DOI:
10.1039/C9RA00357F
(Paper)
RSC Adv., 2019,
9, 5009-5024
Interior engineering of seaweed-derived N-doped versatile carbonaceous beads with CoxOy for universal organic pollutant degradation†
Received
15th January 2019
, Accepted 25th January 2019
First published on 11th February 2019
Abstract
The rational optimization of catalytic composites with excellent catalytic activities and long-term cycling stabilities for environmental remediation is still maintained as highly desired but is an ongoing challenge. Here, seaweed-derived N-doped versatile carbonaceous beads with CoxOy (Co-NC-0.25-700 °C) are employed as a novel catalyst to activate peroxymonosulfate (PMS) for methylene blue (MB) degradation. Profiting from the improved structure–activity relationship and the synergistic effects between the “egg-box” structure and the CoxOy loaded on the N-doped carbonaceous beads, Co-NC-0.25-700 °C exhibited relatively high performance and comparative long-term stability. The universal applicability of Co-NC-0.25-700 °C was investigated by degrading other types of organic pollutants in various systems. For this type of newly fabricated high-performance versatile composites, structure–property relationships were plausibly proposed. Notably, the degradation efficiency and the catalyst structure could be tailored by the amount of polyethyleneimine (PEI) introduced in the preparation process and by the pyrolysis temperature. More favorably, the coupling of the magnetic properties and bead-like shape endows the resultant composites with remarkable reusability and recyclability, as compared to powder state materials. Another interesting finding is that MB degradation over Co-NC-0.25-700 °C is minimally affected by common ions (Cl−, NO3−, SO42−, etc.), and holds a certain catalytic activity under the background conditions of two simulated real water conditions (running water and seawater). Of particular interest, a microreactor filled with Co-NC-0.25-700 °C was utilized as a verification model for practical applications of the reaction in continuous-flow. More far-reaching, the simulations of actual water conditions and the design of a continuous-flow reactor represent a giant step towards universal applications for organic pollution treatment.
1. Introduction
Due to the emission of a large number of organic pollutants, including organic dyes, antibiotic compounds, and phenolic compounds, serious environmental problems have drawn increasing public concerns in recent years.1–4 Currently, advanced oxidation processes (AOPs) are considered effective remediation methods owing to their advantages of generating highly reactive oxygen species such as sulfate radicals (SO4˙−), hydroxyl radicals (˙OH) and superoxide radicals (O2˙−, HO2˙, and 1O2) for the degradation of organic pollutants into harmless products.5–7 Among various AOPs, considerable research efforts have been devoted to the typical Fenton reaction (Fe2+/H2O2) in virtue of the strong oxidative potential (1.8–2.7 V vs. NHE) of the generated ˙OH.8,9 However, its practical applications are prominently limited by the low operation pH (2–4) and a large amount of sludge production.4,10 Notably, SO4˙− radicals have been introduced as an alternative to ˙OH radicals as a result of their high redox potential (2.5–3.1 V vs. NHE),5,11 long half-life time period (30–40 ms),10,12 widely suitable range of pH (2–9),13 and being more selective.14 In this case, multifarious transition metals (Fe,15 Co,16 Mn,5 etc.) have exhibited superiority for the activation of peroxymonosulfate (PMS) compared with carbon materials. Particularly, Co-based materials have attracted significant attention due to the unique valence electron layer structure of cobalt (3d74s2) and the comparatively high standard redox potential of Co2+/Co3+ (1.92 V vs. NHE),1,17 which lead to the rapid transfer of electrons and exhibit good catalytic effects. However, Liu et al.18 and Song et al.19 have already revealed that transition metals (Fe2+, Co2+) can cause the adverse loss of dissolved metal ions, poor stability, and reusability. To alleviate these problems, various supported cobalt nanocomposites12,20 and mixed transition-metal oxides7,8 have been extensively investigated. Nevertheless, catalytic efficiency for PMS activation still needs to be further improved due to the slow conversion rate of Co2+/Co3+ redox pairs.10 In contrast, an ultrafine Co3O4-incorporated carbon composite synthesized by Luo et al. shows high removal efficiency, but still, there have been limited studies on the use of it to overcome the drawbacks of leaching.13 Moreover, metal–carbon based hybrids have also been investigated as a potential method to enhance the degradation performance.9 As we all know, much work hitherto has focused on support materials for cobalt oxides and metallic cobalt including metal–organic frameworks (MOFs),21 covalent organic frameworks (COFs),22 SBA-15, etc. Although, these materials exhibit large specific surface areas (SSA), excellent chemical stability and low cost,12 their powder states make them difficult to separate from solution, limiting their applications. Therefore, enhancing the catalytic activity, cycle stability, and recoverability of cobalt-based heterogeneous catalyst/PMS systems is highly desirable but a huge challenge.
To meet these challenges, it is feasible to develop an effective pathway for the preparation of catalysts and to assemble them into a matrix with exhibited relatively high performance and comparative long-term stability. Biomass, as a source of renewable energy, has received increasing attention for the preparation of useful carbon materials.23,24 Meanwhile, N-doped carbon (NC) matrices have been proven to be fascinating catalytic materials owing to their excellent features of high conductivity and environmental friendliness.25,26 The C atoms in the carbon matrix can be substituted for doped N atoms, providing optimized interfacial properties for the carbon atoms for catalysis, which can not only enhance the π-bonding ability and improve basicity but also undermine the electroneutrality of adjacent C atoms and provide more active sites for nucleophilic and electrophilic attack.3,27,28 Alginate, with abundant hydroxyl and carboxyl groups in its polymeric carbon matrix, is a high-modulus natural polysaccharide isolated from brown algae, whose macromolecules can be easily used for the chelation of metal ions (Ca2+, Fe3+, Co2+, and Ni2+) via ion exchange to form a so-called “egg-box” structure.23,29 After pyrolysis in an inert gas atmosphere, the polymeric carbon matrix is naturally converted into a carbon material such that the metal nanoparticles (NPs) are well encapsulated in carbon shells.23 That makes alginate an appropriate precursor for large-scale manufacture of porous metal/C nanocomposites via an environmentally friendly pathway. Accordingly, alginate is widely utilized in electromagnetic wave absorption,30 heavy metal ion adsorption,31 lithium batteries,23 etc. Polyethyleneimine (PEI), as a kind of amine-rich agent, can not only chelate heavy metal ions but also easily reacts with carbonyl groups. Bertagnolli et al.32 and Yan et al.31 utilized cross-linked PEI followed by glutaraldehyde (GA) to internally and externally modify the preformed Ca2+-alginate beads for adsorption of heavy metal ions, respectively. However, previous publications regarding the application of high performance environmentally friendly catalysts in the degradation of organics are very scarce.
With the above considerations, seaweed-derived N-doped versatile magnetic carbonaceous beads with CoxOy were prepared for universal organic pollutant degradation. The 3D interpenetrating co-network polymer was easily formed by using sol–gel assembly and pyrolysis processes, employing sodium alginate (SA), PEI, cobalt, and GA as a carbon source, a nitrogen source, a precursor and a cross-linking agent, respectively. It is of particular interest that better self-assembly and dispersion of Co2+ at the molecular level can be obtained due to the co-complexation between Co2+ and SA/PEI, which can control the aggregation of CoxOy NPs during pyrolysis. Furthermore, the “egg-box” structure and the “channel confinement effect” can steadily anchor CoxOy NPs and form a shelter for them, leading to reduced leaching of the metal. More favorably, the coupling of magnetic properties and a bead-like shape endow the resultant composites with remarkable reusability and recyclability. Based on the numerous advantages, the catalyst (Co-NC-0.25-700 °C) exhibits relatively high-efficiency degradation of methylene blue (MB) on the basis of SO4˙− and has comparative long-term stability. The degradation of p-nitrophenol (PNP) is mostly carried out with precious metals in NaBH4 systems, and it is rarely reported for Co-based/PMS systems. This research has achieved relatively high catalytic activity for PNP degradation with Co-based/PMS and Co-based/NaBH4, which provides a guideline for substituting precious metal catalysts to degrade PNP. Besides, due to the well-developed interpenetrating co-network structure, unconventional electronic distribution and magnetic properties, this type of semiconductor-like Co-NC-0.25-700 °C could possibly reveal unique potential in other fields. More fascinatingly, it is of significance to highlight that the simulation of real water conditions (running water and seawater) and the column reactor design for continuous flow reaction illustrate a giant step towards the practical application of PMS AOPs.
2. Experimental section
2.1. Materials and chemicals
Materials and chemicals for the preparation procedures are described in Text S1.†
2.2. Preparation of catalysts
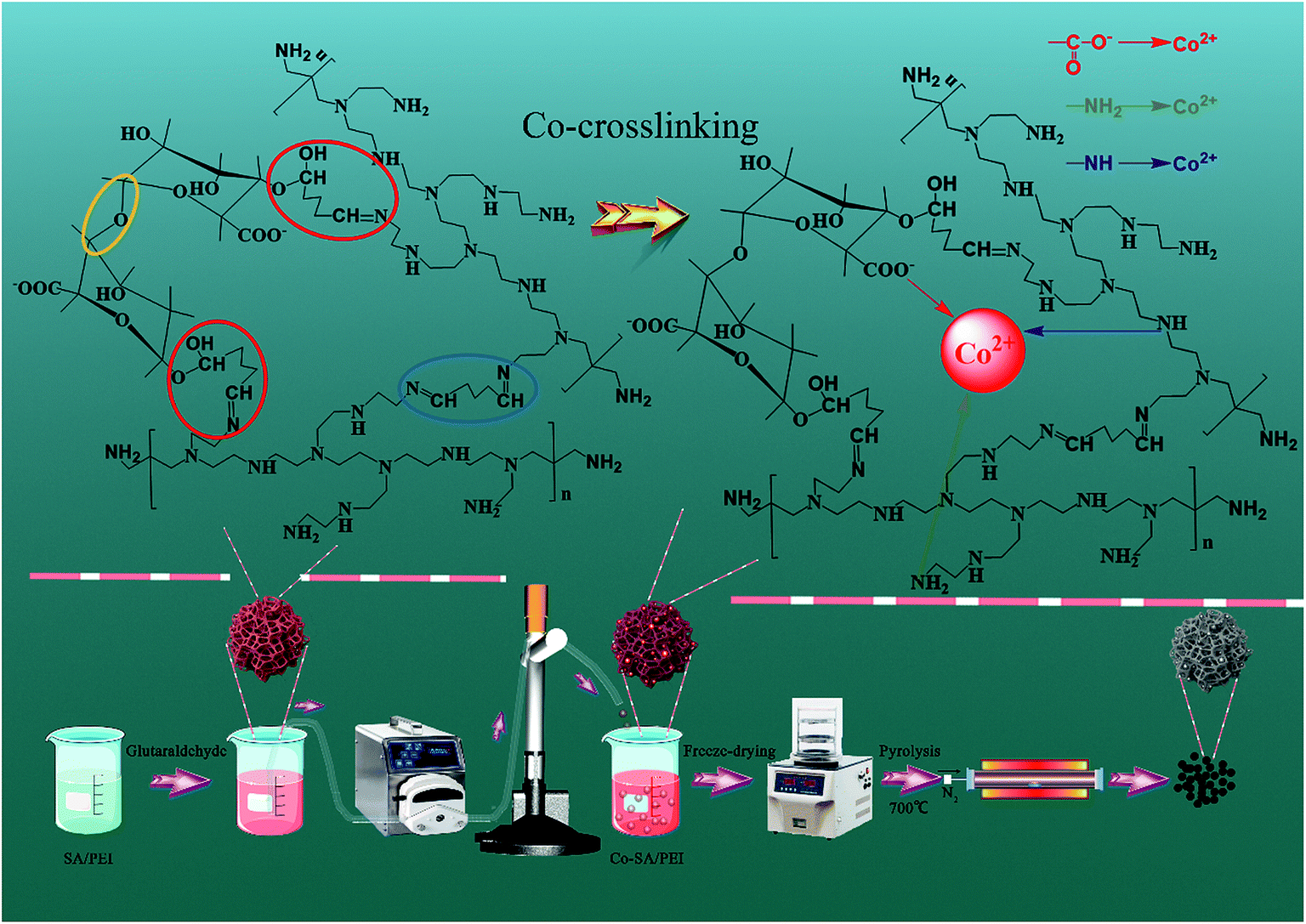
2.2.1 Preparation of Co-NC-x-700 °C. The preparation process is shown in Scheme 1. Firstly, sodium alginate powder (2.0 g) with different amounts of PEI (x represents the content of PEI; x = 0, 0.1, 0.2, 0.25, 0.3, and 0.4 g) were dissolved in deionized water (100 mL) and magnetically stirred for 2 h until the initial SA/PEI was dispersed (Fig. S1a†). Into the resultant dispersion, glutaraldehyde solution (2% (mass ratio) of glutaraldehyde/PEI, 50 wt%) was added consecutively for the cross-linking and curing reaction, and the mixed dispersion was mechanically stirred for 24 h at room temperature until a pink and viscous solution formed (Fig. S1b†). Subsequently, the “box” SA/PEI solution was dripped into a cobalt nitrate hexahydrate (100 mL, 1.5 wt%) solution by a peristaltic pump to conduct the co-chelation of Co2+ with the SA/PEI macromolecules, confining the Co2+ within the “box”. In these circumstances, the SA/PEI dispersion was converted to hydrogel beads since the Co2+ cations coordinated with SA/PEI for another 6 h to completely cross link (Fig. S1c†). Finally, the immobilized Co-SA/PEI hydrogel beads were washed several times with deionized water and ethanol to remove extra Co2+ and NO3− ions, and were dehydrated via a freeze-drying process to produce highly porous and 3D interconnected Co-SA/PEI aerogel beads. After calcination at 700 °C under a N2 atmosphere, the acquired materials were marked as Co-NC-x-700 °C, with x corresponding to the amount of PEI.
