
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of D-glyco-alkynone derivatives via carbonylative Sonogashira reaction†
Mariana P. Darbema,
C. Henrique A. Estevesa,
Isadora M. de Oliveirab,
Joel S. Reisa,
Daniel C. Pimentac and
Hélio A. Stefani *a
*a
aFaculdade de Ciências Farmacêuticas, Universidade de São Paulo, São Paulo, SP, Brazil. E-mail: hstefani@usp.br; Tel: +55 11 3091-3654
bInstituto de Química, Universidade de São Paulo, São Paulo, SP, Brazil
cInstituto Butantã, São Paulo, SP, Brazil
First published on 25th March 2019
Abstract
A carbonylative Sonogashira coupling approach to the synthesis of glyco-alkynones is described. Eighteen examples were obtained in moderate do nearly quantitative yields under mild conditions employing Mo(CO)6 as a safe carbon monoxide source. Functionalization of the alkynyl moiety via cycloaddition with organic azides provided six examples of glyco-triazoles.
Introduction
Alkynones are attractive motifs in organic chemistry involved in the synthesis of medicinally valuable heteroaromatic compounds.1 These molecules are also important intermediates in the synthesis of natural products2 and as part of biologically active molecules (Fig. 1).3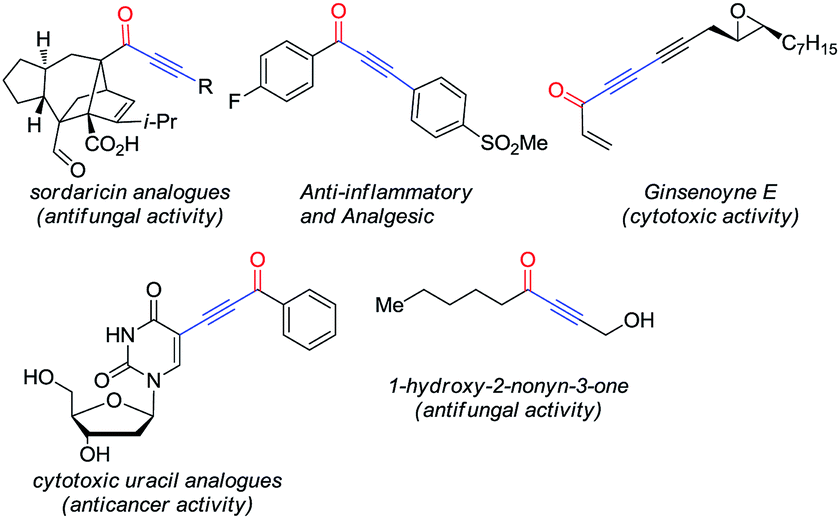 | ||
| Fig. 1 Alkynones in biologically active compounds. | ||
Consequently, a rich variety of methodologies targeting their synthesis has been reported, some of which involve the addition of borylated terminal alkynes to acyl chlorides,4 the addition of hypervalent alkynyl iodides to aldehydes via C–C bond cleavage, metal-catalyzed C–H bond activation of aldehydes5 or the oxidation of propargylic alcohols.6 While impressive, these methodologies present some drawbacks, such as excessive generation of chemical waste, instability of some of the substrates required and poor functional group tolerance. The Pd-catalyzed carbonylative Sonogashira coupling, on the other hand, offers a route to alkynones that is mild, atom-economical and functional-group-tolerant.7 Attracted by these features, we decided to explore the construction of glyco-alkynones relying on this reaction as part of our ongoing research interest in the synthesis of functionalized glycals.8
In a previous report,8a we explored the synthesis of amidoglucals and glucal esters via the carbonylative coupling reaction of 2-iodo-D-glucal. Herein, we describe the synthesis of glyco-alkynones via carbonylative Sonogashira coupling reaction, expanding the spectrum of reactions involving this important substrate (Scheme 1).
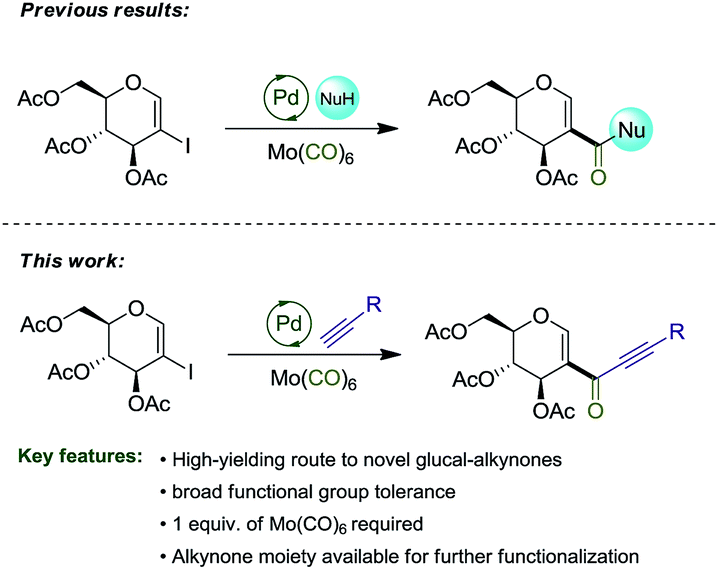 | ||
| Scheme 1 2-Iodoglucal carbonylative coupling reactions. | ||
Taking advantage of the alkynyl group readily installed by this reaction, we also explored the synthesis of glyco-substituted triazoles via click chemistry. This approach has been of pivotal importance for carbohydrate chemistry as a tool to efficiently connect a sugar moiety to a molecule of interest via a triazole linker, improving the hydrophilicity, bioavailability and chemical profile of these fragments.9 Moreover, the biological activity demonstrated by several alkynone derivatives (e.g. triazoles) make new routes to these structures synthetically relevant (Fig. 2).10
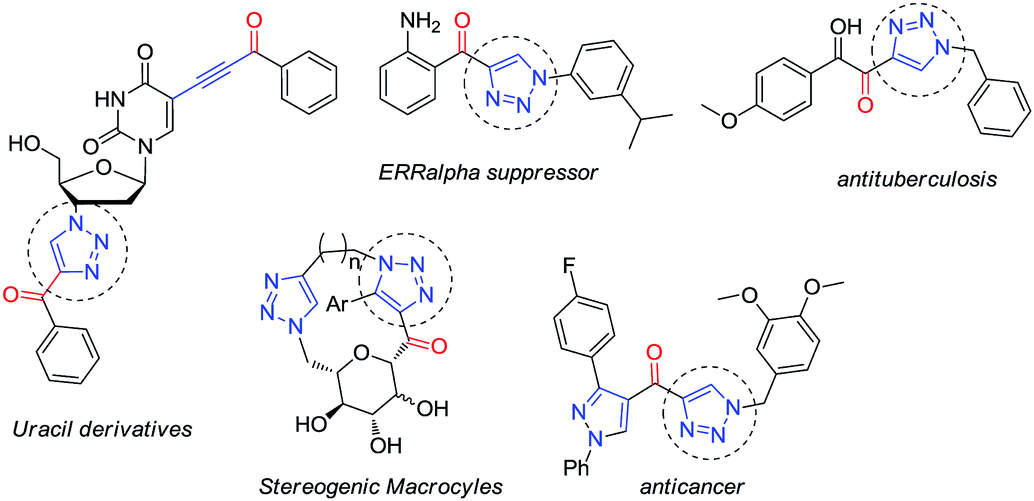 | ||
| Fig. 2 Examples of biologically active triazolic alkynones. | ||
Results and discussion
We commenced our study by synthesizing 2-iodo-tri-O-acetyl-D-glucal (1) from tri-O-acetyl-D-glucal, N-iodosuccinimide (NIS) and AgNO3 (Scheme 2).11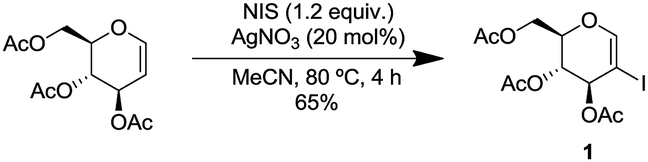 | ||
| Scheme 2 Synthesis of 2-iodo-tri-O-acetyl-D-glucal. | ||
With substrate 1 in hand, we next screened the reaction conditions for the carbonylation of 2-iodo-tri-O-acetyl-D-glucal (1) with Mo(CO)6 and 4-ethynyltoluene. Reactions were followed by TLC to ensure full conversion of the starting material 1 (Table 1).
|
|||||
|---|---|---|---|---|---|
| Entrya | Catalyst/ligand | Base(3.0 equvi.) | Solvent | Reaction time (h) | Yield (%) |
| a Reaction condition: 1 (0.2 mmol), catalyst (5 mol%), ligand (5 mol%), 4-ethynyltoluene (1.5 equvi.), base (3.0 equvi.), solvent (3 mL). | |||||
| Effect of catalyst | |||||
| 1 | PdCl2 | Et3N | 1,4-Dioxane | 12 | 66 |
| 2 | Pd(PhCN)2Cl2 | Et3N | 1,4-Dioxane | 12 | 58 |
| 3 | Pd(PhCN)2Cl2/xantphos | Et3N | 1,4-Dioxane | 12 | 73 |
| 4 | Pd(Prol)2 | Et3N | 1,4-Dioxane | 12 | 63 |
| 5 | PEPPSI-IPr | Et3N | 1,4-Dioxane | 12 | 75 |
| 6 | PdCl2/xantphos | Et3N | 1,4-Dioxane | 2 | 99 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Effects of base | |||||
| 7 | PdCl2/xantphos | DIPEA | 1,4-Dioxane | 16 | 55 |
| 8 | PdCl2/xantphos | DBU | 1,4-Dioxane | 16 | 43 |
| 9 | PdCl2/xantphos | NaOAc | 1,4-Dioxane | 16 | 32 |
| 10 | PdCl2/xantphos | K2CO3 | 1,4-Dioxane | 16 | 25 |
|
|||||
| Effect of solvent | |||||
| 11 | PdCl2/xantphos | Et3N | Toluene | 16 | 55 |
| 12 | PdCl2/xantphos | Et3N | THF | 16 | 43 |
| 13 | PdCl2/xantphos | Et3N | DMF | 16 | 32 |
| 14 | PdCl2/xantphos | Et3N | MeCN | 16 | 25 |

We started by screening the effect of the catalyst on the reaction outcome. PdCl2, Pd(PhCN)2Cl2, and Pd(Prol)2 (Table 1, entries 1, 2 and 4, respectively) led to the formation of alkynone 3a in moderate yields. Catalysts containing ligands that are at the same time electron-rich and sterically demanding, such as xantphos and PEPPSI,12 delivered alkynone 3a in good to nearly quantitative yield (Table 1, entries 3, 5 and 6), with the combination PdCl2/xantphos being the best. In order to seek other high-yielding set of conditions, the effect of the base was next examined. Organic and inorganic bases such as DIPEA, DBU, NaOAc and K2CO3 gave 3a in lower yields, with inorganic K2CO3 delivering the desired product in only 25%. Different solvents were also screened, however, only poor to modest yields of 3a were obtained.
With the optimized reaction conditions in hand, we set out to investigate the generality of this reaction (Scheme 3).
 | ||
| Scheme 3 Sonogashira carbonylative coupling reaction of 2-iodo-D-glucal and terminal alkynes. | ||
Terminal alkynes bearing electron-neutral and electron-donating groups delivered the desired alkynones in good to excellent yields (3a–g). Electron-withdrawing groups such as the difluorinated moiety present in 2h and meta-chloro substituted 2i gave 3h and 3i in good yields, while meta-fluorinated 2j gave 3j in 67%. Incorporation of an heteroaromatic substituent was also tolerated, and alkynone 3k was obtained in 65% yield. Pleasingly, both cyclopropyl and TMS groups proved to be stable under the reaction conditions, with products 3l and 3m being isolated in 72% and 80%, respectively, both leaving useful handles for further functionalization (see Scheme 4).13 Incorporation of terminal alkynes bearing alkyl moieties provided mixed results, with 2n and 2o delivering alkynones in moderate yields, while 2p and 2q, bearing a tertiary alcohol, provided 3p and 3q in good yields. 1,4-Diethynylbenzene 2r was subjected to the reaction conditions, giving the symmetrical alkynone 3r in 70%. Finally, the reaction with 2a was repeated on a gram scale, providing 3a in 80% isolated yield (Scheme 4).
 | ||
| Scheme 4 Gram-scale reaction. | ||
In order to demonstrate the usefulness of this methodology, we decided to explore the formation of 1,2,3-triazoles via click chemistry. An in situ-generated terminal alkyne provided the desired triazoles 5a–f in the presence of organic azides, PMDTA and copper iodide (conditions found after a quick screening).14 A variety of moieties were tolerated at the position 1 of the newly formed ring: a benzylic substituent (5a, 67%), heteroaromatic substituents (5b, 72% and 5c, 70%) and unactivated (5d, 65% and 5e, 58%) and activated aromatic rings (5f, 86%) (Scheme 5).
 | ||
| Scheme 5 Synthesis of D-glyco-1,2,3-triazoles. | ||
Conclusions
In conclusion, we have described a convenient palladium-catalyzed Sonogashira carbonylative coupling reaction for the synthesis of D-glyco-alkynones. This approach permitted the synthesis of 18 examples in moderate to nearly quantitative yields under mild conditions, employing Mo(CO)6 as a safe carbon monoxide source. Further functionalization of a masked terminal alkynone allowed the synthesis of D-glyco-1,2,3-triazoles in moderate yields, demonstrating one of the potential applications of the alkynones described herein.Experimental section
General considerations
The compounds were all identified by usual analytical methods: 1H NMR, 13C NMR, IR, and HR-MS (ESI). 1H and 13C NMR spectra were measured in CDCl3, in a Bruker DPX-300 instrument. 1H chemical shifts were reported in ppm referenced relative to TMS internal standard (0.00 ppm) or the residual chloroform peak (7.26 ppm). Abbreviations to denote the multiplicity of a particular signal are: m (multiplet), s (singlet), d (doublet), t (triplet) and dd (doublet of doublets). 13C chemical shift were reported in ppm relative to the CDCl3 triplet (77.16 ppm). IR spectra were measured on an Agilent Technologies Cary 630 and were reported in wavenumbers (cm−1). High-resolution mass spectra (HRMS) were recorded on a Shimadzu LCMS-TOF, using ESI with 50% solution of acetonitrile/H2O and 0.1% formic acid as ionization method. Thin layer chromatography (TLC) was performed using silica gel UV254 0.20 mm thickness. For visualization, TLC plates were either placed under ultraviolet light, or stained with iodine or acidic vanillin solution. The solvents were purified by distillation or used without any purification in the case of HPLC-grade material. All other compounds were used as received.General procedure for the synthesis of 3a–r
To a vial equipped with a magnetic stirrer bar and sealed with a rubber septum connected to a deflated balloon with a needle were added the tri-O-acetylated iodoglucal (0.2 mmol), 1,4-dioxane (3.0 mL), PdCl2 (5 mol%), xantphos (5 mol%), Mo(CO)6 (0.2 mmol, 1 equiv.), the alkyne (0.3 mmol, 1.5 equiv.) and Et3N (0.6 mmol, 3 equiv.). The reaction mixture was vigorously stirred at 70 °C for 2 to 4 h. The resulting mixture was washed with water and extracted with ethyl acetate. The organic layers were then combined and evaporated. The crude products were purified by flash chromatography using hexane and ethyl acetate as eluent (7:3).
General procedure for the synthesis of 5a–f
To a vial (20 mL) equipped with a magnetic stirrer bar under a nitrogen atmosphere containing CuI (0.25 mmol, 1 equiv.), THF (4 mL), an organic azide (0.3 mmol, 1.2 equiv.) and 3m (0.25 mmol, 1 equiv.) was added PMDETA (0.3 mmol, 1.2 equiv.) and the reaction mixture was stirred at 0 °C for 2 h. After this period, the reaction mixture was diluted with ethyl acetate and washed with aqueous NaCl. The organic phase was collected, dried over MgSO4, filtered and the solvent was evaporated under reduced pressure. Purification was performed using flash chromatography (ethyl acetate/hexane, 4:6).
Analytical data of compounds 3a–r/5a–f
Product 3a was obtained as a yellow oil (83 mg, 0.20 mmol, 99%). 1H NMR (300 MHz, CDCl3): δ = 8.00 (s, 1H), 7.40 (d, J = 7.9 Hz, 2H), 7.12 (d, J = 7.8 Hz, 2H), 5.73 (dd, J = 3.1, 1.6 Hz, 1H), 5.15 (t, J = 3.0 Hz, 1H), 4.70–4.51 (m, 1H), 4.40 (dd, J = 12.1, 7.8 Hz, 1H), 4.14 (dd, J = 12.1, 4.4 Hz, 1H), 2.31 (s, 3H), 2.15–1.89 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 174.5, 170.2, 169.4, 169.1, 160.7, 141.3, 132.8, 129.4, 116.7, 114.9, 91.4, 84.8, 75.6, 65.6, 61.2, 60.9, 21.6, 20.7, 20.6, 20.6. IR (ν, cm−1) = 2877, 2112, 1685, 1564, 1177, 1328, 1197, 1154, 1143, 991. HRMS (ESI-TOF) calc. [C22H22O8Na+] 437.1212, found 437.1212.Product 3b was obtained as a yellow oil (72 mg, 0.18 mmol, 92%). 1H NMR (300 MHz, CDCl3): δ = 8.08 (s, 1H), 7.65–7.55 (m, 2H), 7.49–7.34 (m, 3H), 5.82 (d, J = 1.8 Hz, 1H), 5.23 (t, J = 3.0 Hz, 1H), 4.74–4.60 (m, 1H), 4.48 (dd, J = 12.1, 7.8 Hz, 1H), 4.21 (dd, J = 12.1, 4.5 Hz, 1H), 2.18–2.01 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 174.4, 170.2, 169.4, 169.1, 160.9, 132.8, 130.6, 128.6, 119.8, 114.9, 90.8, 84.9, 75.6, 65.6, 61.2, 60.9, 20.7, 20.6, 20.6. IR (ν, cm−1) = 2959, 2864, 2127, 1682, 1566, 1324, 1266, 1175, 1151, 992. HRMS (ESI-TOF) calc. [C21H20O8Na+] 423.1056, found 423.1051.
Product 3c was obtained as a yellow oil (84 mg, 0.18 mmol, 88%). 1H NMR (300 MHz, CDCl3): δ = 8.10 (s, 1H), 7.74–7.53 (m, 6H), 7.49–7.36 (m, 3H), 5.93–5.79 (m, 1H), 5.24 (t, J = 3.1 Hz, 1H), 4.75–4.64 (m, 1H), 4.49 (dd, J = 12.0, 7.8 Hz, 1H), 4.22 (dd, J = 12.1, 4.5 Hz, 1H), 2.19–1.98 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 174.4, 170.2, 169.4, 169.1, 160.8, 143.5, 139.7, 133.3, 128.9, 128.1, 127.3, 127.1, 118.5, 114.9, 90.9, 85.7, 75.7, 65.6, 61.2, 60.9, 20.7, 20.6, 20.6. IR (ν, cm−1) = 2959, 2931, 2123, 1685, 1566, 1438, 1324, 1264, 1175, 1151, 991. HRMS (ESI-TOF) calc. [C27H24O8Na+] 499.1363, found 499.1361.
Product 3d was obtained as a yellow oil (78 mg, 0.18 mmol, 90%). 1H NMR (300 MHz, CDCl3): δ = 8.06 (s, 1H), 7.54 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 5.87–5.78 (m, 1H), 5.23 (t, J = 3.0 Hz, 1H), 4.72–4.62 (m, 1H), 4.47 (dd, J = 12.1, 7.9 Hz, 1H), 4.21 (dd, J = 12.2, 4.5 Hz, 1H), 3.84 (s, 3H), 2.19–1.99 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 174.5, 170.2, 169.4, 169.1, 161.6, 160.3, 134.8, 114.7, 114.4, 111.6, 91.9, 84.8, 75.5, 65.7, 61.3, 60.9, 55.4, 20.7, 20.6, 20.6. IR (ν, cm−1) = 2866, 2747, 2119, 1685, 1549, 1460, 1175, 1151, 1134, 991. HRMS (ESI-TOF) calc. [C22H22O9Na+] 453.1156, found 453.1159.
Product 3e was obtained as a yellow oil (88 mg, 0.19 mmol, 99%). 1H NMR (300 MHz, CDCl3): δ = 7.88 (s, 1H), 7.29 (d, J = 8.5 Hz, 1H), 6.61–6.47 (m, 2H), 5.67–5.59 (m, 1H), 5.05 (t, J = 3.4 Hz, 1H), 4.54–4.41 (m, 1H), 4.33–4.23 (m, 1H), 4.12–3.95 (m, 1H), 3.62 (s, 3H), 2.29 (s, 3H), 1.96–1.80 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 174.5, 170.2, 169.4, 169.1, 161.5, 160.4, 144.1, 135.2, 115.4, 115.0, 111.8, 111.6, 90.8, 88.4, 75.6, 65.6, 61.2, 60.9, 55.3, 21.0, 20.7, 20.6, 20.5. IR (ν, cm−1) = 2821, 2756, 2112, 1685, 1566, 1549, 1324, 1259, 1179, 1151, 992. HRMS (ESI-TOF) calc. [C23H24O9Na+] 467.1313, found 453.1311.
Product 3f was obtained as a yellow oil (91 mg, 0.19 mmol, 95%). 1H NMR (300 MHz, CDCl3): δ = 8.05 (s, 1H), 7.98 (s, 1H), 7.64 (m, 2H), 7.45 (d, J = 8.4 Hz, 1H), 7.11 (dd, J = 9.0, 2.4 Hz, 1H), 7.04 (d, J = 2.6 Hz, 1H), 5.85–5.70 (m, 1H), 5.28–5.06 (m, 1H), 4.64–4.56 (m, 1H), 4.52–4.35 (m, 1H), 4.24–4.11 (m, 1H), 3.85 (s, 3H), 2.19–1.96 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 174.5, 170.2, 169.4, 169.2, 160.7, 159.3, 135.4, 133.9, 129.7, 128.9, 128.1, 127.2, 119.9, 114.9, 114.4, 105.9, 92.1, 85.0, 75.6, 65.7, 61.3, 61.0, 55.4, 20.7, 20.6, 20.6. IR (ν, cm−1) = 2913, 2866, 2117, 1680, 1560, 1436, 1324, 1177, 1151, 991. HRMS (ESI-TOF) calc. [C26H24O9Na+] 503.1313, found 503.1312.
Product 3g was obtained as a yellow oil (73 mg, 0.17 mmol, 85%). 1H NMR (300 MHz, CDCl3): δ = 9.21 (s, 1H), 7.70 (s, 1H), 7.55–7.33 (m, 4H), 5.93 (s, 1H), 5.37 (s, 2H), 5.23 (t, J = 3.4 Hz, 1H), 4.60–4.52 (m, 1H), 4.47 (dd, J = 11.6, 7.7 Hz, 1H), 4.20 (dd, J = 11.8, 4.1 Hz, 1H), 2.12–2.03 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 171.5, 170.3, 169.6, 169.3, 154.2, 144.0, 131.9, 130.9, 128.3, 128.0, 120.4, 114.5, 96.0, 90.3, 74.3, 73.9, 66.2, 62.5, 61.0, 20.7, 20.7, 20.6. IR (ν, cm−1) = 2861, 2080, 1680, 1574, 1527, 1324, 1177, 1145, 981, 732. HRMS (ESI-TOF) calc. [C22H22O9Na+] 453.1156, found 453.1156.
Product 3h was obtained as a yellow oil (67 mg, 0.15 mmol, 77%). 1H NMR (300 MHz, CDCl3): δ = 7.93 (s, 1H), 7.45–7.30 (m, 1H), 6.80–6.68 (m, 2H), 5.63–5.56 (m, 1H), 5.05 (t, J = 2.8 Hz, 1H), 4.54–4.45 (m, 1H), 4.34–4.21 (m, 1H), 4.13–3.94 (m, 1H), 2.01–1.79 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 173.9, 170.2, 169.3, 169.1, 164.4, 164.3 (dd, J = 253.5 Hz, J = 7.5 Hz), 161.6, 135.7, 135.6, 115.0, 112.3, 112.2 (dd J = 22.2 Hz, 3.3 Hz), 105.2 (dd, J = 3.7 Hz), 104.7 (t, J = 24.7 Hz) 89.4, 75.7, 65.5, 61.0, 60.9, 20.7, 20.6. IR (ν, cm−1) = 2976, 2136, 1685, 1560, 1456, 1326, 1175, 1151, 992, 937, 711. HRMS (ESI-TOF) calc. [C21H18F2O8Na+] 436.0862, found 436.0869.
Product 3i was obtained as a yellow oil (74 mg, 0.17 mmol, 85%). 1H NMR (300 MHz, CDCl3): δ = 7.57 (s, 1H), 7.51–7.39 (m, 1H), 7.35 (d, J = 7.8 Hz, 2H), 5.85–5.75 (m, 1H), 5.23 (t, J = 3.0 Hz, 1H), 4.70–4.68 (m, 1H), 4.56–4.42 (m, 1H), 4.23 (d, J = 4.5 Hz, 1H), 2.22–1.92 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 174.1, 170.2, 169.3, 169.1, 161.2, 134.5, 132.4, 130.9, 130.8, 129.9, 121.5, 114.9, 88.7, 85.5, 75.8, 65.5, 61.0, 60.9, 20.7, 20.6, 20.6. IR (ν, cm−1) = 2975, 2130, 1682, 1562, 1426, 1365, 1266, 1173, 1151, 991. HRMS (ESI-TOF) calc. [C21H19ClO8Na+] 457.0661, found 457.0660.
Product 3j was obtained as a yellow oil (56 mg, 0.13 mmol, 67%). 1H NMR (300 MHz, CDCl3): δ = 8.07 (s, 1H), 7.43–7.37 (m, 2H), 7.32–7.23 (m, 1H), 7.24–7.07 (m, 1H), 5.88–5.78 (m, 1H), 5.23 (t, J = 3.0 Hz, 1H), 4.82–4.64 (m, 1H), 4.49 (dd, J = 12.1, 7.9 Hz, 1H), 4.21 (dd, J = 12.2, 4.5 Hz, 1H), 2.18–2.01 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 174.1, 170.2, 169.3, 169.12, 162.53 (d, J = 246.7 Hz), 161.1, 130.4 (d, J = 8.5 Hz), 128.7 (d, J = 3.2 Hz), 121.6 (d, J = 9.3 Hz), 119.4 (d, J = 23.3 Hz), 118.1 (d, J = 21.2 Hz), 114.9, 88.9 (d, J = 3.4 Hz), 85.2, 75.8, 65.5, 61.0, 60.9, 20.7, 20.6, 20.5. IR (ν, cm−1) = 2970, 2132, 1685, 1564, 1326, 1268, 1177, 1151, 1113, 985, 849. HRMS (ESI-TOF) calc. [C21H19FO8Na+] 441.0956, found 441.0956.
Product 3k was obtained as a yellow oil (52 mg, 0.13 mmol, 65%). 1H NMR (300 MHz, CDCl3): δ = 8.74 (s, 1H), 8.60 (d, J = 4.0 Hz, 1H), 8.01 (s, 1H), 7.87–7.73 (m, 1H), 7.28 (dd, J = 7.9, 5.0 Hz, 1H), 5.74 (s, 1H), 5.16 (s, 1H), 4.68–4.58 (m, 1H), 4.42 (dd, J = 12.2, 7.9 Hz, 1H), 4.13 (dd, J = 12.1, 4.5 Hz, 1H), 2.09–1.90 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 173.9, 170.2, 169.3, 169.1, 161.3, 153.0, 150.6, 139.6, 123.2, 117.2, 114.9, 87.5, 86.8, 75.8, 65.5, 61.0, 60.8, 20.7, 20.6, 20.6. IR (ν, cm−1) = 2859, 2119, 1680, 1566, 1326, 1181, 992. HRMS (ESI-TOF) calc. [C20H19NO8Na+] 424.1003, found 444.1002.
Product 3l was obtained as a yellow oil (53 mg, 0.14 mmol, 72%). 1H NMR (300 MHz, CDCl3): δ = 7.90 (s, 1H), 5.72 (s, 1H), 5.18 (s, 1H), 4.64–4.54 (m, 1H), 4.44 (dd, J = 12.1, 7.8 Hz, 1H), 4.19 (d, J = 4.5 Hz, 1H), 2.19–1.91 (m, 9H), 1.45–1.42 (m, 1H), 1.07–0.86 (m, 4H). 13C NMR (75 MHz, CDCl3): δ = 174.8, 174.8, 170.5, 169.7, 169.5, 160.8, 115.1, 98.5, 75.8, 66.0, 61.5, 61.3, 21.0, 21.0, 20.9, 9.8, 9.8. IR (ν, cm−1) = 2915, 2138, 1682, 1566, 1365, 1175, 1149, 991, 864. HRMS (ESI-TOF) calc. [C18H20O8Na+] 387.1050, found 387.1051.
Product 3m was obtained as a yellow oil (64 mg, 0.16 mmol, 80%). 1H NMR (300 MHz, CDCl3): δ = 7.82 (s, 1H), 5.60–5.48 (m, 1H), 5.02 (t, J = 3.1 Hz, 1H), 4.51–4.39 (m, 1H), 4.26 (dd, J = 12.2, 7.9 Hz, 1H), 4.01 (dd, J = 12.2, 4.5 Hz, 1H), 1.99–1.81 (m, 9H), 0.08 (s, 9H). 13C NMR (75 MHz, CDCl3): δ = 174.8, 170.9, 170.03, 169.8, 162.1, 115.6, 99.7, 98.6, 76.4, 66.3, 61.7, 61.7, 21.4, 21.3, 21.3, 0.0. IR (ν, cm−1) = 2864, 2028, 1914, 1685, 1566, 1324, 1261, 1175, 1151, 987, 817. HRMS (ESI-TOF) calc. [C18H24O8SiNa+] 419.1133, found 419.1135.
Product 3n was obtained as a yellow oil (53 mg, 0.14 mmol, 70%). 1H NMR (300 MHz, CDCl3): δ = 7.95 (s, 1H), 5.73 (dd, J = 3.1, 1.7 Hz, 1H), 5.19 (t, J = 3.1 Hz, 1H), 4.69–4.58 (m, 1H), 4.45 (dd, J = 12.1, 7.8 Hz, 1H), 4.17 (dd, J = 12.1, 4.5 Hz, 1H), 2.39 (t, J = 7.0 Hz, 2H), 2.18–1.96 (m, 9H), 1.69–1.36 (m, 4H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ = 174.7, 170.2, 169.3, 169.1, 160.7, 114.9, 94.1, 77.0, 75.5, 65.6, 61.1, 60.9, 29.7, 22.0, 20.7, 20.6, 20.6, 18.6, 13.4. IR (ν, cm−1) = 2838, 2862, 2147, 1685, 1566, 1365, 1324, 1175, 1149, 991, 864. HRMS (ESI-TOF) calc. [C19H24O8Na+] 403.1363, found 403.1361.
Product 3o was obtained as a yellow oil (45 mg, 0.12 mmol, 62%). 1H NMR (300 MHz, CDCl3): δ = 7.76 (s, 1H), 5.53 (dd, J = 3.1, 1.7 Hz, 1H), 4.99 (t, J = 3.1 Hz, 1H), 4.49–4.37 (m, 1H), 4.25 (dd, J = 12.1, 7.8 Hz, 1H), 3.97 (dd, J = 12.1, 4.5 Hz, 1H), 2.17 (t, J = 7.1 Hz, 2H), 1.94–1.81 (m, 9H), 1.44 (h, J = 7.2 Hz, 2H), 0.84 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ = 174.7, 170.2, 169.3, 169.1, 160.7, 114.9, 93.9, 77.8, 75.5, 65.6, 61.1, 60.9, 21.2, 20.8, 20.7, 20.6, 20.6, 13.5. IR (ν, cm−1) = 2916, 2879, 1680, 1560, 1141989, 836, 724. HRMS (ESI-TOF) calc. [C18H22O8Na+] 389.1207, found 403.1361.
Product 3p was obtained as a yellow oil (67 mg, 0.15 mmol, 78%). 1H NMR (300 MHz, CDCl3): δ = 7.61 (s, 1H), 7.32–7.11 (m, 5H), 5.65–5.52 (m, 1H), 5.16–5.04 (m, 1H), 4.57–4.49 (m, 1H), 4.36 (dd, J = 11.8, 7.8 Hz, 1H), 4.07 (dd, J = 12.0, 4.5 Hz, 1H), 2.84 (t, J = 7.3 Hz, 2H), 2.63 (t, 2H), 2.05–1.94 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 174.5, 170.2, 169.3, 169.1, 161.1, 139.5, 128.5, 128.3, 126.7, 114.9, 92.7, 78.3, 75.4, 65.6, 60.9, 60.8, 33.8, 21.0, 20.7, 20.6, 20.6. IR (ν, cm−1) = 2926, 2840, 2149, 1685, 1566, 1324, 1261, 1175, 1151, 1017, 991, 678. HRMS (ESI-TOF) calc. [C23H24O8Na+] 451.1363, found 451.1361.
Product 3q was obtained as a yellow oil (73 mg, 0.17 mmol, 86%). 1H NMR (300 MHz, CDCl3): δ = 7.91 (s, 1H), 5.68 (d, J = 2.3 Hz, 1H), 5.12 (t, J = 3.0 Hz, 1H), 4.62–4.52 (m, 1H), 4.37 (dd, J = 12.2, 7.8 Hz, 1H), 4.12 (dd, J = 12.1, 4.4 Hz, 1H), 2.68 (s, 1H), 2.09–1.98 (m, 9H), 1.93–1.84 (m, 2H), 1.72–1.43 (m, 8H). 13C NMR (75 MHz, CDCl3): δ = 174.3, 170.2, 169.5, 169.1, 161.0, 114.7, 95.5, 79.9, 75.6, 65.5, 61.1, 60.9, 39.1, 39.1, 24.9, 22.9, 20.7, 20.6, 20.6. IR (ν, cm−1) = 3363, 2840, 2766, 2136, 1691, 1568, 1326, 1182, 1156, 996. HRMS (ESI-TOF) calc. [C21H26O9Na+] 445.1469, found 445.1467.
Product 3r was obtained as a yellow oil (101 mg, 0.14 mmol, 70%). 1H NMR (300 MHz, CDCl3): δ = 8.07 (s, 2H), 7.61 (d, J = 2.6 Hz, 4H), 5.95–5.73 (m, 2H), 5.29–5.10 (m, 2H), 4.77–4.58 (m, 2H), 4.58–4.46 (m, 2H), 4.23 (dd, J = 9.7, 5.6 Hz, 2H), 2.25–1.97 (m, 18H). 13C NMR (75 MHz, CDCl3): δ = 174.0, 170.2, 169.3, 169.1, 161.2, 132.8, 122.0, 114.9, 89.0, 87.0, 75.8, 65.5, 61.0, 60.8, 20.7, 20.6, 20.6. IR (ν, cm−1) = 2870, 2129, 1685, 1564, 1324, 1264, 1177, 1151, 989. HRMS (ESI-TOF) calc. [C36H34O16Na+] 745.1739, found 745.1735.
Product 5a was obtained as a yellow oil (61 mg, 0.13 mmol, 67%). 1H NMR (300 MHz, CDCl3): δ = 9.15 (s, 1H), 8.02 (s, 1H), 7.46–7.34 (m, 5H), 5.96–5.85 (m, 1H), 5.55 (d, J = 1.4 Hz, 1H), 5.30 (s, 2H), 4.64 (d, J = 5.3 Hz, 1H), 4.54–4.44 (m, 1H), 4.23 (dd, J = 12.2, 4.7 Hz, 1H), 2.19–2.05 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 180.3, 169.3, 168.4, 168.2, 161.0, 132.6, 128.3, 128.1, 127.3, 126.5, 126.5, 111.5, 73.6, 64.9, 60.5, 60.1, 53.4, 19.7, 19.6, 19.6. IR (ν, cm−1) = 3261, 2866, 2836, 1680, 1560, 1475, 1324, 1179, 1149, 989, 706. HRMS (ESI-TOF) calc. [C22H23N3O8Na+] 480.1377, found 480.1375.
Product 5b was obtained as a yellow oil (69 mg, 0.14 mmol, 72%). 1H NMR (300 MHz, CDCl3): δ = 9.15 (s, 1H), 8.67 (s, 1H), 8.45 (s, 1H), 7.87 (s, 1H), 7.55–7.42 (m, 2H), 7.30 (d, J = 2.8 Hz, 1H), 6.68–6.52 (m, 1H), 5.97–5.84 (m, 1H), 5.23 (t, J = 2.9 Hz, 1H), 4.65–4.56 (m, 1H), 4.46 (m, 1H), 4.21 (m, 1H), 2.06–1.97 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 181.5, 170.3, 169.5, 169.3, 162.0, 148.1, 135.90, 128.1, 126.7, 126.2, 126.2, 115.5, 113.5, 112.6, 112.1, 103.5, 74.7, 66.0, 61.6, 61.2, 20.8, 20.7, 20.6. IR (ν, cm−1) = 2834, 2862, 2779, 1682, 1560, 1475, 1460, 1324, 1162, 1011, 989, 700. HRMS (ESI-TOF) calc. [C23H22N4O8Na+] 505.1330, found 505.1329.
Product 5c was obtained as a yellow oil (70 mg, 0.14 mmol, 70%). 1H NMR (300 MHz, CDCl3): δ = 9.13 (s, 1H), 9.07 (s, 1H), 8.58 (s, 1H), 8.37 (t, 1H), 8.24 (d, J = 8.0 Hz, 1H), 7.82 (d, J = 8.9 Hz, 1H), 5.93–5.89 (m, 1H), 5.24–5.22 (m, 1H), 4.64–4.57 (m, 1H), 4.47 (dd, J = 12.0, 7.8 Hz, 1H), 4.20 (dd, J = 12.0, 4.5 Hz, 1H), 2.08–1.94 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 181.1, 170.3, 169.4, 169.2, 162.2, 162.1, 156.2, 148.6, 135.2, 126.0, 124.9, 120.0, 119.3, 115.1, 112.7, 74.8, 65.9, 61.5, 61.1, 20.8, 20.7, 20.6. IR (ν, cm−1) = 2985, 2864, 2037, 1687, 1562, 1186, 1154, 994, 838, 855. HRMS (ESI-TOF) calc. [C22H20N4O8SNa+] 523.0894, found 523.0890.
Product 5d was obtained as a yellow oil (89 mg, 0.13 mmol, 65%). 1H NMR (300 MHz, CDCl3): δ = 9.09 (s, 1H), 8.48 (s, 1H), 7.95 (d, J = 8.4 Hz, 1H), 7.49 (s, 1H), 7.40–7.21 (m, 11H), 6.28 (d, J = 3.5 Hz, 1H), 5.98–5.85 (m, 1H), 5.29 (s, 2H), 5.18 (s, 2H), 4.68–4.54 (m, 1H), 4.50–4.41 (m, 1H), 4.18 (dd, J = 12.1, 4.5 Hz, 1H), 2.10–1.92 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 201.9, 180.2, 170.4, 169.8, 169.1, 164.9, 163.5, 159.3, 139.6, 135.6, 135.5, 133.7, 128.7, 128.5, 128.2, 128.2, 128.2, 127.2, 125.6, 121.5, 113.7, 112.7, 111.6, 105.9, 71.8, 71.1, 67.1, 65.3, 61.1, 60.5, 20.8, 20.6, 20.5. IR (ν, cm−1) = 2967, 2931, 1687, 1559, 1195, 1169, 1046, 998. HRMS (ESI-TOF) calc. [C36H33N3O11Na+] 706.2007, found 706.2004.
Product 5e was obtained as a yellow oil (57 mg, 0.12 mmol, 58%). 1H NMR (300 MHz, CDCl3) δ = 9.07 (s, 1H), 8.69–8.59 (m, 2H), 8.30 (d, J = 8.0 Hz, 1H), 8.11 (d, J = 8.5 Hz, 1H), 7.74 (t, J = 8.1 Hz, 1H), 6.28 (d, J = 3.5 Hz, 1H), 5.92–5.83 (m, 1H), 4.66–4.57 (m, 1H), 4.52–4.41 (m, 1H), 4.19 (dd, J = 12.1, 4.5 Hz, 1H), 2.09–1.98 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 180.1, 170.4, 169.8, 169.1, 163.6, 137.0, 131.2, 126.0, 125.7, 123.9, 115.8, 115.8, 113.7, 112.7, 74.9, 71.8, 61.5, 61.1, 20.7, 20.7, 20.6. IR (ν, cm−1) = 2868, 1687, 1562, 1486, 1309, 1188, 1171, 994, 717. HRMS (ESI-TOF) calc. [C21H20N4O10Na+] 511.1072, found 511.1071.
Product 5f was obtained as a yellow oil (81 mg, 0.17 mmol, 86%). 1H NMR (300 MHz, CDCl3): δ = 9.11 (s, 1H), 8.41 (s, 1H), 7.58 (d, J = 9.0 Hz, 2H), 6.98 (d, J = 8.9 Hz, 2H), 5.89 (t, 2H), 5.21 (t, J = 3.1 Hz, 1H), 4.64–4.55 (m, 1H), 4.44 (dd, J = 12.1, 7.7 Hz, 1H), 4.19 (dd, J = 12.0, 4.8 Hz, 3H), 2.08–1.94 (m, 9H). 13C NMR (75 MHz, CDCl3): δ = 181.3, 170.3, 169.4, 169.2, 162.0, 160.3, 129.6, 127.8, 125.7, 122.3, 115.0, 112.6, 74.7, 65.9, 61.6, 61.1, 55.6, 20.7, 20.7, 20.6. IR (ν, cm−1) = 2902, 2875, 1687, 1564, 1471, 1326, 1262, 1184, 1153, 998, 838. HRMS (ESI-TOF) calc. [C22H23N3O9Na+] 496.1327, found 496.1329.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the São Paulo Research Foundation, FAPESP for financial support (grant 2017/24821-4) and fellowships to MPD (2016/24396-9), CHAE (2017/26673-2), IMO (2016/04289-3), JSR (2016/02392-1). They are also thankful for the financial support provided by The National Council for Scientific and Technological Development (CNPq) for the fellowship to HAS (306119/2014-5).Notes and references
- (a) M. C. Bagley, J. W. Dale and J. Bower, Synlett, 2001, 7, 1149–1151 CrossRef; (b) C. G. Lee, K. Y. Lee, S. Lee and J. N. Kim, Tetrahedron, 2005, 61, 8705–8710 CrossRef; (c) H. Liu, H. Jiang, M. Zhang, W. Yao, Q. Zhu and Z. Tang, Tetrahedron Lett., 2008, 49, 3805–3809 CrossRef CAS; (d) J. D. Kirkham, S. J. Edeson, S. Strokes and J. P. Harrity, Org. Lett., 2012, 14, 5354–5357 CrossRef CAS PubMed; (e) M. S. Mohamed Ahmed, K. Kobayashi and A. Mori, Org. Lett., 2005, 7, 4487–4489 CrossRef PubMed.
- (a) A. S. Karpov, E. Merkul, F. Rominger and T. J. Müller, Angew. Chem., Int. Ed., 2005, 44, 6951–6956 CrossRef CAS PubMed; (b) K. T. Neumann, S. R. Laursen, A. T. Lindhardt, B. Bang-Andersen and T. Skrydstrup, Org. Lett., 2014, 16, 2216–2219 CrossRef CAS PubMed; (c) A. Linda, N. Patrik, W. Matyas, R. O. Luke and L. Mats, J. Org. Chem., 2015, 80, 1464–1471 CrossRef PubMed; (d) I. Muneaki and K. Yoshinori, Eur. J. Org. Chem., 2007, 5180–5182 Search PubMed; (e) Y. Tu, Z. Zhang, T. Wang, J. Ke and J. Zhao, Org. Lett., 2017, 19, 3466–3469 CrossRef CAS PubMed; (f) J. D. Kirkham, S. J. Edeson, S. Stokes and J. P. A. Harrity, Org. Lett., 2012, 14, 5354–5357 CrossRef CAS PubMed; (g) L. F. Tietze, R. R. Singidi, K. M. Gericke, H. Böckemeier and H. Laatsch, Eur. J. Org. Chem., 2007, 5875–5878 CrossRef CAS.
- (a) N. G. Kundu, J. S. Mahanty, C. Chowdhury, S. K. Dasgupta, B. Das, C. P. Spears, J. Balzarini and E. De Clercq., Eur. J. Med. Chem., 1999, 34, 389–398 CrossRef CAS; (b) C. A. Quesnelle, P. Gill, M. Dodier, D. S. Laurent, M. Serrano-Wu, A. Marinier, A. Martel, C. E. Mazzucco, T. M. Stickle, J. F. Barret, D. M. Vyas and B. N. Balasubramanian, Bioorg. Med. Chem. Lett., 2003, 13, 519–524 CrossRef CAS PubMed; (c) D. V. Kuklev, A. J. Domb and V. M. Dembitsky, Phytomedicine, 2013, 20, 1145–1159 CrossRef CAS PubMed; (d) G. Prashant, J. T. Neelam and M. B. Bhalchandra, ACS Omega, 2019, 4, 1560–1574 CrossRef.
- C. Taylor and Y. Bolshan, Org. Lett., 2014, 16, 488–491 CrossRef CAS PubMed.
- W. Ai, Y. Wu, H. Tang, X. Yang, Y. Yang, Y. Li and B. Zhou, Chem. Commun., 2015, 51, 7871–7874 RSC.
- Y. Maeda, N. Kakiuchi, S. Matsumura, T. Nishimura, T. Kawamura and S. Uemura, J. Org. Chem., 2002, 67, 6718–6724 CrossRef CAS PubMed.
- (a) K. T. Neumann, S. R. Laursen, A. T. Lindhardt, B. Bang-Andersen and T. Skrydstrup, Org. Lett., 2014, 16, 2216–2219 CrossRef CAS PubMed; (b) X.-F. Wu, H. Neumann and M. Beller, Chem.–Eur. J., 2010, 16, 12104–12107 CrossRef CAS PubMed; (c) A. Arcadi, S. Cacchi, F. Marinelli, P. Pace and G. Sanzi, Synlett, 1995, 823–824 CrossRef CAS; (d) L. Delaude, A. M. Masdeu and H. Alper, Synthesis, 1994, 11, 1149–1151 CrossRef.
- (a) M. P. Darbem, K. S. Kanno, I. M. de Oliveira, C. H. A. Esteves, D. C. Pimenta and H. A. Stefani, New J. Chem., 2019, 43, 696–699 RSC; (b) A. Shamim, S. N. S. Vasconcelos, I. M. Oliveira, J. S. Reis, D. C. Pimenta, J. Zukerman-Schpector and H. A. Stefani, Synthesis, 2017, 49, 5183–5196 CrossRef CAS; (c) A. Shamim, F. B. Souza, S. N. S. Vasconcelos and H. A. Stefani, Tetrahedron Lett., 2017, 58, 884–888 CrossRef CAS; (d) A. Shamim, C. S. Barbeiro, B. Ali and H. A. Stefani, ChemistrySelect, 2016, 1, 5653–5659 CrossRef CAS; (e) A. Shamim, S. N. S. Vasconcelos, B. Ali, L. S. Madureira, J. Zukerman-Schpector and H. A. Stefani, Tetrahedron Lett., 2015, 56, 5836–5842 CrossRef CAS; (f) A. Shamim, F. B. Souza, G. H. G. Trossini, F. M. Gatti and H. A. Stefani, Mol. Diversity, 2015, 19, 423–434 CrossRef CAS PubMed; (g) A. S. Vieira, P. F. Fiorante, T. L. S. Hough, F. P. Ferreira, D. S. Ludtke and H. A. Stefani, Org. Lett., 2008, 10, 5215–5218 CrossRef CAS PubMed.
- For reviews see: (a) A. L. Garner, Chem. Commun., 2018, 54, 6531–6539 RSC; (b) V. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS PubMed; (c) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed; (d) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- (a) A. Palasz and D. Ciez, Eur. J. Med. Chem., 2015, 97, 582–611 CrossRef CAS PubMed; (b) A. Ajay, S. Sharma, M. P. Gupt, V. Bajpai, B. Kumar, M. P. Kaushik, R. Konwar, R. S. Ampapathi and R. P. Tripathi, Org. Lett., 2012, 14, 4306–4309 CrossRef CAS PubMed; (c) J. Thomas, V. Goyvaerts, S. Liekens and W. Dehaen, Chem.–Eur. J., 2016, 22, 9966–9970 CrossRef CAS PubMed.
- S. Dharuman and Y. D. Vankar, Org. Lett., 2014, 16, 1172–1175 CrossRef CAS PubMed.
- N. Hadei, E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Org. Lett., 2005, 7, 3805–3807 CrossRef CAS PubMed.
- For reviews, see: (a) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS PubMed; (b) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804–818 RSC; (c) Z.-B. Zhu, Y. Wei and M. Shi, Chem. Soc. Rev., 2011, 40, 5534–5563 RSC; (d) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS PubMed.
- (a) H. A. Stefani, N. C. S. Silva, F. Manarin, D. S. Lüdtke, J. Zukerman-Schpector, L. S. Madureira and E. R. T. Tiekink, Tetrahedron Lett., 2012, 53, 1742–1747 CrossRef CAS; (b) H. A. Stefani, S. N. S. Vasconcelos, F. Manarin, D. M. Leal, F. B. Souza, L. S. Madureira, J. Zukerman-Schpector, M. N. Eberlin, M. N. Godoi and R. S. Galaverna, Eur. J. Org. Chem., 2013, 3780–3785 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00523d |
| This journal is © The Royal Society of Chemistry 2019 |