DOI:
10.1039/C9RA00483A
(Paper)
RSC Adv., 2019,
9, 15224-15228
Design and synthesis of acetaminophen probe APAP-P1 for identification of the toxicity targets thioredoxin reductase-1 in HepaRG cells†
Received
20th January 2019
, Accepted 21st April 2019
First published on 15th May 2019
Abstract
Drug-induced liver injury is one of the main causes of drug non-approval and drug withdrawal by the Food and Drug Administration (FDA). Acetaminophen (APAP) is a widely used non-steroidal anti-inflammatory drug for treating fever and headache. APAP is considered safe at therapeutic doses; however, there have been reports of acute liver injury following the administration of APAP. To explore APAP hepatotoxicity and its mechanisms, we designed and synthesized a new click chemistry probe, APAP-P1, in our current study. We introduced the PEG-azide probe linker into the acetyl group of acetaminophen. First, we evaluated the probe toxicity in HepaRG cells and found that it still retained hepatotoxicity. We also found that this probe APAP-P1 can be metabolized by HepaRG cells. This demonstrated that the APAP-P1 probe still kept its metabolism characteristics. Using this probe, we pulled down its potential targets in vivo and in vitro. APAP can directly target TrxR1; thus, we tested for this interaction by Western blotting of pull-down proteins. The results showed that APAP-P1 can pull down TrxR1 in vivo and in vitro.
Introduction
Drug-induced liver injury (DILI) is an unpredictable adverse drug reaction that affects only a small subset of treated patients and remains an increasingly recognized cause of hepatotoxicity and liver failure worldwide.1–3 Acetaminophen (APAP) is a widely used agent for the treatment of pain and fever around the world and is considered safe at therapeutic doses; however, overdose can cause severe hepatotoxicity leading to acute liver failure, which is the most common cause of drug-induced liver damage in the world.4,5 APAP causes potentially fatal hepatocellular necrosis when taken in nontherapeutic doses.6 APAP can undergo metabolism to form the reactive electrophile N-acetyl-p-benzoquinone imine (NAPQI), which causes extensive and rapid glutathione depletion, mitochondrial dysfunction, oxidant stress, peroxynitrite formation and nuclear DNA fragmentation in hepatocytes.1,7–9 Its molecular toxicology was proposed to be initiated by reactive oxygen species (ROS) formation in the mitochondria, leading to the subsequent activation of apoptosis signalling-regulating kinase 1 (ASK1) and the c-Jun N-terminal kinase (JNK) pathway.10,11 Thioredoxin reductase-1 (TrxR1), encoded by the Txnrd1 gene, is a NADPH-dependent enzyme and is known to be related to oxidative stress and the regulation of cell growth and development. Some studies have found that the GSH- or TrxR1-dependent redox pathway can independently support hepatocyte proliferation during liver growth.12 Previous research has found that NAPQI directly inactivates TrxR1, which may be associated with enhanced Nrf2 activity.13,14 TrxR1 is important for attenuating the activation of apoptosis signaling-regulating kinase 1 (ASK1) and the c-JunN-terminal kinase (JNK) pathway, which is caused by high doses of APAP.15 The Trx system impacts bioenergetics and drug metabolism, and some studies have indicated it to be a potential marker of DILI, as in the case of anti-tuberculosis drugs.16,17
HepaRG cells express most of the enzymes and transcription factors involved in xenobiotic biotransformation and transport and have been shown to be a valuable model for studying the mechanism of hepatotoxicity induced by drugs and toxins.18 Research has demonstrated that HepaRG cells can be used to explore the mechanisms of APAP hepatotoxicity.19 APAP is converted to the reactive intermediate NAPQI in HepaRG cells, which has been thought to be the toxin for hepatotoxicity.20 In our research, we chose the HepaRG cell line as our in vitro model to evaluate the liver damage caused by APAP.
Chemical proteomics is a growing area of chemical biology that seeks to design small molecule probes to understand protein function.21 Activity-based protein profiling (ABPP) has emerged as a powerful functional chemical proteomic strategy that enables global profiling of proteome reactivity toward bioactive small molecules in complex biological and/or pathological processes.22 Activity-based probes (ABPs) usually consist of three fundamental building blocks with distinct functions: (1) an active warhead (WH) that contains reactive groups that target the conserved mechanistic or structural feature of a set of proteins; (2) a reporter tag for identification and purification of modified proteins; and (3) a linker region that can modulate the reactivity and specificity of the reactive group and binding group while providing enough space between the reporter and the reactive or binding group.23,24 In this study, we designed and synthesized a new ABPP probe, APAP-P1, for the identification of APAP toxicity targets in HepaRG cells.
Results and discussion
Chemistry
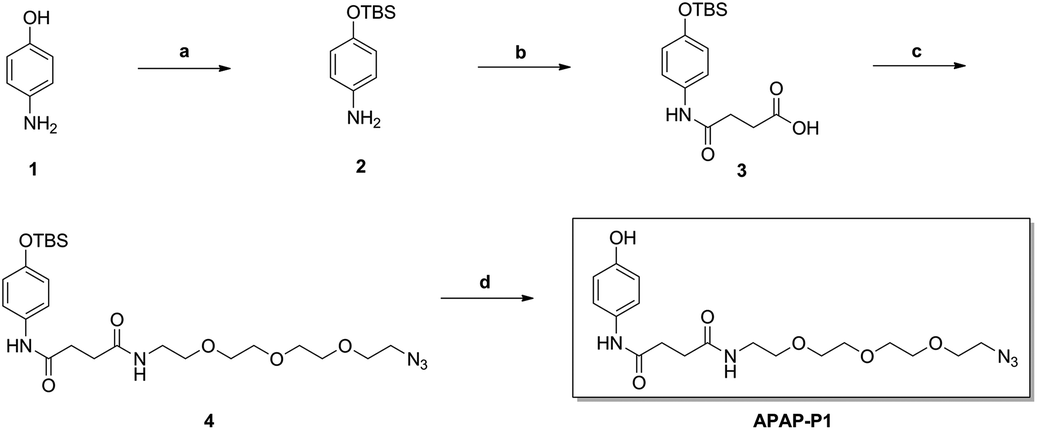
As illustrated in Scheme 1, commercially available p-aminophenol (1) was treated with tert-butyldimethylsilyl chloride (TBSCl) and imidazole in dry tetrahydrofuran (THF) to obtain a good yield of intermediate 2. Intermediate 2 reacted with succinic anhydride (SAA) in organic alkali conditions with dimethylaminopyridine (DMAP) to provide compound 3, which was subjected to an acylation reaction to obtain compound 4 in the presence of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) and DMAP. Then, APAP-P1 was obtained via the deprotection reaction of intermediate 4 with tetrabutylammonium fluoride in THF solution.
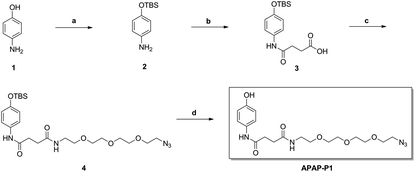 |
| | Scheme 1 The synthesis of toxic probe APAP-P1. Reagents and conditions: (a) TBSCl, imidazole, THF; (b) succinic anhydride (SAA), DMAP, DCM; (c) EDCI, DMAP, DCM; (d) TBAF, THF. | |
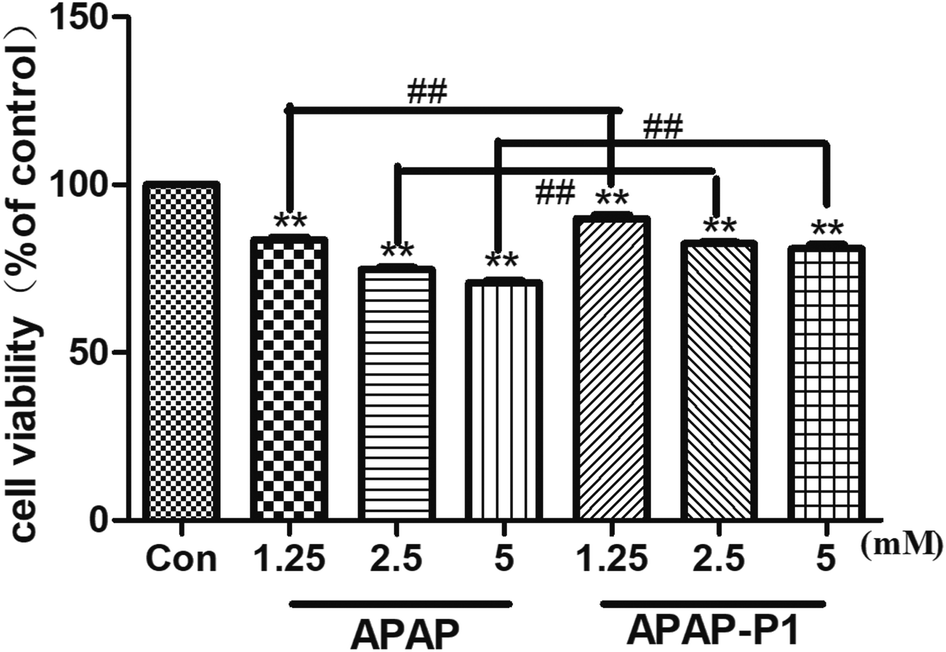
Cytotoxicity of APAP-P1 Administered to HepaRG cell line. To study the mechanism of APAP hepatotoxicity in humans, the cell line HepaRG is a useful human model for the study of APAP-induced liver injury.19 After the probe design, we needed to evaluate its associated activities to ensure that this linker didn't change its hepatotoxicity, so the probe could be used for further experiments. First, to investigate the cytotoxic effects of various concentrations of APAP and APAP-P1 in the HepaRG cell line, we performed MTT assays after 24 h of treatment, and the results showed that both APAP and APAP-P1 inhibited cell viability in a dose-dependent manner (Fig. 1). The results showed that APAP-P1 cytotoxicity was lower than that of APAP at the same concentration; however, APAP-P1 had similar effects as APAP. This demonstrated that APAP-P1 can still maintain cytotoxicity after inducing the PEG-azide probe linker into the acetyl group of APAP. With this probe generated, we moved forward to APAP-P1 cytotoxicity target identification.
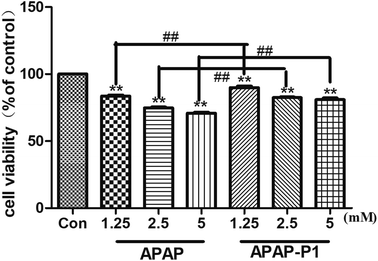 |
| | Fig. 1 Evaluation of the cytotoxicity of APAP and APAP-P1 in HepaRG cells. Cell viability of HepaRG cells incubated with different concentrations (1.25, 2.5, 5 mM) of APAP and the same concentrations (1.25, 2.5, 5 mM) of APAP-P1 for 24 h. The data are expressed as the mean ± SD from three independent experiments. ##P < 0.01 versus the same concentration group, **P < 0.01 versus control group. | |
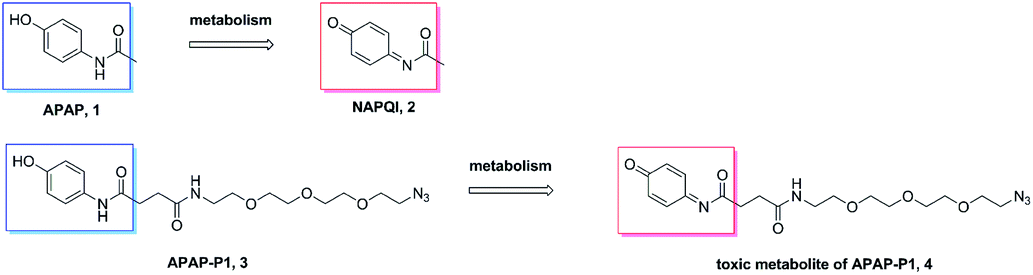
Metabolism of APAP-P1. A previous study demonstrated that APAP can be metabolized to NAPQI in HepaRG cells,20 NAPQI was the main hepatotoxic intermediate. To explore whether APAP-P1 can be metabolized to NAPQI analogue by HepaRG cells, we incubated 5 mM APAP-P1 with HepaRG cells for 24 h and then collected the cells and medium for UPLC-Q-TOF/MS analysis. Our results showed that the APAP-P1 (3) probe was metabolized to the toxic metabolite APAP-P1 (4) (Scheme 2). This also demonstrated that the APAP-P1 probe retained toxic characteristics after introduction of the PEG-azide probe linker. So these above results demonstrated APAP-P1 can be used for hepatotoxic targets fishing and identification.
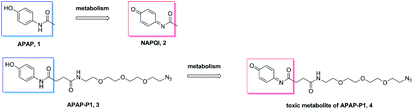 |
| | Scheme 2 APAP and APAP-P1 metabolism pathway. | |
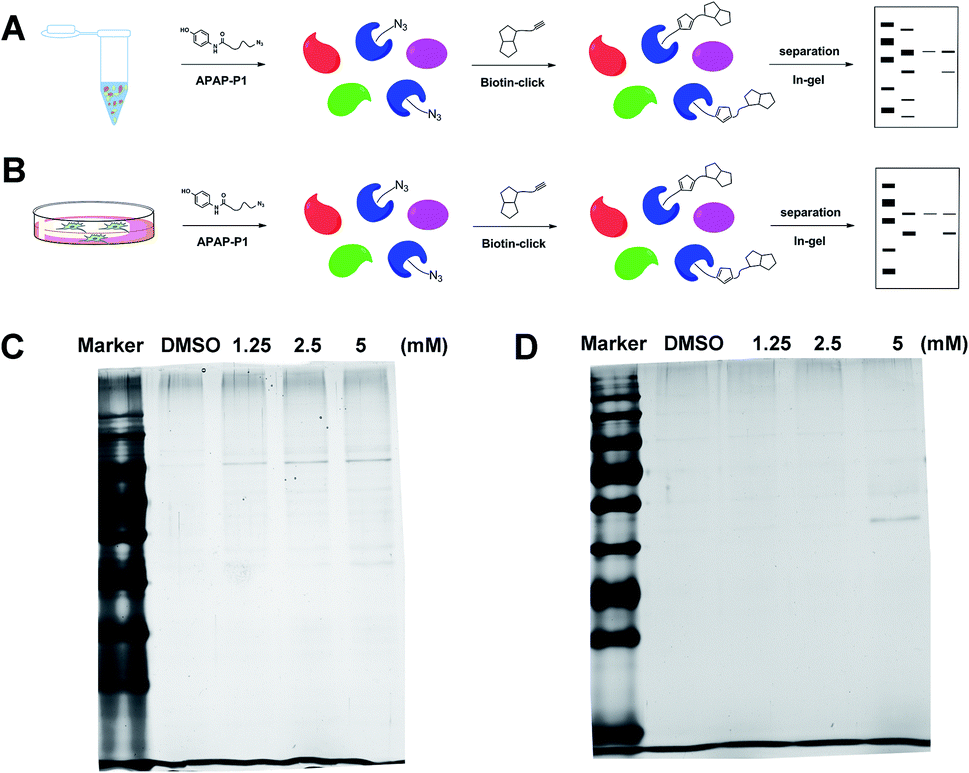
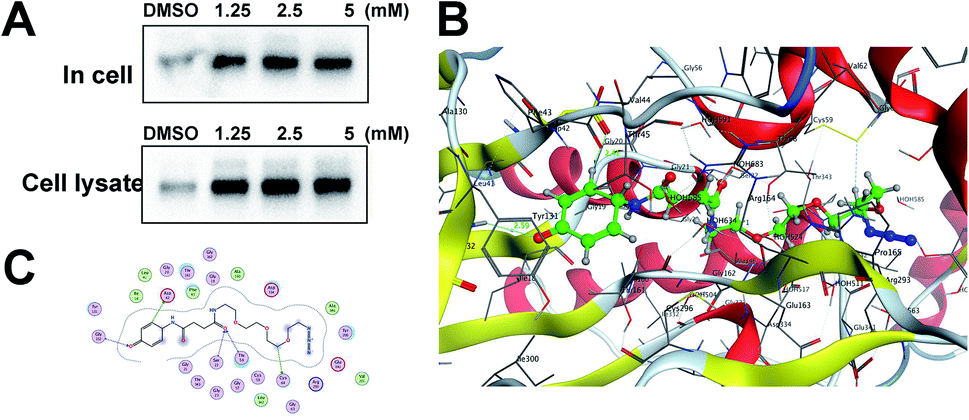
Gel-based CC-ABPP profiling of APAP-interacting proteins in cell lysate and living cells. With the APAP-P1 probe in hand, we then evaluated its labelling efficiency in cell lysate and living cells by silver staining. The workflow in Fig. 2A lists the APAP-P1 CC-ABPP experiment step-by-step in HepaRG cell lysate. HepaRG cell lysates were treated with different concentrations of APAP-P1 (1.25, 2.5, 5 mM) for 1 h to form the probe–protein complex. The labelled cell lysates were conjugated with biotin-alkynyl via CuAAC and separated by SDS-PAGE. The results revealed clear and distinct labelling protein profiling for different concentration probes (Fig. 2C), which suggested that the APAP-P1 probe could pull down the proteomes in a dose-dependent manner. APAP can be converted to the reactive intermediate NAPQI. To explore its protein profiling in cells, we also designed a CC-ABPP experiment in living HepaRG cells, as shown in Fig. 2B. We first incubated the probe with HepaRG cells for 24 h, then the labelled protein–probe complex were conjugated with biotin-alkynyl via CuAAC and the proteins were pulled down and separated by SDS-PAGE. Finally, we found that 5 mM APAP-P1 pulled down a specific 42–55 kDa band.
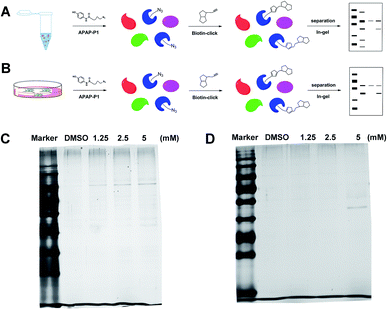 |
| | Fig. 2 Gel-based CC-ABPP profiling of APAP-interacting proteins in cell lysate and living cells. (A) Workflow for the gel-based CC-ABPP profiling of APAP-P1 in HepaRG cell lysate. (B) Workflow for the gel-based CC-ABPP profiling of APAP-P1 in living HepaRG cells. (C) Protein profiling of the APAP-P1 probe in HepaRG cell lysates was separated by SDS-PAGE and stained by silver staining. (D) Protein profiling of the APAP-P1 probe in living HepaRG cells was separated by SDS-PAGE and stained by silver staining. Identification of TrxR1 as the APAP potential toxicity target. | |
From our results, we found that APAP-P1 has a similar protein profiling in HepaRG cell lysates and in HepaRG cells. APAP-P1 can pull down 42–55 kDa bands; however, there are obvious differences. For the cell lysate, the probe mainly pulled down the 100 kDa protein bands, but 42–55 kDa bands were more obvious in cells. These results demonstrated that after APAP-P1 metabolism, its interaction protein profile was changed. This suggests that we should focus more on the APAP metabolism for hepatotoxicity. With this useful probe, we will perform additional in vivo research in the future.
Identification of TrxR1 as the APAP potential toxicity target. Previous studies have shown that TrxR1 is a potential target for NAPQI.15,25 To explore whether the probe could be used for further in vivo research, we tested the APAP-P1 pull-down proteins TrxR1. APAP-P1 pulled down TrxR1 both in cells and in cell lysate (Fig. 3A). We used the docking assay to predict the binding model. APAP-P1 was docked into the active pocket of 3QFA. The most reasonable binding mode was chosen based on the compatibility with the reported protein structure and binding data, as well as energetic considerations. The in silico-predicted data showed that APAP-P1 was located in the pocket of the TrxR1 3D structure (Fig. 3B), and the position of APAP-P1 in the pocket of TrxR1 is depicted. The surface of APAP-P1 possesses the same orientation, which could exactly map the pocket. Hydrogen bonds were formed at Ser-22, Asp-42, Thr-58, Cys-64, and Gly-132 in TrxR1 (Fig. 3C), which might be the key residues in the interaction between APAP-P1 and TrxR1.
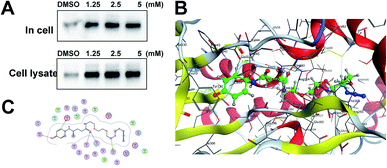 |
| | Fig. 3 Identification of Txnrd1 as the APAP potential toxicity target. (A) Western blotting validation of the APAP-P1 target TrxR1 in cell lysates and in cells after performing a pull-down assay. (B) Binding mode of APAP-P1 in the 3D structure of TrxR1. (C) Ser-22, Asp-42, Thr-58, Cys-64, and Gly-132 may be the key residues in the interaction between APAP-P1 and TrxR1. | |
Experimental
Chemistry
Procedure for the synthesis of intermediate 2. To a solution of p-aminophenol (1) (3.0 g, 27.5 mmol) in dry DCM (20 mL), TBSCl (5.4 g, 36.1 mmol) and imidazole (2.96 g, 43.5 mmol) were added at 0 °C. Then the reaction mixture was stirred at room temperature for 3 h. Reaction was monitored by TLC. The crude mixture was separated and the water layer was extracted with DCM (3 × 20 mL). The combined organic layer was evaporated and purified through column chromatography (eluent![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PE-EtOAc, 1:1) to offer pure white solid compound 2, 93% yield; 1H-NMR (600 MHz, CDCl3) δ: 6.69–6.68 (m, 2H, Ph-H), 6.58–6.56 (m, 2H, Ph-H), 3.45 (s, 2H, NH2), 1.01 (s, 9H, C-(CH3)3), 0.19 (m, 6H, Si-(CH3)2); 13C-NMR (150 MHz, CDCl3) δ: 148.0, 140.4, 120.6, 116.3, 25.8, 18.2, −4.48; HRMS (ESI): calcd for [M + H]+ C12H22NOSi: 224.1471, found 224.1484.
:PE-EtOAc, 1:1) to offer pure white solid compound 2, 93% yield; 1H-NMR (600 MHz, CDCl3) δ: 6.69–6.68 (m, 2H, Ph-H), 6.58–6.56 (m, 2H, Ph-H), 3.45 (s, 2H, NH2), 1.01 (s, 9H, C-(CH3)3), 0.19 (m, 6H, Si-(CH3)2); 13C-NMR (150 MHz, CDCl3) δ: 148.0, 140.4, 120.6, 116.3, 25.8, 18.2, −4.48; HRMS (ESI): calcd for [M + H]+ C12H22NOSi: 224.1471, found 224.1484.
Procedure for the synthesis of intermediate 3. To a solution of compound 2 (0.5 g, 2.25 mmol) in dry DCM (10 mL), and succinic anhydride (0.25 g, 2.48 mmol), catalytic amount of DMAP (27.5 mg, 0.225 mmol) and triethylamine (0.62 mL, 4.5 mmol) were added. Reaction was monitored by TLC. Then the reaction mixture was stirred at room temperature for 12 h until its completion. The crude mixture was evaporated and purified through column chromatography (eluent:EtOAc) to offer pure white solid compound 3, 81% yield; 1H-NMR (600 MHz, CDCl3) δ: 8.29 (s, 1H, NH), 7.32–7.31 (m, 2H, Ph-H), 6.72–6.70 (m, 2H, Ph-H), 2.63 (m, 2H, CH2COOH), 2.53 (m, 2H, CH2CONH), 0.95 (s, 9H, C-(CH3)3), 0.14 (m, 6H, Si-(CH3)2); 13C-NMR (150 MHz, CDCl3) δ: 177.9, 170.9, 152.5, 131.5, 122.0, 120.4, 31.8, 30.1, 25.8, 18.3, −4.35; HRMS (ESI): calcd for [M + Na]+ C16H25NNaO4Si: 346.1451, found 346.1476.
Procedure for the synthesis of intermediate 4. A mixture of compound 3 (0.23 g, 0.68 mmol), DMAP (8.3 mg, 0.068 mmol) and EDCI (0.19 g, 1 mmol) was stirred in dry DCM 10 mL, then 198 μL amine (1 mmol) was dropped, and stirred under N2 until completion. The mixture was evaporated and purified through column chromatography (eluent:DCM-MeOH, 20:1) to offer pure white solid compound 4, 65% yield; 1H-NMR (600 MHz, CDCl3) δ: 8.66 (s, 1H, CONHPh), 7.37–7.35 (m, 2H, Ph-H), 6.75–6.73 (m, 2H, Ph-H), 6.67 (t, J = 5.7 Hz, 1H, CH2CONH), 3.66–3.57 (m, 10H, CH2(OCH2CH2)2), 3.53–3.51 (m, 2H, CH2O), 3.44–3.42 (m, 2H, CH2N3), 3.38–3.37 (m, 2H, CONHCH2), 2.67–2.65 (m, 2H, CONHCH2), 2.61–2.59 (m, 2H, CH2CONH), 0.95 (s, 9H, C-(CH3)3), 0.15 (m, 6H, Si-(CH3)2); 13C-NMR (150 MHz, CDCl3) δ: 172.7, 170.5, 152.0, 132.2, 121.3, 120.3, 70.7, 70.7, 70.6, 70.3, 70.1, 69.8, 50.7, 39.5, 32.9, 31.7, 25.8, 18.3, −4.38; HRMS (ESI): calcd for [M + Na]+ C24H41N5NaO6Si: 546.2724, found 546.2753.
Procedure for the synthesis of intermediate APAP-P1. To a solution of compound 4 (0.24 g, 0.47 mmol) in dry tetrahydrofuran (THF) 5 mL, TBAF (321 μL, 1.17 mmol) was slowly dropped at 0 °C and reacted for 2 h. When complete, the crude product was evaporated and subjected to column chromatography (eluent:EtOAc), to offer pure white solid APAP-P1, 90% yield; 1H-NMR (600 MHz, CDCl3) δ: 8.68 (s, 1H, Ph-OH), 7.92 (s, 1H, CONHPh), 7.20–7.19 (m, 2H, Ph-H), 6.74 (t, J = 5.3 Hz, 1H, CH2CONH), 6.69–6.68 (m, 2H, Ph-H), 3.63–3.55 (m, 10H, CH2(OCH2CH2)2), 3.48–3.47 (m, 2H, CH2O), 3.38–3.33 (m, 4H, CH2N3, CONHCH2), 2.64–2.62 (m, 2H, CONHCH2), 2.56–2.54 (m, 2H, CH2CONH); 13C-NMR (150 MHz, CDCl3) δ: 172.9, 170.9, 153.4, 130.5, 122.5, 115.7, 70.7, 70.7, 70.6, 70.3, 70.1, 69.7, 50.8, 39.6, 32.5, 31.5; HRMS (ESI): calcd for [M + Na]+ C18H27N5NaO6: 432.1859, found 432.1875.
Metabolism research by UPLC-Q-TOF/MS analysis. To explore the metabolism of APAP-P1 in HepaRG cells, we incubated DMSO or 5 mM APAP-P1 with HepaRG cells for 24 h, then collected the cells and medium.20 All of the in vitro incubation samples were prepared by the same methods as follows. To a 200 μL aliquot of the in vitro incubations, 200 μL of ethanol was added. This sample was vortex-mixed and centrifuged at 14000g for 5 min. The supernatant was transferred into a glass tube, evaporated to dryness under a stream of nitrogen.26 Then the dried metabolites extract was dissolved in 300 μL of methanol, and an aliquot of 2 μL was injected for UPLC/MS analysis. The lysate and cell samples were analysed on a Waters Acquity™ Ultra Performance LC system (Waters Corporation, Milford, MA, USA) equipped with a BEH C18 column (100 mm × 2.1 mm, 1.7 μm). The autosampler temperature was kept at 4 °C and the column compartment was set at 40 °C. The mobile phase was composed of solvents A (2 mM ammonium acetate in 95% H2O/5% acetonitrile + 0.1% acetic acid) and B (2 mM ammonium acetate in 95% acetonitrile/5% H2O + 0.1% acetic acid). A Waters SYNAPT G2 HDMS (Waters Corp., Manchester, UK) was used to carry out the mass spectrometry with an electrospray ionization source (ESI) operating in positive ion modes. The UPLC-Q-TOF/MS raw data were processed by MarkerLynx Applications Manager version 4.1 (Waters, Manchester, UK). The procedure included integration, normalization, and alignment of peak intensities, and a list of m/z and retention times with corresponding peaks was provided for all metabolites in every sample in the data set.
Docking research. Docking experiment was employed to explore the binding modes of APAP-P1 with TrxR1 by MOE software (Chemical Computing Group, Inc). The 3D structure of APAP-P1 was energy minimized by MOE using the MMFF94x force field, and the protein TrxR1 was energy minimized by MOE using the Amber 10 EHT force field. The interactive forces between the ligand and receptor primarily included hydrogen bonding, aromatic interactions and hydrophobic interactions. The residues within eight Å from the ligand were set to be flexible. No explicit water molecules were added during the docking simulation, and the solvation and entropic effects were not taken into account.
Biological studies
Cell culture and treatment. HepaRG cells were obtained from Guan Dao Biotechnology (Shanghai, China) and cultured in RPMI-1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. The media and supplements were purchased from Gibco containing 5% CO2 at 37 °C. APAP and APAP-P1 were dissolved in DMSO and incubated in FBS-free complete medium with a final DMSO concentration of 1%.
Cell viability. Cell viability was determined using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium, Amresco, 0973) assay as previously described.27 Briefly, HepaRG cells were plated on 96-well plates at a density of 8 × 104 cells per well and then grown at 37 °C for 24 h. The cells were pre-treated with APAP or APAP-P1 with different concentrations for 24 h. Twenty microliters of MTT (5 mg mL−1) was added to each well and incubated for 4 h. The medium was removed, and the formazan crystals were dissolved with dimethyl sulfoxide (DMSO). The absorbance was measured at 570 nm on a microplate reader (TECAN Infinite M1000, Grödig, Austria).
Probe labelling of HepaRG cell lysate for biotin–streptavidin pull-down assay. HepaRG at 100% confluence were lysed in PBS buffer, and the protein concentration was adjusted to 2 mg mL−1. The experimental and control samples were incubated with 5 μL of 125, 250, 500 mmol L−1 APAP-P1 or 5 μL DMSO into 500 μL proteomes at room temperature for 1 h. Then, the proteomes were labelled with biotin-alkynyl (100 μM), TCEP (1 mM), TBTA (100 μM), and CuSO4·5H2O (1 mM) for 2 h. Seven hundred fifty microliters of cold MeOH were added and sonicated for 3–4 s using a probe sonicator (30% power level) at 4 °C to re-suspend the protein. The samples were then centrifuged for 10 min at 6500 × g at 4 °C and the supernatant was removed. The pellets were dissolved in PBS containing 1.2% SDS via sonication and then diluted with PBS containing 0.2% SDS. The samples were incubated with streptavidin beads for 2 h at room temperature and washed with PBS several times. Samples were denatured by heating in 2 × SDS-loading buffer and analysed by SDS-PAGE. The resulting bands were visualized by silver staining.
Probe labelling in HepaRG cells for biotin–streptavidin pull-down assay. HepaRG at 80% confluence were treated with 0, 1.25, 2.5, 5 mmol L−1 APAP-P1 for 24 h. Then the cells were lysed in PBS buffer, and the concentration was adjusted to 2 mg mL−1 for biotin–streptavidin pull-down assay just like cell lysate.
Western blotting. Proteomes from pull-down assay were used for Western blot experiment. 10 μL samples were subjected to SDS-PAGE electrophoresis and transferred to NC membranes. The membranes were blocked with 5% (w/v) non-fat dried milk for 2 h and incubated with various specific primary antibodies at 4 °C overnight. The membranes were subsequently washed with TBST and incubated with the appropriate secondary antibody at room temperature for 2 h. Following washes with TBST, the protein bands were visualized using an ECL system (CW Biotech, Beijing, China) and imaged using a Bio-Rad imaging system (Bio-Rad, Hercules, CA, USA).
Statistics. All data are expressed as the means ± SD of at least three independent experiments. Significant differences between the groups were determined by ANOVA. Data analyses were performed using GraphPad Prism 5 (GraphPad Prism Software, 2012, La Jolla, CA, USA).
Conclusions
In conclusion, we designed and synthesized one new powerful probe APAP-P1, introducing the PEG-azide probe linker into the acetyl group of acetaminophen. This probe maintained hepatotoxicity in HepaRG cells by MTT assay, and demonstrated that the PEG-azide probe linker does not change APAP hepatotoxicity. Metabolite of APAP-P1 by HepaRG cells showed this probe still can be metabolised to hepatotoxic intermediate. Results above proved APAP-P1 worked well in HepaRG cells, so we can use this probe for target fishing and identification. By CC-ABPP pull-down assays in both cell lysate and cells, we identified their protein profiles. There was some difference under the two conditions. This change is due to the metabolism of APAP-P1. This is similar to the process of APAP in vivo. TrxR1 has been proved NAPQI hepatotoxicity target in previous research, finally, we also identified that APAP-P1 can target TrxR1 by pull-down protein Western blot assay and thus APAP-P1 can be used for further research in vivo.
Conflicts of interest
The authors confirm that there are no conflicts of interest.
Acknowledgements
This work was supported by the National Science and Technology Major Project (Grant No. 2015ZX09501004-001-003), the Special Research Project for TCM (Grant No. 201507004), and the CAMS Innovation Fund for Medical Science (CIFMS) (Grant No. 2016-I2M-1-012), Beijing Natural Science Foundation (Grant No. 7192129).
Notes and references
- R. Chan and L. Z. Benet, Chem. Res. Toxicol., 2016, 30, 1017–1029 Search PubMed.
- G. A. Kullak-Ublick, R. J. Andrade, M. Merz, P. End, A. Benesic, A. L. Gerbes and G. P. Aithal, Gut, 2017, 66, 1154–1164 CrossRef CAS PubMed.
- D. Bissell, Hepatology, 2001, 33, 1009–1013 CrossRef CAS PubMed.
- W. M. Lee, J. Hepatol., 2017, 67, 1324–1331 CrossRef CAS.
- D. Bartlett, J. Emerg. Nurs., 2004, 3, 281–283 CrossRef.
- M. R. McGill and H. Jaeschke, Pharm. Res., 2013, 30, 2174–2187 CrossRef CAS PubMed.
- M. L. Bajt, Toxicol. Sci., 2004, 80, 343–349 CrossRef CAS PubMed.
- J. A. Hinson, A. B. Reid, S. S. McCullough and L. P. James, Drug Metab. Rev., 2004, 36, 805–822 CrossRef CAS.
- H. Jaeschke, C. D. Williams, A. Ramachandran and M. L. Bajt, Liver Int., 2012, 32, 8–20 CrossRef CAS.
- K. Du, Y. Xie, M. R. McGill and H. Jaeschke, Expert Opin. Drug Metab. Toxicol., 2015, 11, 1769–1779 CrossRef CAS PubMed.
- J. A. Hinson, D. W. Robert and L. P. James, Handb. Exp. Pharmacol., 2010, 196, 369–405 CAS.
- J. R. Prigge, S. Eriksson, S. V. Iverson, T. A. Meade, M. R. Capecchi, E. S. J. Arnér and E. E. Schmidt, Free Radical Biol. Med., 2012, 52, 803–810 CrossRef CAS PubMed.
- S. V. Iverson, S. Eriksson, J. Xu, J. R. Prigge, E. A. Talago, T. A. Meade, E. S. Meade, M. R. Capecchi, E. S. J. Arnér and E. E. Schmidt, Free Radical Biol. Med., 2013, 63, 369–380 CrossRef CAS PubMed.
- Y. Jan, D. E. Heck, A. Dragomir, C. R. Gardner, D. L. Laskin and J. D. Laskin, Chem. Res. Toxicol., 2014, 27, 882–894 Search PubMed.
- M. Saitoh, H. Nishitoh, M. Fujii, K. Takeda, K. Tobiume, Y. Sawada, M. Kawabata, K. Miyazono and H. Ichijo, EMBO J., 1998, 17, 2596–2606 CrossRef CAS PubMed.
- J. Kwon, E. Shin, J. Lee, S. Kim, S. Kim, Y. Jee, Y. Kim, H. Park, K. Min and H. Park, Allergy, Asthma Immunol. Res., 2012, 4, 132 CrossRef CAS PubMed.
- B. Fu, W. Meng, X. Zeng, H. Zhao, W. Liu and T. Zhang, BioMed Res. Int., 2017, 2017, 1–8 Search PubMed.
- A. Michaut, D. Le Guillou, C. Moreau, S. Bucher, M. R. McGill, S. Martinais, T. Gicquel, I. Morel, M. Robin, H. Jaeschke and B. Fromenty, Toxicol. Appl. Pharmacol., 2016, 292, 40–55 CrossRef CAS.
- M. R. McGill, H. Yan, A. Ramachandran, G. J. Murray, D. E. Rollins and H. Jaeschke, Hepatology, 2011, 53, 974–982 CrossRef CAS PubMed.
- Y. Xie, M. R. McGill, S. F. Cook, M. R. Sharpe, R. D. Winefield, D. G. Wilkins, D. E. Rollins and H. Jaeschke, Xenobiotica, 2015, 45, 921–929 CrossRef CAS PubMed.
- M. H. Wright and S. A. Sieber, Nat. Prod. Rep., 2016, 5, 681–708 RSC.
- J. Krysiak and R. Breinbauer, Top. Curr. Chem., 2012, 43–84 CAS.
- A. E. Speers and B. F. Cravatt, Curr. Protoc. Chem. Biol., 2009, 1, 29–41 Search PubMed.
- A. B. Berger, P. M. Vitorino and M. Bogyo, Am. J. PharmacoGenomics, 2004, 4, 371–381 CrossRef CAS PubMed.
- A. D. Patterson, B. A. Carlson, F. Li, J. A. Bonzo, M. Yoo, K. W. Krausz, M. Conrad, C. Chen, F. J. Gonzalez and D. L. Hatfield, Chem. Res. Toxicol., 2013, 26, 1088–1096 Search PubMed.
- L. Li, X. Chen, D. Li and D. Zhong, Drug Metab. Dispos., 2011, 39, 472–483 CrossRef CAS.
- S. Wang, M. Wang, M. Wang, Y. Tian, X. Sun, G. Sun and X. Sun, Toxins, 2018, 10, 154 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00483a |
| ‡ These two authors contributed equally to this paper. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abcde,
Yu Tian‡abcde,
Shan Luabcde,
Ruiying Wangabcde,
Hai Shangabcde,
Xuelian Zhangabcde,
Chenyang Zhangabcde,
Guibo Sun*abcde,
Xudong Xu*abcde and
Xiaobo Sun*abcde
abcde,
Yu Tian‡abcde,
Shan Luabcde,
Ruiying Wangabcde,
Hai Shangabcde,
Xuelian Zhangabcde,
Chenyang Zhangabcde,
Guibo Sun*abcde,
Xudong Xu*abcde and
Xiaobo Sun*abcde