
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic bactericide and antibiotic effects of dimeric, tetrameric, or palindromic peptides containing the RWQWR motif against Gram-positive and Gram-negative strains
Yerly Vargas-Casanovaa,
Andrea Verónica Rodríguez-Mayor b,
Karen Johanna Cardenasc,
Aura Lucía Leal-Castrod,
Liliana Constanza Muñoz-Molinae,
Ricardo Fierro-Medinab,
Zuly Jenny Rivera-Monroyb and
Javier Eduardo García-Castañeda*e
b,
Karen Johanna Cardenasc,
Aura Lucía Leal-Castrod,
Liliana Constanza Muñoz-Molinae,
Ricardo Fierro-Medinab,
Zuly Jenny Rivera-Monroyb and
Javier Eduardo García-Castañeda*e
aBiotechnology Institute, Universidad Nacional de Colombia, Carrera 45 No 26-85, Bogotá, 11321, Colombia
bChemistry Department, Universidad Nacional de Colombia, Carrera 45 No 26-85, Building 450, Office 213, Bogotá, 11321, Colombia
cPharmacy Department, Universidad Nacional de Colombia, Carrera 45 No 26-85, Building 450, Office 213, Bogotá, 11321, Colombia
dMedicine Faculty, Universidad Nacional de Colombia, Carrera 45 No 26-85, Building 450, Office 213, Bogotá, 11321, Colombia
eUniversidad Colegio Mayor de Cundinamarca, Calle 28 No. 5B-02, Bogotá, 110311, Colombia. E-mail: jaegarciaca@unal.edu.co; Tel: +57-1-3165000 extn 14436
First published on 5th March 2019
Abstract
Dimeric and tetrameric peptides derived from LfcinB (20–25): RRWQWR, LfcinB (20–30): RRWQWRMKKLG, LfcinB (17–31): FKARRWQWRMKKLGA, or the palindromic sequence LfcinB (21–25)Pal: RWQWRWQWR were obtained by means of the SPPS-Fmoc/tBu methodology. The antibacterial activity of these molecules was evaluated against Escherichia coli (ATCC 25922 and ATCC 11775), Staphylococcus aureus (ATCC 25923), Enterococcus faecalis (ATCC 29212), and Pseudomonas aeruginosa (ATCC 27853). The dimer LfcinB (20–25)2: (RRWQWR)2K-Ahx, the tetramer LfcinB (20–25)4: (RRWQWR)4K2-Ahx2-C2, and the palindromic sequence LfcinB (21–25)Pal exhibited the highest antibacterial activity against the tested bacterial strains. In all cases, the antibacterial activity was dependent on peptide concentration. The polyvalent molecules LfcinB (20–25)2 and LfcinB (20–25)4 exhibited bacteriostatic and bactericidal activity against E. coli, P. aeruginosa, and S. aureus strains; additionally, this dimer and this tetramer combined with ciprofloxacin exhibited a synergistic antibacterial effect against E. coli ATCC 25922 and P. aeruginosa, respectively. Furthermore, the peptides LfcinB (20–30)4, LfcinB (20–25)4, and LfcinB (21–25)Pal combined with vancomycin exhibited a synergistic antibacterial effect against S. aureus and E. faecalis, respectively. This study showed that polyvalent peptides derived from LfcinB exhibit significant antibacterial activity, suggesting that these peptides could have a therapeutic application. Furthermore, our results suggest that polyvalent peptide synthesis could be considered as an innovative and viable strategy for obtaining promising antimicrobial molecules.
1 Introduction
According to the World Health Organization (WHO), antibiotic resistance (AR) is a great threat to global health, affecting people regardless of age, race, or geographic region. AR occurs naturally due to the misuse of antibiotics in humans and animals, and as a result it increases hospital stays, medical costs, and mortality.1 Currently, AR bacteria associated with opportunistic infections are: (i) E. coli, which mainly causes three syndromes: enteric/diarrheal disease, urinary tract infection, and sepsis/meningitis.2 (ii) P. aeruginosa, which affects immunocompromised patients, especially those using medical equipment such as catheters or mechanical ventilators. It causes respiratory tract infections and urinary infections, and affects the skin and soft tissue. This bacterium can migrate through blood circulation, causing septicemia and high mortality rates.3 (iii) S. aureus, which causes bacteremia, infective endocarditis, skin and soft tissue infections (impetigo, folliculitis, boils, carbuncles, and cellulitis), lung infections (pneumonia and empyema), gastroenteritis, meningitis, toxic shock syndrome, and urinary tract infections.4 (iv) E. faecalis is an important nosocomial pathogen, and infects the urinary tract, the bloodstream, the endocardium, the abdomen, the biliary tract, and burn wounds. E. faecalis forms a biofilm in external medical devices and has been implicated in endodontic infections.5Infections caused by pathogens resistant to conventional antibiotics limit therapeutic options. It is necessary to identify promising molecules for developing new drugs against bacterial infections.6,7 Chemical synthesis is considered to be a feasible and novel tool for designing and obtaining therapeutic peptides.8 Synthetic peptides are safe, since their amino acid sequence is determined, and they can be obtained quickly and with high purity. Solid-phase peptide synthesis (SPPS) allows obtaining short peptides with high purity and enough quantity for preclinical and clinical phases in drug development. However, synthetic peptides are susceptible to proteases and can be rapidly eliminated by the body.9 To overcome these drawbacks, various strategies have been designed, such as (i) inclusion of non-natural amino acids, and (ii) conjugation with sugars, lipids, and proteins, and (iii) polyvalent peptide synthesis (dimers, tetramers, etc.).10
Bovine lactoferricin (LfcinB: 17FKCRRWQWRMKKLGAPSITC-VRRAF41) is a 25-amino acid peptide with antibacterial, antifungal, antiparasitic, antiviral, and antitumoral activity.11–16 LfcinB-derived short peptides have exhibited higher antibacterial activity than LfcinB and Lactoferrin themselves.10,17–21 Previous studies have shown that the inclusion of non-natural amino acids in some LfcinB-derived sequences improved their antibacterial activity13,17–20 Furthermore, dimeric and tetrameric peptides containing the RRWQWR motif exhibited higher antibacterial activity than Lactoferrin or LfcinB. In like manner, tetrameric and dimeric peptides containing 20RRWQWR25 or 17FKARRWQWRMKKLGA31 sequences exhibited higher antibacterial activity against E. coli strains than their monomeric peptide analogues.13,17–20 High antibacterial activity was also found when the palindromic sequence RWQWRWQWR was tested.13,17,19 In the present study, the antibacterial activity of polyvalent peptides derived from LfcinB was evaluated against E. coli ATCC 43827, S. aureus ATCC 25923, E. faecalis ATCC 29212, and P. aeruginosa ATCC 27853. Our results indicate that polyvalence of LfcinB-derived sequences significantly enhances antibacterial activity. The tetrameric peptide containing the RRWQWR motif exhibited a bactericidal effect against the evaluated strains. Additionally, the peptides LfcinB (20–25)4 or LfcinB (20–25)2 combined with vancomycin or ciprofloxacin exhibited a significant synergistic antibacterial effect.
2 Experimental details
2.1 Reagents
Muller–Hinton, Agar SPC, Muller Hinton Broth (MHB), ciprofloxacin (Bayer), vancomycin (Hospira. Inc- Pfizer). Bacterial strains (E. coli ATCC 43827, S. aureus ATCC 25923, E. faecalis ATCC 29212, and P. aeruginosa ATCC 27853) were obtained from ATCC (Manassas, VA, USA). N,N-Diisopropylethylamine (DIPEA), triisopropylsilane (TIPS), 1,2-ethanedithiol (EDT), 4-methylpiperidine, pyridine, and ninhydrin were obtained from Sigma-Aldrich (St. Louis, MO, USA). Rink amide resin, Fmoc-amino acids, 6-chloro-1-hydroxybenzotriazole (6-Cl-HOBt), and N,N-dicyclohexylcarbodiimide (DCC) were purchased from AAPPTec (Louisville, KY, USA). Methanol, diethyl ether, N,N-dimethylformamide (DMF), absolute ethanol, dichloromethane (DCM), acetonitrile (ACN), isopropylalcohol (IPA), and trifluoroacetic acid (TFA) were obtained from Merck (Darmstadt, Germany). All reagents were used without further purification. Silicycle® SiliaPrep™ C18 cartridges were kindly donated by EcoChem Especialidades Químicas.2.2 Solid-phase peptide synthesis
Peptides were synthesized using the manual SPPS-Fmoc/tBu methodology.18,22 Briefly, Rink amide resin (100 mg, 0.46 meq g−1) was swilled in DCM for 2 h at room temperature (RT). (a) Fmoc group removal was carried out with 20% 4-methylpiperidine in DMF, for 10 min at RT, twice. (b) Amino acid coupling reaction was carried out as follows: the Fmoc-amino acid (0.23 mmol) was mixed with DCC/6-Cl-HOBt (0.23/0.23 mmol) in 2 mL of DMF, and the mixture was gently shaken for 15 min at RT. Then preactivated Fmoc-amino acid was added to the free amino resin and the reaction mixture was stirred at RT for 3 h. (c) Side-chain deprotection reactions and peptide separation from solid support were carried out via the treatment of the resin-peptide with a solution containing TFA/water/TIPS/EDT (93/2/2.5/2.5 v/v/v) for 4 h at RT. Afterward, the resin was filtrated and cooled. Ethyl ether was added to the solution for precipitating the peptide, and then the solid was washed five times with cool ethyl ether and dried at RT.2.3 Reverse-phase HPLC
Crude peptides were analysed using an Agilent 1200 liquid chromatograph (Omaha, Nebraska, USA) with UV-Vis detector (210 nm) and a Merck Chromolith® C18 (50 × 4.6 mm) column. For the analysis, 10 μL of peptide solution (1.0 mg mL−1) was injected and eluted with a linear gradient, ranging from 5% to 70% of solvent B in solvent A. The gradient time was 11.5 min., and the analysis was performed at a flow rate of 2.0 mL min−1 at RT.13,17 Solvent B was 0.05% TFA in ACN and solvent A was 0.05% TFA in water.2.4 Peptide purification