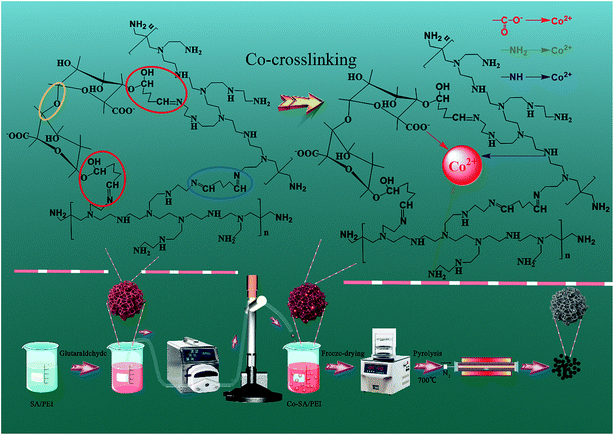 |
| | Scheme 1 Illustration of the preparation processes of Co-NC-x-700 °C and Co-NC-0.25-y. | |
2.2.2 Preparation of Co-NC-0.25-y. 3D Co-SA/PEI-0.25 aerogel beads were calcined for 2 h to reach a set temperature (y is the pyrolysis temperature; y = 500, 600, 700, and 800 °C) at a ramp rate of 5 °C min−1 under a N2 atmosphere in a tube furnace. Then, the 3D Co-SA/PEI aerogel beads were changed into 3D CoxOy-NC beads, in which the SA/PEI “box” was converted to a high-density N-doped amorphous carbon shell to encapsulate the CoxOy NPs. After being cooled down to room temperature under flowing N2, the final materials were recorded as Co-NC-0.25-y, with y corresponding to the pyrolysis temperature.In contrast, Ca-SA and Ca-SA/PEI-0.25 were treated at 700 °C for 2 h with a heating rate of 5 °C min−1 in a N2 atmosphere and the resulting catalysts were denoted as Ca-C-700 °C and Ca-NC-0.25-700 °C, respectively.
2.2.3 Quantification of the co-crosslinking degree of Co2+ and SA/PEI. Quantification of the co-crosslinking degree of Co2+ and SA/PEI macromolecules is expounded in Text S2.†
2.3. Characterization
Characterization of the catalysts is presented in Text S3.†
2.4. Catalytic activity measurements
Catalytic activity measurements are given in Text S4.†
2.5. Column catalysis experiments
Dynamic catalysis experiments were conducted in a glass column with an internal diameter of 6 mm and a height of 8 cm, which was filled with the Co-NC-0.25-700 °C (0.2 g) composite. Moreover, a little gauze was placed on both sides of the fixed-bed reactor to make the catalysts stabilized before each experiment (Fig. S3†). An MB solution (240 mg L−1) and a PMS solution (6 mM) were mixed into the column at an equal flow rate (1 mL min−1) by two identical peristaltic pumps. Meanwhile, the reacted liquid was collected at decided intervals and measured using a UV-vis spectrophotometer. A flow chart of the column catalysis experiments is exhibited in Scheme S1.†
3. Results and discussion
3.1. The formation mechanism of Co-NC-0.25-700 °C
In the weakly alkaline SA/PEI system, the aldehyde groups at both ends of glutaraldehyde allow not only the interconnection of PEI macromolecules to each other by aldimine condensation but also the formation of hemiacetals by the bonding of SA with PEI macromolecules, and this may result in the formation of interpenetrating polymer co-network materials. The carbonyl group of glutaraldehyde is a strongly polar group, and the carbon is strongly positively charged, so it is easy to react this group with a nucleophile. The nucleophilic addition reaction occurs between the aldehyde groups at both end of glutaraldehyde and PEI that contains a large amount of amino groups. PEI is therefore utilized as a nucleophilic reagent. The nitrogen atom with a lone pair of electrons in the amino structure of PEI attacks a positively charged carbon atom on the carbonyl group of glutaraldehyde. Thereby, the nucleophilic addition reaction is completed to form an intermediate α-hydroxylamine compound, which is then further dehydrated to form a Schiff base. Meanwhile, the oxygen in the hydroxyl group of the alginate molecules has a lone pair of electrons, making it highly nucleophilic. Under alkaline conditions, the hydroxyl group is converted to an alkoxy anion which enhances its nucleophilic capacity, and the oxygen with a lone pair of electrons attacks the carbonyl carbon to form the hemiacetal. More importantly, the alginate molecules themselves are linked by ether bonds, and each molecule is interconnected through interactions of chemical bonds to form an interpenetrating SA/PEI co-network. With the introduction of Co2+, the resulting Co-SA/PEI complex structure has been described as a so-called “egg-box” model, in which Co2+ is coordinated by the carboxylate groups of the alginate macromolecules and the amino groups of the PEI macromolecules to form a restricted “box”. Subsequently, the obtained Co-SA/PEI hydrogel beads were converted to Co-SA/PEI aerogel beads via a freeze-drying process. The 3D porous skeleton in the aerogel bead initially took shape through the removal of adsorbed water around the interpenetrating SA/PEI co-network. Finally, the “egg-box” confined Co2+ could be first converted to CoxOy@C core/shell NPs after carbonization in a N2 atmosphere. Furthermore, the novel 3D N-doped carbon nanomaterial with multimodal pores furnished nucleation sites for in situ growth and repression of CoxOy NPs.
3.2. Catalyst characterization
3.2.1 FT-IR. The functional components of the Co-SA and Co-SA/PEI formed by co-complexation of Co2+ and alginate/polyethyleneimine were assessed by obtaining FT-IR spectra (Fig. S5†). The characteristic peaks of Co-SA (Fig. S5a†) at 3423, 1286 and 2938 (723) cm−1 were attributed to the stretching vibration of O–H, C–O and C–H, respectively.33 The bands located at 1419, 1067 and 881 cm−1 related to the bending modes of –OH, ether bond (C–O–C) ring skeletal vibrations of SA and the β-glucosidic linkage between the sugar units in SA, respectively.34 The peak at 1633 cm−1 was due to the bending vibration of absorbed water. Compared with Co-SA, the peak positions and intensities of Co-SA/PEI at 3423 and 1068 cm−1 have a certain degree of offset, respectively, which could be ascribed to the overlap of the stretching vibrations of O–H from SA and N–H from PEI. Obviously, the characteristic absorption peak of the strong C–N bond at 1385 cm−1 further proves the successful grafting of PEI in the Co-SA/PEI.Furthermore, the characteristic absorption peak of a newly emerging C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N at 1722 cm−1 may be attributed to the Schiff base formed between the aldehyde group of the crosslinking agent GA and the remaining free amino group of PEI. In addition, the increase of the broad peak intensity at about 3431 cm−1 may be due to the formation of a hemiacetal between the aldehyde group of the GA and the hydroxyl groups of SA. Convincingly, due to the presence of C–O–C, CN, C–N and C–O, the existence of a co-crosslinked structure between Co2+ and SA/PEI is perfectly proved.
N at 1722 cm−1 may be attributed to the Schiff base formed between the aldehyde group of the crosslinking agent GA and the remaining free amino group of PEI. In addition, the increase of the broad peak intensity at about 3431 cm−1 may be due to the formation of a hemiacetal between the aldehyde group of the GA and the hydroxyl groups of SA. Convincingly, due to the presence of C–O–C, CN, C–N and C–O, the existence of a co-crosslinked structure between Co2+ and SA/PEI is perfectly proved.
3.2.2 SEM and TEM. The morphologies of the as-prepared Co-C-700 °C and Co-NC-0.25-700 °C were checked by SEM, as shown in Fig. 1a and b. An overall view demonstrates that Co-NC-0.25-700 °C maintains a unique 3D interpenetrating polymer co-network structure, which is composed of SA/PEI composites with CoxOy NPs. Moreover, the CoxOy NPs are distributed homogeneously on the nodes of the co-network structure, which can neatly shorten the electron transport pathways and effectively enhance the mass transfer of reactants. The co-complexation leads to an enhanced self-assembly and dispersion of Co2+ at the molecular level, which efficiently controls the aggregation of CoxOy NPs during pyrolysis. Compared with Co-C-700 °C, doping with electron-rich nitrogen modifies the surface structure of the Co-NC-0.25-700 °C composites, providing enhanced-binding ability between the active sites and the 3D N-doped carbon, which slightly reduces the particle size of the nanoparticles due to the asymmetric structure of CN. These results are accordant with the TEM and BET information. Fig. S6a–d† show SEM images of Co-NC-0.25-y. Compared with the Co-NC-0.25-k (k = 500 and 600 °C), it can be seen that the Co-NC-0.25-700 °C exhibits more abundant pores and a more developed co-network structure with the rise of temperature. However, another interesting finding is that they exhibit fewer pores, which is attributed to the aggregation of the CoxOy NPs at high temperature (800 °C). Fig. 1d shows a typical TEM image of Co-C-700 °C, and further indicates that CoxOy NPs were loaded on the C–N skeleton of SA/PEI and were well-dispersed after carbonization. More fascinatingly, the CoxOy NPs were encapsulated by a N-doped amorphous carbon (SA/PEI) shell that resulted from the carbonization of the alginate “egg-box”, and the average size of the CoxOy NPs was approximately 110 nm (Fig. 1c and e).
 |
| | Fig. 1 SEM images of (a) Co-C-700 °C and (b) Co-NC-0.25-700 °C; TEM images of (c and e) Co-NC-0.25-700 °C and (d) Co-C-700 °C; (f) HRTEM image of Co-NC-0.25-700 °C; (g) SEM image of Co-NC-0.25-700 °C and the corresponding EDS mapping for C, N, O and Co elements. | |
From the TEM images (Fig. S6e and f†), it can be found that the Co-NC-0.4-700 °C possesses a much more irregular morphology and large CoxOy agglomerated NPs that are worse than Co-NC-0.1-700 °C. This might be attributed to the presence of more of the N heteroatom, which leads to an increase in the surface energy of the CoxOy NPs, making them easier to aggregate. Fig. 1f shows a clear HRTEM image of the sample which exhibits a set of fringe spacings of 0.338 nm and 0.227 nm, corresponding to the (002) plane of graphite carbon and the (200) plane of CoO NPs. Moreover, EDS elemental mapping clearly shows that C, N, O, and Co are homogeneously distributed, further revealing the uniform dispersion of CoxOy NPs. All of the above can contribute synergistically to high degradation efficiency.
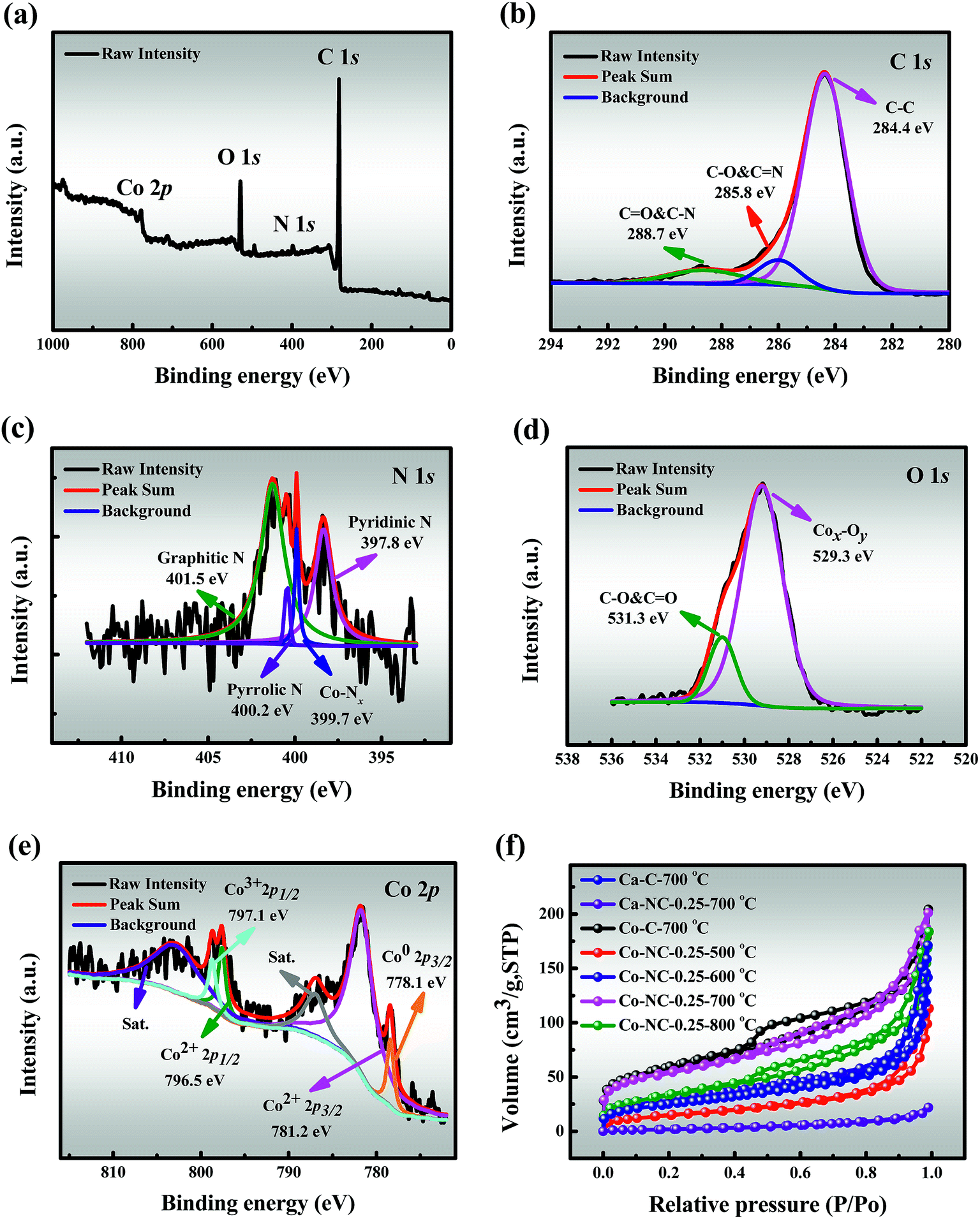
3.2.3 XPS. The elemental composition and the valence states of Co-NC-0.25-700 °C were further investigated by XPS. The full-survey spectrum demonstrates the existence of C (82.64 at%), N (2.64 at%), O (13.15 at%) and Co (1.57 at%) elements as shown in Fig. 2a, as expected. The C 1s spectrum can be fitted into three peaks corresponding to CC (284.4 eV), C–C & CO (285.8 eV) and C–O & CN (288.7 eV) respectively, which indicates the existence of heteroatoms (Fig. 2b).35,36 The four deconvoluted peaks (Fig. 2c) of N 1s at 394.8 eV (pyridinic N), 400.4 eV (Co–Nx), 400.2 eV (pyrrolic N) and 401.5 eV (graphitic N) suggest successful N-doping and Co–Nx site formation in the carbon framework, which not only serve as anchoring sites for the formation of Co–Nx sites, but also improve the catalytic activity.37,38 Fig. 2d shows two peaks in the spectrum of O 1s, corresponding to Cox–Oy (529.3 eV) and C–O & CO (531.3 eV), and this is consistent with the O mapping shown in Fig. 1g, which further indicates the presence of CoxOy NPs on the surface of Co-NC-0.25-700 °C. It is significant to highlight that the Co spectrum (Fig. 2e) is fitted with four components corresponding to Co0, Co2+, Co3+ and the satellites peaks. The binding energies of Co2+ 2p3/2 and Co2+ 2p1/2 are observed at 781.2 and 796.5 eV, which can be ascribed to the surface Co combined with oxygen from the atmosphere during the catalyst preparation and storage process.9 Besides, the surface Co2+ species can be further evidenced by satellite peaks at approximately 5eV above the main peaks.39 Moreover, other peaks at the binding energies of 778.1 and 797.1 eV correspond to Co0 2p3/2 and Co3+ 2p1/2.35,40 Inspiringly, the existence of three different valence states of cobalt effectively accelerates the initial conversion rate of cobalt electrons pair, which leads to enhanced catalytic effects.
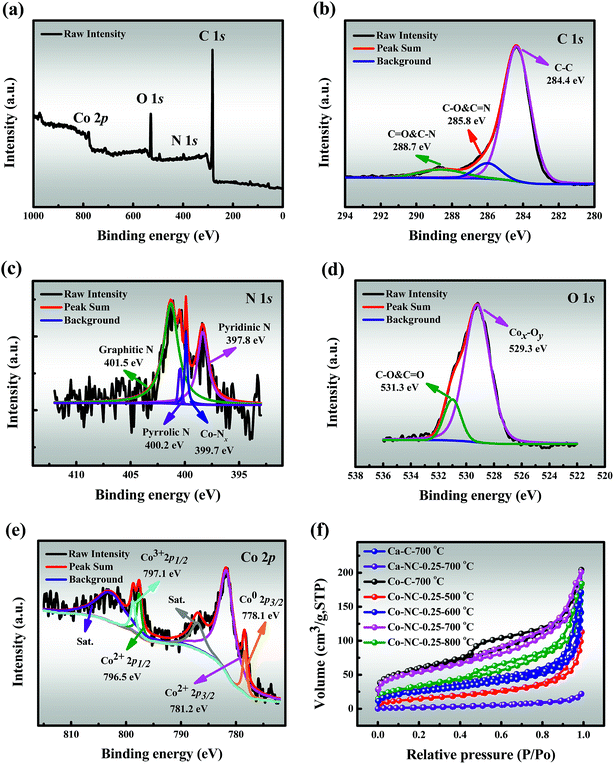 |
| | Fig. 2 XPS spectra of Co-NC-0.25-700 °C: (a) full-survey spectrum, (b) C 1s, (c) N 1s, (d) O 1s and (e) Co 2p; (f) N2 adsorption/desorption isotherms of the different as-prepared catalysts. | |
3.2.4 BET. The N2 adsorption/desorption isotherms (Fig. 2f) show that the prepared catalysts except Ca-NC-0.25-700 °C reveal a typical type-IV profile with a distinct hysteresis loop, indicating the existence of mesopores, as confirmed by the corresponding pore distribution curve (Fig. S7†).16 Meanwhile, a large SSA benefits from the high pyrolysis temperatures of Co-NC-0.25-l (l = 500, 600, and 700 °C), which might be due to the release of H2O and CO2 during the thermal decomposition of SA/PEI.29 The maximal SSA (191.240 m2g−1) and pore volume (0.304 cm3g−1) were recorded for Co-NC-0.25-700 °C, while there was a significant decline in SSA (125.128 m2g−1) for Co-NC-0.25-800 °C, given that the SA/PEI skeleton collapsed at a higher temperature.It is noteworthy that the SSA (7.725 m2g−1) of Ca-NC-0.25-700 °C has a sharp drop from that of Ca-C-700 °C (89.499 m2g−1), which seems to be originated from the strong bonding ability between Ca2+ and SA. Furthermore, the SSA of Co-C-700 °C is 201.480 m2g−1 in contrast to that of Ca-C-700 °C, since the binding ability of Co2+ and SA/PEI is slightly weaker. However, the SSA (191.240 m2g−1) of Co-NC-0.25-700 °C is similar to that of Co-C-700 °C, which may be ascribed to the following two phenomena: (i) a better cooperative interaction between SA/PEI and Co2+ in the self-assembly process, and (ii) the deposition of CoxOy NPs on the pores of SA/PEI.38,41 The relatively large specific surface area and pore volume accelerate the degradation rate by improving the mass transfer driving force. Textural characteristics of the prepared materials are listed in Table S1.†
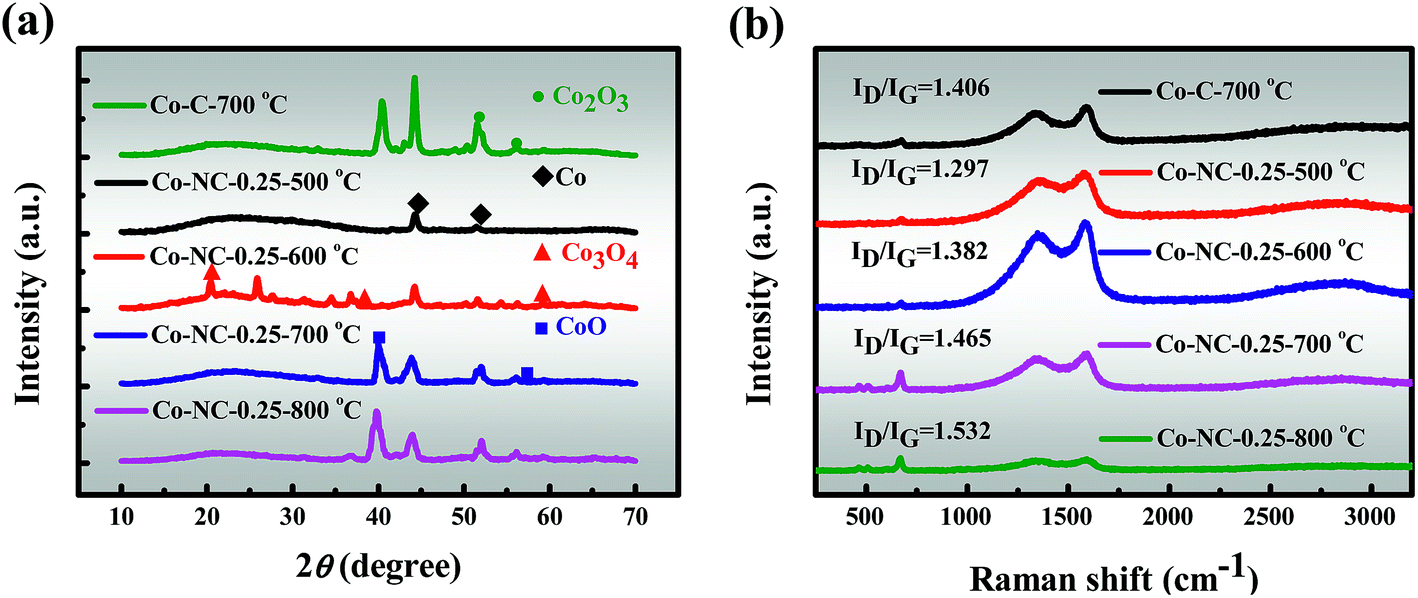
3.2.5 XRD. The crystal structures of the as-prepared catalysts were investigated by XRD and the crystal phase conversion pattern is revealed in Fig. 3a. The major peaks for Co-C-700 °C can be assigned to Co (46.2° peak, JCPDS 15-0806), CoO (39.6° peak, JCPDS 42-1300) and Co2O3 (51.3 and 56.4° peaks, JCPDS 02-0770), and the results are slightly different from the previously reported examples.41,42 Almost the same XRD pattern is exhibited for Co-NC-0.25-700 °C. However, as the content of CoO increases, the diffraction signal intensity for Co-NC-0.25-700 °C decreases slightly in comparison with that of Co-C-700 °C, which suggests the likely fact that the interaction between the CoxOy NPs and the N heteroatoms is most likely to form Co–Nx, which corresponds with the XPS findings.37 In addition, distinct peaks at 44.2° (111 plane) and 51.5° (200 plane) are also observed in Co-NC-0.25-700 °C, which are attributed to the formation of metallic Co with a cubic structure.13,43 At a higher temperature of 600 °C, the cobalt species is mainly present as highly dispersed cobalt oxides and Co, marking the initial development of CoxOy NPs in a better direction. Interestingly, both Co-NC-0.25-700 °C and Co-NC-0.25-800 °C show a stronger peak, implying a higher crystallinity of the CoxOy NPs. Moreover, there are two possible reasons for the formation of Co0 diffraction peaks: (i) Co0 nanoparticles themselves grow at high temperatures; (ii) C and N are released from the precursor to facilitate Co0 generation during the carbonization, indicating that CoxOy and Co–Nx are formed first.16,41 The most obvious peaks are weak and wide diffraction peaks at 24°, which could be assigned to amorphous carbon, coinciding with the results of Raman spectroscopy.9
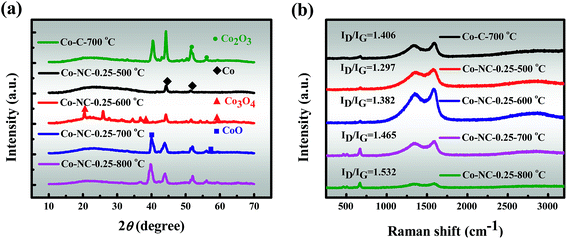 |
| | Fig. 3 (a) XRD patterns and (b) Raman spectra of the as-prepared catalysts. | |
3.2.6 Raman. Consequently, the effect of the introduced PEI to SA was further proved using Raman spectra (Fig. 3b), in which the G band (1580 cm−1) reveals the in-plane vibration of the sp2 carbon atoms and the D band (1340 cm−1) indicates structural defects and amorphous carbon.13 The ID/IG (peak area ratio) increases from 1.406 to 1.465 after the introduction of N, indicating the generation of more structural defects within Co-NC-0.25-700 °C. Furthermore, the ID/IG of the samples increases continuously with increasing carbonization temperature, indicating an increasing amorphous carbon ratio, and such a trend accords well with the observations of the Co-NC-x-700 °C with varying N contents (Fig. S8†). It should be pointed out that a low-intensity band centered at 665 cm−1 was detected, which can be unambiguously attributed to the CoxOy species.9,16
3.2.7 VSM. As is known to all, magnetic properties have a key role in the recyclability of magnetic catalysts. Thus, the magnetic properties of Co-NC-0.25-700 °C were investigated by VSM, and the results are shown in Fig. S9.† Clearly, the Co-NC-0.25-700 °C exhibits typical ferromagnetic behavior with S-like shaped hysteresis loops, demonstrating the presence of magnetic CoxOy NPs. As shown, the coercivity (Hc), saturation magnetization (Ms) and remnant magnetization (Mr) are 214.695 Oe, 38.768 emu g−1 and 3.514 emu g−1 for Co-NC-0.25-700 °C. The excellent Ms ensures the magnetic response of the Co-NC-0.25-700 °C to external magnetic fields, so that the Co-NC-0.25-700 °C can be easily separated from MB solutions (Fig. S2†) and secondary pollution can be reduced.
3.3. Catalytic activity evaluation
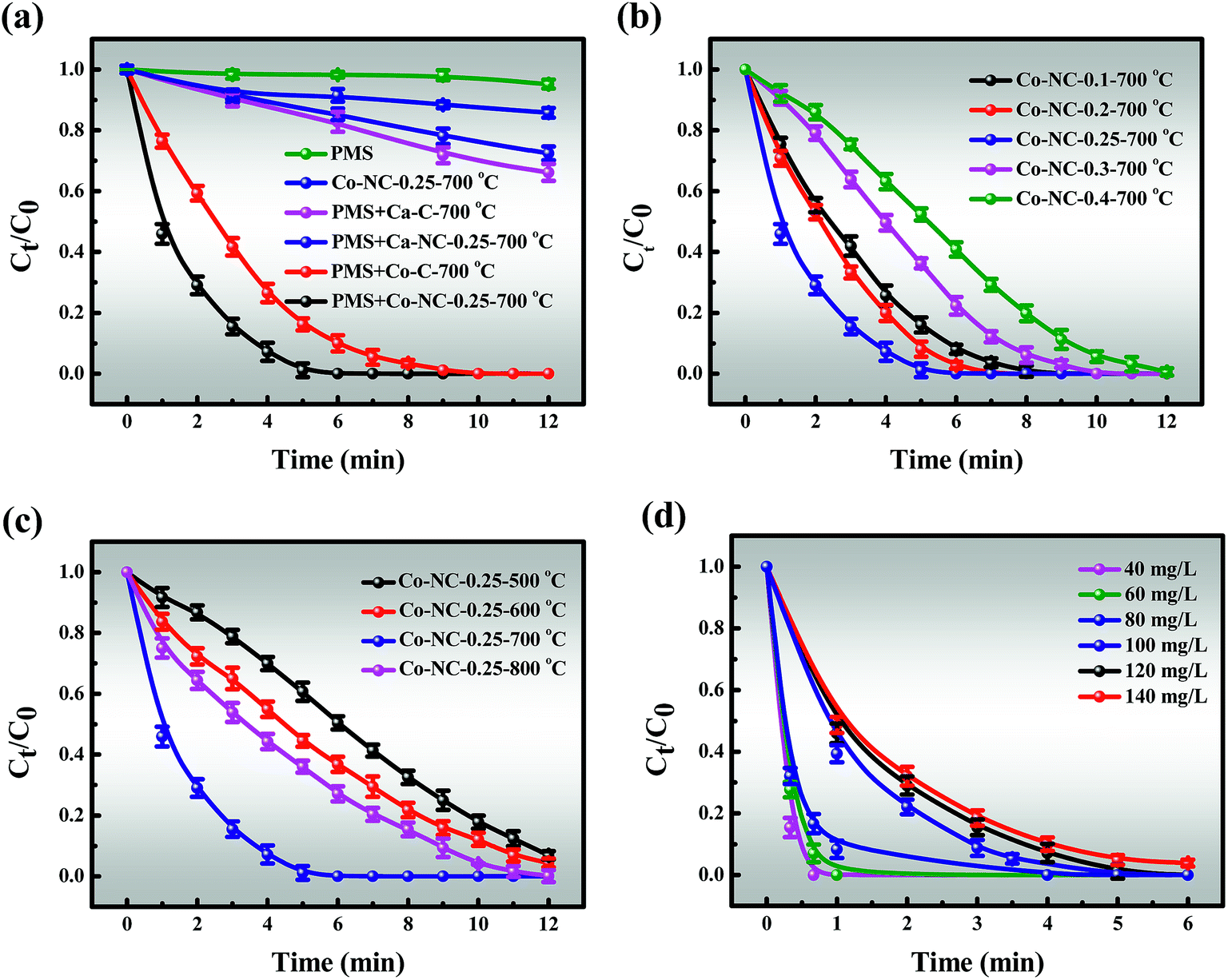
3.3.1 Catalytic activity of Co-NC-0.25-700 °C. In view of the above efforts in the in-depth study of the Co-NC-0.25-700 °C structure, we next explain the relationship between structure and catalytic performance. The removal efficiencies of MB in different systems are presented in Fig. 4a. It must also be mentioned that the effect of static adsorption on removal efficiency cannot be ignored in the degradation process. About 14.23% of MB could be absorbed by Co-NC-0.25-700 °C within 12 min, which may be attributed to the large SSA and pore volume. Besides, PMS alone was also evaluated for MB degradation, and less than 5% of MB was decomposed, indicating the weak self-decomposition ability of PMS in the absence of catalysts.44 In contrast, the catalytic activity was significantly enhanced in the Co-C-700 °C/PMS systems, and the removal efficiency was 97.59% within 9 min. Encouragingly, the degradation efficiency was optimized in the Co-NC-0.25-700 °C/PMS systems.
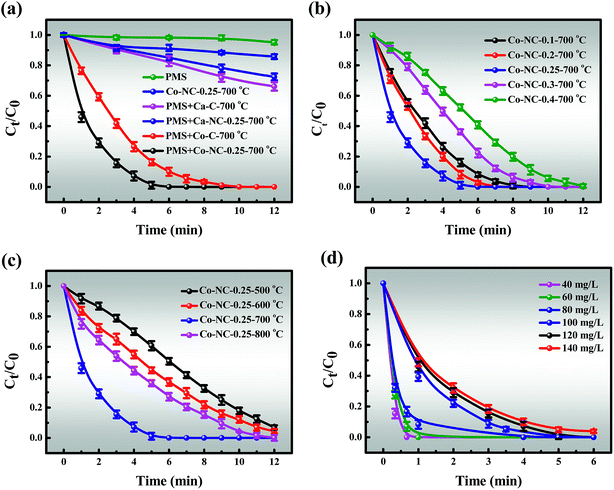 |
| | Fig. 4 (a) The removal efficiency of MB in different systems within 12 min; the removal efficiency of MB using catalysts with different (b) content of introduced PEI and (c) pyrolysis temperature; (d) the effect of MB concentration on the removal efficiency. Reaction conditions: [MB] = 120 mg L−1, [PMS] = 3 mM, catalyst = 0.4 g L−1, T = 298 K, initial solution pH 7.62. | |
In addition, the MB removal efficiency of different previously reported catalysts by PMS activation were compared (Table S2†).1,2,15,45–48 There is no denying that carbon-based materials are widely reported as highly efficient catalysts.2 Little is known, however, about Ca2+ as a catalyst for activating PMS. An identified Ca-C-700 °C that degrades MB in Ca-C-700 °C/PMS systems with 34.88% of MB decomposed within 12 min can be shown as a contrast. It is possible to envision, but it is yet to be demonstrated, for example, that the catalytic efficiency of Ca2+ is much lower than that of Co2+/Co3+. It was reported that heteroatom doping can effectively improve catalytic effect.27 On the contrary, Ca-NC-0.25-700 °C exhibits lower catalytic performance than Ca-SA-700 °C. These results may be generated due to the synergies of the following properties: (i) Ca-NC-0.25-700 °C has a smaller SSA than Ca-C-700 °C, which is not conducive to mass transfer, and this could become dominant. This is consistent with the observation from BET; (ii) the active sites of the C–N and C–C, CC bonds were formed by introducing N; (iii) there is rapid transfer of Co2+/Co3+ valence electrons.
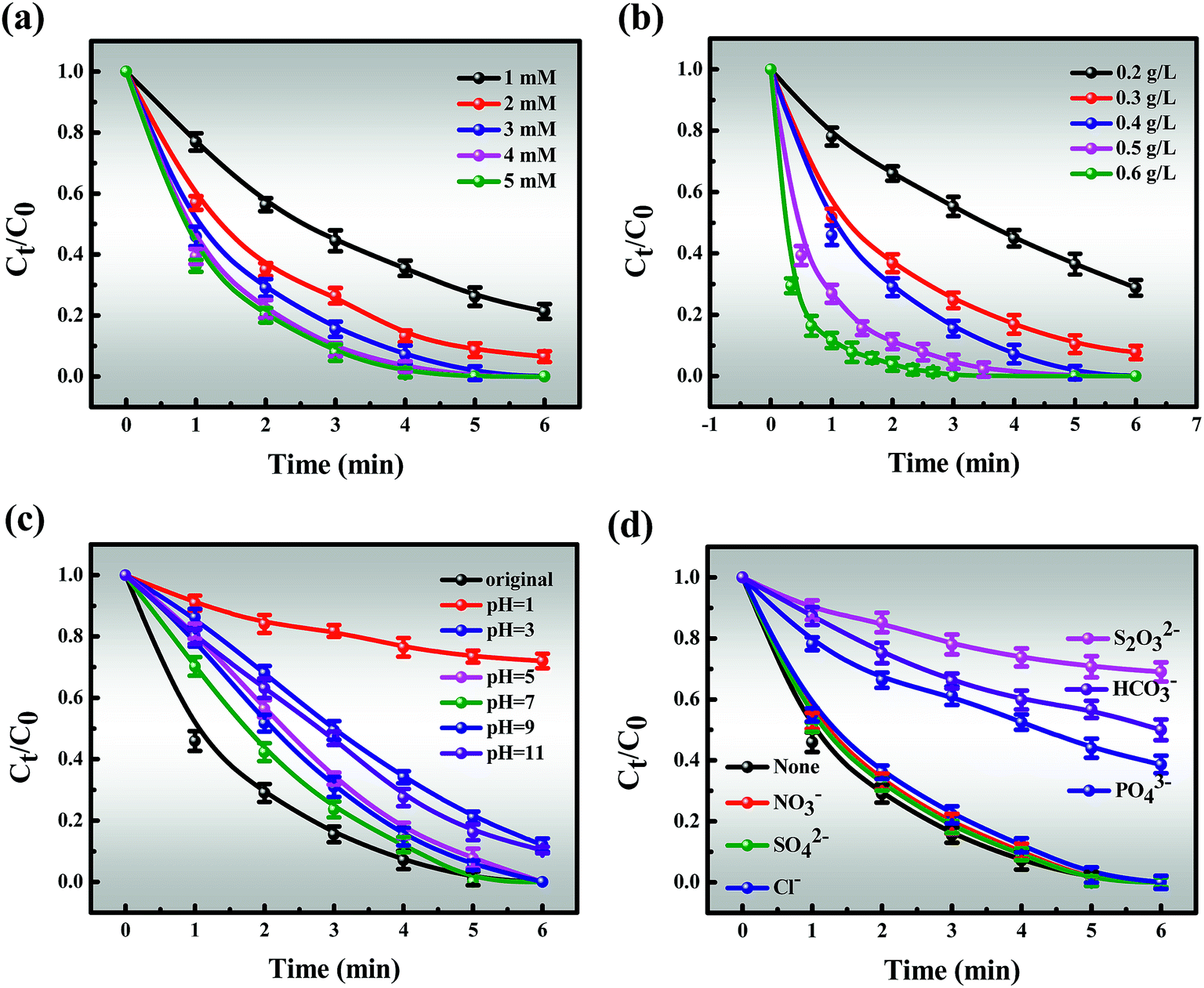
In contrast, it is momentous to highlight that the amount of PEI introduced has an obvious impact on the catalytic performance of the Co-NC-x-700 °C microspheres (Fig. 4b). The improvement of the removal efficiency followed the order of Co-NC-0.25-700 °C > Co-NC-0.2-700 °C > Co-NC-0.1-700 °C > Co-SNC-0.3-700 °C > Co-NC-0.4-700 °C. Increasing the amount of electron-rich N introduced at the beginning (0.1, 0.2, and 0.25 g) could further modify the surface structure of the C,27 which could improve the catalytic performance. Nevertheless, excessive PEI introduction (0.3 and 0.4 g) may lead to aggregation of CoxOy NPs, resulting in fewer active sites and smaller SSA, thus reducing the degradation efficiency.49 In addition, a much more irregular and disordered morphology for Co-NC-0.4-700 °C is also proved in Fig. S1f.†
However, another fascinating finding is that there is a similar trend to that of the PEI content with the increase of pyrolysis temperature. That is, the pyrolysis temperature has a greater influence on the degradation efficiency (Fig. 4c). The results revealed that Co-NC-0.25 obtained at 700 °C afforded the best catalytic activity and it was suitable for large-scale production for subsequent experiments. In comparison, the ideal catalytic performance of the catalysts could not be achieved at other pyrolysis temperatures, obviously, Co-NC-0.25-500 °C, Co-NC-0.25-600 °C, and Co-NC-0.25-800 °C exhibited removal efficiencies of 50.57%, 63.24% and 73.91% for MB degradation within 6 min, respectively. This is probably due to the fact that active sites don’t grow well at lower pyrolysis temperatures (600 and 700 °C).16 While when the carbonization temperature is increased to 800 °C, the collapsed C–N skeleton leads to a decrease in catalytic activity. Such results make it clear that the N heteroatom in the carbon framework and the pyrolysis temperature play critical roles in the degradation experiments.
3.3.2 Effect of parameters on catalytic activity.
3.3.2.1 Effect of MB concentration, PMS doses and catalyst doses. The effects of reaction parameters were investigated to study the synergistic removal of MB under different conditions in the Co-NC-0.25-700 °C/PMS system. As illustrated in Fig. 4d, the removal efficiency of MB decreases from 100% to 95.31% with an increase in the initial concentration of MB from 40 to 140 mg L−1, and the corresponding apparent rate constant (k1) drops from 3.9822 to 0.5537 (R2 > 0.99) (Fig. S10†). Obviously, the degradation efficiency of MB is inversely proportional to the initial concentration. All of the above demonstrates that the time-consuming nature of MB removal at high concentration is possibly due to the following two reasons: (i) the production of large amounts of intermediates adsorbed on the surface of Co-NC-0.25-700 °C with an abundant pore structure results in the covering of active sites and the hindrance of the formation of active free radicals.4,45 (ii) Since more free MB molecules compete for free radicals, the decomposition of PMS to generate free radicals is limited to a certain extent. On the basis of these results, 120 mg L−1 of MB solution was utilized for subsequent experiments due to the ultrafast removal of MB at low concentration.Moreover, the MB removal efficiency can reach about 78.68%, 92.49% and 100% at the PMS concentrations of 1, 2, and 3 mM, respectively. However, the MB removal efficiency is not significantly affected with further increases in the PMS loading (4 and 5 mM) (Fig. 5a). This might be explained by the fact that unfavorable consumption of SO4˙−/˙OH by excess HSO5− gives rise to the scavenging of SO4˙−/˙OH (eqn (1)).8,48,50
| | |
HSO5− + SO4˙−/˙OH → SO5˙− + SO42− + H+/OH−
| (1) |
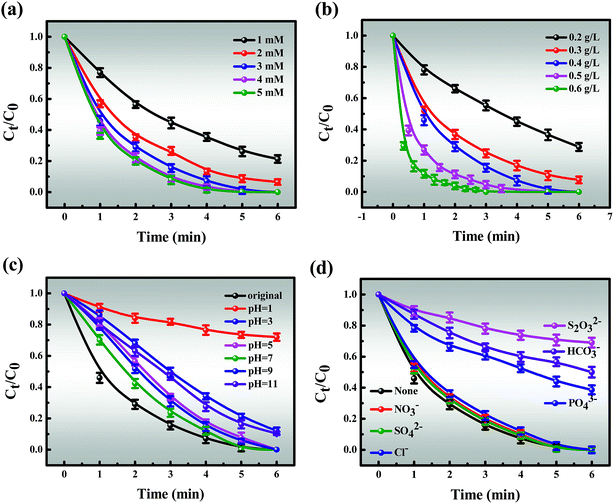 |
| | Fig. 5 Effects of different (a) PMS doses, (b) catalyst doses, (c) initial pH and (d) coexisting ions on the MB removal efficiency. Reaction conditions: [MB] = 120 mg L−1 (for a, b, c and d), [PMS] = 3 mM (for b, c and d), catalyst = 0.4 g L−1 (for a, c and d), T = 298 K (for a, b, c and d), initial solution pH 7.62 (for a, b and d). | |
Also, free radical self-reaction may take precedence over free radical/MB reactions (eqn (2)).51
| | |
SO4˙− + SO4˙− → S2O82−
| (2) |
Similarly, the catalyst dosage also acts as a major factor for MB removal since it could activate PMS to generate SO4˙− and ˙OH, thus resulting in improved removal efficiency. As depicted in Fig. 5b, the removal efficiency increases from 71.22% to 100% with an increase in the catalyst dosage from 0.2 to 0.4 g L−1. However, further increase in the catalyst dosage to 0.6 g L−1 only slightly affected the catalytic activity. Given removal efficiency and cost factors, 3 mM PMS and 0.4 g L−1 of catalyst were chosen for the following experiments.
3.3.2.2 Effect of initial pH. In the MB oxidation process, the pH of the MB solution should significantly affect the production of free radicals. Fig. 5c shows that the MB degradation rate maintains a high level over an initial broad pH range from 5.0 to 9.0, which covers the pH scope of most wastewater. Considering that the MB solution is naturally weakly alkaline, it is of particular concern whether the MB degradation efficiency achieved is optimal under these conditions. Therefore, the natural pH of MB was selected in the subsequent experiments. With the pH ranging from 7 to 9, HSO5− is the main species instead of H2SO5 due to the dissociation of PMS and further self-reactivity of PMS free radicals (SO5˙−), resulting in lower pH of the solution and higher efficiency (eqn (3) and (4)).13,52However, a further increase of the pH to 11 leads to the removal efficiency decreasing to 88.9% within 6 minutes, and the possible reasons for this are shown below (eqn (5)–(10)):
| | |
HSO5− → SO4˙− + OH− (ref. 4)
| (5) |
| | |
SO4˙− + OH− → SO42− + ˙OH
| (6) |
| | |
SO4˙− + ˙OH → HSO4− + 1/2O2
| (7) |
| | |
Co2+ + 2OH− = Co(OH)2↓
| (10) |
(i) Alkaline conditions are disadvantageous to the oxidation processes proceeding according to Le Chatelier’s principle as shown in eqn (5). Notably, there is a problem that deserves our consideration. Where the domination of ˙OH may lead to a slight decrease in degradation efficiency; (ii) OH− was oxidized to ˙OH by SO4˙−, and simultaneously, SO4˙− could further react with the generated ˙OH. The reduction of free radicals leads to a decrease in degradation efficiency (eqn (6) and (7));8,48 (iii) a large amount of OH− covering the Co-NC-0.25-700 °C surface promotes the reaction between Co2+ and OH− (H2O), resulting in loss of active sites (eqn (8) and (9));11,53 (iv) the Co-NC-0.25-700 °C is significantly reduced since Co(OH)2 precipitate is formed. These results indicate that Co-NC-0.25-700 °C performed under neutral or alkaline conditions (eqn (10)).12
In contrast, about 87.6% MB could be decomposed within 6 min at pH 3 while only 27.9% could be decomposed at pH 1. Thus PMS activation is slower at relatively lower pH values, following two aspects (eqn (11) and (12)):
| | |
SO4˙− + H+ + e− → HSO4˙−
| (12) |
(i) PMS may be more stable under acidic conditions;54 (ii) SO4˙− and ˙OH could be quenched by H+, which reduces the catalytic performance.55
3.3.2.3 Effect of coexisting ions and illumination. To some extent, the presence of inorganic anions (Cl−, NO3−, SO42−, HCO3−, PO43−, and S2O32−) influences the generation of radicals in MB degradation. Hence, an anion concentration of 5 mM was selected as representative to evaluate these effects, which is a better fit for actual sewage solution (Fig. 5d). Overall, the fact is that all added inorganic anions have a negative impact on the degradation tests following the order of S2O32− > HCO3− > PO43− > Cl− > NO3− > SO42−,11,14,56 which is a slight deviation from previous reports.9,57 It is clear that the degradation curves of MB partially overlap with the control experiment, indicating that the presence of NO3− and SO42− show the tiniest inhibitory impacts on catalytic efficiency.58 Compared to NO3− and SO42−, Cl− shows more of a repressive effect at low concentration, which is attributed to the conversion of Cl− to the chloride radical (Cl˙) with the assistance of SO4˙− (eqn (13) and (14)).52| | |
SO4˙− + Cl− → SO42− + Cl˙
| (13) |
Although a detailed investigation of various concentrations of Cl− is beyond the scope of this work, we acknowledge that the effect of Cl− is altered from repressive to mildly promotional when Cl− is added at the appropriate concentration, which may be due to the generation of high levels of Cl˙.8,12,59 Moreover, the inhibitory effect is significant when PO43− and HCO3− exist in the degradation system, which may be due to their reaction with free radicals to produce PO4˙− and HCO3˙.12,55,60 It is of importance to highlight that the most inhibition in MB degradation was observed when S2O32− existed, and the main reason for this is due to the competitive effects of absorption of S2O32− and MB onto the surface of the catalyst, resulting in a decline in degradation efficiency.56
Under normal conditions, light intensity has a certain influence on the degradation of dyes. Therefore, there is a need to investigate the effect of illumination on the MB degradation procedure. The MB degradation curve in the presence of strong light is almost completely coincident with that of light-shielded conditions (Fig. S11†), showing only a slight promotion. Although further studies are required to illuminate this question, a possible tentative explanation is that only a small amount of methylene blue is demethylated and decomposed under intense natural light, and its effect is negligible compared to the strong oxidation of sulfate radicals and hydroxyl radicals.
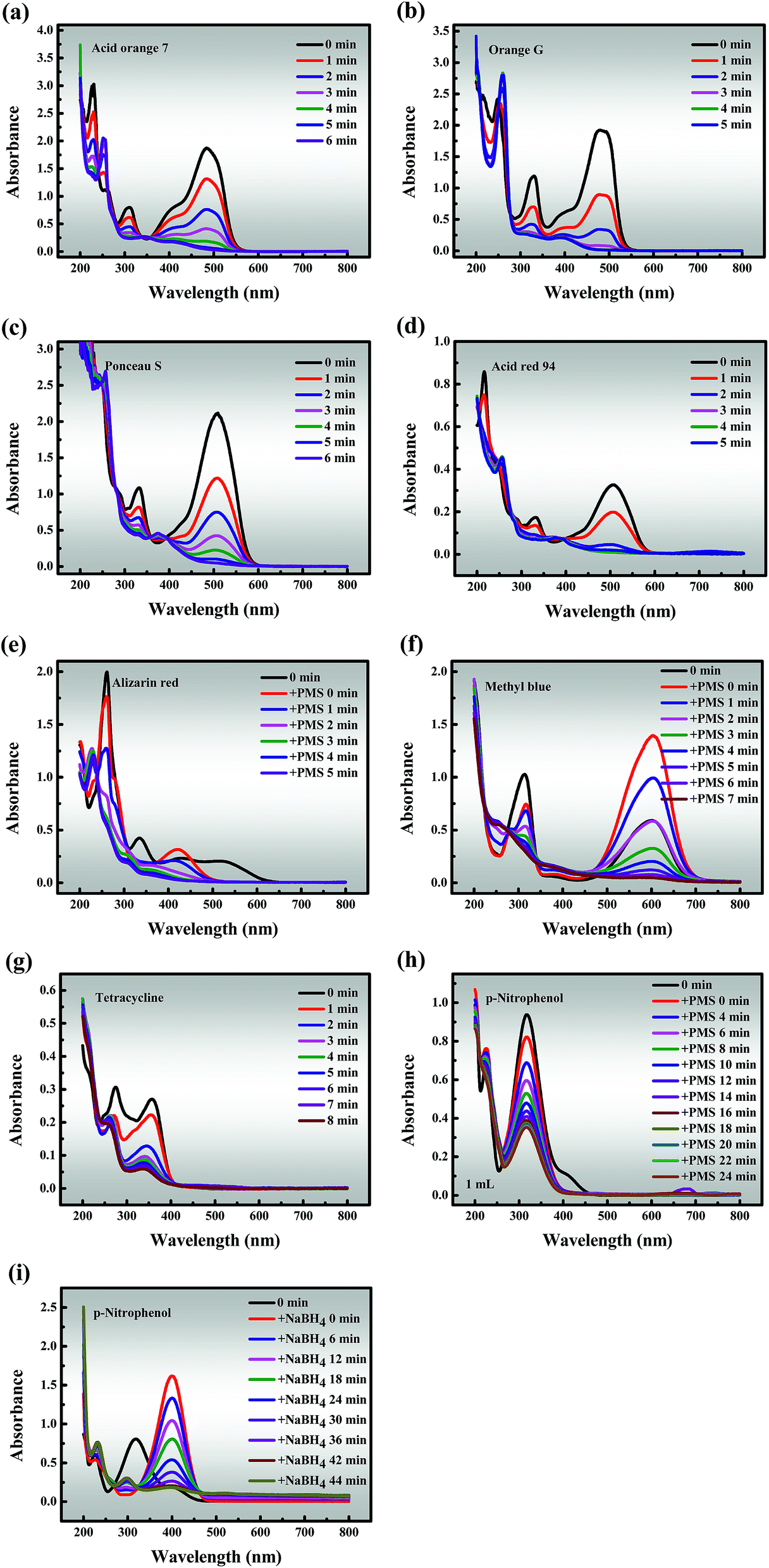
3.3.3 Universal applicability. To investigate the general applicability, Co-NC-0.25-700 °C was utilized as a heterogeneous catalyst to degrade six other organic dyes (AO7, OG, PS, AR94, AR and MeB), an antibiotic compound (TC) and a phenolic compound (PNP) in Co-NC-700 °C/PMS systems (Fig. 6a–h). Fascinatingly, all pollutant concentrations were adjusted to 3, 4, 8, 12, 2, 6, 1 and 1 times the selected concentration to better distinguish the degradation ability of Co-NC-0.25-700 °C, respectively. Almost all of the dyes can be ultimately removed within 6 min, and the removal efficiency increased following the order of AR94 > PS > MeB > OG > AO7 > AR. It should be pointed out that the peaks of AR and MeB underwent peak shifts and peak intensity mutations when PMS was added, separately. This may be attributed to the self-decomposition of the PMS, leading to changes in the pH of the systems, but this possibility remains to be explored. On the contrary, the degradation efficiency of TC and PNP only reached about 94.86% and 68.86% within 8 min and 24 min, respectively. There is a relatively reasonable explanation for the above phenomenon: p-nitrophenol is relatively difficult to degrade with sulfate radicals due to the nitro substituent.61 As a comparison, the reduction of PNP to p-aminophenol (PAP) was evaluated in Co-NC-0.25-700 °C/NaBH4 systems under the same conditions, and the conversion ratio exceeded 95% within 44 min (Fig. 6i). Furthermore, the formation of a characteristic absorption peak at 400 nm is observed, owing to the formation of p-nitrophenolate ions under the increasing alkalinity.3 These findings lead us to a preliminary conclusion that Co-NC-0.25-700 °C could effectively degrade the organic pollutants, and the degradation efficiencies follow the order of organic dyes > antibiotic compounds > phenolic compounds,1–4 which is hardly reported in previous work.
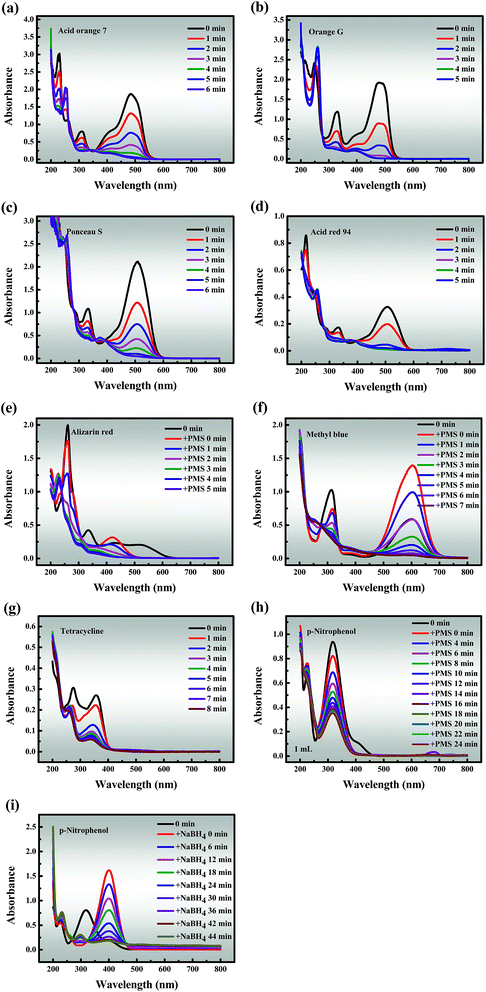 |
| | Fig. 6 Degradation of various pollutants: (a) Acid Orange 7; (b) Orange G; (c) Ponceau S; (d) Acid Red 94; (e) Alizarin Red; (f) Methyl Blue; (g) tetracycline; (h and i) p-nitrophenol. Reaction conditions: [PMS] = 0.6 mM (for a, b, c, d, e, f, g and h), [NaBH4] = 0.6 mM (for i), catalyst = 0.3 g L−1 (for a, b, c, d, e, f, g, h and i), T = 298 K (for a, b, c, d, e, f, g, h and i). | |
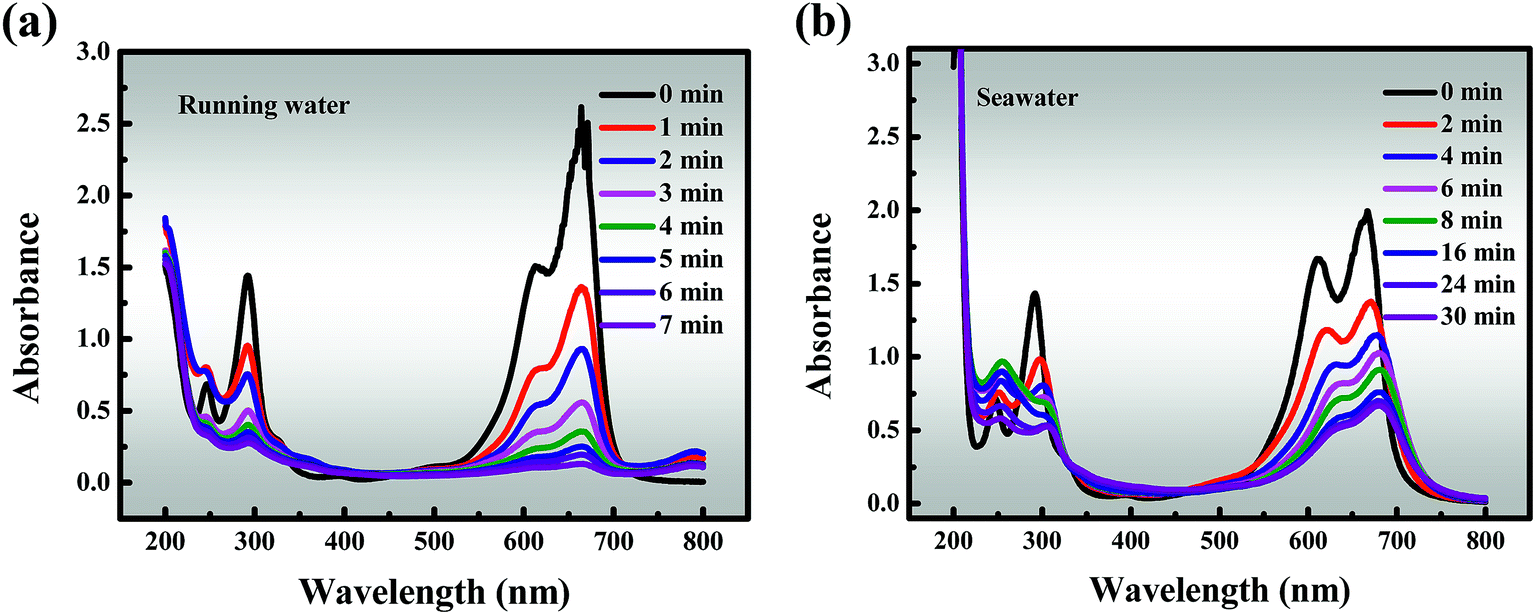
In addition, the results of studies on running water and seawater are shown in Fig. 7a and b, which are invaluable because they represent simulated actual conditions. The removal efficiency (96.68%) of MB within 7 min is slightly reduced in running water compared to that in deionized water, but it still remains at a high level. This may be due to the running water containing low levels of quenching free radicals, and proves that the material has good degradation properties. It must also be pointed out that seawater is a very complex multi-component aqueous solution in which various anions are dissolved, of which about 90% are Cl−, and it also contains SO42−, Br−, HCO3−, CO32−, F−, etc. As described above, these anions have a negative impact on the catalytic activity, resulting in degradation efficiency of about 50% in 8 minutes and a slightly offset of the characteristic absorption peak in the initial stages of degradation.
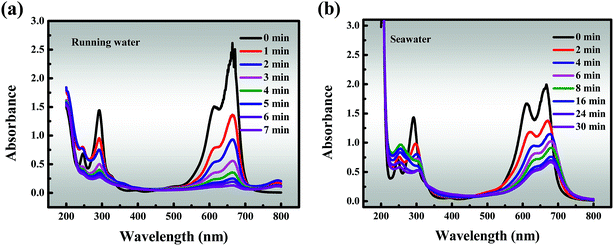 |
| | Fig. 7 Effects of different solvents: (a) running water, and (b) seawater. Reaction conditions: [MB] = 120 mg L−1, [PMS] = 3 mM, catalyst = 0.4 g L−1, T = 298 K, initial solution pH 7.62. | |
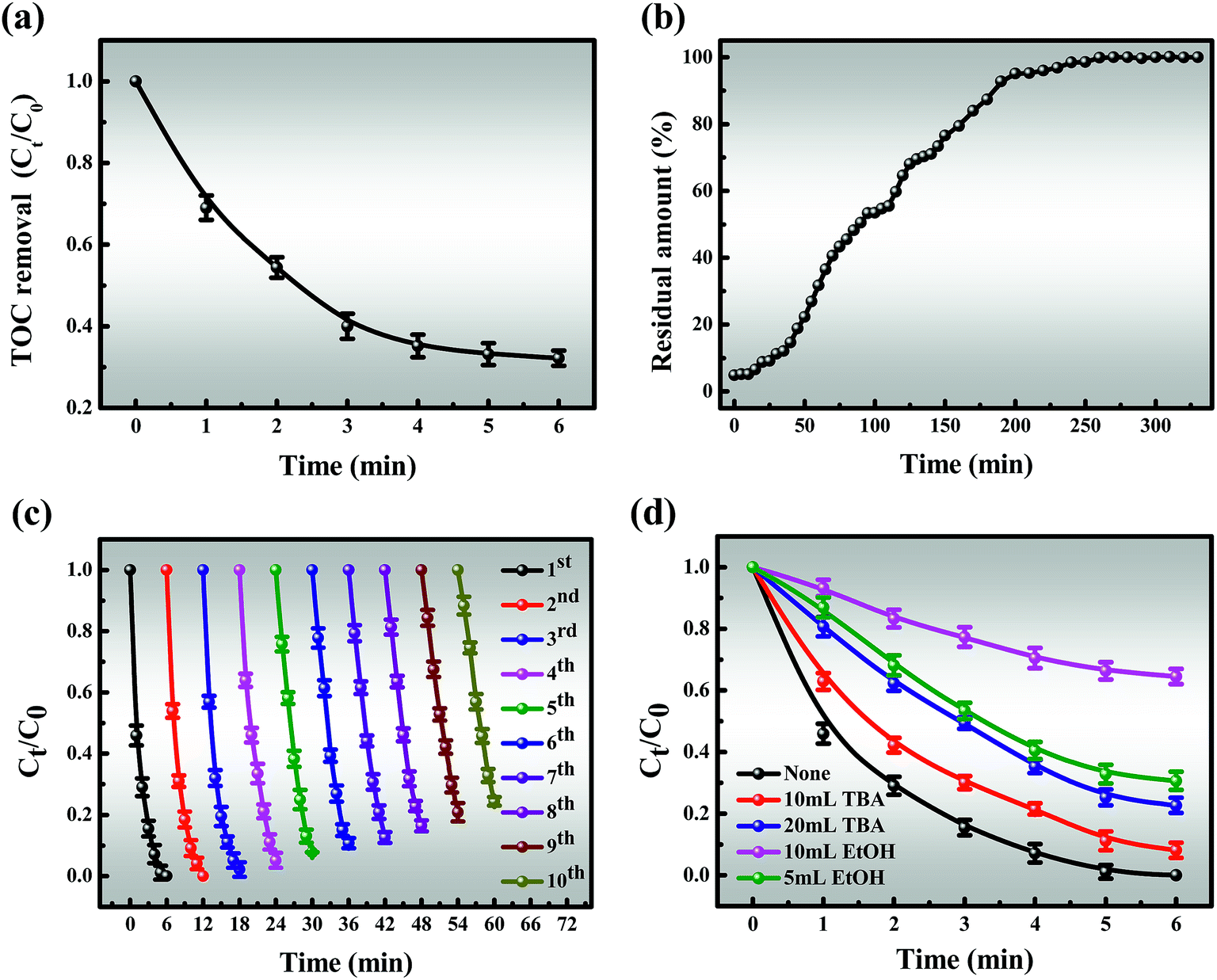
3.3.4 TOC and HPLC analysis for MB degradation. From Fig. 8a, it is indicated that the TOC removal efficiency is 68.8% and the slope of the TOC curve gradually decreases after 6 min of the reaction, which is similar to the degradation curve. The elimination of TOC is very efficient in the early stages of the reaction and significantly slows down afterward until it reaches a balance, which may be due to the following two points: (i) the generation of a series of intermediates; (ii) useless side reactions of Co2+/Co3+ and oxone. Moreover, the tailing of the TOC curve is likely attributed to the depletion of oxone. Undoubtedly, all of the above results are important to understanding the mechanisms of degradation.
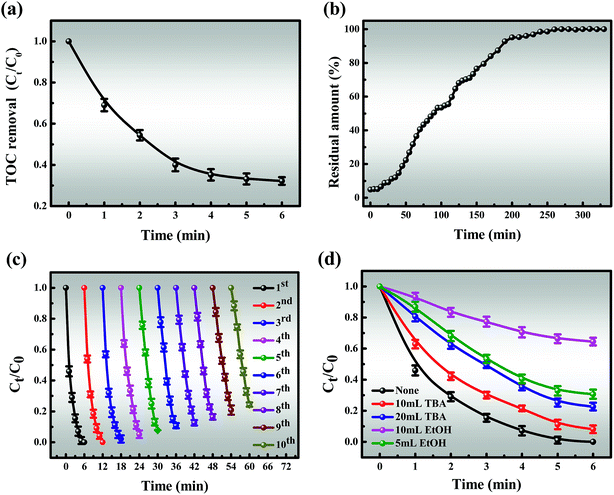 |
| | Fig. 8 (a) TOC removal of MB in the Co-NC-0.25-700 °C/PMS system; (b) column catalytic efficiency; (c) recyclability of the catalyst; (d) effect of radical scavengers on MB degradation. Reaction conditions: [MB] = 120 mg L−1 (for a, b, c and d), [PMS] = 3 mM (for a, b, c and d), catalyst = 0.4 g L−1 (for a, c and d), T = 298 K (for a, b, c and d), initial solution pH 7.62 (for a, b, c and d). | |
Notably, HPLC detection has great significance for the degradation of MB. Complete details are provided in Fig. S12,† and the characteristic absorption peak generated at the residual time of 2.9 minutes is attributed to the MB in the Co-NC-0.25-700 °C/PMS system, and its concentration is at a higher level (Fig. S12a†). As the reaction proceeds, the superior oxidizing agent produced by PMS can despoil benzene electrons and destroy original paired electrons to cause an electron transfer reaction. This is consistent with the observations from Fig. S12b–d,† where the MB absorption peak intensity gradually decreases, accompanied by the production of certain intermediates, indicating that the degradation process of MB is not a simple self-decomposition, but a complex advanced oxidation process that gradually evolves and decomposes layers. Finally, MB is almost completely decomposed and converted into a certain amount of intermediates, and H2O and CO2 are present at the end of the reaction (Fig. S12e†), which is highly consistent with UV-vis data. However, what the intermediate produced by the decomposition of MB is, and whether it is still harmful to the water system is still unclear, and we need to further explore this in future work.
3.4. Column catalytic efficiency and stability of the catalyst
The ability of continuous degradation is an indispensable factor in the practical application of the Co-NC-0.25-700 °C/PMS coupled process. As shown in Fig. 8b, the degradation rate is quicker in the initial stages of the column catalytic process, which can be attributed to the availability of abundant active sites on the surface of the catalyst. This is conducive to rapid contact between MB and the active sites. As the reaction proceeds, more active sites are covered so that the catalytic activity is slightly reduced, corresponding to the slope of the curve increasing. This, in turn, is attributed to the repulsion between MB molecules in the solid and bulk phases, resulting in a low degradation rate until equilibrium at 260 minutes. This is consistent with the kinetics of the reaction that take the shape of an inverted “S” after the reaction is finished.
The stability of the catalyst is a crucial estimate for its practical application, which was evaluated through consecutive operations, with the results shown in Fig. 8c. It was observed that even after 10 cycles, the MB degradation efficiency exceeded 86% within 6 min, which may give credit to the fact that the intermediates formed on the catalyst surface could not be cleaned, and masked the active sites. Furthermore, the active component may slightly leach from the catalyst, which leads to a decrease in catalytic activity. As a result, it can be seen that catalysts with low ion leaching are responsible for water treatment (Fig. S13†). The excellent stability of this egg-box structure, which can encapsulate CoxOy NPs in situ into C–N co-networks rather than on the surface, was further demonstrated. A possible tentative explanation is the fact that glutaraldehyde makes the intermolecular connection stronger and the dual action between SA/PEI and Co2+ makes the cobalt nanoparticles more stable in the catalyst.
3.5. Probable degradation and synergistic mechanism
In the Co-NC-0.25-700 °C/PMS coupled activation process, we follow a dual-reaction-site mechanism for the Fenton-like reaction with fast electron accumulation and depletion.14,62 Firstly, Co2+ can be released via aerobic oxidation of Co0 and reaction of Co0 with PMS.12,43 ˙OH and SO4˙− are generated with the cleavage of the peroxide bond (–O–O–) of PMS by accepting one electron from the Co2+. Meanwhile, Co3+ can be reduced back to Co2+ via a one-electron reduction process by PMS producing SO4˙− and can be deposited on the Co0 surfaces to release Co2+. Besides, ˙OH comes into being with the reaction between SO4˙− and water or OH−. Finally, SO4˙−/˙OH with powerful oxidizing properties can oxidize the target organic pollutant, MB, to the intermediates, innocuous CO2 and H2O. And the reaction mechanism might be as follows: (eqn (15)–(25))| | |
Co0 → Co2+ + 2e− E = 0.28 V
| (15) |
| |
2![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Co0 + O2 + 2H2O → 2Co2+ + 4OH− Co0 + O2 + 2H2O → 2Co2+ + 4OH−
| (16) |
| | |
Co0 + 2HSO5− → Co2+ + 2SO4˙− + 2OH−
| (17) |
| | |
Co0 + HSO5− + 2H+ → Co2+ + HSO4− + H2O
| (18) |
| | |
Co2+ + HSO5− → Co3+ + SO42− + ˙OH
| (19) |
| | |
Co2+ + HSO5− → Co3+ + SO4˙− + OH−
| (20) |
| | |
Co3+ + HSO5− → Co2+ + SO5˙− + H+
| (21) |
| | |
Co0 + 2Co3+ → 3 Co2+
| (22) |
| | |
SO4˙− + OH− → ˙OH + SO42−
| (23) |
| | |
SO4˙− + H2O → ˙OH + HSO4− → SO42− + ˙OH + H+
| (24) |
| | |
SO4˙−/˙OH + MB → intermediates + H2O + CO2
| (25) |
Co based catalysts demonstrate efficient degradation via PMS activation and the high standard redox potential of Co2+/Co3+ (E0 = +l.92 V vs. NHE) enables Co2+/Co3+ to effectively self-recycle, allowing the reaction to continue until all PMS is depleted. The possible synergistic mechanism of MB degradation in the Co-NC-0.25-700 °C/PMS system is illustrated in Scheme 2. At the very beginning, MB and PMS enter the highly porous and 3D interconnected carbonaceous beads by diffusion, which conforms to a “donor–acceptor complex” mechanism.62 A mass transfer driving force arises from the concentration gradient, which can be ascribed to the confinement effect of the “egg-box” structure as the reaction progresses.14 The unique “egg-box” structure effectively controls the aggregation and leaching of CoxOy NPs. Furthermore, the formation of amorphous carbon is advantageous for the transfer of electrons, since it will lead to a faster increase of the degradation rate with increasing SO4˙−/˙OH (eqn (26)–(28)).63
| | |
HSO5− + C → OH− + SO4˙− + AC+
| (26) |
| | |
HSO5− + C → ˙OH + SO42−+ AC+
| (27) |
| | |
HSO5− + C+ → H+ + SO5˙− + AC
| (28) |
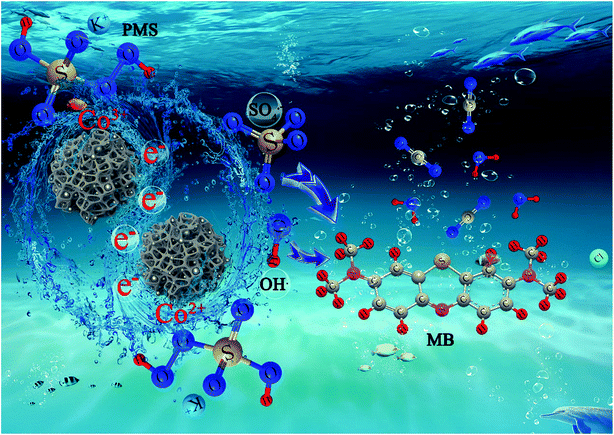 |
| | Scheme 2 The possible synergistic mechanisms of MB degradation with the Co-NC-0.25-700 °C system. | |
It should be pointed out that the substantial combination of N-doping and heat for modification of the C further increases the catalytic activity, for instance: (i) the graphitic nitrogen with higher electronegativity and a smaller covalent radius than the C atoms can not only improve the ability to adsorb HSO5− and break the O–O bond (HO–SO4−) but also induces electron transfer from adjacent carbon atoms to nitrogen;64,65 (ii) the pyridinic and pyrrolic N can be considered as Lewis basic reaction sites on account of the generation of SO4˙−and ˙OH.66,67
Classical radical scavenger experiments were performed by using TBA without α-H and EtOH with α-H as scavengers to confirm the contributions of ˙OH and SO4˙− during the degradation process. EtOH reacts with both ˙OH and SO4˙−at a similar rate (eqn (29) and (30)), but the reaction rate between TBA and ˙OH is over 1000-fold faster than that of TBA and SO4˙− (eqn (31) and (32)).11,50
| | |
EtOH + ˙OH → products k = (1.2–2.8) × 108 M−1 s−1
| (29) |
| | |
EtOH + SO4˙− → products k = (1.6–7.7) × 108 M−1 s−1
| (30) |
| | |
TBA + ˙OH → products k = (3.8–7.6) × 108 M−1 s−1
| (31) |
| | |
TBA + SO4˙− → products k = (4.0–9.1) × 105 M−1 s−1
| (32) |
As shown in Fig. 8d, the degradation efficiency for MB is slightly affected when adding 10 mL TBA after 6 min, revealing that the activation of PMS is slightly inhibited by TBA and ˙OH might be involved in MB degradation. In addition, the presence of 5 mL EtOH significantly suppresses the degradation process and only about 69.4% MB could be removed after 6 min. More noticeable inhibition effects are attributed to higher TBA and EtOH concentrations. Of particular interest is that the degradation efficiency decreased from 69.4 to 35.5% when the amount of EtOH was increased to 20 mL. As observed above, ˙OH and SO4˙− are the primary reactive species, but SO4˙− plays a major role in MB degradation, corresponding to the apparent rate constant.
4. Conclusion
In summary, seaweed-derived N-doped versatile carbonaceous beads with CoxOy were successfully prepared through simple sol–gel assembly and pyrolysis procedures for the activation of PMS. With the improved structure–activity relationship and the synergistic effects between the C–N skeleton and the CoxOy nanoparticles, Co-NC-0.25-700 °C exhibited remarkable degradation performance for a variety of organic pollutants such as organic dyes, antibiotic compounds, and phenolic compounds. Fascinatingly, MB degradation based on Co-NC-0.25-700 °C/PMS was minimally affected by a wide pH range (4–10) and common ions (Cl−, NO3−, SO42−, etc.). Radical quenching experiments revealed that SO4˙− plays a more crucial role in the Co-NC-0.25-700 °C/PMS system than ˙OH. Cycle and cobalt ion leaching test results indicated the outstanding stability and recoverability of Co-NC-0.25-700 °C. Of particular interest is that the as-prepared catalysts, when implemented in simulated actual conditions and in a column reactor designed for a continuous flow reaction, exhibited relatively excellent catalytic activity and long-term operational stability. This study will provide a new avenue, on the basis of PMS AOPs, for a further step towards the universal application of organic pollutant treatment.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21676039), State Key Laboratory of Bio-Fibers and Eco-Textiles (2017kfkt12), and Innovative Talents in Liaoning Universities and Colleges (LR2017045) are highly appreciated.
References
- Q. X. Yang, X. F. Yang, Y. Yan, C. Sun, H. J. Wu, J. He and D. S. Wang, Chem. Eng. J., 2018, 348, 263–270 CrossRef CAS.
- W. J. Ren, J. K. Gao, C. Lei, Y. B. Xie, Y. R. Cai, Q. Q. Ni and J. M. Yao, Chem. Eng. J., 2018, 349, 766–774 CrossRef CAS.
- J. P. Sheng, L. Q. Wang, L. Deng, M. Zhang, H. C. He, K. Zeng, F. Y. Tang and Y. N. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 7191–7200 CrossRef CAS PubMed.
- N. Wang, W. Ma, Z. Q. Ren, Y. C. Du, P. Xu and X. J. Han, J. Mater. Chem. A, 2018, 6, 884–895 RSC.
- A. Khan, H. B. Wang, Y. Liu, A. Jawad, J. Ifthika, Z. W. Liao, T. Wang and Z. Q. Chen, J. Mater. Chem. A, 2018, 6, 1590–1600 RSC.
- A. Khan, S. H. Zou, T. Wang, J. Ifthikar, A. Jawad, Z. W. Liao, A. Shahzad, A. Ngambia and Z. Q. Chen, Phys. Chem. Chem. Phys., 2018, 20, 13909–13919 RSC.
- D. N. Zhou, L. Chen, J. J. Li and F. Wu, Chem. Eng. J., 2018, 346, 726–738 CrossRef CAS.
- Y. Q. Pi, L. J. Ma, P. Zhao, Y. D. Cao, H. Q. Gao, C. F. Wang, Q. L. Li, S. Y. Dong and J. H. Sun, J. Colloid Interface Sci., 2018, 526, 18–27 CrossRef CAS PubMed.
- D. M. Wu, P. Ye, M. Y. Wang, Y. Wei, X. X. Li and A. H. Xu, J. Hazard. Mater., 2018, 352, 148–156 CrossRef CAS PubMed.
- S. Lu, G. l. Wang, S. Chen, H. T. Yu, F. Ye and X. Quan, J. Hazard. Mater., 2018, 353, 401–409 CrossRef CAS PubMed.
- Y. X. Liu, R. Luo, Y. Li, J. W. Qi, C. H. Wang, J. S. Li, X. Y. Sun and L. J. Wang, Chem. Eng. J., 2018, 347, 731–740 CrossRef CAS.
- G. Farshid and M. Mahsa, Chem. Eng. J., 2017, 310, 41–62 CrossRef.
- R. Luo, C. Liu, J. S. Li, C. H. Wang, X. Y. Sun, J. Y. Shen, W. Q. Han and L. J. Wang, J. Mater. Chem. A, 2018, 6, 3454–3461 RSC.
- M. Zhang, C. H. Wang, C. Liu, R. Luo, J. S. Li, X. Y. Sun, J. Y. Shen, W. Q. Han and L. J. Wang, J. Mater. Chem. A, 2018, 6, 11226–11235 RSC.
- X. Zhao, Q. D. An, Z. Y. Xiao, S. R. Zhai and Z. Shi, Chin. J. Catal., 2018, 39, 1842–1853 CrossRef CAS.
- H. H. Yang, R. F. Nie, W. Xia, X. L. Yu, D. F. Jin, X. H. Lu, D. Zhou and Q. H. Xia, Green Chem., 2017, 19, 5714–5722 RSC.
- X. Zhao, Q. D. An, Z. Y. Xiao, S. R. Zhai and Z. Shi, Chem. Eng. J., 2018, 353, 746–759 CrossRef CAS.
- Y. D. Liu, A. G. Zhou, Y. Q. Gan and X. Q. Li, J. Hazard. Mater., 2018, 344, 98–103 CrossRef CAS PubMed.
- F. Song, H. Y. Zhang, S. B. Wang, L. H. Liu, X. Y. Tan and S. M. Liu, Chem. Commun., 2018, 54, 4919–4922 RSC.
- R. Y. Xiao, Z. H. Luo, Z. S. Wei, S. Luo, R. Spinney, W. C. Yang and D. D. Dionysiou, Curr. Opin. Chem. Eng., 2018, 19, 51–58 CrossRef.
- P. Q. Yin, T. Yao, Y. E. Wu, L. R. Zheng, Y. Lin, W. Liu, H. X. Ju, J. F. Zhu, X. Hong, Z. X. Deng, G. Zhou, S. Q. Wei and Y. D. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- J. Sun, C. Lv, F. Lv, S. Chen, D. H. Li, Z. Q. Guo, W. Han, D. J. Yang and S. J. Guo, ACS Nano, 2017, 11, 6186–6193 CrossRef CAS PubMed.
- M. B. Ahmed, J. L. Zhou, H. H. Ngo, W. S. Guo and M. F. Chen, Bioresour. Technol., 2016, 214, 836–851 CrossRef CAS PubMed.
- L. Jiao, G. Wan, R. Zhang, H. Zhou, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 8525–8529 CrossRef CAS PubMed.
- E. K. Rideal and W. M. Wright, J. Chem. Soc., 1926, 129, 1813–1821 RSC.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- L. Liu, X. F. Yang, N. Ma, H. T. Liu, Y. Z. Xia, C. M. Chen, D. J. Yang and X. D. Yao, Small, 2016, 12, 1295–1301 CrossRef CAS PubMed.
- N. Zhou, Q. D. An, W. Zheng, Z. Y. Xiao and S. R. Zhai, RSC Adv., 2016, 6, 98128–98140 RSC.
- Y. Z. Yan, Q. D. An, Z. Y. Xiao, W. Zheng and S. R. Zhai, Chem. Eng. J., 2017, 313, 475–486 CrossRef CAS.
- C. Bertagnolli, A. Grishin, T. Vincent and E. Guibal, Ind. Eng. Chem. Res., 2016, 55, 2461–2470 CrossRef CAS.
- M. Monier, D. M. Ayad, Y. Wei and A. A. Sarhan, J. Hazard. Mater., 2010, 177, 962–970 CrossRef CAS PubMed.
- D. M. Guo, Q. D. An, Z. Y. Xiao, S. R. Zhai and D. J. Yang, Carbohydr. Polym., 2018, 202, 306–314 CrossRef CAS PubMed.
- Y. L. Niu, X. Q. Huang, X. S. Wu, L. Zhao, W. H. Hu and C. M. Li, Nanoscale, 2017, 9, 10233–10239 RSC.
- H. Y. Zhao and L. Zhao, Dalton Trans., 2018, 47, 3321–3328 RSC.
- Y. N. Duan, T. Song, X. S. Dong and Y. Yang, Green Chem., 2018, 20, 2821–2828 RSC.
- S. Nandi, P. Patel, N.-u. H. Khan, A. V. Biradar and R. I. Kureshy, Inorg. Chem. Front., 2018, 5, 806–813 RSC.
- L. H. Ai, X. Y. Gao and J. Jiang, J. Power Sources, 2014, 257, 213–220 CrossRef CAS.
- Z. Z. Liang, C. C. Zhang, H. T. Yuan, W. Zhang, H. Q. Zheng and R. Cao, Chem. Commun., 2018, 54, 7519–7522 RSC.
- W. J. Tian, H. Y. Zhang, Z. Qian, T. H. Ouyang, H. Q. Sun, J. Y. Qin, M. O. Tadé and S. B. Wang, Appl. Catal., B, 2018, 225, 76–83 CrossRef CAS.
- J. Yu, G. Chen, J. Sunarso, Y. L. Zhu, R. Ran, Z. H. Zhu, W. Zhou and Z. P. Shao, Adv. Sci., 2016, 3, 1600060 CrossRef PubMed.
- Y. J. Yao, C. Xu, J. C. Qin, F. Y. Wei, M. N. Rao and S. B. Wang, Ind. Eng. Chem. Res., 2013, 52, 17341–17350 CrossRef CAS.
- R. Luo, C. Liu, J. S. Li, J. Wang, X. R. Hu, X. Y. Sun, J. Y. Shen, W. Q. Han and L. J. Wang, J. Hazard. Mater., 2017, 329, 92–101 CrossRef CAS PubMed.
- Z. G. Zhu, C. H. Ji, L. L. Zhong, S. Liu, F. Y. Cui, H. L. Sun and W. Wang, J. Mater. Chem. A, 2017, 5, 18071–18080 RSC.
- Y. P. Zhu, T. Z. Ren and Z. Y. Yuan, Nanoscale, 2014, 6, 11395–11402 RSC.
- F. Yang, B. B. Wang, H. Su, S. J. Zhou and Y. Kong, Mater. Chem. Front., 2017, 1, 2065–2077 RSC.
- C. Cai, H. Zhang, X. Zhong and L. W. Hou, J. Hazard. Mater., 2015, 283, 70–79 CrossRef CAS PubMed.
- P. H. Shi, X. F. Dai, H. A. Zheng, D. X. Li, W. F. Yao and C. Y. Hu, Chem. Eng. J., 2014, 240, 264–270 CrossRef CAS.
- G. Wang, S. Chen, X. Quan, H. T. Yu and Y. B. Zhang, Carbon, 2017, 115, 730–739 CrossRef CAS.
- X. Wang, L. G. Wang, J. B. Li, J. J. Qiu, C. Cai and H. Zhang, Sep. Purif. Technol., 2014, 122, 41–46 CrossRef CAS.
- K. Y. A. Lin and Z. Y. Zhang, Chem. Eng. J., 2017, 313, 1320–1327 CrossRef CAS.
- Z. Z. Liu, S. J. Yang, Y. N. Yuan, J. Xu, Y. F. Zhu, J. J. Li and F. Wu, J. Hazard. Mater., 2017, 324, 583–592 CrossRef CAS PubMed.
- W. L. Guo, S. N. Su, C. L. Yi and Z. M. Ma, Environ. Prog. Sustainable Energy, 2013, 32, 193–197 CrossRef CAS.
- Y. H. Huang, Y. F. Huang, C. i. Huang and C. Y. Chen, J. Hazard. Mater., 2009, 170, 1110–1118 CrossRef CAS PubMed.
- Y. J. Yao, H. Chen, J. C. Qin, G. D. Wu, C. Lian, J. Zhang and S. B. Wang, Water Res., 2016, 101, 281–291 CrossRef CAS PubMed.
- A. H. Xu, X. X. Li, S. Ye, G. C. Yin and Q. F. Zeng, Appl. Catal., B, 2011, 102, 37–43 CrossRef CAS.
- Y. R. Wang and W. Chu, J. Hazard. Mater., 2011, 186, 1455–1461 CrossRef CAS PubMed.
- L. M. Hu, G. S. Zhang, M. Liu, Q. Wang and P. Wang, Chem. Eng. J., 2018, 338, 300–310 CrossRef CAS.
- X. P. Tian, P. P. Gao, Y. L. Nie, C. Yang, Z. X. Zhou, Y. Li and Y. X. Wang, Chem. Commun., 2017, 53, 6589–6592 RSC.
- S. Luo, Z. S. Wei, D. D. Dionysiou, R. Spinney, W. P. Hu, L. Y. Chai, Z. H. Yang, T. T. Ye and R. Y. Xiao, Chem. Eng. J., 2017, 327, 1056–1065 CrossRef CAS.
- X. N. Li, X. Huang, S. B. Xi, S. Miao, J. Ding, W. Z. Cai, S. Liu, X. L. Yang, H. B. Yang, J. J. Gao, J. H. Wang, Y. Q. Huang, T. Zhang and B. Liu, J. Am. Chem. Soc., 2018, 140(39), 12469–12475 CrossRef CAS PubMed.
- X. G. Duan, Z. M. Ao, D. G. Li, H. Q. Sun, L. Zhou, A. Suvorova, M. Saunders, G. X. Wang and S. B. Wang, Carbon, 2016, 103, 404–411 CrossRef CAS.
- X. G. Duan, Z. M. Ao, H. Q. Sun, S. Indrawirawan, Y. X. Wang, J. Kang, F. L. Liang, Z. H. Zhu and S. B. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 4169–4178 CrossRef CAS PubMed.
- D. H. Deng, X. L. Pan, L. Yu, Y. Cui, Y. P. Jiang, J. Qi, W. X. Li, Q. Fu, X. C. Ma, Q. K. Xue, G. Q. Sun and X. H. Bao, Chem. Mater., 2011, 23, 1188–1193 CrossRef CAS.
- Z. S. Wu, S. B. Yang, Y. Sun, K. Parvez, X. L. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- S. Z. Liu, W. C. Peng, H. Q. Sun and S. B. Wang, Nanoscale, 2014, 6, 766–771 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00357f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Junmei Luo,
Zuoyi Xiao
*,
Junmei Luo,
Zuoyi Xiao