PubMed.
- F. Cui, J. Drobeniuc, S. C. Hadler, Y. J. Hutin, F. Ma, S. Wiersma, F. Wang, J. Wu, H. Zheng, L. Zhou and S. Zuo, Vaccine, 2013, 31(Suppl 9), J79–J84 CrossRef PubMed.
- W. Cao, Z. Qiu, T. Zhu, Y. Li, Y. Han and T. Li, J. Clin. Virol., 2014, 61, 40–46 CrossRef CAS PubMed.
- E. F. Duffell and M. J. van de Laar, Euro Surveill., 2015, 20, 17–24 CAS.
- Y. H. Hsieh, I. J. Su, H. C. Wang, W. W. Chang, H. Y. Lei, M. D. Lai, W. T. Chang and W. Y. Huang, Carcinogenesis, 2004, 25, 2023–2032 CrossRef CAS PubMed.
- Y. Li, Q. Cai, Q. Xie, Y. Zhang, X. Meng and Z. Zhang, Med. Sci. Monit., 2017, 23, 1385–1393 CrossRef CAS PubMed.
- T. C. Tseng, C. J. Liu, H. C. Yang, T. H. Su, C. C. Wang, C. L. Chen, S. F. Kuo, C. H. Liu, P. J. Chen, D. S. Chen and J. H. Kao, Gastroenterology, 2012, 142, 1140–1149 CrossRef CAS PubMed.
- S. Fourati and J. M. Pawlotsky, F1000Research, 2016, 5, 2243 Search PubMed.
- H. Ward, L. Tang, B. Poonia and S. Kottilil, Future Microbiol., 2016, 11, 1581–1597 CrossRef CAS PubMed.
- V. Soriano, P. Barreiro, L. Benitez, J. M. Pena and C. de Mendoza, Expert Opin. Invest. Drugs, 2017, 26, 843–851 CrossRef CAS PubMed.
- S. W. Santoro and G. F. Joyce, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4262–4266 CrossRef CAS PubMed.
- N. Singh, A. Ranjan, S. Sur, R. Chandra and V. Tandon, J. Biosci., 2012, 37, 493–502 CrossRef CAS PubMed.
- D. Kumar, I. Chaudhury, P. Kar and R. H. Das, J. Gastroenterol. Hepatol., 2009, 24, 872–878 CrossRef CAS PubMed.
- V. Sood, N. Gupta, A. S. Bano and A. C. Banerjea, Oligonucleotides, 2007, 17, 113–121 CrossRef CAS PubMed.
- H. Unwalla, S. Chakraborti, V. Sood, N. Gupta and A. C. Banerjea, Antiviral Res., 2006, 72, 134–144 CrossRef CAS PubMed.
- W. Hou, J. E. Wo, K. Z. Liu, M. W. Li, L. W. Chen, Z. R. Hu, R. H. Liu and M. J. Hu, Zhonghua Neike Zazhi, 2006, 45, 396–399 CAS.
- Y. K. Kim, A. Minai-Tehrani, J. H. Lee, C. S. Cho, M. H. Cho and H. L. Jiang, Int. J. Nanomed., 2013, 8, 1489–1498 Search PubMed.
- J. T. Kwon, H. L. Jiang, A. Minai-Tehrani, C. Gyu Woo, M. Choi, C. S. Cho, Y. S. Kim and M. H. Cho, Iran. J. Pharm. Res., 2013, 12, 281–286 CAS.
- V. Gopal, J. Xavier, G. H. Dar, M. Jafurulla, A. Chattopadhyay and N. M. Rao, Int. J. Pharm., 2011, 419, 347–354 CrossRef CAS PubMed.
- S. Falsini and S. Ristori, Methods Mol. Biol., 2016, 1445, 33–43 CrossRef CAS PubMed.
- M. Saunders, K. M. Taylor, D. Q. Craig, K. Palin and H. Robson, Pharm. Res., 2007, 24, 1954–1961 CrossRef CAS PubMed.
- S. Mao, W. Sun and T. Kissel, Adv. Drug Delivery Rev., 2010, 62, 12–27 CrossRef CAS PubMed.
- S. R. Ryoo, H. Jang, K. S. Kim, B. Lee, K. B. Kim, Y. K. Kim, W. S. Yeo, Y. Lee, D. E. Kim and D. H. Min, Biomaterials, 2012, 33, 2754–2761 CrossRef CAS PubMed.
- A. Tahamtan, A. Ghaemi, A. Gorji, H. R. Kalhor, A. Sajadian, A. Tabarraei, A. Moradi, F. Atyabi and M. Kelishadi, J. Biomed. Sci., 2014, 21, 69 CrossRef PubMed.
- Y. K. Kim, M. Zhang, J. J. Lu, F. Xu, B. A. Chen, L. Xing and H. L. Jiang, J. Drug Targeting, 2016, 24, 457–467 CrossRef CAS PubMed.
- J. J. Deng, Y. F. Zhou, B. Xu, K. J. Mai, Y. B. Deng and L. M. Zhang, Biomacromolecules, 2011, 12, 642–649 CrossRef CAS PubMed.
- B. Layek, L. Lipp and J. Singh, J. Controlled Release, 2015, 207, 143–153 CrossRef CAS PubMed.
- E. Baghdan, S. R. Pinnapireddy, B. Strehlow, K. H. Engelhardt, J. Schafer and U. Bakowsky, Int. J. Pharm., 2018, 535, 473–479 CrossRef CAS PubMed.
- X. Huang, X. Huang, X. H. Jiang, F. Q. Hu, Y. Z. Du, Q. F. Zhu and C. S. Jin, J. Microencapsulation, 2012, 29, 1–8 CrossRef CAS PubMed.
- F. Q. Hu, W. W. Chen, M. D. Zhao, H. Yuan and Y. Z. Du, Gene Ther., 2013, 20, 597–606 CrossRef CAS PubMed.
- J. J. Yao, Y. Z. Du, H. Yuan, J. You and F. Q. Hu, Int. J. Nanomed., 2014, 9, 2993–3003 Search PubMed.
- Y. Y. Xu, Y. Z. Du, H. Yuan, L. N. Liu, Y. P. Niu and F. Q. Hu, J. Drug Targeting, 2011, 19, 344–353 CrossRef CAS PubMed.
- J. Yan, Y. Z. Du, F. Y. Chen, J. You, H. Yuan and F. Q. Hu, Mol. Pharmaceutics, 2013, 10, 2568–2577 CrossRef CAS PubMed.
- C. M. Beddoes, C. P. Case and W. H. Briscoe, Adv. Colloid Interface Sci., 2015, 218, 48–68 CrossRef CAS PubMed.
- R. L. Juliano and K. Carver, Adv. Drug Delivery Rev., 2015, 87, 35–45 CrossRef CAS PubMed.
- J. Miao, X. G. Zhang, Y. Hong, Y. F. Rao, Q. Li, X. J. Xie, J. E. Wo and M. W. Li, Carbohydr. Polym., 2012, 87, 1342–1347 CrossRef CAS.
- J. J. Zhang, X. Y. Sun, R. Shao, W. Q. Liang, J. Q. Gao and J. L. Chen, J. Drug Delivery Sci. Technol., 2016, 31, 187 CrossRef.
- M. Saunders, K. M. G. Taylor, D. Q. M. Craig, K. Palin and H. Robson, Pharm. Res., 2007, 24, 1954–1961 CrossRef CAS PubMed.
- S. R. Mao, W. Sun and T. Kissel, Adv. Drug Delivery Rev., 2010, 62, 12–27 CrossRef CAS PubMed.
- Q. Li, Y. Z. Du, H. Yuan, X. G. Zhang, J. Miao, F. D. Cui and F. Q. Hu, Eur. J. Pharm. Sci., 2010, 41, 498–507 CrossRef CAS PubMed.
- Z. X. Du, M. Y. Chen, Q. Q. He, Y. Zhou and T. Jin, Int. J. Pharm., 2012, 434, 437–443 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |