
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetformin promotes proliferation and suppresses apoptosis in Ox-LDL stimulated macrophages by regulating the miR-34a/Bcl2 axis†
Liuliu Feng,
Tianhua Liu,
Yuya Yang,
Wenying Xiao,
Jun Shi,
Xiang Mei,
Songmei Tian,
Xinbing Liu,
Hongman Huang and
Yanyan Bai *
*
Department of Cardiology, Shidong Hospital, Yangpu District, Shanghai, 200438, China. E-mail: baiyysdhdc04@126.com; Tel: +86-021-25066666-13101
First published on 10th May 2019
Abstract
Background: Metformin, an antidiabetic drug, has been reported to be involved in atherosclerosis (AS). In this study, the effects of metformin on oxidized low-density lipoprotein (Ox-LDL)-induced macrophage apoptosis were investigated, and the mechanisms involved in this process were examined. Methods: qRT-qPCR analysis was performed to detect the expression of miR-34a in macrophage cells. Cell proliferation was determined by MTT assays and colony formation assays. Cell apoptosis was assessed by the detection of apoptotic rate and caspase 3 activity. Western blot analysis was performed to evaluate the expression of Bcl2 protein. Results: Metformin treatment promoted proliferation and suppressed apoptosis in macrophages following the treatment of oxidized low-density lipoprotein (Ox-LDL). Metformin could inhibit miR-34a in macrophages. miR-34a overexpression could reverse the effect of metformin on proliferation and apoptosis in Ox-LDL-treated macrophages. Moreover, metformin could increase the expression of the miR-34a target gene Bcl2. Furthermore, metformin treatment exerted the pro-proliferation and anti-apoptosis effect through regulating Bcl2 expression in Ox-LDL-stimulated macrophages. Conclusion: Metformin facilitated proliferation and inhibited apoptosis of macrophages treated with Ox-LDL through the miR-34a/Bcl2 axis, indicating the potential value of metformin in AS therapy.
Introduction
Cardiovascular diseases (CVDs) are a primary cause of death with more than 17.3 million deaths per year worldwide.1 Atherosclerosis (AS), a major underlying basis of heart attacks and strokes, accounts for a large proportion of cardiovascular diseases. AS is an inflammatory disease with the characteristics of lipid accumulation, fibrous cap formation and necrotic core generation.2 Multiple cell types such as smooth muscle cells (SMCs), endothelial cells (ECs) and macrophages are involved in atherosclerotic lesion formation.3 Moreover, previous studies showed that abnormal proliferation, apoptosis and migration of macrophages were closely associated with AS progression.4,5 Oxidized low-density lipoprotein (Ox-LDL), a critical risk factor in AS progression, was reported to be able to affect macrophages' apoptosis.6 Thus, in the present study, we employed Ox-LDL-treated macrophages as an AS cell model to investigate the regulatory mechanisms involved in AS.Metformin is a synthetic dimethyl biguanide and has been a mainstay of therapy for patients with type 2 diabetes mellitus (T2DM).7 Metformin could suppress the expression of proinflammatory factors, and reduce oxidative stress.8–10 Several researchers have reported that metformin can slow or reverse the progress of AS.10–13 However, the underlying mechanisms of the antiatherogenic role of metformin remain not well elucidated.
MicroRNAs, a class of small non-coding RNA of 22–25 nucleotides, are implicated in the regulation of genes at posttranscriptional level by repressing translation or promoting RNA degradation.14 In AS, many microRNAs have been confirmed as vital modulators of pathological processes, such as immune responses, endothelial cell biology, cholesterol and lipid biosynthesis, and lipoprotein metabolism and cholesterol efflux.15 Increasing evidence indicated that miR-34a acted as a tumor suppressor in many cancers such as gastric cancer,16 breast cancer17 and non-small cell lung cancer.18 Previous studies revealed that miR-34a was upregulated in atherosclerotic samples and could contribute to AS progression.19–21 Nevertheless, how miR-34a was regulated and its functional role in Ox-LDL stimulated macrophages are still unclear. Previous studies reported that metformin could inhibit miR-34a expression in nonalcoholic fatty liver disease and in rat mesangial cells treated with high glucose.22,23 However, whether metformin ameliorates the progression of AS through regulating miR-34a remains uncertain.
In our study, we aimed to further explore the underlying roles and molecular mechanisms of metformin in Ox-LDL stimulated macrophages to discover potential therapy targets of AS.
Materials and methods
Cell culture
The THP1 macrophage cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in RPMI-1640 culture medium supplemented with 10% FBS (Invitrogen), and 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (Sigma-Aldrich, St. Louis, MO, USA) in a humidified incubator with 5% CO2 at 37 °C. The cells were treated with 0.5 mM metformin or 50 μg ml−1 Ox-LDL for 24 h for further experiments. Metformin (1,1-dimethylbiguanide, 99.6% purity) was obtained from Spectrum Chemical manufacturing Corp (New Brunswick, NJ).Cell transfection and treatment
Small interference RNA (siRNA) against Bcl2 (si-Bcl2) and its scramble control (si-con), miR-34a mimic (miR-34a) and its negative control (miR-con), miR-34a inhibitor (anti-miR-34a) and its negative control (anti-miR-con) were designed and synthesized by GenePharma (Guangzhou, China). Cell transfection was performed using lipofectamine 2000 reagent (Invitrogen).RT-qPCR assays
Total RNA was extracted using TRIzol® reagent (Invitrogen) and reversely transcribed into cDNA with M-MLV (Invitrogen). Then, the SYBR Green Realtime PCR Master Mix (TOYOBO, Osaka, Japan) were used to detect the expression of miR-34a and Bcl2. U6 snRNA and GAPDH served as endogenous controls for miR-34a and Bcl2. The primer sequences were as follows: for miR-34a 5′-TGGCAG TGTCTTAGCTGGTTGT-3′ (forward) and 5′-GCGAGCACAGAATTAATACGAC-3′ (reverse); for Bcl2 5′-CTGGCATCTTCTCCTTCCAGC-3′ (forward) and 5′-ACCTACCCAGCCTCCGTTATC-3′ (reverse). The relative expression of miR-34a and Bcl2 was calculated using the 2−ΔΔCt method.Western blot assays
Western blot analysis was performed as previously described.24 Briefly, the proteins were extracted using RIPA buffer (KeyGEN Biotech, Nanjing, China) and separated by SDS-PAGE gel and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). Then, the membranes were incubated with primary antibodies against Bcl2 (sc-783, rabbit polyclonal) and β-actin (sc-1615, goat polyclonal) from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Membranes were washed three times prior to incubation with HRP-conjugated anti-mouse secondary antibodies from Santa Cruz Biotechnology. Finally, the protein bands were detected by ECL chemiluminescent regent (Pierce, Rockford, IL, USA).MTT assays
Viable macrophages were determined by MTT at 0, 24, 48, or 72 h post-transfection. Briefly, the cells were seeded at density of 2.0 × 103 cells per well into 96-well plates. Following treatment with 50 μg ml−1 Ox-LDL and 0.5 mM metformin, 10 μl 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich, USA) was added into each well of 96-well plates. The plates were incubated for 4 h at 37 °C. Then the absorbance at 490 nm was determined to evaluate cell proliferation.Colony formation assays
Transfected macrophages (2.0 × 104 cells per well) were seeded in a 12-well plate and cultured for 15 days in complete medium. Then cells were fixed with methanol and stained with 0.1% crystal violet solution (Sigma-Aldrich). At last, the total number of colonies with a minimum of 50 cells was detected using a microscope.Flow cytometric analysis
The apoptosis rate of macrophages was evaluated using Annexin V-FITC/PI Apoptosis Detection Kit (KeyGEN Biotech, Nanjing, China) as described previously.25 Briefly, macrophages were stained with 5 μl Annexin V-FITC (1 μg ml−1) for 10 min and 5 μl propidium iodide (PI) (1 μg ml−1) for 5 min in the dark at room temperature. The apoptotic rates were analyzed using CellQuest software (BD Biosciences, Franklin Lakes, NJ, USA).Caspase 3 activity detection
Macrophages were treated with 50 μg ml−1 Ox-LDL and 0.5 mM metformin for 24 h. Cells were lysed, and the protein concentration was measured using the Bradford Protein Assay Kit (Beyotime Biotechnology, Shanghai, China). Then, equal amount of proteins was incubated with reaction buffer containing enzyme-specific substrates for caspase 3 at 37 °C for 1 h. The absorbance of 405 nm was detected using a microplate reader (Molecular Devices, Sunnyvale, California, USA) using the Caspase 3 Assay Kit (Calbiochem, San Diego, California, USA).Statistical analysis
All data were presented as mean ± standard deviation (SD). The comparisons of data in different groups were performed using Student's t-test or one-way variance analysis. P < 0.05 was statistically significant.Results
Metformin promoted proliferation and suppressed apoptosis in Ox-LDL-treated macrophages
To investigate the functional role of metformin in AS process, we explored the effect of metformin on proliferation and apoptosis in Ox-LDL-treated macrophages. Macrophages were treated with 50 μg ml−1 Ox-LDL and with or without 0.5 mM metformin for 24 h, followed by measurement of cell proliferation ability and apoptotic rate. MTT assays revealed that Ox-LDL could suppress macrophages proliferation, however, metformin treatment weakened the inhibitory effect of Ox-LDL on macrophages proliferation (Fig. 1A). Moreover, colony formation assays also demonstrated that metformin significantly improved colony formation potential of Ox-LDL-treated macrophages (Fig. 1B). Additionally, flow cytometry assays showed that Ox-LDL could induced macrophages apoptosis, nevertheless, metformin markedly lowered the apoptotic rate of Ox-LDL-treated macrophages (Fig. 1C). Consistently, metformin treatment resulted in a remarkable reduction of caspase 3 activity in Ox-LDL-treated macrophages (Fig. 1D). In summary, these results suggested that metformin facilitated proliferation and suppressed apoptosis in Ox-LDL-treated macrophages.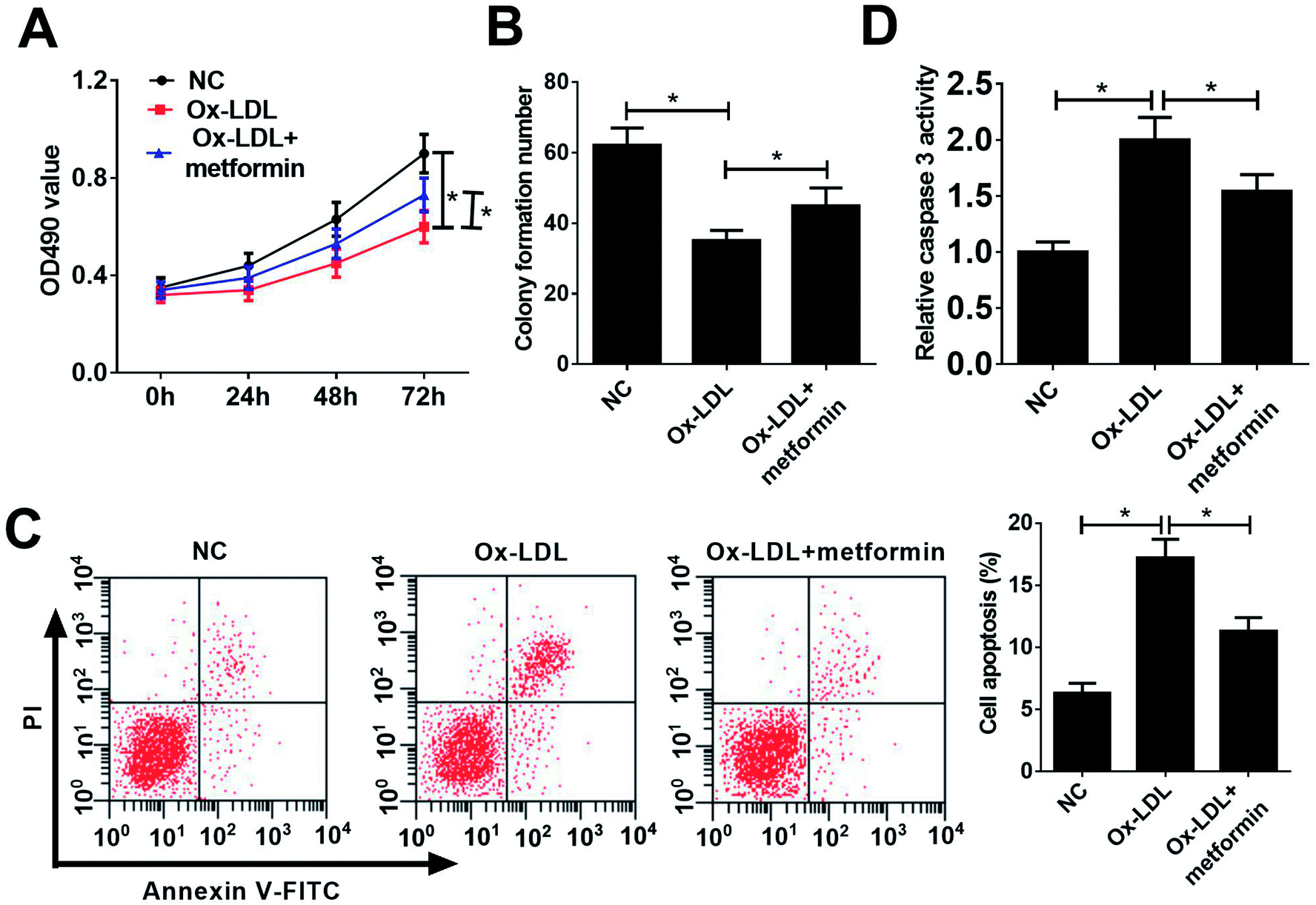 | ||
| Fig. 1 Metformin promoted proliferation and suppressed apoptosis in Ox-LDL-treated macrophages. Macrophages were treated with 50 μg ml−1 Ox-LDL or 0.5 mM metformin for 24 h, followed by the detection of proliferation capacity (A), clone formation ability (B), apoptotic rate (C), and caspase 3 activity (D). *P < 0.05. | ||
miR-34a overexpression reversed the effects of metformin on proliferation and apoptosis of Ox-LDL-treated macrophages
Previous studies reported that metformin could down-regulate miR-34a expression in high glucose condition and downregulation of miR-34a could suppress Ox-LDL-induced apoptosis.26,27 Hence, we presumed that metformin may promote proliferation and suppressed apoptosis in Ox-LDL stimulated macrophages through regulating miR-34a expression. To confirm whether the pro-proliferation and anti-apoptosis effect of metformin was mediated by miR-34a, we first detected miR-34a expression in macrophages with Ox-LDL and metformin treatment. As expected, Ox-LDL could increase miR-34a expression, which was weakened by metformin treatment (Fig. 2A). Then, we treated Ox-LDL stimulated macrophages with metformin or metformin + miR-34a. qRT-PCR analysis indicated that introduction of miR-34a mimics could abolish the inhibitory effect of metformin on miR-34a expression (Fig. 2B). MTT and colony formation assays further showed that the enhancive effect of metformin on cell growth was greatly abrogated by miR-34a overexpression (Fig. 2C and D). As expected, miR-34a overexpression markedly abated metformin-mediated apoptosis inhibition (Fig. 2E and F). Taken together, these results indicated that metformin exerted its pro-proliferation and anti-apoptosis effect in Ox-LDL-treated macrophages through inhibiting miR-34a expression.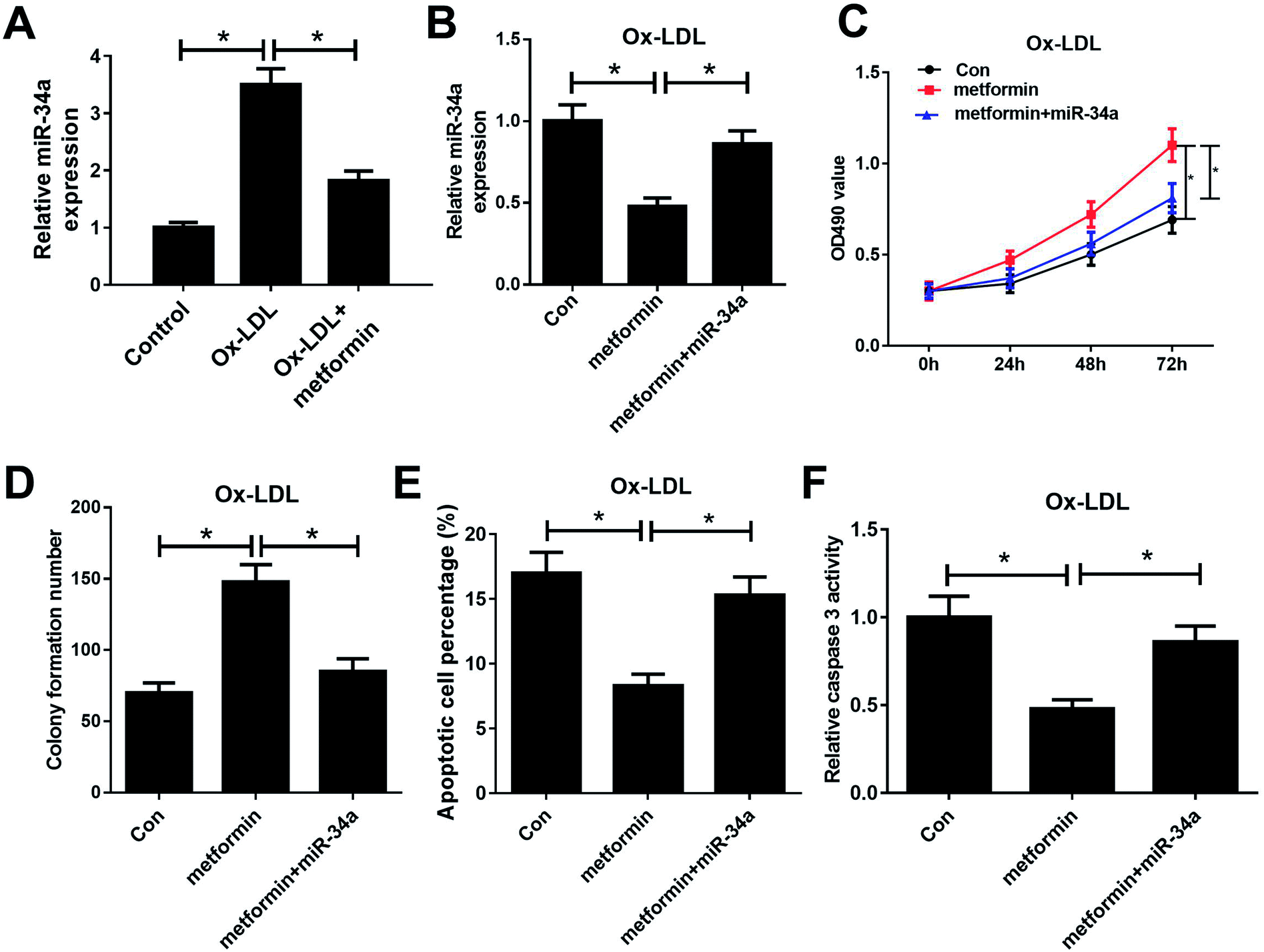 | ||
| Fig. 2 miR-34a overexpression reversed the effects of metformin on proliferation and apoptosis of Ox-LDL-treated macrophages. (A) qRT-PCR analysis was performed to detect miR-34a expression in Ox-LDL, or Ox-LDL + metformin treated macrophages. Macrophages stimulated with 50 μg ml−1 Ox-LDL were treated with Con, metformin, or metformin + miR-34a were for 24 h, followed by the measurement of miR-34a expression (B), cell proliferation capability (C), colony formation ability (D), apoptotic rate (E), and caspase 3 activity (F). *P < 0.05. | ||
Metformin elevated the expression of miR-34a target Bcl2
It is well known that miRNAs exert their functions through targeting special mRNAs. Bcl2, a known target of miR-34a, has been reported to be implicated with AS progression.28 Hence, we further explored whether metformin positively regulated Bcl2 through inhibiting miR-34a expression. As displayed in Fig. 3A and B, miR-34a inhibition could elevate the reduced Bcl2 mRNA and protein expression caused by Ox-LDL in macrophages. Consistently, metformin increased Bcl2 mRNA and protein levels in Ox-LDL treated macrophages (Fig. 3C and D). Additionally, miR-34a overexpression relieved metformin-mediated increase on Bcl2 mRNA and protein levels in Ox-LDL treated macrophages (Fig. 3E and F). Collectively, these data indicated that metformin could elevate Bcl2 expression through suppressing miR-34a in Ox-LDL treated macrophages.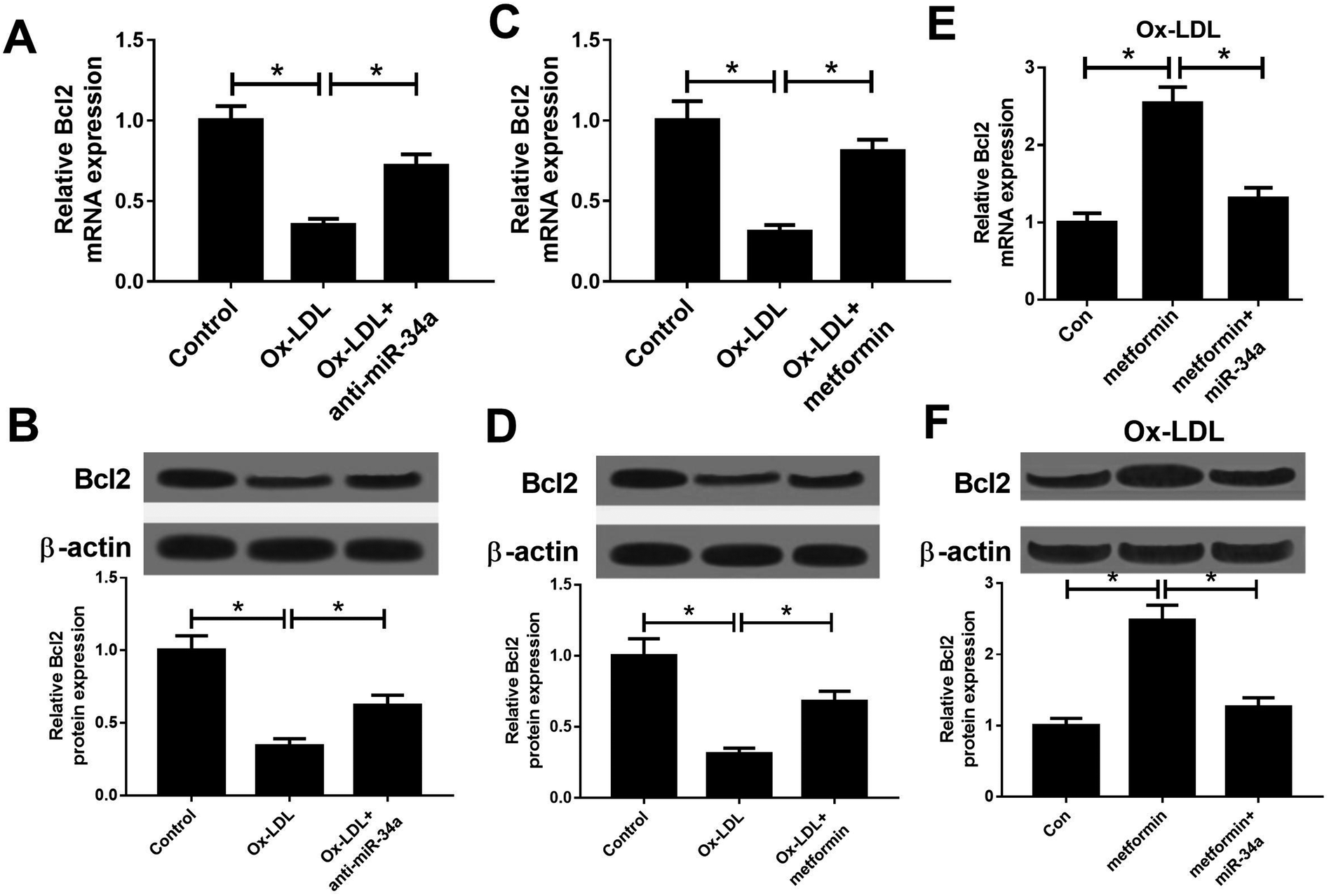 | ||
| Fig. 3 Metformin promoted the expression of miR-34a target Bcl2. (A and B) The Bcl2 mRNA and protein expression was determined in macrophages treated with Ox-LDL or Ox-LDL + anti-miR-34a. (C and D) The Bcl2 mRNA and protein expression was determined in macrophages treated with Ox-LDL or Ox-LDL + metformin. (E and F) The Bcl2 mRNA and protein expression was determined in Ox-LDL treated macrophages treated with Con, metformin, or metformin + miR-34a. *P < 0.05. | ||
Metformin promoted by regulating miR-34a/Bcl2 axis in Ox-LDL-treated macrophages
To further examine whether the effect of metformin on proliferation and apoptosis was mediated by miR-34a/Bcl2 axis in Ox-LDL-stimulated macrophages, Ox-LDL treated macrophages were treated with Con, metformin or metformin + si-Bcl2. Western bolt analysis revealed that introduction of si-Bcl2 weakened the increased Bcl2 mRNA and protein levels caused by metformin treatment in macrophages (Fig. 4A and B). Moreover, metformin-mediated enhancement on cell growth was greatly declined by Bcl2 knockdown in macrophages (Fig. 4C and D). Furthermore, Bcl2 silencing markedly abated metformin-mediated apoptosis inhibition macrophages (Fig. 4E and F). These data demonstrated that metformin exerted its pro-proliferation and anti-apoptosis effect by regulating miR-34a/Bcl2 axis in Ox-LDL-treated macrophages.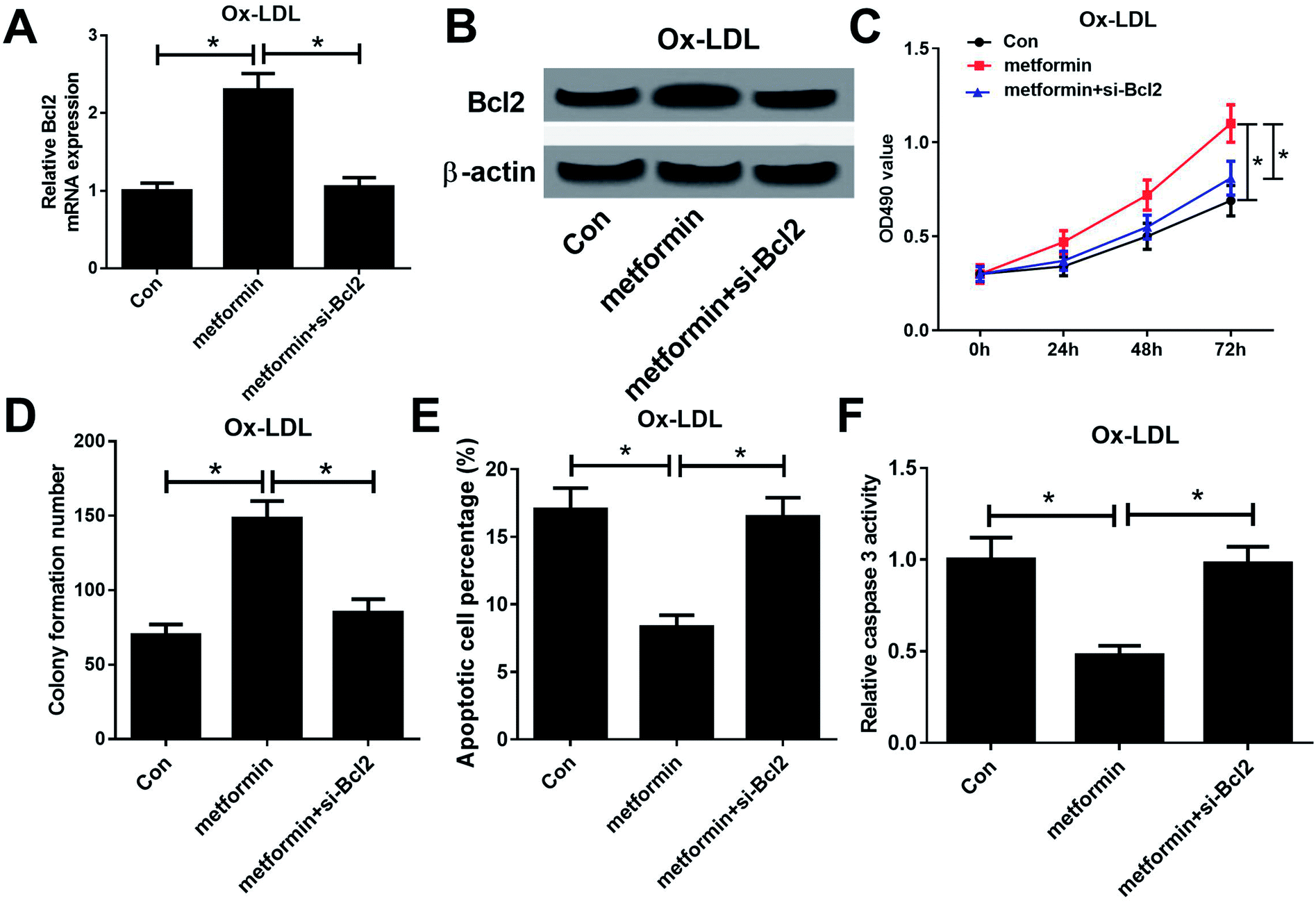 | ||
| Fig. 4 Metformin promoted proliferation and inhibited apoptosis by regulating miR-34a/Bcl2 axis in Ox-LDL-treated macrophages. Macrophages were treated with 50 μg ml−1 Ox-LDL and Con, metformin, or metformin + si-Bcl2 for 24 h, followed by the determination of Bcl2 mRNA and protein levels (A and B), cell proliferation ability (C), colony formation capability (D), apoptotic rate (E), and caspase 3 activity (F). *P < 0.05. | ||
Discussion
Although much progress has been made in the pathogenesis and therapeutic options of AS, AS is still a major threaten of human health with a high morbidity and mortality worldwide.29 Previous studies have demonstrated that metformin, an antidiabetic drug, has obvious anti-atherogenic properties,12,30–32 but how metformin exerts these effects is unclear. In the present study, we investigated the effects of metformin treatment on the progression of atherogenesis using an Ox-LDL-induced atherosclerotic cell model. Our results suggested that metformin significantly attenuated the progression of atherosclerosis by promoting proliferation and suppressing apoptosis in Ox-LDL stimulated macrophages through miR-34a/Bcl2 axis.In recent years, many studies have revealed that metformin has many excess effects on blood vessels besides its hypoglycemic effect.33,34 Metformin was identified to be the only hypoglycemic drug lowering the risk of cardio-cerebrovascular complications of T2DM patients.35 Increasing evidence has suggested that metformin is implicated with the progression of AS. For example, metformin could suppress diabetes-related atherosclerosis via inhibiting Drp1-mediated mitochondrial fission.13 Furthermore, metformin could ameliorate the pro-inflammatory state in patients with carotid artery atherosclerosis.36 Metformin could also reduce lipid accumulation in macrophages by inhibiting FOXO1-mediated transcription of fatty acid-binding protein 4.37 Another study demonstrated that metformin inhibits macrophage cholesterol biosynthesis rate through attenuated oxidative stress.38 Since macrophages play a vital role in the progress of atherosclerosis, we assume that metformin attenuated atherosclerosis may be related to the inhibition of apoptosis in macrophages. Our findings revealed that metformin treatment promoted proliferation and suppressed apoptosis in Ox-LDL stimulated macrophages, which suggested that metformin could attenuate AS progression.
In this study, we further investigate the mechanism underlying the inhibition of AS progression by metformin. Previous studies reported metformin could regulate the expression of miRNAs, which exerts vital roles in various diseases. Therefore, we presumed that metformin may attenuate AS through modulating AS-related miRNAs. A previous study reported that metformin could regulate miR-34a expression in rat mesangial cells cultured with high glucose.26 Moreover, downregulation of miR-34a suppressed Ox-LDL-induced apoptosis and oxidative stress in human umbilical vein endothelial cells.27 However, the role of miR-34a in Ox-LDL stimulated macrophages is still unclear. Hence, we further explored whether metformin promoted proliferation and suppressed apoptosis in Ox-LDL stimulated macrophages through regulating miR-34a expression. Our study found that metformin treatment decreased miR-34a expression in macrophages. Furthermore, miR-34a overexpression could reverse the inductive effect of metformin on proliferation and the inhibitory effect of metformin on apoptosis in Ox-LDL stimulated macrophages. Similarly, a previous study revealed that downregulation of miR-34a promoted endothelial cell growth and suppresses apoptosis in Ox-LDL-treated human aortic endothelial cells.27 All these data demonstrated that metformin ameliorated AS through inhibiting miR-34a expression.
Emerging evidence shows that miRNAs exert functions by regulating translation or stability of target mRNAs. Hence, the potential target mRNAs of miR-34a was further investigated. Bcl2, an anti-apoptotic gene, was reported to be a target of miR-34a,27 which has been reported to be involved in AS progress.39,40 In this work, we found that metformin could elevate Bcl2 expression through inhibiting miR-34a expression. Furthermore, Bcl2 knockdown could abolished the inhibitory effect of metformin treatment on AS. In a word, metformin ameliorated AS through modulating miR-34a/Bcl2 axis.
In summary, our study demonstrated that metformin promoted proliferation and suppressed apoptosis in ox-LDL-treated macrophages by elevating Bcl2 expression via inhibiting miR-34a. This finding elucidated the involvement of metformin/miR-34a/Bcl2 regulatory axis in AS progression, which provided a promising target for AS treatment.
Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The research was approved by the Project of Shanghai Municipal Commission of Health and Family Planning.References
- L. J. Laslett, P. Alagona, B. A. Clark, J. P. Drozda, F. Saldivar, S. R. Wilson, C. Poe and M. Hart, J. Am. Coll. Cardiol., 2012, 60, S1–S49 CrossRef.
- S. Carr, A. Farb, W. H. Pearce, R. Virmani and J. S. T. Yao, J. Vasc. Surg., 1996, 23, 755 CrossRef CAS.
- A. C. Doran, M. Nahum and C. A. Mcnamara, Arterioscl. Throm. Vas., 2008, 28, 812 CrossRef CAS PubMed.
- B. Wang, Z. Ge, Z. Cheng and Z. Zhao, Biol. Open, 2017, 6, 489–495 CrossRef CAS PubMed.
- X. Zhuang, R. Li, A. Maimaitijiang, R. Liu, F. Yan, H. Hu, X. Gao and H. Shi, J. Cell. Biochem., 2018, 120, 6304–6314 CrossRef PubMed.
- Y. Chen, M. Li, Y. Zhang, M. Di, W. Chen, X. Liu, F. Yu, H. Wang, X. Zhen and M. Zhang, Biochem. Biophys. Res. Commun., 2018, 501, 336–342 CrossRef CAS.
- C. J. Bailey and R. C. Turner, N. Engl. J. Med., 1996, 334, 574–579 CrossRef CAS PubMed.
- W. Xu, Y. Y. Deng, L. Yang, S. Zhao, J. Liu, Z. Zhao, L. Wang, P. Maharjan, S. Gao and Y. Tian, Transl. Res., 2015, 166, 451–458 CrossRef CAS PubMed.
- D. Pavlović, R. Kocić, G. Kocić, T. Jevtović, S. Radenković, D. Mikić, M. Stojanović and P. B. Djordjević, Diabetes, Obes. Metab., 2010, 2, 251–256 CrossRef.
- J. C. Mamputu, N. F. Wiernsperger and G. Renier, Diabetes Metab., 2003, 29, 6S71–6S76 CrossRef CAS.
- F. Kathleen, A. Suhny, L. Hang, S. Eleni, S. Rachel, M. Theresa, H. Linda, T. Martin and G. Steven, AIDS, 2012, 26, 587–597 CrossRef PubMed.
- L. Fei, G. Yuan, G. Ruan, J. Long, X. Zheng, X. Qin, S. Zhao, D. Peng, Z. Fang and X. Li, Sci. Rep., 2017, 7, 2169 CrossRef PubMed.
- Q. Wang, M. Zhang, G. Torres, S. Wu, C. Ouyang, Z. Xie and M. H. Zou, Diabetes, 2017, 66, 193–205 CrossRef CAS PubMed.
- M. Georges and C. Charlier, Curr. Opin. Genet. Dev., 2007, 17, 166–176 CrossRef CAS PubMed.
- H. Giral, A. Kratzer and U. Landmesser, Best Pract. Res., Clin. Endocrinol. Metab., 2016, 30, 665–676 CrossRef CAS PubMed.
- Y. Peng, J. J. Guo, Y. M. Liu and X. L. Wu, Biosci. Rep., 2014 DOI:10.1042/BSR20140020.
- M. Xu, D. Li, C. Yang and J. S. Ji, Cell. Physiol. Biochem., 2018, 46, 1286–1304 CrossRef CAS PubMed.
- J. Y. Zhou, X. Chen, J. Zhao, Z. Bao, X. Chen, P. Zhang, Z. F. Liu and J. Y. Zhou, Cancer Lett., 2014, 351, 265–271 CrossRef CAS PubMed.
- E. Raitoharju, L.-P. Lyytikäinen, M. Levula, N. Oksala, A. Mennander, M. Tarkka, N. Klopp, T. Illig, M. Kähönen and P. J. Karhunen, Atherosclerosis, 2011, 219, 211–217 CrossRef CAS PubMed.
- X. Zhong, P. Li, J. Li, R. He, G. Cheng and Y. Li, Int. J. Mol. Med., 2018, 42, 1134–1144 CAS.
- G. Su, G. Sun, H. Liu, L. Shu and Z. Liang, Heart Vessels, 2018, 33, 1–10 CrossRef PubMed.
- K. Akiko, M. Asahiro, I. Hisakazu, T. Joji, S. Teppei, T. Miwa, T. Yuka, F. Koji, K. Kiyohito and M. Emiko, Int. J. Mol. Med., 2015, 35, 877–884 CrossRef PubMed.
- C. Wu, N. Qin, H. Ren, M. Yang, S. Liu and Q. Wang, Int. J. Endocrinol., 2018, 6462793, DOI:10.1155/2018/6462793.
- K. F. Lei, Y. F. Wang, X. Q. Zhu, P. C. Lu, B. S. Sun, H. L. Jia, N. Ren, Q. H. Ye, H. C. Sun and L. Wang, BMC Cancer, 2007, 7, 172 CrossRef PubMed.
- W. J. Wang, Y. Yao, L. L. Jiang, T. H. Hu, J. Q. Ma, Z. J. Liao, J. T. Yao, D. F. Li, S. H. Wang and K. J. Nan, PLoS One, 2013, 8, e76596 CrossRef CAS PubMed.
- C. Wu, N. Qin, H. Ren, M. Yang, S. Liu and Q. Wang, Int. J. Endocrinol., 2018, 2018, 1–15 Search PubMed.
- X. Zhong, P. Li, J. Li, R. He, G. Cheng and Y. Li, Int. J. Mol. Med., 2018, 42, 1134–1144 CAS.
- E. Thorp, Y. Li, L. Bao, P. M. Yao, G. Kuriakose, J. Rong, E. A. Fisher and I. Tabas, Arterioscler., Thromb., Vasc. Biol., 2009, 29, 169–172 CrossRef CAS PubMed.
- W. Christian and N. Heidi, Nat. Med., 2011, 17, 1410–1422 CrossRef PubMed.
- F. Farshad, S. Gloria, P. Nikolay, X. Shiqin, H. Lula, F. Baowei and A. R. Wayne, J. Am. Heart Assoc., 2014, 3, A2040 Search PubMed.
- K. Adriaan, D. J. Jolien, L. Philippe, B. Daniel, M. G. Wulffelé, A. J. M. Donker and C. D. A. Stehouwer, Arch. Intern. Med., 2009, 169, 616 CrossRef.
- P. M. Ridker, Lancet, 2014, 384, 607–617 CrossRef CAS.
- A. R. Cameron, V. L. Morrison, D. Levin, M. Mohan, C. Forteath, C. Beall, A. D. Mcneilly, D. J. K. Balfour, T. Savinko and A. K. F. Wong, Circ. Res., 2016, 119, 652–665 CrossRef CAS PubMed.
- S. Gillessen, C. Gilson, N. James, A. Adler, M. R. Sydes and N. Clarke, Eur. Urol., 2016, 70, 906–908 CrossRef PubMed.
- A. S. G. W. Committee, Diabetes Care, 2016, 39, 701 CrossRef PubMed.
- W. Xu, Y. Y. Deng, L. Yang, S. Zhao, J. Liu, Z. Zhao, L. Wang, P. Maharjan, S. Gao and Y. Tian, Transl. Res., 2015, 166, 451–458 CrossRef CAS PubMed.
- J. Song, P. Ren, L. Zhang, X. L. Wang, L. Chen and Y. H. Shen, Biochem. Biophys. Res. Commun., 2010, 393, 89–94 CrossRef CAS PubMed.
- M. Korengluzer, M. Aviram and T. Hayek, Biochem. Biophys. Res. Commun., 2013, 439, 396–400 CrossRef CAS PubMed.
- Y. Yu, Q. Liu, S. Guo, Q. Zhang, J. Tang, G. Liu, D. Kong, J. Li, S. Yan, R. Wang, P. Wang, X. Su and Y. Yu, J. Cell. Mol. Med., 2017, 21, 3540–3551 CrossRef CAS PubMed.
- R. Wu, S. Tang, M. Wang, X. Xu, C. Yao and S. Wang, PLoS One, 2016, 11, e0167052 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00705a |
| This journal is © The Royal Society of Chemistry 2019 |