The peptides were purified using solid-phase extraction columns (Silicycle® SiliaPrep™ C18 cartridges, 2.0 g resin). Columns were activated and equilibrated in accordance with supplier recommendations. The peptides were eluted with a solvent B gradient and the fractions were analysed using RP-HPLC (as described above), and those that contained the pure peptide were collected and lyophilized.17,18,222.5 MALDI-TOF MS
The peptide (1 mg mL−1) was mixed with the matrix (1.0 mg mL−1 of 2,5-dihydroxybenzoic acid, or sinapinic acid) (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18, v/v), and then 1 μL of this mixture was seeded on a steel target. The experiment was carried out on a Microflex TOF mass spectrometer (Bruker Daltonics, Bremen, Germany), laser: 250 shots and 25–30% power.17,18,22
:18, v/v), and then 1 μL of this mixture was seeded on a steel target. The experiment was carried out on a Microflex TOF mass spectrometer (Bruker Daltonics, Bremen, Germany), laser: 250 shots and 25–30% power.17,18,22
2.6 Antibacterial activity assays
The minimal inhibitory concentration (MIC) was determined using a microdilution assay.17–19 In brief, using a 96-well microtiter plate, 90 μL of peptide (200, 100, 50, 25, 12.5, and 6.2 μg mL−1) and 10 μL of inoculum (5 × 106 CFU mL−1) were added to each well. Afterward, the bacteria were incubated for 24 h at 37 °C and absorbance at 620 nm was measured. The minimum bactericidal concentration (MBC) was determined as follows: a small sample was taken from each well where there was no visible growth, using an inoculation loop, which was then spread on MHA plates and incubated overnight at 37 °C. MBC was considered to be the plate that exhibited no bacterial growth. Each of these tests was performed twice (n = 2).2.7 Time-kill curve
The time-kill curve was constructed using the CLSI protocol, with some modifications (CLSI, 1999). 270 μL of peptide (final concentrations 0.25 MIC, MIC, 2 MIC and 4 MIC), and 30 μL inoculum (5 × 105 CFU mL−1) were added to a 100-well microtiter plate. The samples were incubated in Bioscreen equipment for 48 h at RT, and the absorbance (600 nm) was measured every hour.232.8 Synergy test
The synergy test was performed according to the checkerboard method.23,24 25 μL of peptide and 25 μL of antibiotic were mixed (final concentrations: 0; 0.06; 0.12; 0.25; 0.50; 1 and 2 times the MIC) in order to obtain all possible combinations between them. Then, 50 μL of inoculum (5 × 105 CFU mL−1) was added and incubated at 37 °C for 16–20 h, and the new MIC was established. The fractional inhibitory concentration (FIC) index was calculated as follows: [A/MICA] + [P/MICP] = FIC, where MICA and MICP are the MICs of the antibiotic and peptide, respectively. The MICs of the antibiotic and the peptide determined in combination correspond to A and P, respectively. Combinations were classified as synergistic (FIC ≤ 0.5), indifferent (0.5 < FIC < 4), and antagonist (FIC > 4).2.9 Haemolysis assay
5 mL of heparinized peripheral blood was centrifuged at 1000g for 7 min. The erythrocyte fraction was suspended in 10 mL of saline solution (SS) and washed twice by centrifugation at 1000g for 7 min. The erythrocytes (2% hematocrit) were incubated with peptide (ranging from 6.2 to 200 μg mL−1), for 2 h at 37 °C. SS was used as negative control, while distilled water was used as positive control. The mixtures were centrifuged, the supernatants were collected, and the absorbance was determined to be 540 nm.253 Results and discussion
Linear, dimeric, and tetrameric peptides containing sequences derived from LfcinB (Table 1) were designed and synthesized through SPPS, using the Fmoc/tBu strategy. Dimeric peptides ((sequence)2-K-Ahx) were synthesized using the MAPs (multiple antigen peptides) methodology, and tetrameric peptides were obtained by oxidation of a precursor dimeric peptide whose sequence contained a C-terminal cysteine residue ((sequence)2-K-Ahx-C).17,26 The linear, dimeric, and tetrameric peptides were obtained with high chromatographic purity, and the final products had the expected monoisotopic mass.17| Peptide code | Sequence | Antibacterial activity, MIC (MBC) μM | |||||
|---|---|---|---|---|---|---|---|
| E. coli | P. aeruginosa | E. faecalis | S. aureus | ||||
| ATCC 11775 | ATCC 25922 | ATCC 43827 | ATCC 27853 | ATCC 29212 | ATCC 25923 | ||
| a Data reported by ref. 17. | |||||||
| LfcinB (20–25) | 20RRWQWR25 | 203 (203)a | 203 (203)a | 102 (203) | 203 (>203) | >203 (>203) | 203 (>203) |
| LfcinB (20–25)2 | (RRWQWR)2K-Ahx | 22 (22)a | 6 (11)a | 11 (91) | 23 (91) | 91 (>91) | 91 (91) |
| LfcinB (20–25)4 | ((RRWQWR)2K-Ahx-C)2 | 22 (44)a | 22 (22)a | 5 (44) | 11 (22) | 44 (>44) | 22 (44) |
| LfcinB (20–30) | 20RRWQWRMKKLG30 | 130 (130)a | 130 (130)a | 65 (130) | >130 (>130) | >130 (>130) | 130 (>130) |
| LfcinB (20–30)2 | (RRWQWRMKKLG)2K-Ahx | 60 (60)a | 30 (60)a | 8 (15) | 15 (30) | 60 (60) | 60 (>60) |
| LfcinB (20–30)4 | ((RRWQWRMKKLG)2K-Ahx-C)2 | 15 (15)a | 15 (15)a | 7 (15) | 15 (30) | 30 (30) | 30 (>30) |
| [Ala19]-LfcinB (17–31) | 17FKARRWQWRMKKLGA31 | >102 (>102)a | 102 (102)a | 102 (>102) | 102 (>102) | >102 (>102) | >102 (>102) |
| [Ala19]-LfcinB (17–31)2 | (FKARRWQWRMKKLGA)2K-Ahx | >48 (>48)a | 24 (24)a | 12 (24) | 48 (48) | 48 (>48) | >48 (>48) |
| LfcinB (21–25)Pal | RWQWRWQWR | 17 (17) | 17 (34) | 17 (67) | 67 (135) | 67 (67) | 135 (>135) |
| LfcinB (21–25)Pal2 | (RWQWRWQWR)2K-Ahx | 31 (63) | 16 (63) | 63 (63) | 63 (>63) | 31 (>63) | 63 (>63) |
| LfcinB (21–25)Pal4 | ((RWQWRWQWR)2K-Ahx-C)2 | >30 (>30) | >30 (>30) | >30 (>30) | >30 (>30) | >30 (>30) | >30 (>30) |
Previously we reported the antibacterial activity of peptides containing LfcinB (20–25), LficnB (20–30), and [Ala19]-LfcinB (17–31) sequences against two E. coli strains (ATCC 25922 and 11775).17 It was established that dimeric and tetrameric peptides exhibited higher antibacterial activity than their monomeric analogue peptides. In the present investigation, the antibacterial activity of LfcinB-derived monomeric, dimeric, and tetrameric peptides was evaluated against E. coli ATCC 43827, P. aeruginosa ATCC 27853, E. faecalis ATCC 29212, and S. aureus ATCC 25923. Our results confirm that dimeric and tetrameric peptides derived from LfcinB (20–25) exhibited higher antibacterial activity against the evaluated strains than their monomeric counterpart. Furthermore, the tetrameric and dimeric peptides LfcinB (20–25)4 and LfcinB (20–25)2 exhibited high antibacterial activity against all the tested E. coli strains; specifically, their MICs ranged from 5 to 22 μM. Additionally, LfcinB (20–25)4 exhibited the highest antibacterial activity against S. aureus (MIC 22 μM) and P. aeruginosa (MIC 11 μM), so this molecule could be considered to be a promising candidate for drug development. On the other hand, LfcinB (20–30)2 and LficnB (20–30)4 exhibited high antibacterial activity against P. aeruginosa and E. coli ATCC 43827. The dimeric peptide [A19]-LfcinB (17–31)2 exhibited significant antibacterial activity against E. coli ATCC 43827 (MIC 12 μM), while it did not exhibit high antibacterial activity against S. aureus, P. aeruginosa, and E. faecalis.
The results obtained in this investigation, together with our previous reports, indicate that polyvalence enhanced the antibacterial activity of LfcinB-derived sequences. The increase in antibacterial activity caused by polyvalence could be due to: (i) a greater number of both positively-charged (Arg) and hydrophobic (Trp) side chains. Dimeric peptides have two chains containing the RRWQWR sequence (six Arg and four Trp residues), while tetrameric peptides have four chains (twelve Arg and eight Trp residues). It has been established that Arg and Trp residues are found in unusually high proportion in many antimicrobial peptides.27 It is common knowledge that positively-charged side chains are involved in the interaction between the peptide and the bacterial surface, while the hydrophobic side chains interact with the lipidic bilayer, causing their disruption, which causes cellular lysis. (ii) Complexity of peptide structure and/or presence of non-natural amino acid (Ahx). It has been reported that peptides containing non-natural amino acids are less susceptible to enzymatic degradation and in some cases the potency of the biological activity. On the other hand, dimeric and tetrameric peptides could have better stability and resistance to proteolytic degradation caused by bacterial enzymes than monomeric sequences. Linear peptides are often easily proteolytically degraded.27–30
Our results agree with previous reports, which showed that LfcinB polyvalent peptides (dimers, trimers, tetramers, octamers, etc.) exhibited significant antibacterial activity.20,31 It has been suggested that the MAP structure increases the antibiotic peptides' activity to an extent that makes them comparable to classical antibiotics.31 The antibacterial activity of dimeric and tetrameric peptides is in agreement with both (i) the mechanism suggested for LfcinB, which involves electrostatic interaction between positively-charged side chains of LfcinB and negatively-charged molecules on the bacterial surface,14,32,33 and (ii) studies that suggest that LfcinB peptides self-assemble, forming polymeric structures that are requisite for the interaction with molecules on the bacterial surface.14,32,33
On the other hand, the palindromic monomeric peptide LfcinB (21–25)Pal exhibited antibacterial activity against the tested bacterial strains, with MICs ranging from 17 to 135 μM. Its highest activity was against E. coli strains, while, the dimeric and tetrameric peptides containing a palindromic sequence did not exhibit significant antibacterial activity against the strains evaluated. Interestingly, the tetrameric peptide LfcinB (21–25)Pal4 practically did not exhibit antibacterial activity at the tested concentrations (Table 1). Peptides containing RWQWRWQWR exhibited a behaviour opposite from peptides containing LfcinB (20–25), LfcinB (20–30), and [Ala19]-LfcinB (17–31) sequences. The antibacterial activity of the peptide LfcinB (21–25)Pal suggests that the amphipathic properties of the RWQWRWQWR sequence are relevant for the antibacterial activity. The cationic nature and the hydrogen bonding geometry of Arg and the complex properties of Trp seem to complement each other well for antibacterial activity. The cationic charge of Arg is essential for electrostatic interaction, and hydrogen bonding facilitates its interaction with negatively-charged surfaces such as LPS, teichoic acid, or phosphatidyl glycerol phospholipid head groups.32–35 Studies have shown that Trp residues penetrating into the interfacial layer of the membrane could be associated as well with the positively-charged choline headgroups of the lipid bilayer. Thus it has been proposed that the complex electrostatic nature of the interfacial region of lipid bilayers is ideal for accommodating Trp residues. Trp enables the prolonged association of the peptide with the membrane. Furthermore, it has been proposed that Trp solely acts as an anchoring force, possibly allowing the peptide to penetrate through membrane towards an intracellular target.27,36,37
For the sequence RWQWRWQWR, polyvalence does not increase antibacterial activity, indicating that the number of positive charges is not the unique fact that influences this activity. Also, it is possible that the palindromic sequence polyvalence diminished the peptide amphipathicity caused by chain aggregation.
The haemolysis assays and time-kill curves were carried out with peptides that exhibited the greatest antibacterial activity against each strain. Thus LfcinB (20–25)Pal, LfcinB (20–25)2, LfcinB (20–25)4, and LfcinB (20–30)4 were selected. The peptides LfcinB (20–25)Pal and LfcinB (20–25)2 did not exhibit a high haemolytic effect at the evaluated concentrations. It was ≤5% (Table 2), indicating that these peptides have a high therapeutic index. In contrast, the tetrameric peptides LfcinB (20–25)4 and LfcinB (20–30)4 exhibited a haemolytic effect ranging from 14% to 26% at concentrations corresponding to the MICs (Table 2). The dimeric and tetrameric peptides showed higher haemolytic affect than monomeric peptides, suggesting that the increase of positive charges in the molecule could cause the haemolytic effect. These results are according with previous reports where it is stated that positively charged dendrimers are mostly toxic both in vitro and in vivo.38 Drug development based on these kinds of molecules requires pharmaceutical technology in order to design strategies for the administration of the drugs.
| Bacterial strain | Peptide (MIC) | Haemolysis (%) | Effect (μM) | |
|---|---|---|---|---|
| Bacteriostatic | Bactericide | |||
| E. coli ATCC 25922 | LfcinB (20–25)2 (6 μM) | 1 | 6 | >24 |
| E. coli ATCC 11775 | LfcinB (20–25)Pal (17 μM) | 5 | 34 | >68 |
| P. aeruginosa ATCC 47853 | LfcinB (20–25)4 (11 μM) | 14 | 11 | 22 |
| S. aureus ATCC 25923 | LfcinB (20–25)4 (22 μM) | 22 | 22 | 44 |
| E. faecalis ATCC 29212 | LfcinB (20–30)4 (30 μM) | 26 | 30 | 120 |
The time kill curves show that at tested concentrations (MIC, 2 MIC, and 4 MIC), the tetrameric peptides LfcinB (20–25)4 and LfcinB (20–30)4 exhibit a bactericidal effect, and for LfcinB (20–25)2 and LfcinB (21–25)Pal, a bacteriostatic effect was found (Table 2).
As an example, Fig. 1 shows the time-kill curve of S. aureus 25923 treated with tetramer LfcinB (20–25)4 used at 22, 44, and 88 μM. As can be seen, when the peptide concentration was 22 μM (MIC), the bactericidal effect was observed only up to 23 h of incubation, while at higher concentrations, 2 MIC and 4 MIC, the bactericidal effect was observed all along the 48 h. These results confirm the potentiating effect of polyvalence on the antibacterial action of peptides derived from LfcinB.
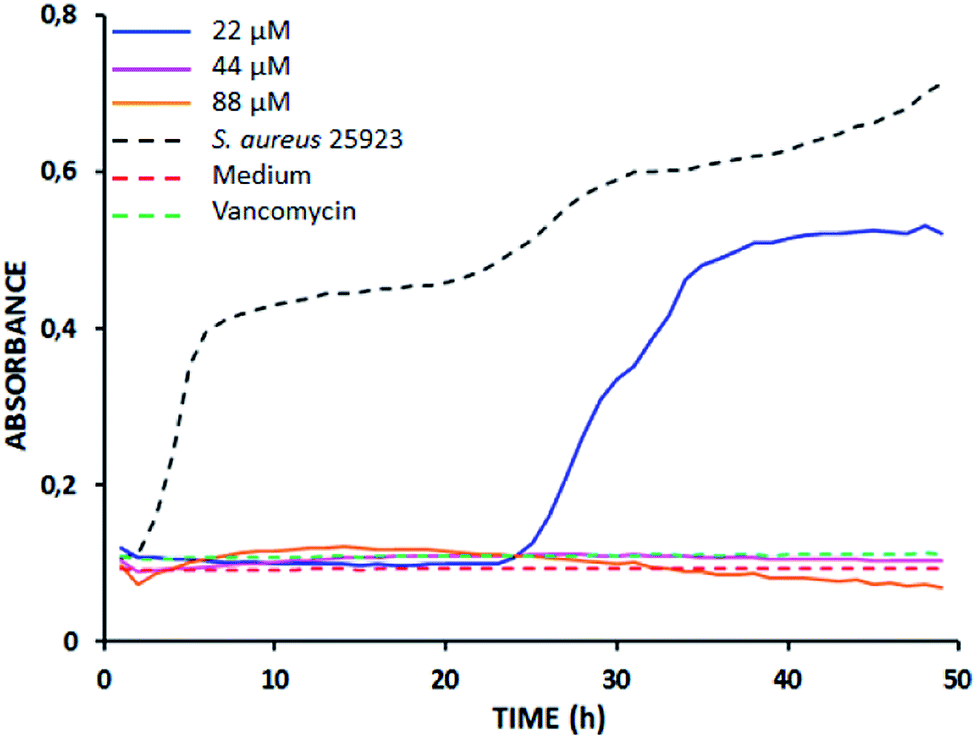 | ||
| Fig. 1 Time-kill curve plot. Peptide LfcinB (20–25)4 against S. aureus ATCC 25923. The peptide was tested at concentrations corresponding to MIC (blue line), 2 MIC (pink line) and 4 MIC (orange line) values. | ||
Finally, the synergy test was performed by means of the checkerboard method. For each bacterial strain, the peptides with antibacterial activity were selected (Table 1), and antibiotics were selected according to the CLSI protocol, specifically ciprofloxacin and vancomycin for Gram-negative and Gram-positive bacteria, respectively. The results are summarized in Table 3. The peptide LfcinB (20–25)2 exhibited synergy with ciprofloxacin against E. coli 25922. Ciprofloxacin antibacterial activity increased up to 15 times (MICA/A) when it was combined with this dimeric peptide.
| Bacterial strain | Antibiotic | Peptide | MICA | MICP | A | P | FIC, index | MICA/A | Activity | |
|---|---|---|---|---|---|---|---|---|---|---|
| a MICA, MICP,A and P in μg mL−1. S: synergy; I: indifference. | ||||||||||
| Gram negative | E. coli 25922 |
C | LfcinB (20–25)2 | 0.09 | 25 | 0.006 | 0.75 | 0.09 | 15 | S |
| E. coli 11775 |
C | LfcinB (21–25)Pal | 0.04 | 50 | 0.020 | 50 | 1.50 | 2 | I | |
| P. aeruginosa 27853 |
C | LfcinB (20–25)4 | 0.40 | 100 | 0.020 | 3.1 | 0.09 | 20 | S | |
| Gram positive | E. faecalis 29212 |
V | LfcinB (20–30)4 | 6.4 | 400 | 0.200 | 12.5 | 0.06 | 32 | S |
| LfcinB (21–25)Pal | 6.4 | 200 | 0.100 | 25 | 0.14 | 64 | S | |||
| S. aureus 25923 |
V | LfcinB (21–25)Pal | 6.4 | 400 | 0.060 | 0.06 | 0.01 | 107 | S | |
| LfcinB (20–25)4 | 6.4 | 100 | 0.100 | 3.1 | 0.04 | 64 | S | |||
The peptide LfcinB (20–25)4 exhibited an antibacterial effect synergistically with ciprofloxacin and vancomycin (P. aeruginosa and S. aureus) (Table 3). These results indicate that ciprofloxacin or vancomycin antibacterial activity increased by 20 and 64 times, respectively, when they were combined with tetramer LfcinB (20–25)4. Interestingly, the tetrameric peptide exhibited synergism with ciprofloxacin and vancomycin independently of the mechanism associated with these antibiotics. It is possible that this peptide affects membrane permeability, facilitating the internalization of ciprofloxacin or vancomycin. In a similar manner, the tetrameric molecule LfcinB (20–30)4 exhibited synergy when it was mixed with vancomycin, and it enhanced the activity of this antibiotic by 30 times against E. faecalis. Furthermore, vancomycin antibacterial activity against S. aureus and E. faecalis increased by 107 and 64 times, respectively, when this antibiotic was mixed with the palindromic peptide. Additionally, the antibacterial activity of the tetrameric peptides LfcinB (20–30)4 and LfcinB (20–25)4 increased by at least 30 times when combined with vancomycin or ciprofloxacin. This implies that tetrameric peptides could be used in low concentrations, which will not induce a haemolytic effect. The palindromic peptide exhibited synergism only with vancomycin, suggesting that this peptide acts on the bacterial surface. On the other hand, when the palindromic peptide was combined with ciprofloxacin, a synergistic effect was not observed, indicating that the peptide does not cause membrane permeation.
These results agree with other studies, which indicate that many antimicrobial peptides increase their antibiotic action, since they induce membrane perturbation or pore formation on the bacterial cell wall, and this enhances the uptake of antibiotics.39 It has been reported that LfcinB mixed with each of the following antibiotics: penicillin G, vancomycin, gentamicin, cycloserine, or erythromycin, did not exhibit a synergistic effect against S. aureus. In the present investigation, it was possible to identify two LfcinB-derived peptides that exhibited a synergistic effect with vancomycin against a S. aureus strain. On the other hand, our results are in accordance with reports that showed that LfcinB combined with conventional antibiotics exhibits a synergic effect against Gram-negative and Gram-positive strains.40–43
In summary, our results indicate that polyvalence of LfcinB-derived sequences increases their antibacterial activity. The tetrameric peptide LficnB (20–25)4 combined with vancomycin or ciprofloxacin exhibited a synergistic effect for antibacterial activity. The palindromic peptide LfcinB (21–25)Pal exhibited significant antibacterial activity and a synergistic effect with vancomycin. The obtained results suggest that the peptides LfcinB (20–25)2, LfcinB (20–25)4, LfcinB (20–30)4, and LfcinB (21–25)Pal could be considered as candidates for drug development.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was conducted with the financial support of División de Investigación y Extensión sede Bogotá (DIEB), Universidad Nacional de Colombia (Project code 41520). Yerly Vargas thanks COLCIENCIAS for financing her Master studies. Convocatoria para la Formación de Capital Humano de Alto Nivel para el Departamento del Putumayo. Number: 457.Notes and references
- WHO, available online in: http://www.who.int/mediacentre/factsheets/antibiotic-resistance/en/, accessed on October 2018.
- J. P. Nataro, T. Steiner and R. L. Guerrant, Emerging Infect. Dis., 1998, 4, 251–261 CrossRef CAS PubMed.
- K. Streeter and M. Katouli, Infect., Epidemiol. Microbiol., 2016, 2, 25–32 CrossRef.
- O. Akanbi, H. Njom, J. Fri, A. Otigbu and A. Clarke, Int. J. Environ. Res. Public Health, 2017, 14, 1001 CrossRef PubMed.
- G. Kayaoglu and D. Ørstavik, Crit. Rev. Oral Biol. Med., 2004, 15, 308–320 CrossRef.
- J. M. Sierra, E. Fusté, F. Rabanal, T. Vinuesa and M. Viñas, Expert Opin. Biol. Ther., 2017, 17, 663–676 CrossRef PubMed.
- K. Radek and R. Gallo, Semin. Immunopathol., 2007, 29, 27–43 CrossRef CAS PubMed.
- N. J. Afacan, A. T. Y. Yeung, O. M. Pena and R. E. W. Hancock, Curr. Pharm. Des., 2012, 18, 807–819 CrossRef CAS PubMed.
- A. Giuliani, G. Pirri and S. Nicoletto, Open Life Sci., 2007, 2, 1–33 CAS.
- Y. Hao, N. Yang, D. Teng, X. Wang, R. Mao and J. Wang, BioMetals, 2018, 31, 331–341 CrossRef CAS PubMed.
- K. Yamauchi, M. Tomita, J. T. Giehl and R. T. Ellison, Infect. Immun., 1993, 61, 719–728 CAS.
- M. Sinha, S. Kaushik, P. Kaur, S. Sharma and T. Singh, Int. J. Pept., 2013, 2013, 1–12 CrossRef PubMed.
- M. León, A. Leal, G. Almanzar, J. Rosas, J. García and Z. Rivera, BioMed Res. Int., 2015, 2015, 1–8 Search PubMed.
- S. Farnaud and R. W. Evans, Mol. Immunol., 2003, 40, 395–405 CrossRef CAS PubMed.
- M. Bellamy, M. Takase, H. Wakabayashi, K. Kawase and M. Tomita, J. Appl. Bacteriol., 1992, 73, 472–479 CrossRef PubMed.
- H. Vorland, H. Ulvatne, J. Andersen, H. Haukland, O. Rekdal, J. Svendsen and T. Gutteberg, Infect. Dis., 1998, 30, 513–517 Search PubMed.
- Y. Vargas, G. Rodríguez, Y. Umaña, A. Leal, G. Almanzar, J. E. García and Z. J. Rivera, Molecules, 2017, 22, E1641 CrossRef PubMed.
- N. J. Huertas, A. J. Rivera, R. F. Medina and J. E. García, Molecules, 2017, 22, Ep87 CrossRef PubMed.
- N. J. Huertas, Y. Vargas, A. Gómez, E. Hernández, A. L. Leal, J. Melo, Z. J. Rivera and J. E. García, Molecules, 2017, 22, E452 CrossRef PubMed.
- S. C. Vega, D. A. Martínez, M. Chalá, H. A. Vargas and J. E. Rosas, Front. Microbiol., 2018, 9, 329 CrossRef PubMed.
- Ch. Sun, Y. Li, S. Cao, H. Wang, Ch. Jiang, Sh. Pang, M. Hussain and J. Hou, Int. J. Mol. Sci., 2018, 19, 2951 CrossRef PubMed.
- C. Vergel, Z. J. Rivera, J. E. Rosas and J. E. García, J. Mex. Chem. Soc., 2014, 58, 386–392 Search PubMed.
- SEIMC, Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica, available online in: https://seimc.org/contenidos/documentoscientificos/procedimientosmicrobiologia/seimc-procedimientomicrobiologia12.pdf.2017, accessed on July 2018 to January 2019.
- S. Sanchez-Gomez, B. Japelj, R. Jerala, L. Moriyon, M. Fernandez Alonso, J. Leiva, S. Blondelle, J. Andra, K. Brandenburg, K. Lohner and G. Martinez de Tejada, Antimicrob. Agents Chemother., 2010, 55, 218–228 CrossRef PubMed.
- V. A. Solarte, J. E. Rosas, Z. J. Rivera, M. L. Arango-Rodríguez, J. E. García and J. P. Vernot, BioMed Res. Int., 2015, 2015, 630179 Search PubMed.
- Z. J. Rivera, G. Granados, M. Pinto, D. Varón, C. Carvajal, F. Chaves, J. Calvo, R. Rodríguez, F. Guzmán and M. E. Patarroyo, J. Pept. Res., 2002, 59, 62–70 CrossRef CAS PubMed.
- D. I. Chan, E. J. Prenner and H. I. Vogel, Biochim. Biophys. Acta, 2006, 1758, 1184–1202 CrossRef CAS PubMed.
- R. Bottger, R. Hoffmann and D. Knappe, PLoS One, 2017, 12, e0178943 CrossRef PubMed.
- K. Hamamoto, Y. Kida, Y. Zhang, T. Shimizu and K. Kuwano, Microbiol. Immunol., 2002, 46, 741–749 CrossRef CAS PubMed.
- C. Falciani C, L. Lozzi, A. Pini, F. Corti, M. Fabbrini, A. Bernini, B. Lelli, N. Niccolai and L. Bracci, Chem. Biol. Drug Des., 2007, 69, 216–221 CrossRef PubMed.
- M. Azuma, T. Kojima, I. Yokoyama, H. Tajiri, K. Yoshikawa, S. Saga and C. A. Del Carpio, J. Pept. Res., 1999, 54, 237–241 CrossRef CAS PubMed.
- D. S. Chapple, R. Hussain, C. l. Joannou, R. E. Hancock, E. Odell and R. W. Evans, Antimicrob. Agents Chemother., 2004, 48, 2190–2198 CrossRef CAS PubMed.
- S. Farnaud, C. Spiller, L. C. Moriarty, A. Patel, V. Gant, E. W. Odell and R. W. Evans, FEMS Microbiol. Lett., 2004, 233, 193–199 CrossRef CAS PubMed.
- E. Elass-Rochard, A. Roseanu, D. Legrand, M. Trif, V. Salmon, C. Motas, J. Montreuil and G. Spik, Biochem. J., 1995, 312, 839–845 CrossRef CAS PubMed.
- L. H. Vorland, H. Ulvatne, O. Rekdal and J. S. Svendsen, Scand. J. Infect. Dis., 1999, 31, 467–473 CrossRef CAS PubMed.
- H. H. Haukland, H. Ulvatne, K. Sandvik and L. H. Vorland, FEBS Lett., 2001, 508, 389–393 CrossRef CAS PubMed.
- M. Moniruzzaman, M. Z. Islam, S. Sharmin, H. Dohra and M. Yamazaki, Biochemistry, 2017, 56, 4419–4431 CrossRef CAS PubMed.
- M. Niederhafner, L. Bednárová, M. Buděšínský, M. Safařík, S. Ehala, J. Ježek, L. Borovičková, V. Fučík, V. Ceřovský and J. Slaninová, Amino Acids, 2010, 39, 1553–1561 CrossRef PubMed.
- S. Nuding, T. Frasch, M. Schaller, E. F. Stange and L. Zabel, Antimicrob. Agents Chemother., 2014, 58, 5719–5725 CrossRef PubMed.
- H. Wakabayashi H, S. Teraguchi and Y. Tamura, Biosci., Biotechnol., Biochem., 2002, 66, 2161–2167 CrossRef PubMed.
- Y. Liu, F. Han, Y. Xie and Y. Wang, BioMetals, 2011, 24, 1069–1078 CrossRef CAS PubMed.
- S. Sánchez-Gómez, B. Japelj, R. Jerala, I. Moriyón, M. Fernández Alonso, J. Leiva, S. E. Blondelle, J. Andrä, K. Brandenburg, K. Lohner and G. Martínez de Tejada, Antimicrob. Agents Chemother., 2011, 55, 218–228 CrossRef PubMed.
- T. Z. Oo, N. Cole, L. Garthwaite, M. D. Willcox and H. Zhu, J. Antimicrob. Chemother., 2010, 65, 1243–1251 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |