
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of methylpyrimidine ring-fused diterpenoid analogs as a novel testosterone synthesis promoter†
Jie Bai‡
a,
Jia Xie‡b,
Li-Ting Wanga,
Yajing Xingb,
Qian-Ru Jianga,
Fan Yang a,
Jie Tanga,
Zhengfang Yi*b and
Wen-Wei Qiu*a
a,
Jie Tanga,
Zhengfang Yi*b and
Wen-Wei Qiu*a
aShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, School of Chemistry and Molecular Engineering, East China Normal University, 3663 North Zhongshan Road, Shanghai 200062, China. E-mail: wwqiu@chem.ecnu.edu.cn; Tel: +86-21-54340103
bShanghai Key Laboratory of Regulatory Biology, Institute of Biomedical Sciences, School of Life Sciences, East China Normal University, Shanghai 200241, China. E-mail: zfyi@bio.ecnu.edu.cn; Tel: +86-21-54345016
First published on 27th March 2019
Abstract
Herein we screened our small synthetic library of diterpenoid analogs for hit compounds on promoting testosterone synthesis and the methylpyrimidine ring-fused diterpenoid analog 7 was obtained as the hit. Based on the hit, a series of derivatives were designed, synthesized and evaluated for their effects on testosterone secretion in mouse Leydig TM3 cells. Most of the derivatives showed better activity in promoting testosterone synthesis than the positive control compound icariin, among which compound 17 has optimal activity and little cytotoxicity. Preliminary mechanism studies indicated that 17 significantly promoted the expression of testosterone synthesis-related marker genes (StAR, 3β-HSD and CYP11A1). Further studies showed that 17 provided sufficient steroid materials for testosterone synthesis by stimulating autophagy in Leydig cells. Thus compound 17 emerged as a potential lead compound for further development of therapeutics for late onset of hypogonadism (LOH).
Introduction
Diterpenoids are a major branch of natural products which exert many different biological activities.1 For instance, antiproliferative,2 multi-drug resistance-reversing,3 antimicrobial,4 vasoactive,5 antidiabetic,6 anti-osteoporosis7 immunomodulatory,8 and anti-inflammatory9 effects. Therefore, diterpenoids play an important role in the discovery of novel bioactive agents.Testosterone, which is primarily produced in Leydig cells as the major circulating androgen, plays important roles in sexual differentiation, secondary sex characteristics, reproductive function and sexual function.10,11 Serum testosterone deficiency causes late onset of hypogonadism (LOH), which is a clinical and biochemical syndrome.12 The incidence rates of LOH are 13%, 30% and 47% for men aged 40–49, 50–59 and over 70 respectively.13 Previous studies indicated that LOH-related hypogonadal symptoms included sexual desire decrease, muscle mass and strength reduction, osteoporosis, increased central body fat, forgetfulness, loss of memory, difficulty in concentration, insomnia and erectile dysfunction (ED).14,15
Testosterone replacement therapy (TRT), as the main clinical treatment method for LOH at present, provides a wide range of benefits for hypogonadism, improving libido and sexual function, fertility, bone density, muscle mass, and quality of life.16,17 However the clinical efficacy and long-term safety of TRT remains controversial. TRT involves the direct administration of an exogenous hormone, and with this treatment, the androgen level in the serum is superphysiological and unstable.13 So, it has considerable side effects, for instance, testicular atrophy, erythropoiesis, prostate cancer, cardiovascular events, intrahepatic cholestasis, sleep apnea, liver failure and worsening of lower urinary tract symptoms from an enlarged prostate.18,19 Therefore, the discovery of novel agents as anti-LOH agents is urgently needed.
Testosterone production is regulated by luteinizing hormone (LH) and LH induces cAMP synthesis in Leydig cells by binding to the membrane LH receptors.20 cAMP catalyzes the synthesis of PKA, which transports cholesterol from the cytoplasmic pool to mitochondria and promotes steroidogenesis by steroid-generating enzymes (CYP11A1, 3β-HSD) and steroidogenic acute regulatory (StAR) protein21 Additionally, autophagy in Leydig cells promotes lipid metabolism and provides sources such as triglycerides (TGs) and cholesterol for testosterone production, which plays an important role in testosterone synthesis.22,23
Herein, we report the discovery of a hit compound as testosterone synthesis promoter by screening our small synthetic library of diterpenoid analogs. The methylpyrimidine ring-fused diterpene analogue 7 was selected as the hit. Then a series of analogs were synthesized based on the hit and their activities in promoting testosterone production in mouse Leydig TM3 cells were evaluated. Results showed that compound 17 (SH380) was the most potent testosterone synthesis promoter. The mechanism by which the 17 increases testosterone production was preliminary explored.
Results and discussion
Chemistry
The methylpyrimidine acyl derivatives 7–14 were synthesized as shown in Scheme 1. Compound 6 was prepared according to our previously reported procedure24,25 and details were as follows. Coupling reaction of 6,7-epoxygeranyl acetate with (4-methoxybenzyl) magnesium chloride yielded compound 1, Li2CuCl4 was added as catalyst. Key intermediate 2 was obtained by the cyclization of 1 in presence of boron trifluoride etherate, and is a racemate according to the Stork–Eschenmoser hypothesis.26,27 Therefore, all the synthesized diterpenoids herein belong to racemates. Compound 3 was obtained by oxidation of 2 with 2-iodoxybenzoic acid (IBX). Claisen condensation of 3 with ethyl formate in the presence of NaH provided compound 4. Compound 6 was furnished by reaction of 4 with piperidine in EtOH and then condensation with acetamidine hydrochloride. Compound 7–14 were prepared by Friedel–Crafts acylation from 6 with various acyl chlorides respectively.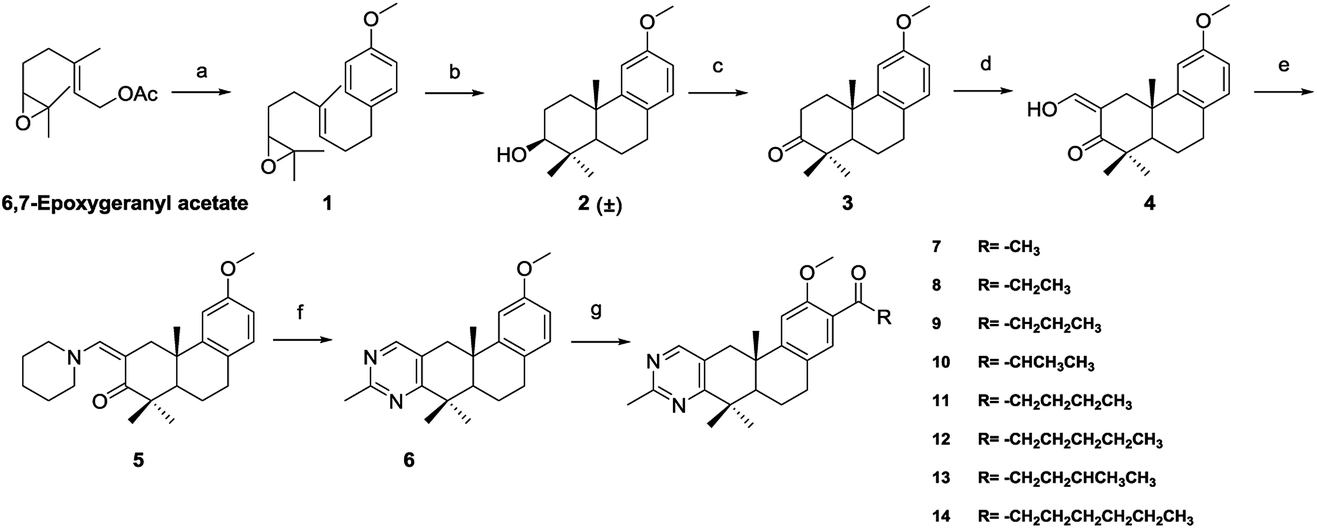 | ||
| Scheme 1 Reagents and conditions: (a) (4-methoxybenzyl) magnesium chloride, LiCuCl4, anhydrous THF, 0 °C; (b) boron trifluoride etherate, anhydrous CH2Cl2, −78 °C; (c) IBX, THF, DMSO, rt; (d) ethyl formate, NaH, anhydrous THF, rt; (e) piperidine, EtOH, reflux; (f) acetamidine hydrochloride, sodium methoxide, EtOH, reflux; (g) acyl chloride, AlCl3, DCM, 0 °C. | ||
The synthetic route of 2-methylpyrimidine derivatives 15–22 is outlined in Scheme 2. Compound 15 was prepared from 7 with hydroxylamine hydrochloride in the presence of K2CO3. Condensation of 7 with hydrazine hydrate (85%) yielded 16. Reaction of 7 with methylmagnesium chloride yielded 17. Compound 18 was produced by Wolff–Kishner–Huang Min-lon reduction of 7 with hydrazine hydrate (85%) in the presence of NaOH. Reduction of 7 with sodium borohydride produced compound 19, and 20 was provided by dehydration of 19 with p-toluenesulfonic acid. Oxidation of 7 with CrO3 in AcOH produced 21.
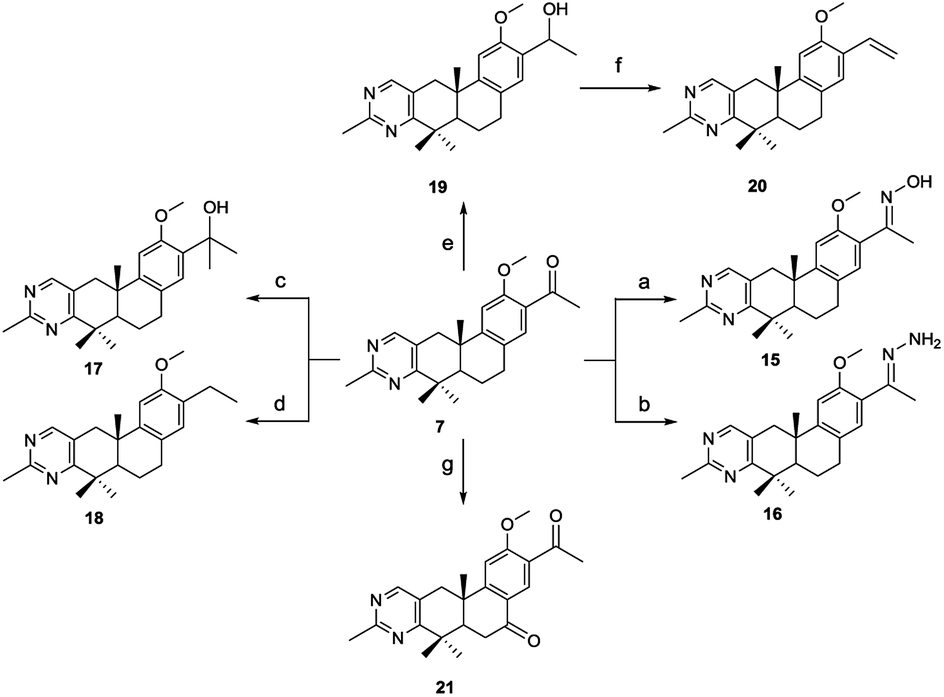 | ||
| Scheme 2 Reagents and conditions: (a) hydroxylamine hydrochloride, K2CO3, EtOH, rt; (b) hydrazine hydrate (85%), TsOH, EtOH, reflux; (c) methylmagnesium chloride, anhydrous THF, 0 °C; (d) hydrazine hydrate (85%), NaOH, triglycol, 120 °C to 190 °C; (e) NaBH4, DCM, MeOH, 0 °C; (f) TsOH, anhydrous THF, 60 °C; (g) CrO3, AcOH, rt. | ||
Activity and cytotoxicity evaluation of compound 7 and its derivatives
A hit was obtained by screening our small synthetic diterpenoids library. Mouse TM3 Leydig cells were treated with each tested compound at concentration of 20 μM for 24 h, and the testosterone levels in the culture medium were measured by ELISA (enzyme-linked immuno sorbent assay) kit. Among those analogs, compound 7 (QB364) was determined as a testosterone synthesis promoter with a testosterone concentration of 271.7 μM L−1 and was more potent than the positive icariin.28 Therefore, it was selected as the hit compound for further structure–activity relationship (SAR) optimization.The first-round synthetic compounds 8–14 were obtained by Friedel–Crafts acylation with various acyl chlorides respectively and their activity in promoting testosterone production on mouse Leydig TM3 cells was determined by ELISA kit. As shown in Table 1, the activity decreased significantly when the acetyl group (7) was replaced by various acyl substituents. Thus, compound 7 was selected for the second-round structure modification.
|
||
|---|---|---|
| Compd | R1 | Testosterone Levelsb (μM L−1) |
| a See Experimental section.b Testosterone levels in the culture medium are measured by ELISA kit; data are expressed as mean ± SD of three independent assays. | ||
| Control | — | 164.5 ± 0.2 |
| Icariin | — | 229.8 ± 4.8 |
| 7 | 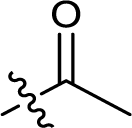 |
271.7 ± 10.0 |
| 8 | 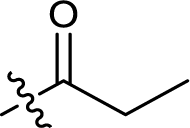 |
233.0 ± 0.5 |
| 9 | 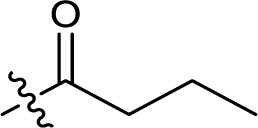 |
252.3 ± 2.4 |
| 10 | 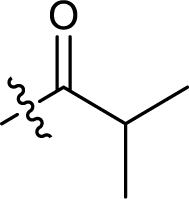 |
242.6 ± 6.0 |
| 11 | 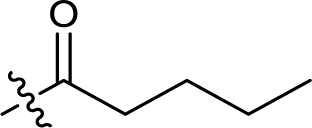 |
245.4 ± 4.9 |
| 12 | 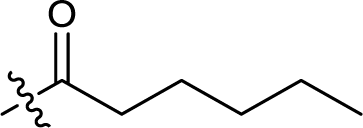 |
217.1 ± 9.9 |
| 13 | 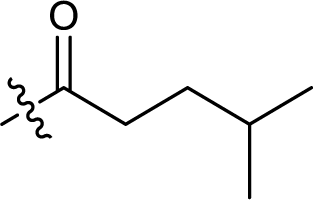 |
174.1 ± 0.5 |
| 14 | 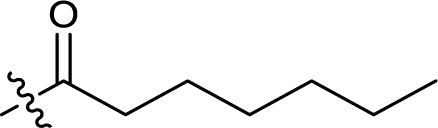 |
204.2 ± 6.0 |
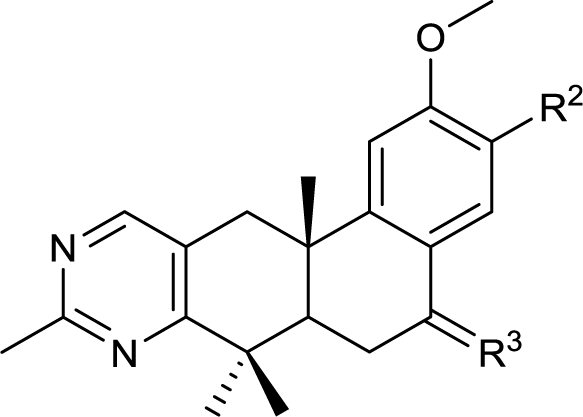
The second-round synthetic analogs 15–21 were obtained by modification of the acetyl group of 7 and their activity in promoting testosterone production was determined using ELISA kit. The results (Table 2) showed that the activity increased when the carbonyl group of 7 was substituted by oxime (15), while decreased as hydrazine substituent (16) was introduced into. If the carbonyl group of 7 was reduced to hydroxy (19) the activity decreased obviously. To our delight, the testosterone level in TM3 was increased significantly as the acetyl group was replaced by tert-butanol group (17). If the acetyl group was converted to the alkyl substituent such as ethyl (18) or vinyl (20), the activity of testosterone production was also decreased. For derivative modified at the C-6 and C-7 positions, the activity decreased as the carbonyl (21) substituent was introduced into C7 position. Further cytotoxicity evaluation in mouse TM3 cells with the SRB assay disclosed that all these analogs showed very little cytotoxicity (IC50 > 100 μM). Considering the above results, the safe compound 17 exhibited most potent activity in promoting testosterone production, thus it was selected for further studies.
|
||||
|---|---|---|---|---|
| Compd | R2 | R3 | Testosterone Levelsb (μM/L) | IC50c (μM) |
| a See Experimental section.b Testosterone levels in the culture medium are measured by ELISA kit; data are expressed as mean ± SD of three independent assays.c Activities of compounds against the growth of TM3 cells are detected with the SRB assay and their IC50 data are an average of at least three independent experiments. | ||||
| 7 | 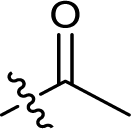 |
— | 271.7 ± 10.0 | >100 |
| 15 | 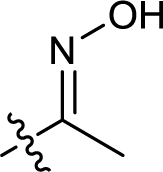 |
— | 282.2 ± 5.5 | >100 |
| 16 | 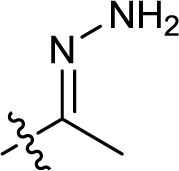 |
— | 188.5 ± 5.5 | >100 |
| 17 | 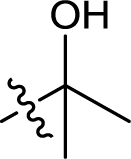 |
— | 303.5 ± 5.5 | >100 |
| 18 | 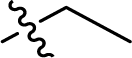 |
— | 237.0 ± 4.0 | >100 |
| 19 | 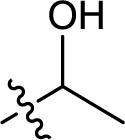 |
— | 222.8 ± 7.0 | >100 |
| 20 | 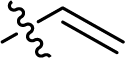 |
— | 227.1 ± 3.0 | >100 |
| 21 | 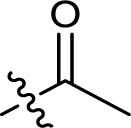 |
O | 259.5 ± 6.5 | >100 |
Compound 17 has little cytotoxicity on TM3 and other normal cells within the effective concentrations
To further determination of the in vitro safety of 17, we detected its cytotoxicity in TM3 and several human normal cells including NCM460, HAF, L-02 and PTN1A by SRB assay. As shown in Table 3, compound 17 showed very little toxicity on cell proliferation of TM3 (IC50 = 112.6 μM) and HAF (IC50 = 112.6 μM) cells. In addition, 17 has almost no toxic effects on NCM460, L-02 and PTN1A cells (IC50 > 200 μM). Therefore, compound 17 is a safe compound in vitro.| Leydig cell | Normal cellb | ||||
|---|---|---|---|---|---|
| a See Experimental section.b HAF, human fibroblast cell line; NCM460, human colon mucosal epithelial cell line; L-02, human fetal hepatocyte cell line; PTN1A, human prostate epithelial cell line.c Various cells were treated with indicated concentrations of compound 17 for 24 h; cell proliferation was determined by SRB assay; data were expressed as IC50 of three independent assays.d Variation ± 10%. | |||||
| Cell line | TM3 | HAF | NCM460 | L-02 | PTN1A |
| IC50 (μM)c | 112.6d | 111.8d | >200 | >200 | >200 |
Compound 17 enhanced the expression of testosterone synthesis-regulating gens
For determination of the primary mechanism of 17 on testosterone synthesis, the mRNA and protein expression levels of testosterone synthetase, including StAR, 3β-HSD and CYP11A1 were analyzed. The qPCR analysis showed that the mRNA expression levels of StAR, 3β-HSD and CYP11A1 were increased significantly in a concentration-dependent manner (Fig. 1). Furthermore, the protein expression levels of the three key enzymes in TM3 cells were validated by western blotting. Results indicated that 17 also increased the protein expression levels of StAR, 3β-HSD and CYP11A1 obviously as the concentration rised from 10 μM to 100 μM (Fig. 2). These primary mechanism studies indicated that 17 increased testosterone levels in Leydig cells via promoting testosterone synthesis.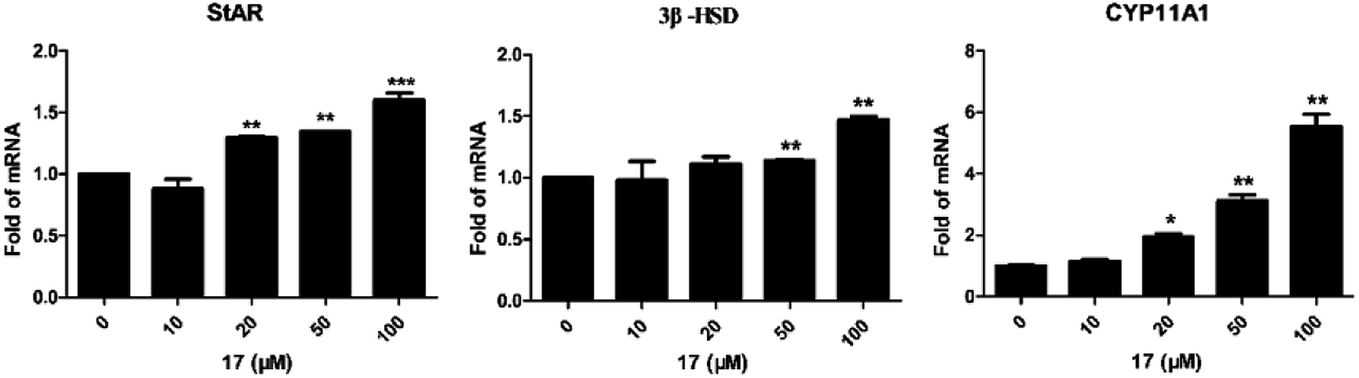 | ||
| Fig. 1 Compound 17 advanced the mRNA expression levels of testosterone synthetase in Leydig cells.a aSee Experimental section. Mouse Leydig TM3 cells were treated with different concentrations of compound 17 for 24 h. mRNA levels of steroidogenesis-related genes StAR, 3β-HSD and CYP11A1 were measured by quantitative-PCR. Data were expressed as mean ± SD of three independent assays; Student's t-tests were performed; *p < 0.05, **p < 0.01, ***p < 0.001. | ||
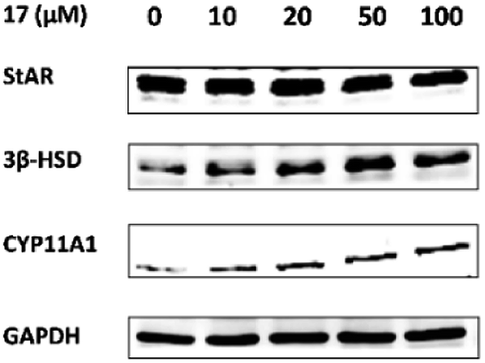 | ||
| Fig. 2 Compound 17 advanced the protein expression levels of testosterone synthetase in Leydig cells.a aSee Experimental section. Mouse Leydig TM3 cells were treated with different concentrations of 17 for 24 h and the protein levels of StAR, 3β-HSD and CYP11A1 were measured by Western blotting. GAPDH was used as a control. | ||
Compound 17 regulates the expression of autophagy-related proteins in Leydig cells
Autophagy dysfunction played a key role in the loss of testosterone production in some LOH patients.21 Therefore, we tried to identify if 17 promoted testosterone production via induction of autophagy. LC3-II serves as a reliable marker for mature autophagosomes and ATG5 is a gene product required for the formation of autophagosomes. We assessed the levels of the autophagy-related proteins by western blotting. As shown in Fig. 3, the expressions of ATG5, LC3-II and the ratio of LC3-II/LC3-I were increased significantly after treatment with 17. It suggested that the compound could promoted testosterone synthesis via activating autophagy in Leydig cells.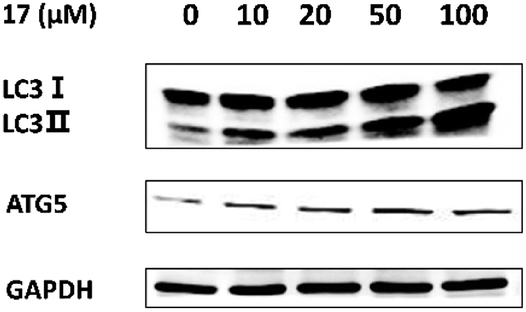 | ||
| Fig. 3 Compound 17 promotes the autophagy-related protein expression levels in TM3 cells.a aSee Experimental section. Mouse Leydig TM3 cells were treated with different concentrations of 17 for 24 h. The expression of LC3 I/II and ATG5 were detected by western blotting. GAPDH was used as a control. | ||
Conclusions
In this study, we designed and synthesized a number of 15 methylpyrimidine ring-fused tricyclic diterpene analogs. Most of the derivatives showed potent activity in promoting testosterone production in mouse Leydig TM3 cells, which was much better than that of the positive control icariin. Among these analogs, compound 17 exhibited the most potent activity and almost no toxic effect. Preliminary mechanism studies indicated that 17 could significantly enhance the expression of testosterone synthesis-related marker genes (StAR, 3β-HSD and CYP11A1) in a concentration-dependent manner. Further studies disclosed that 17 could also stimulate autophagy in Leydig cells and promote the expression of autophagy-related genes (LC3 and ATG 5). In conclusion, compound 17 was discovered as a promising compound, which could be further developed as a potential therapeutic agent for LOH.Experimental section
Chemistry
Testosterone assays
TM3 cells were seeded in 24-well plates at a density of 1.0 × 105 cells per well and incubated overnight. The culture medium was then replaced with fresh medium containing various compounds (20 μM) respectively. After incubation for 24 h, the culture medium was collected and centrifuged at 1000 × g for 5 min. Testosterone concentrations in the supernatant were determined with enzyme-linked immunosorbent assay (EISA) kits (Shanghai XinFan Biological Technology Co Ltd, Shanghai, China).Cell lines and culture conditions
The cell lines used in this study were obtained from the American Type Culture Collection (ATCC). NCM460, L-02 and PTN1A cell lines were cultured in RPMI 1640 medium containing 10% foetal bovine serum (FBS), and TM3 mouse Leydig cell line was cultured in Dulbecco's modified eagle's medium (DMEM)/F12 medium, which was supplemented with 2.5% FBS and 5% horse serum. Moreover, HAF cell line was cultured in DMEM medium with 1% glutamine and 10% FBS. All cells were incubated at 37 °C and 5% CO2 incubator.Cell viability assay
The cell viability of cell lines in the presence of this series of compounds was determined by SRB (Sigma Aldrich) assay. In brief, cells were plated at the appropriate cell densities in 96-well plates during the experiment. After incubation for 24 h, the cells were treated with different concentrations of various compounds for 24 h. Control group were exposed to DMSO at a concentration equivalent to that of the compound-treated cells. After treatment, 25 μL of 50% TCA was added for cell fixation at 4 °C. After 1 hour or more, the plates were washed by water for five times. The plates were allowed to dry using hair dryer followed by being dyed with 100 μL 0.4% SRB for 10 min. After dying, the plates were washed by 1% acetic acid to remove the dye and allowed to dry using hair dryer. 100 μL of 10 mM Tris-based solution was added to each well, and absorbance was measured using a 96-well plate reader at 515 nm. Three independent experiments were carried out in triplicate. The IC50 was calculated using GraphPad Software.Western blot analysis
Cells were exposed to various concentrations of compounds for indicated time, and total cellular protein was lysed in RIPA buffer [50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1% sodium deoxycholic acid, 0.1% SDS, 2 mM phenylmethanesulfonyl fluoride (PMSF), 30 mM Na2HPO4, 50 mM NaF, 1 mM Na3VO4] containing protease/phosphatase inhibitors (Roche). Lysates were combined with sample loading buffer and heated at 100 °C. Protein concentrations were measured by a Bicinchoninic acid assay (Thermo Scientific). Lysates were mixed with sample loading buffer and heated at 100 °C. for 15 min. After separated by 8–15% SDS-PAGE, extracted protein were transferred to nitrocellulose membranes. Membranes were incubated in 5% (w/v) bovine serum albumin (TBST/BSA) and stored overnight at 4 °C on a shaker with specific primary antibodies (1/1000 in TBST/BSA). Then membranes were washed with TBST and incubated for 45 min with secondary antibody (1/10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 in TBST/BSA) at room temperature. Immunoreactive proteins were visualized using the Odyssey Fluorescence Scanner (LI-COR Bioscience, Inc., Lincoln, NE, USA). The StAR (ab133657; 1:3000), HSD3B1 (ab65156; 1:1000) and CYP11A1 (BS6578; 1:1000) antibodies were purchased from Abcam (Cambridge, MA) and Bioworld Technology, Inc (St. Paul, MN). LC3A/B (12741; 1:3000) and Atg5 (12994; 1:3000) antibodies were purchased from Cell Signaling Technology (Danvers, MA). The β-actin and GAPDH antibody (1:10000) was purchased from Sigma (St Louis, MO). The secondary antibody was conjugated with IRDye 680/800 (Millennium Science; 926-32221, 926-32210; 1:10000).
000 in TBST/BSA) at room temperature. Immunoreactive proteins were visualized using the Odyssey Fluorescence Scanner (LI-COR Bioscience, Inc., Lincoln, NE, USA). The StAR (ab133657; 1:3000), HSD3B1 (ab65156; 1:1000) and CYP11A1 (BS6578; 1:1000) antibodies were purchased from Abcam (Cambridge, MA) and Bioworld Technology, Inc (St. Paul, MN). LC3A/B (12741; 1:3000) and Atg5 (12994; 1:3000) antibodies were purchased from Cell Signaling Technology (Danvers, MA). The β-actin and GAPDH antibody (1:10000) was purchased from Sigma (St Louis, MO). The secondary antibody was conjugated with IRDye 680/800 (Millennium Science; 926-32221, 926-32210; 1:10000).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from cells with TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocols. Total RNA (1 μg) was converted to cDNA using a PrimeScript reverse transcription Master Mix kit (TaKaRa, Japan) according to the manufacturer's instructions. Quantitative RT-PCR was performed using SYBR Premix Ex Taq™ Kit (TaKaRa, Japan). The relative expression of StAR, 3β-HSD and CYP11A1 were analyzed by RT-PCR with β-actin as an internal control. Three independent experiments were carried out in triplicate. The gene-specific primers are listed in Table S1.†Statistical analysis
Grouped data are expressed as mean ± s.d. Significance between groups was analysed by one-way analysis of variance or Student's t-test using GraphPad Prism 5.0 (GraphPad Software). Data were expressed as mean and S.D., and P < 0.05 was considered significant. All experiments were performed at least three times except for animal experiments.Conflicts of interest
There are no conflicts of interest to declare.Abbreviations
| NPL | Natural product-like |
| LOH | Late-onset hypogonadism |
| StAR | Steroidogenic acute regulatory |
| 3β-HSD | 3β-Hydroxysteroid dehydrogenase |
| CYP11A1 | Cytochrome P450 family 11 subfamily A member 1 |
| TRT | Testosterone replacement therapy |
| ED | Erectile dysfunction |
| LH | Luteinizing hormone |
| cAMP | Cyclic adenosine monophosphate |
| PKA | Protein kinase A |
| TG | Triglyceride |
| IBX | 2-Iodoxybenzoic acid |
| THF | Tetrahydrofuran |
| SAR | Structure–activity relationship |
| Rt | Room temperature |
| ELISA | Enzyme-linked immuno sorbent assay |
| IC50 | Half maximal inhibitory concentration |
| SRB | Sulforhodamine B |
| RT-PCR | Real-time polymerase chain reaction |
| NMR | Nuclear magnetic resonance |
| GAPDH | Reduced glyceraldehyde-phosphate dehydrogenase |
| LC3 | Light chain 3 |
| ATG5 | Autophagy related 5 |
| H&E | Haematoxylin and eosin |
| IHC | Immunohistochemical |
| TMS | Tetramethylsilane |
| IF | Immunofluorescence |
| PMSF | Phenylmethanesulfonyl fluoride |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| TBST | Tris buffered saline tween |
| BSA | Bovine serum albumin |
| FITC | Fluorescein isothiocyanate |
Acknowledgements
This work was supported by Shanghai Science and Technology Council (Grant 18ZR1411200), National Natural Science Foundation of China (No. 81773204, No. 81472788) and Major State Basic Research Development Program of China (2015CB910400).References
- A. Vasas and J. Hohmann, Chem. Rev., 2014, 114, 8579–8612 CrossRef CAS PubMed.
- X. Rao, X. Huang, L. He, J. Song, Z. Song and S. Shang, Comb. Chem. High Throughput Screening, 2012, 15, 840–844 CrossRef CAS PubMed.
- N. Duarte, A. Varga, G. Cherepnev, R. Radics, J. Molnar and M. J. U. Ferreira, Bioorg. Med. Chem., 2007, 15, 546–554 CrossRef CAS PubMed.
- K. Yoshikawa, N. Kokudo, M. Tanaka, T. Nakano, H. Shibata, N. Aragaki, T. Higuchi and T. Hashimoto, Chem. Pharm. Bull., 2008, 56, 89–92 CrossRef CAS.
- Y. Tian, W. Xu, C. Zhu, S. Lin, Y. Li, L. Xiong, S. Wang, L. Wang, Y. Yang, Y. Guo, H. Sun, X. Wang and J. Shi, J. Nat. Prod., 2011, 74, 1221–1229 CrossRef CAS PubMed.
- C. Lipina and H. S. Hundal, Cell. Signalling, 2014, 26, 2343–2349 CrossRef CAS PubMed.
- T. T. Dao, K. Y. Lee, H. M. Jeong, P. H. Nguyen, T. L. Tran, P. T. Thuong, B. T. Nguyen and W. K. Oh, J. Nat. Prod., 2011, 74, 2526–2531 CrossRef CAS PubMed.
- M. Shahlaei, S. M. Ghanadian, A. M. Ayatollahi, M. A. Mesaik, O. M. Abdalla, S. Afsharypuor and M. Rabbani, Med. Chem. Res., 2003, 22, 1795–1803 CrossRef.
- C. S. Kim, L. Subedi, S. Y. Kim, S. U. Choi, K. H. Kim and K. R. Lee, J. Nat. Prod., 2016, 79, 387–394 CrossRef CAS PubMed.
- E. J. Richmond and A. D. Rogol, Nat. Clin. Pract. Endocrinol. Metab., 2007, 3, 338–344 CrossRef CAS PubMed.
- S. G. Haider, Endocrinology, 2007, 148, 2581–2582 CrossRef CAS PubMed.
- C. Wang, E. Nieschlag, R. Swerdloff, H. M. Behre, W. J. Hellstorm, L. J. Gooren, J. M. Kaufman, J. Legros, B. Lunenfeld, A. Morales, J. E. Morley, C. Schulman, I. M. Thompson, W. Weidner and F. C. W. Wu, Eur. Urol., 2009, 55, 121–130 CrossRef PubMed.
- D. Sun, Y. Cui, B. Jin, X. Zhang, X. Yang and C. Gao, Evid. Based Complement. Alternat. Med., 2012, 2012, 1–10 Search PubMed.
- B. Lunenfeld, F. Saad and C. E. Hoesl, Aging Male, 2005, 8, 59–74 CrossRef CAS PubMed.
- C. H. Liao, Y. N. Wu, F. Y. Lin, W. K. Tsai, S. P. Liu and H. S. Chiang, Andrology, 2013, 1, 563–569 CrossRef CAS PubMed.
- N. Bassil, S. Alkaade and J. E. Morley, Ther. Clin. Risk Manage., 2009, 5, 427–448 CAS.
- S. Kliesch, Urologe A, 2010, 49, 32–36 CrossRef CAS PubMed.
- J. I. Makinen and I. Huhtaniemi, Gerontology, 2011, 57, 193–202 CrossRef CAS PubMed.
- R. Vigen, C. I. O'Donnell, A. E. Baron, G. K. Grunwald, T. M. Maddox, S. M. Bradley, A. Barqawi, G. Woning, M. E. Wierman, M. E. Plomondon, J. S. Rumsfeld and P. M. Ho, J. Am. Med. Assoc., 2013, 310, 1829–1836 CrossRef CAS PubMed.
- M. L. Dufau, Annu. Rev. Physiol., 1988, 50, 483–508 CrossRef CAS PubMed.
- K. Henriksen, H. Hakovirta and M. Parvinen, Endocrinology, 1995, 136, 3285–3291 CrossRef CAS PubMed.
- J. D. Rabinowitz and E. White, Science, 2010, 330, 1344–1348 CrossRef CAS PubMed.
- F. Gao, G. Li, C. Liu, H. Gao, H. Wang, W. Liu, M. Chen, Y. Shang, L. Wang, J. Shi, W. Xia, J. Jiao, F. Gao, J. Li, L. Chen and W. Li, J. Cell Biol., 2018, 217, 2103–2119 CrossRef CAS.
- G. Wei, Y. Wu, X. L. He, T. Liu, M. Liu, J. Luo and W. W. Qiu, Eur. J. Med. Chem., 2017, 131, 48–67 CrossRef CAS PubMed.
- L. G. Yu, T. F. Ni, W. Gao, Y. He, Y. Y. Wang, H. W. Cui, C. G. Yang and W. W. Qiu, Eur. J. Med. Chem., 2015, 90, 10–20 CrossRef CAS PubMed.
- A. Eschenmoser and D. Arigoni, Helv. Chim. Acta, 2005, 88, 3011–3050 CrossRef CAS.
- G. Stork and A. W. Burgstaher, J. Am. Chem. Soc., 1955, 77, 5068–5077 CrossRef CAS.
- Z. B. Zhang and Q. T. Yang, Asian J. Androl., 2006, 8, 601–605 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectrum of the corresponding compounds. 1H and 13C NMR spectra of all final compounds. See DOI: 10.1039/c9ra00702d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |