
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilyl diol ester as a new selectivity control agent in MgCl2-supported Ziegler–Natta systems for propylene polymerization: catalyst structure and polymer properties†
Fatemeh Poorsank,
Hassan Arabi * and
Nona Ghasemi Hamedani
* and
Nona Ghasemi Hamedani
Department of Polymerization Engineering, Iran Polymer and Petrochemical Institute, P.O. Box: 14965/115, Tehran, Iran. E-mail: h.arabi@ippi.ac.ir
First published on 6th March 2019
Abstract
In this study, bis(benzoyloxy)dimethylsilane (SDE) was developed as a non-phthalate selectivity control agent (internal donor (ID) and external donor (ED)) in MgCl2-supported Ziegler–Natta (ZN) systems. The impact of SDE as an ID was investigated in regards to chemical composition, morphology, adsorption behavior (using FTIR spectroscopy, WAXD and SEM studies) and the propylene polymerization performance of the catalyst. The results of the adsorption behavior of SDE revealed that SDE, while able to stabilize the electronically unsaturated surfaces of MgCl2 [(104) and (110)], has a rather high tendency to be absorbed on strongly acidic Mg ions at the corners of these crystals. The catalyst with optimum SDE content as an ID afforded a system with reasonable activity and isotacticity (even in the absence of ED) along with broader molecular weight distribution (PDI: 6.2–10) and greater flexural modulus than phthalate-based ZN systems. SDE was also used as a new ED in most widely used fourth- and fifth-generation catalyst systems. The results revealed that in fifth-generation catalyst systems, there was an increase in hydrogen response (>40%) along with an increase of activity without any change in the thermal properties of the final polymer in comparison to a conventional ED (alkoxy silane).
Introduction
Magnesium-based Ziegler–Natta (ZN) catalysts hold the greatest share in the industrial production of isotactic polypropylene. The basis of these catalysts consists of a highly disordered MgCl2 support, on which TiCl4 is adsorbed but these formulations have been further complexed by the incorporation of certain Lewis bases called selectivity control agents (SCA), which have a crucial role in improving the stereoselectivity of propylene polymerization. SCA could be added either during the catalyst synthesis as an internal electron donor (ID) or externally together with an organoaluminum cocatalyst (generally triethylaluminium (TEA)) during the polymerization process as an external electron donor (ED). It has been proposed that SCAs can bind directly to small MgCl2 crystallites, thus controlling the amount and distribution of TiCl4 adsorbed on the support1 or transform atactic centers into isotactic ones.2–4Consequently, the catalyst activity, microstructure of the resulting poly(a-olefin) including stereoregularity, molecular weight (Mw), and polydispersity index (PDI), and the hydrogen response in the polymerization process will be affected by SCAs.5–12 Therefore, the evolution of a new generation of polypropylene catalysts is strongly related to the introduction of new structures and functions that have the potential to act as IDs and EDs.13 In particular, the fourth generation of catalysts incorporating phthalate as the most common commercial ID and alkoxy silane as an ED has been successful in producing highly isotactic polypropylene, but phthalate is frequently criticised for its environmental and health risks.14,15 Therefore, there is an urgent need for efficient organic compounds to replace it.
Different organic structures have been tested as IDs over the past decades, including maleates and malonates,16,17 dibenzoylsulfide,18,19 1,3-diethers,20 phosphites,21 succinates,22 salicylates23 and bio-derived diesters.24 These and other electron donors generally could be separated into two classes.25 The first class consists of structures such as 1,3-diethers, which have a short spacer between their two oxygen atoms and could be adsorbed only at the MgCl2 (110) surface with the chelating mode. The second class contains compounds such as phthalates and succinates, which could be adsorbed at both planes of (100) and (110) MgCl2 with the bridge, chelate or zip coordination modes due to longer spacers between the coordinating O atoms.
On the other hand, EDs as a component of ZN catalyst systems, which can be directly added into the reactor without affecting the polymerization process, have a significant effect on catalyst performance through its coordination with active centers. Therefore, changing the species of ED is a proper approach to producing polypropylene with the proper molecular weight, broad molecular weight distribution and high isotacticity, which could be a convenient method to obtain polypropylene with a high melt flow index (MFI) and good mechanical properties.26
Benzoic ester,27 organic amine,28 ether,29 alkoxysilane30 and amino-silanes28,31 have been studied as EDs. Of course, at present, alkoxy silanes are the most widely used EDs in the polypropylene industry.
It is important to note that in spite of evaluating different compounds as SCAs, satisfactory results have only been obtained with a few combinations of them.21
Recently, silyl diol ester compounds (Fig. 1) have been claimed to be an alternative to the existing internal electron donors for heterogeneous ZN catalysis.32,33 The molecular structure of these compounds is different from conventional 4th- and 5th-generation compounds (phthalates and 1,3-diethers). Thus the assessment of its effect on the properties of polypropylene helps to shed light on the way silyl diol esters interact with the catalyst and modify its structure.
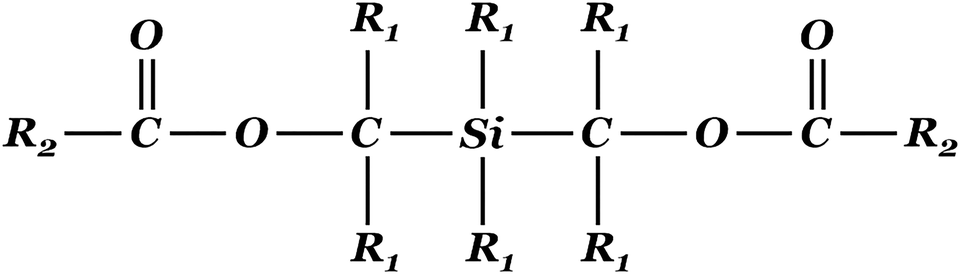 | ||
| Fig. 1 Silyl diol ester structure. R1: hydrogen or hydrocarbonyl group; R2: benzene-containing group. | ||
A step towards analysing these types of donors has been reported, which showed that the systems containing BenMag (benzoate-containing magnesium/titanium) procatalyst and silyl diol ester as the ID, showing reasonable activity and stereoselectivity in comparison to similar systems containing diisobutylphthalate (DIBP).32,33
However, to our knowledge, there are no prior reports related to the study of the role of SDE as an SCA on ZN catalysts based on MgCl2–ethanol adduct support, its possible interactions with the support and the verification of catalyst preparation condition to obtain more satisfactory polymerization results.
In this paper, in order to gain insight into the potential role of SDE as SCA in MgCl2/ID/TiCl4/ED-TEA type ZN catalysts, we have carried out the synthesis of catalysts, taking into account the effect of the amount of SDE as an ID in the catalyst systems and compared the performance of the newly developed systems with the conventional 4th- and 5th-generation catalysts in terms of catalytic activity and polymer properties in propylene polymerization.
The adsorption behavior of SDE on the MgCl2 support was studied using Fourier transform infrared (FTIR) spectroscopy, X-ray diffraction (XRD) analysis and scanning electron microscopy (SEM) techniques. In addition, the effects of SDE as a new ED on catalytic performance were investigated. Our objective in replacing conventional alkoxy silane with SDE is to obtain polymers with high hydrogen response without any negative effects on catalytic performance. The present report is a part of ongoing investigations from our group to understand the role of non-phthalate electron donors in ZN catalyst systems.
Experimental
Materials
Research-grade propylene (purity >99.9%) was supplied by Arak Petrochemical Co (Iran). Nitrogen and hydrogen with a purity of 99.99% were supplied by the Arkan Gas Company (Iran). Triethylaluminum (TEA), titanium tetrachloride (TiCl4), bis(chloromethyl) dimethylsilane and potassium benzoate were purchased from Aldrich Co (Germany). Cyclohexyl-methyl-dimethoxy-silane (C-Donor) was obtained from Arak Petrochemical Co (Iran). N-Heptane and n-hexane as diluents were purchased from Dijung Co. Dimethylformamide (DMF), sodium sulfate, sodium chloride, diethyl ether and diisobutyl phthalate (DIBP) were purchased from the Merck Company. Microspherodial MgCl2·2.1 (C2H5OH), having an average particle size of 28.72 μm, was prepared in accordance with the method described in the literature.34Synthesis of ID
Colorless oil, IR (KBr): 840, 1245, 1602, 1719, 2914.
1H-NMR (500 MHz, CDCl3): δ 0.311 (6H, s, CH2), 4.210 (4H, s, CH2), 7.404 (4H, t, Ar–H), 7.536 (2H, t, Ar–H), 8.34 (4H, d, Ar–H). 13C-NMR (500 MHz, CDCl3): δ 5.692, 56.096, 128.376, 129.530, 130.213, 132.856, 167.246. (Yield: 90%). The results of the GC-mass analysis showed that the purity of synthesized SDE was approximately 96%.
White solid: mp 92–95 °C. 1H-NMR (300 MHz, CDCl3): δ 3.38 (6H, s, CH3), 3.67 (4H, s, CH2), 7.29–7.79 (8H, m, Ar–H). 13C NMR (75 MHz, CDCl3): δ 55.5, 59.6, 74.8, 119.9, 125.3, 127.0, 127.8, 140.6, 146.8. Yield: 60%. Purity: 98% (GC area percentage).
Preparation of SDE-MgCl2 and SDE-TiCl4
The compound SDE-MgCl2 was prepared by dosing silyl diol ester (SDE/Mg ratio of 0.028) to a suspension of MgCl2 in heptane (25 mL g−1 of MgCl2) at 20 °C for 0.5 h and was kept at this temperature for 1 h. The sample was washed once with heptane and the FTIR spectrum of its solid component was recorded. The SDE-TiCl4 was prepared by adding 0.1 mol of SDE to 80 mL of heptane at 40 °C under nitrogen, followed by the dropwise addition of 0.10 mol of TiCl4. After the reaction at 40 °C for 1 h, a yellowish solid product was separated by filtration, washed with heptane and dried in a glove box under nitrogen.Preparation of the solid catalyst
Into a 1 L stainless steel Buchi reactor, purged with nitrogen, 175 mL of TiCl4 were introduced at 0 °C. While stirring, 10.0 g of microspherodial MgCl2·2.1 (C2H5OH) and an amount of ID were added. The Mg/ID molar ratios were varied in every catalyst synthesis experiment to acquire varying degrees of donor incorporation. The temperature was raised to 100 °C within 60 min and kept at this value for 2 h. Then, the stirring was stopped and the liquid was siphoned off. After siphoning, 175 mL of fresh TiCl4 were added, and the temperature was raised to 120 °C within 15 min and kept at this value for 1 h. After sedimentation and siphoning at 120 °C the solid was washed 8 times with anhydrous hexane at 60 °C. Six catalysts were prepared in the same process: catalysts A1–A4 with bis(benzoyloxy)dimethylsilane (SDE) as the ID, Mg/ID molar ratio: 2–10; catalyst B with 9,9-bis(methoxymethyl)-fluorene (BMF) as the ID, Mg/ID molar ratio: 6; and catalyst D with DIBP as the ID, Mg/ID molar ratio: 6. The list of the prepared catalysts and their chemical compositions are described in Table 1.| Catalyst | Internal donor (ID) | Ti (wt%) | Mg (wt%) | Cl (wt%) |
|---|---|---|---|---|
| A3 | SDE | 4.6 | 10.3 | 50.4 |
| B | BMF | 5.4 | 13 | 47 |
| D | DIBP | 4 | 19 | 50 |
Propylene polymerization
Polymerization was carried out in a 1 L stainless steel Buchi reactor. Before starting the reaction, the temperature of the reactor was increased to 125 °C and was simultaneously purged by nitrogen to remove excess oxygen and humidity. In the start-up of the reaction, the reactor was cooled to 0 °C while kept under purging with nitrogen. Next, the reactor was charged with 500 mL of heptane under nitrogen atmosphere, and afterward, the temperature and pressure of the reactor were increased to 20 °C and 2 bar, respectively. Triethylaluminium (TEA) and the catalyst, with and without an ED, were transferred separately to the precontacting vial and after 20 min were injected into the reactor under nitrogen atmosphere. As soon as the injection of the catalyst system was carried out, the propylene valve was opened, and the prepolymerization intended to reach better morphology was started at 2 bar monomer pressure. At the end of the prepolymerization, the polymerization process was continued at 70 °C and at 6 bar monomer pressure for 2 h. After the illustrated polymerization procedure, the polymerization was terminated.The vent valve was opened, and the unreacted monomer was evaporated quickly. The product was dried overnight at room temperature.
Characterization
The contents of titanium and magnesium were determined by atomic absorption spectroscopy. The content of chloride in the catalysts was determined by microwave digestion and potentiometric titration, the UOP 291ctest method. The SEM images were performed using a TESCAN VEGA scanning electron microscope. The distribution of elements (Mg, Cl, Si and Ti) in the catalyst was analyzed using an energy dispersive spectrometer (EDS) on an SEM microscope. Fourier-transform infrared (FTIR) spectra of all samples were recorded on an ABB Bomem MB-100 spectrometer using dry KBr powders with 15 scans in the range of 400–4000 cm−1. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O IR band, in the case of catalysts, is very complex, and to resolve it, deconvolution was performed using a Gaussian-line-shape function. Polypropylene isotacticity was determined using a procedure based on dissolving a shot of polypropylene in o-xylene at 135 °C: cooling the solution to 25 °C under controlled conditions, filtering the solid phase, distilling the o-xylene from the solution, and measuring the mass of the dissolved substances. The average molecular weights (Mn and Mw) and MWD of polypropylene were measured using Polymer Laboratory PL 220 high-temperature gel permeation chromatography (PL 220, Agilent Company) in a nitrogen atmosphere at 160 °C, using 1,2,4-trichlorobenzene as a solvent, within ±0.1 error. The injection volume was 20 μL. The molecular weight distribution curve of catalysts was deconvoluted to Flory components by MATLAB R2014a software. The melt flow index (MFI) is typically stated in terms of grams of polymer passing through a standardized capillary under a standard load over 10 min. The MFI of polypropylene was measured on an MFI-4 (Gottfert Company) at 230 °C and at a constant load of 2.16 kg over 10 min. All differential scanning calorimetry (DSC) measurements were performed with METTLER TOLEDO differential scanning. The sample was heated to 200 °C at a heating rate of 10 °C min−1 and maintained for 5 min to remove its thermal history. Then, it was cooled to 25 °C at a rate of 10 °C min−1 followed by reheating at 10 °C min−1. The melting point and heat of fusion were determined in the second scan. Wide angle X-ray diffraction (XRD) measurements were carried using a SIEMENS, D5000 (Germany) X-ray diffractometer. The step size in the WAXD measurements was 0.02, and the time per step was 1 s. The flexural modulus of the prepared polypropylenes was determined in accordance with ASTM D790.
O IR band, in the case of catalysts, is very complex, and to resolve it, deconvolution was performed using a Gaussian-line-shape function. Polypropylene isotacticity was determined using a procedure based on dissolving a shot of polypropylene in o-xylene at 135 °C: cooling the solution to 25 °C under controlled conditions, filtering the solid phase, distilling the o-xylene from the solution, and measuring the mass of the dissolved substances. The average molecular weights (Mn and Mw) and MWD of polypropylene were measured using Polymer Laboratory PL 220 high-temperature gel permeation chromatography (PL 220, Agilent Company) in a nitrogen atmosphere at 160 °C, using 1,2,4-trichlorobenzene as a solvent, within ±0.1 error. The injection volume was 20 μL. The molecular weight distribution curve of catalysts was deconvoluted to Flory components by MATLAB R2014a software. The melt flow index (MFI) is typically stated in terms of grams of polymer passing through a standardized capillary under a standard load over 10 min. The MFI of polypropylene was measured on an MFI-4 (Gottfert Company) at 230 °C and at a constant load of 2.16 kg over 10 min. All differential scanning calorimetry (DSC) measurements were performed with METTLER TOLEDO differential scanning. The sample was heated to 200 °C at a heating rate of 10 °C min−1 and maintained for 5 min to remove its thermal history. Then, it was cooled to 25 °C at a rate of 10 °C min−1 followed by reheating at 10 °C min−1. The melting point and heat of fusion were determined in the second scan. Wide angle X-ray diffraction (XRD) measurements were carried using a SIEMENS, D5000 (Germany) X-ray diffractometer. The step size in the WAXD measurements was 0.02, and the time per step was 1 s. The flexural modulus of the prepared polypropylenes was determined in accordance with ASTM D790.
Results and discussion
Effect of ID content on the performance of catalyst A
The silyl diol ester compound, SDE, as an ID was synthesized and characterized. The MgCl2·2.1 (C2H5OH)-supported ZN catalysts containing different amounts of SDE (catalysts A1–A4) were prepared and used for propylene polymerization. The content of silyl diol ester, added in the synthetic processes of the catalysts, was optimized in relation to the activities of the catalysts with and without ED. The results of polymerization with catalysts A1–A4 are presented in Table 2. As shown in the table, the activity of the catalysts increases as Mg/SDE increases and reaches a maximum at Mg/SDE = 6. A similar trend was observed for the isotacticity index (I.I), but the decrease is sharper when Mg/SDE is above 6. These results are in accordance with the data reported in the literature for succinate used as the ID.36 To explain this trend, we used the model for active sites proposed by Soga et al.37 According to this model, a non-isospecific active site (site I in Fig. 2) has two vacancies, whereas an isospecific site (site II in Fig. 2) possesses only one vacancy. We predicted that the introduction of SDE excludes part of the unstable non-isospecific active sites. It can selectively replace one of the vacancies of the non-specific sites, the other vacancy reshapes the isospecific sites (as we have proposed in path 1 in Fig. 2) and a new equilibrium is formed among active site I, active site II and SDE electron donor. Nevertheless, Ti content on the catalyst and the sum of active sites decreased, whereas that of active site II increased. So, the addition of SDE as a Lewis base improved isotacticity and productivity as has been shown in Table 2. Introducing excessive amounts of SDE leads to a reduction in the number of isospecific active site II (path 2 in Fig. 2), resulting in lower isotacticity and productivity. Kumawat et al.38 have found that the acidic titanium centre in the active catalytic species and TiCl2Et on the MgCl2 support can participate in undesired, decomposing side reactions with donors. This result provides an explanation for the poisoning of the active site by the addition of excess SDE in the catalyst systems (Mg/SDE < 6).| Catalysta | Mg/SDE | External donorb (mg) | Activity (kg PP gcat−1) | I.Ic (%) |
|---|---|---|---|---|
| a Polymerization condition: 20 mg cat, TEA/cat (weight ratio): 10.b Cyclohexylmethyl-dimethoxysilane (C-donor).c Xylene solubility. | ||||
| A1 | 2 | — | 1.62 | 92 |
| A1 | 2 | 13.5 | 0.5 | 91 |
| A2 | 3 | — | 1.96 | 93.8 |
| A2 | 3 | 13.5 | 0.78 | 93 |
| A3 | 6 | — | 2.47 | 97.6 |
| A3 | 6 | 13.5 | 1.84 | 97.5 |
| A4 | 9 | — | 2.42 | 94.9 |
| A4 | 9 | 13.5 | 1.79 | 96 |
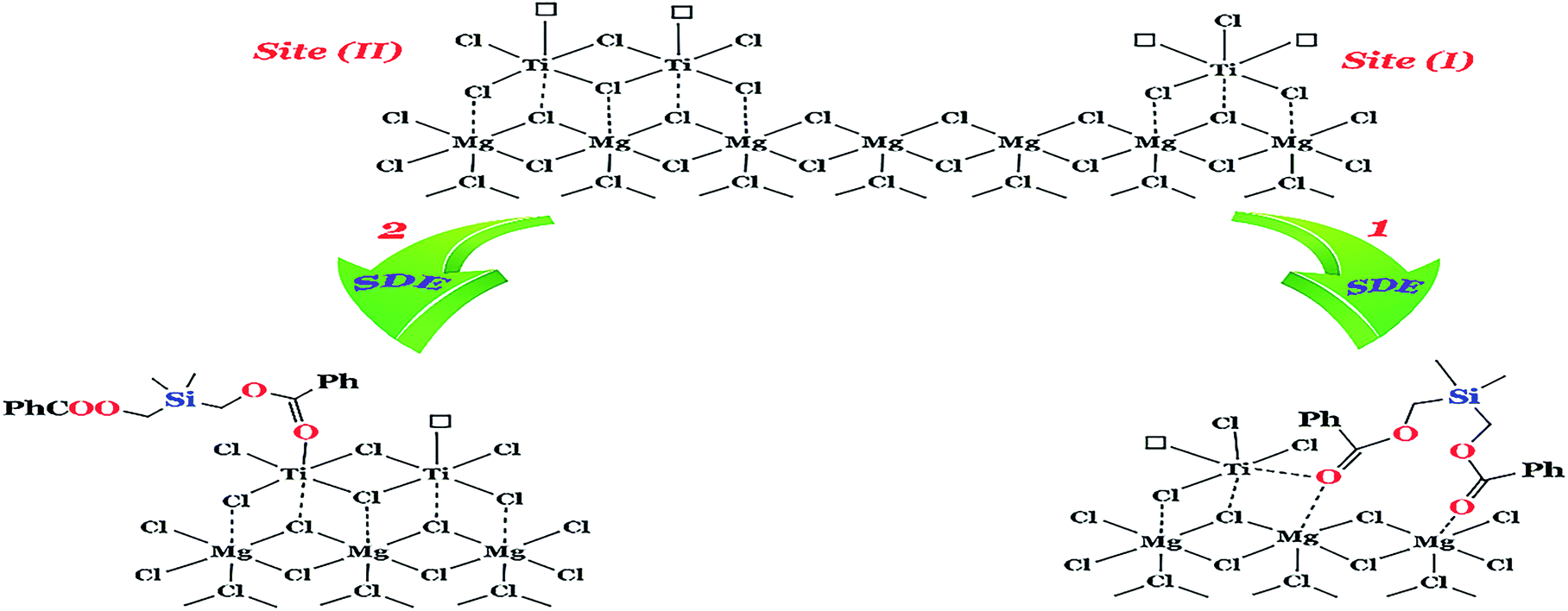 | ||
| Fig. 2 A plausible mechanism of the additive effect of SDE for the aspecific active site (site I) and the isotactic active site (site II). | ||
Elemental distribution in catalyst A3 particles
The distribution of four elements (Mg, Cl, Si and Ti) in the catalyst A3 was analyzed using an energy dispersive spectrometer (EDS) on an SEM microscope (Fig. S3†). The inorganic components of the catalyst can be considered as microcrystals of MgCl2 with Ti compounds embedded into their lattices.39 Judging by the Cl distribution data (the maximum component of the catalyst), the catalyst particle has a uniform apparent density with no voids in the center or edge of the particle. The distribution of Mg atoms is similar to that of Cl atoms, as expected from the MgCl2 crystals. The distribution of Ti atoms is also similar to the distribution of Cl and Mg atoms. Also, the Ti and Si atoms are spread uniformly along the diameter axis, which is very important for propylene polymerization. When the active centers are distributed uniformly, the polymer chains can achieve the same polymerization rate. If not, the stresses in different parts of a polymer particle in the process of polymerization are different, and the particles would break up easily.40,41Effect of ED/cat and TEA/cat ratios on the performance of catalyst A3
The effect of the different amounts of ED (C-donor) on the catalyst activity, I.I and molecular weight was also studied using catalyst A3 (Table S1†). The ED/cat ratio of catalyst A3 has little effect on the polymerization activity. However, I.I increases to 97.4 (ED/cat ratio: 0.66) first and then decreases to 96.3 (ED/cat ratio: 1). This shows that ED, in an optimum amount, has a positive effect on blocking non-isospecific Ti sites.It is important to note that in the absence of ED, the activity and isotacticity value of catalyst A3 are at optimal levels, but the Mw of the polymer decreases in comparison to polymers produced in the presence of ED. The PDI value of the polymers is in the range of 6.2–7.7 (Table S1†). The influence of the TEA/cat ratio was also investigated with the C-donor as the ED (Table S2†). Upon increasing the TEA/cat ratio from 5 to 20, the activity of the catalyst passes through a maximum at 10. This shows that excess TEA can lead to an extensive reduction of active centers (Ti4+ and Ti3+) and turned it to Ti2+ which shows no polymerizing effect.42,43 The excess TEA also reduces the Mw of the polymers obtained with catalyst A3, because it acts as a chain transfer agent (Table S2†). However, the influence of the TEA/cat ratio on the isotacticity is not remarkable.
Effect of ID structure on catalytic performance
The performance of catalysts A3, B and D in the presence and absence of ED are shown in Table 3. In the absence of an ED, the activity of the catalyst decreases in the following order: B > A3 > D. This clearly indicates that the structure of the ID strongly influences the nature and structure of the active centres. Introducing the ED decreases the activity of the A3 and B catalysts, while it enhances the activity of catalyst D. It is well known that the ED has two effects on the ZN catalyst: (a) it poisons the non-stereospecific sites and (b) it activates the isospecific sites.44 When the activation of isospecific sites prevails over the poisoning of non-stereospecific ones, in addition to I.I, the polymer yield also increases. So for catalyst D, the improved catalyst activity is due to more activated isospecific sites induced by the ED (C-donor).| Catalyst | TEA/cat (weight ratio) | TEA/ED (weight ratio) | Activitya (kg PP gcat−1) | I.Ib (%) | Mn | Mw | PDI | Tmc (°C) | Xcd (%) | Flexural modulus (Mpa) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Polymerization condition: 20 mg catalyst, ED = C-donor.b Xylene solubility.c Tm: melting temperature.d Xc: crystallinity degree. | ||||||||||
| A3 | 54 | 15 | 0.8 | 97.1 | ||||||
| A3 | 10 | 15 | 1.54 | 97.4 | 152![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
943000 |
6.22 | 159.95 | 37.69 | 1.57 |
| A3 | 10 | 0 | 2.06 | 97.6 | 84000 |
647000 |
7.71 | 158.18 | 36.46 | |
| B | 54 | 15 | 2.20 | 97.5 | 101000 |
502000 |
4.96 | 160.22 | 43.36 | |
| B | 54 | 0 | 4.40 | 96 | 68500 |
312000 |
4.54 | 159.64 | 39.59 | |
| D | 54 | 15 | 2.20 | 97.5 | 104000 |
614000 |
5.91 | 161.27 | 40.97 | 1.39 |
| D | 54 | 0 | 1.15 | 87.7 | 54000 |
383000 |
7.08 | 158.02 | 29.98 | |
In the absence of ED, catalyst D shows a lower I.I in comparison to catalyst B, which is in agreement with previous reports that claim that about 70 wt% of DIBP will be extracted by TEA, while it is just 2 wt% for diethers (BMF).45,46 For catalyst A3, the I.I is quite high (97.5%) in the absence of the ED. This confirms the reported data that silyl esters are likely to be quite resilient to decomposition by both Al2Et6 and TiCl2Et on the MgCl2 surface.38 Table 3 shows that the catalyst A3 provides polymers with an MWD ranging from 6.2–7.7, which is broader in comparison to catalyst B and catalyst D. It is also broader than polymers prepared with the maleate47 and dibenzoylsulfide18 internal donor and is similar to that prepared by the salicylate23 and succinate48 internal donor. These results could be attributed to the different active sites and/or the numbers of active sites in silyl diol ester-based catalysts.49,50
The molecular weight distribution curves of the polymers obtained with catalysts A3, B and D in the absence/presence of an ED are shown in Fig. 3. It shows that for each of these catalysts, the presence of an ED could not remarkably change the MWD, except for a partial shift of a low molecular weight fraction to a higher region. This change is more obvious for catalyst D, which could be attributed to the replacement of most of the phthalate by an ED.
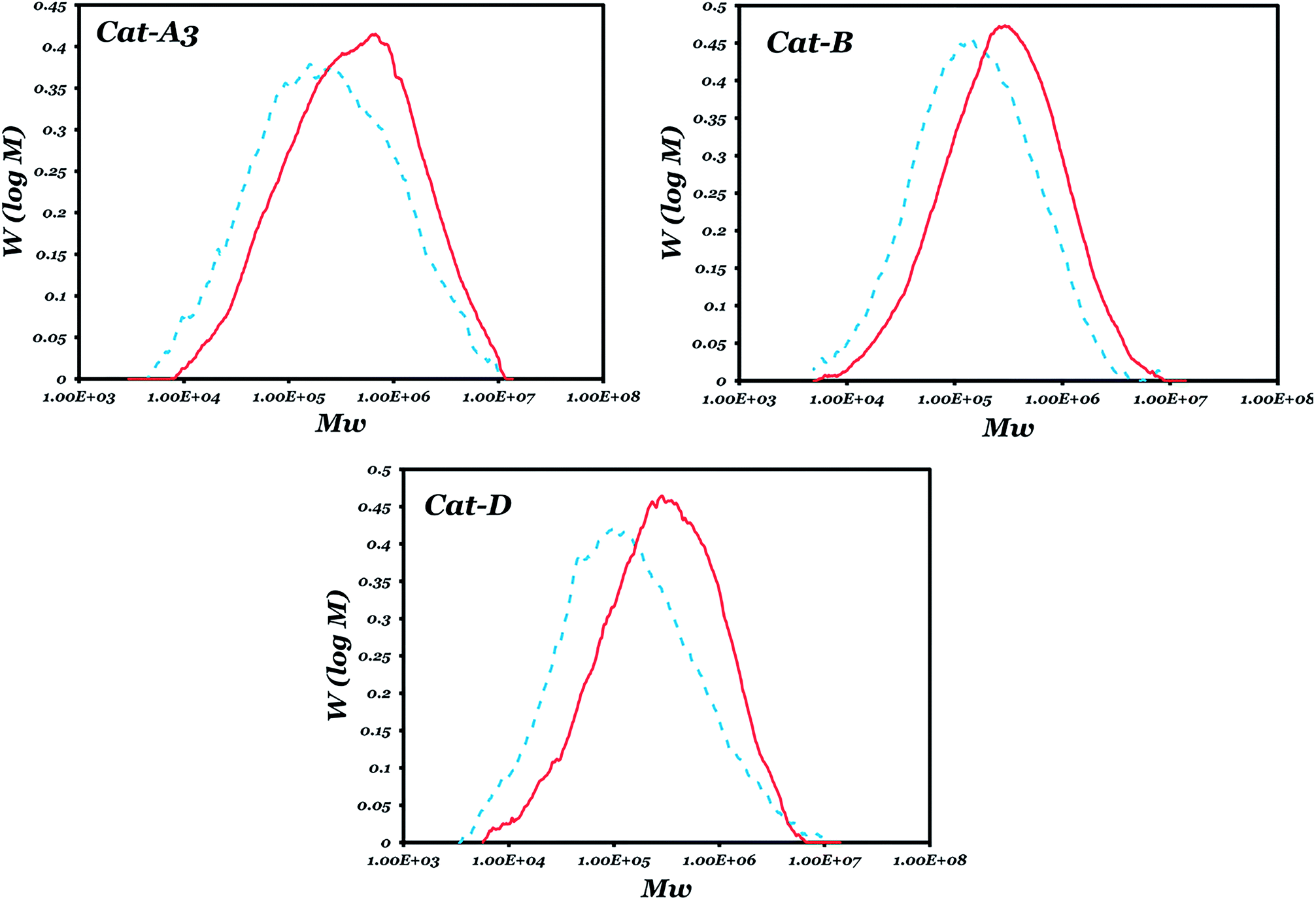 | ||
Fig. 3 Molecular weight distribution curves of the samples obtained with three prepared catalyst in the presence ( ) and absence (----) of an ED. ) and absence (----) of an ED. | ||
The thermal properties of the polymers show that the Tm is in the range of 158.02 to 161.27 °C and that the Xc is in the range of 29.98% to 43.36% (Table 3). The Tm and Xc of the produced PP are influenced by the kind of ID in the catalyst. For all catalysts in Table 3, the Tm and Xc improved with the introduction of an ED. Catalyst B, as we expected,48 showed the highest Tm and Xc values in the absence of an ED, and catalysts A3 and D go to the next positions respectively. The Xc of the catalyst with succinate as the ID,48 both in presence and absence of an ED, is about 47%, which is higher than for catalyst A3. Of course, it was done in completely different polymerization conditions. The addition of ED has no significant effect on the thermal properties of the produced PP using catalyst A3. Conversely, it leads to significant improvements for catalyst D. Table 3 shows that catalyst A3 provides the polymer with an improved flexural modulus (1.57 Mpa) in comparison to catalyst D (1.39 Mpa), which could be attributed to the broader molecular weight of the polymer produced with catalyst A3.
Hydrogen response performance of the catalysts
As can be seen in Fig. 4 and Table S3,† the hydrogen response of the catalysts are in the following order: catalysts A3 < D < B. It is in accordance with literature that among catalysts with diether, succinate and phthalate as the IDs, diether-based catalysts have the highest hydrogen response.13 The trend in Fig. 4 depicts that catalyst A3 has the highest similarity to the succinate-based catalyst. The lower hydrogen response shown by catalysts D and A3 could be attributed to the presence of active species having high stereospecificity and high regiospecificity with the relatively low content of 2,1-inserted species in the isotactic fraction.51,52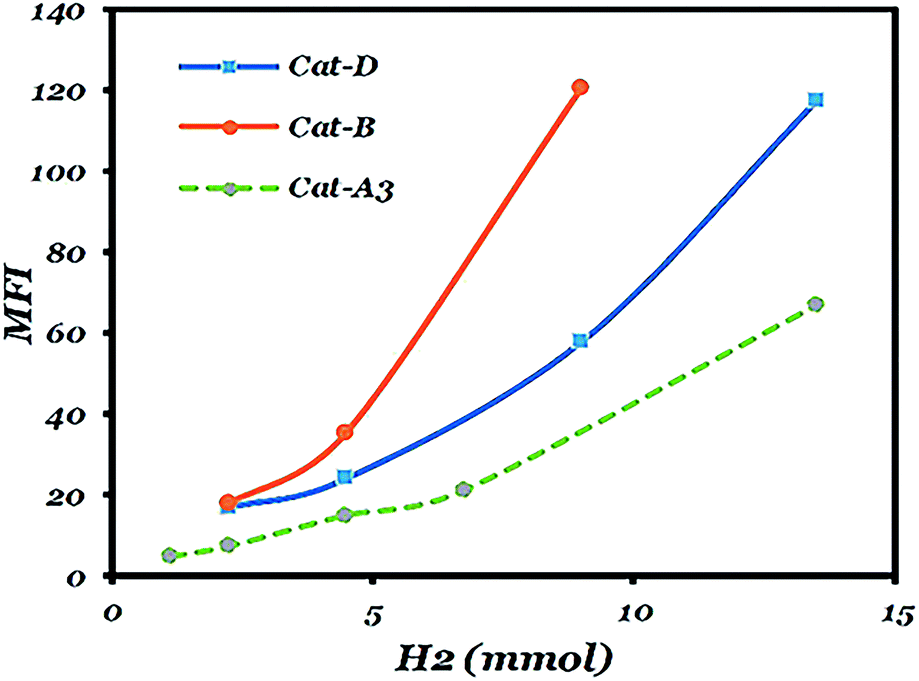 | ||
| Fig. 4 Hydrogen response of the as-prepared catalysts. | ||
As could be seen in Tables 3 and S3,† after the addition of hydrogen, I.I increases and reaches a maximum. The reason is that chain transfer to hydrogen in 2,1-inserted chains leads to the formation of a more isotactic polymer than an atactic one but after reaching a maximum value; the addition of more hydrogen causes an increase in solubility of the very low molecular weight isotactic polymer in xylene and a decrease of I.I.52
Active centers based on polypropylene MWD
Fig. S4† shows the molecular weight distribution curves of catalysts B and A3, which are deconvoluted to four and five Flory components respectively. Each Flory component can be ascribed to a certain type of active centre that has a characteristic molar mass (Mn and Mw), PDI equal to 2 and produces chains, which constitute a certain mass fraction of the polymer (Fr, %).53 Thus, there are four and five types of active centres in catalysts B and A3, respectively, site 1 to site 5 according to the sequence of Mw from low to high. The weight average molecular weight (Mw) and the weight percentage of the fraction produced by a certain active centre (Fr) of catalysts A3 and B are shown in Table S4.† The Mw of polypropylene prepared by each kind of active centre of catalyst A3 is different from B, which indicates that the type of active centres in these catalysts are different from each other. The highest Mw produced with catalyst B (Mw: 46.29 × 104 g mol−1) belongs to site 4 of this catalyst, which is much lower than the highest Mw produced with site 5 of catalyst A3 (Mw: 130.98 × 104 g mol−1).Effects of ED/catalyst A3 ratio on the active centers of the catalysts
The effect of ED/cat ratio on Mw and Fr of different sites of catalyst A3 is shown in Table S5 and Fig. S5.† By introducing ED to catalyst A3, the Mw of the produced polypropylene in all sites increases, but the fraction value Fr of the active centres changes slightly. It seems that ED interacts with the empty active sites of the catalyst and reduces the number of active centres in all sites of catalyst A3, which increases the Mw of the prepared polypropylene and decreases the activity of the catalyst as can be seen in Table S1.†FTIR study of silyl diol ester donor in the supported ZN catalyst
Fig. 5 depicts the FTIR spectra of liquid SDE (Fig. 5a) in comparison with SDE adsorption on TiCl4 (Fig. 5b) within the 1850–1550 cm−1 spectral range, the spectra recorded upon SDE adsorption on activated MgCl2 (Fig. 5c) and solid catalyst A3 (MgCl2/TiCl4/SDE) (Fig. 5d). The characteristic FTIR absorption band of SDE is located at 1719 cm−1, which can be assigned to (CO) as shown in Fig. 5a.
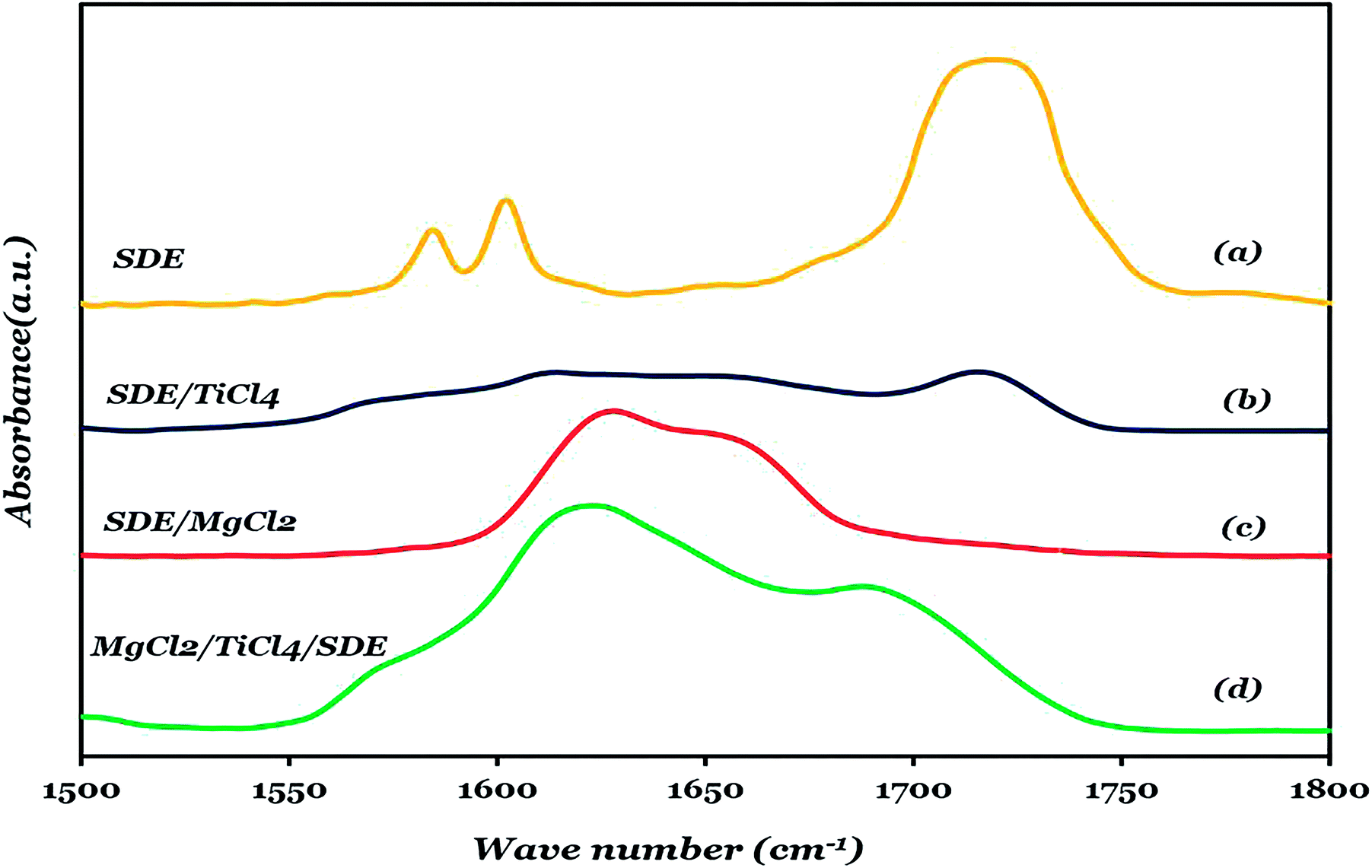 | ||
| Fig. 5 FTIR spectra, in the (CO) vibrational region, liquid SDE (a), SDE/TiCl4 (b), SDE/MgCl2(c), and MgCl2/TiCl4/SDE (catalyst A3) (d). | ||
Fig. 5c shows the absorption spectrum at room temperature of SDE on activated MgCl2. It is worth noticing that the SDE dosage was limited in order to avoid the presence of loosely coordinated SDE. The (CO) absorption peak of SDE in this sample shifts to lower the frequency region and is more broad as compared to the free SDE sample.
It is known that the CO absorption band is very sensitive to coordination to Lewis acid sites.54 The broad asymmetric shape of the CO band in Fig. 5c shows the presence of several low-coordinated Mg sites on the MgCl2 surface with different Lewis acidic properties. The Lewis acidity of these sites increases from 5-coordinated Mg ions of the (104) face (Q5) to 4-coordinated Mg ions of the (110) face (Q4) which contains the most three-coordinated Mg ions at the edges and corners of the MgCl2 crystals (Q3).55,56 The deconvolution of the CO absorption band of the MgCl2/SDE system shows three bands with distinct maxima at ca. 1621, 1633, 1655 cm−1, which are most likely from the complexes of SDE with the surface acidic sites of MgCl2. The FTIR spectrum of catalyst A3 is very complicated with several pronounced maxima (Fig. 5d). These spectra can be best represented as a superposition of at least 5 separate bands at ca. 1578, 1616, 1630, 1656 and 1691 cm−1. The integrated band intensity as determined by the deconvolution of the FTIR spectra for MgCl2/SDE and MgCl2/TiCl4/SDE is shown in Table 4. The most important observation in Table 4 is that in the presence of TiCl4, the IR absorption band related to CO adsorbed on strongly acidic Mg ions (Q3, Q4) moderately loses its intensity, whereas the band assigned to CO adsorbed on weakly acidic Mg ions (Q5) is almost eliminated. In accordance with other reports, the lower content of the ID in catalyst A3 is most likely the result of the competition of TiCl4 and SDE for the same surface adsorption sites.57 The data in Table 4 provides evidence that SDE is preferentially adsorbed on the strongly acidic Mg ions (Q3, Q4) and almost all fractions of the SDE adsorbed on the Q5 is substituted by TiCl4. These data are in accordance with previous reports of the rather high mobility of the electron donors on the Q4 surface and of the relative high stiffness of the same donors on the Q5 surface.4,58–60 The band at 1691 cm−1 regarding its low Δv value, indicates the presence of the free or weakly bound carbonyl groups of the SDE molecules.
There is also a band at 1578 cm−1, which could be attributed to another type of carbonyl species in the catalyst that is the complexes between TiCl4 and SDE. As a comparison, the FTIR spectrum of the (TiCl4-SDE) complex is depicted in Fig. 5b. This spectrum shows a broad asymmetric feature with bands at 1569 and 1584 cm−1. The complexes of TiCl4 and esters of aromatic acids were thoroughly described in the literature,60–64 and it has been reported that the esters of benzoic acid form strong monodentate complexes with TiCl4, the Δv value for which range from 135 to 145 cm−1.63,65,66 Therefore, the broad bands in Fig. 5b is probably an overlap of two closely positioned bands of different monodentate SDE complexes with TiCl4. These data provide evidence of the co-presence of SDE adsorbed on Mg ions and of TiCl4-SDE complexes. The TiCl4-ID complexes loosely attach to the MgCl2 surface through a few bridging chlorine atoms.60 The band at 1714 cm−1 is also related to loosely coordinated SDE (Fig. 5b).
Effect of ID on the crystalline structure of the catalysts
Powder XRD pattern of all catalysts is shown and compared with that of the MgCl2·2.1 (C2H5OH) adduct in Fig. 6. The crystalline structure of the adduct (Fig. 6) is different from what has been reported in the literature for anhydrous MgCl2,67–70 which shows that the transformation of anhydrous MgCl2 to an active form leads to an irregular structure suitable for use as a support for the catalyst. The general comparison of the XRD pattern of catalysts B, D and A3 shows the disordered δ-MgCl2, namely a complex of the α-form (cubic close packing (ccp)) and (β-form hexagonal close packing (hcp)) of MgCl2. These two crystalline forms can be identified from the XRD pattern in the range of 30–35° (Fig. 6). In this range, two peaks near to 34° and 32.0° where considered, which were attributed to the (104) plane of α-MgCl2 and the (011) plane of β-MgCl2, respectively.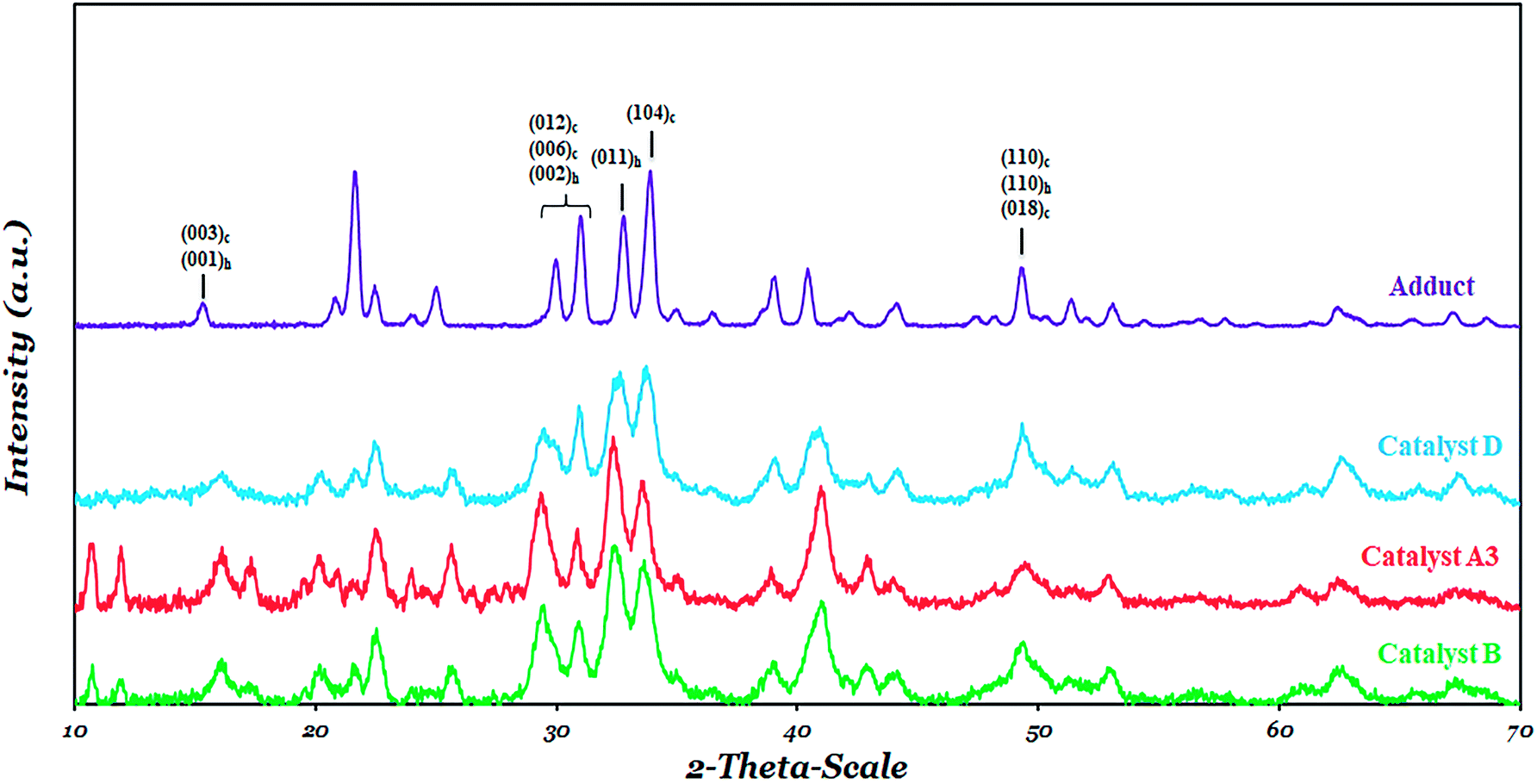 | ||
| Fig. 6 XRD patterns of adduct and catalysts: B, D and A3. | ||
The ratio of the intensities of these peaks was shown in Table 5. There is a significant difference among α/β ratios of catalyst D and catalysts B and A3. Catalyst A3 and catalyst B showed a higher β-MgCl2 phase or hcp packing of MgCl2 triple layers in comparison to catalyst D. This is in agreement with the literature stating that during catalyst preparation, the DIBP electron donor could provide stability to the α-MgCl2 through the zip coordination mode.50,71 This observation indicates that, although all the catalyst were prepared using the same MgCl2-adduct, that the structural differences of the electron donors lead to a noticeable difference in the phase distribution of α-MgCl2 and β-MgCl2 in the catalyst.
As can be seen in Fig. 6, all the catalysts showed a broad peak near 50° which was assigned to the (110) planes of α-MgCl2 and β-MgCl2 and to the (018) plane of the α-MgCl2 phase.70 The full-width at half-maximum (FWHM), an appropriate approximation of the MgCl2 crystalline size, for peaks near 34° and 50° was shown in Table 5. The similarity of the FWHM data for peaks near 34° and 50° for catalyst D indicates that DIBP stabilize both the (104) and (110) faces of MgCl2, while in catalyst B, a higher FWHM at 50° shows the ability of BMF to stabilize the (110) face of MgCl2 more than (104). Catalyst A3 demonstrates behaviour similar to catalyst B, which indicates SDE prefers to sit at the (110) face of MgCl2.
In catalysts A3 and B, the similarity of the α-MgCl2 and β-MgCl2 phase concentrations as well as that of the FWHM at 50° may suggest that the coordination behaviour of the silyl diol ester (SDE) is similar to the diether (BMF), which could stabilize the (110) face of MgCl2 more than (104). This is in agreement with our data reported in the IR section. On the other hand, there is a keynote question that if the diether (BMF) and silyl diol ester (SDE) have the same interact with support, why are their polymer properties, especially their MWD (Table 3), so different? We predict that SDE could stabilize the (110) face of MgCl2 through both the chelate and bridge coordination mode, as has been shown in Fig. 7(a) and (b), which could result in a broader variety of active centers (Fig. S4†), while the structural restrictions of diether imposes just the (110)-chelate coordination mode, creating more uniform active centers.45
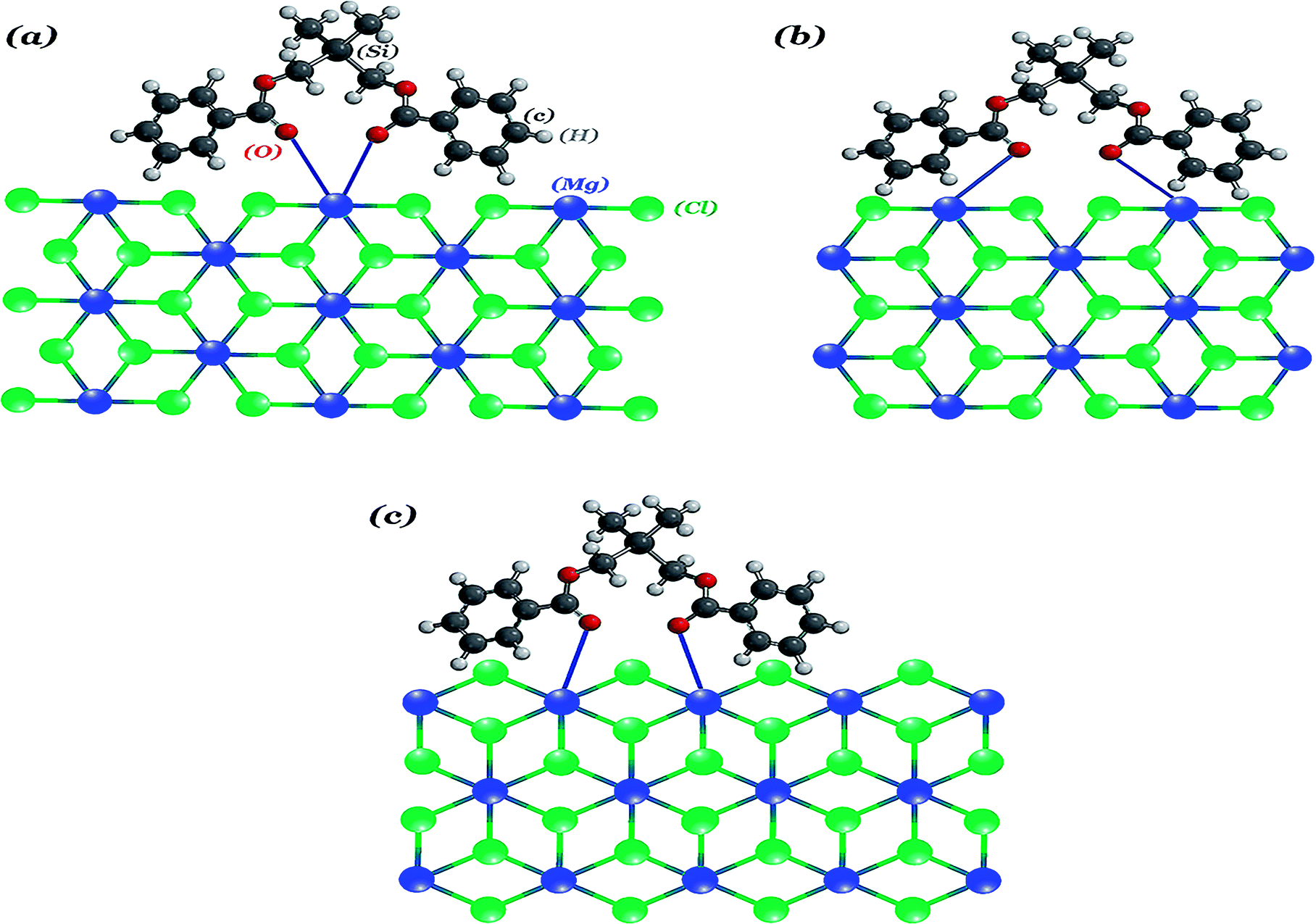 | ||
| Fig. 7 Possible SDE complexes: on the (110) surface (a, b) and on the (104) surface (c). The Mg atoms are colored in blue, the Cl atoms in green, the O atoms in red, the C atoms in dark gray and the H atoms in gray. | ||
Effect of SDE as an ED on the performance of the ZN catalyst
The performance of the catalysts A3, B and D in the presence of SDE, as the ED, in comparison to conventional ED (C-donor), are shown in Table 6. For catalysts D, A3 and B in the presence of SDE, there is a remarkable increase in hydrogen response. This could be explained by the decrease of Mw, as could be seen in Table 6. The activity of catalysts D and A3 in the presence of SDE decreases in comparison to the presence of the C-donor. There is another consideration in regards to the activity, which is contrary to what has been reported for diether as the ED.29 It showed that not only is there no noticeable reduction in the activity of catalysts D and A3 when SDE is added as the ED (Table 6) in comparison to when it is presented as the ID (Table 3), but also there is a two-fold increase of the activity of catalyst B. There is no prior report on the effect of SDE as the ED, but this behaviour could be explained by the mechanistic model reported by Shen et al.72 It includes the competitive and reversible coordination of an ED molecule on either the central Ti of the active center or the Mg adjacent to the central Ti. The former coordination leads to the deactivation of the active centers, while the latter causes an increase in the propagation rate constant of the active center. It has also been reported that this reaction is completely dependent on the chemical composition and textural parameters of the catalysts.71 Thus, the data in Table 6 indicates that SDE in the catalyst systems containing 1,3-diether as the ID (catalyst B) has a stronger tendency to coordinate on the adjacent Mg than the central Ti.| Catalyst | TEA/cat (weight ratio) | TEA/ED (weight ratio) | H2 (mmol) | External donor | Activitya (kg PP gcat−1) | I.Ib (%) | MFR (g/10 min) | Tmc (°C) | Xcd (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Catalyst: 20 mg.b Xylene solubility.c Tm: melting temperature.d Xc: crystallinity degree. | |||||||||
| D | 54 | 15 | 4.5 | SDE | 2.33 | 98.57 | 48 | 159.63 | 42.31 |
| D | 54 | 15 | 4.5 | C-donor | 4.32 | 99.3 | 22.9 | 159.91 | 47.26 |
| A3 | 10 | 15 | 2.25 | SDE | 2.28 | 97.97 | 14 | 161.13 | 42.10 |
| A3 | 10 | 15 | 2.25 | C-donor | 3.48 | 96.66 | 7.18 | 162.06 | 44.14 |
| B | 54 | 15 | 4.5 | SDE | 7.9 | 99.17 | 49.8 | 158.10 | 47.38 |
| B | 54 | 15 | 4.5 | C-donor | 3.75 | 99.2 | 35.37 | 157.73 | 47.26 |
The thermal properties of the polymers were also studied (Table 6). The results revealed that SDE in comparison to the C-donor leads to the decrease of Tm and Xc, with an obvious exception for catalyst B, in which no significant change in the thermal properties was observed. Thus, it could be concluded that the catalyst with optimum SDE content as ID afforded system with a reasonable activity and isotacticity along with broader MWD (PDI: 6.2–10) and improved flexural modulus than phthalate-based ZN systems. Meanwhile, it showed an isotacticity higher than 96% even in the absence of an external donor (ED), which is similar to the most widely used fifth-generation catalyst. On the other hand, developing its application as ED in systems containing 1,3-diether as ID leads to the increase of activity as well as hydrogen response.
Conclusions
SDE compound was synthesized and employed as SCA in the MgCl2-based ZN catalyst for PP polymerization. The catalysts using SDE as the ID were fully investigated. The adsorption behaviour of these systems, using FTIR and WAXD studies, shows that the SDE has quite a high tendency to absorb onto strongly acidic Mg ions (Q3, Q4 (110)) at the edges and corners of the MgCl2 crystals, and polymerization data revealed that the highest activity was reached at molar ratio of MgCl2 to SDE equal to 6 (cat A3). This catalyst also showed a broad MWD (PDI: 6.2–10) and an isotacticity above 96.3% even in the absence of ED, meaning that TEA cannot extract SDE from the catalyst system. This performance was compared with the most widely used fourth- (ID: phthalate) and fifth-generation (ID: 1,3-diether) catalyst systems. Meanwhile, the performance of SDE as ED in the TiCl4/ID/MgCl2 (ID: SDE, 1,3-diether, phthalate) systems was studied. The results showed that in all three systems, the hydrogen response increased (>40%) in comparison to conventional compounds (alkoxysilanes) used as ED, and in fifth-generation catalyst systems, the activity increased significantly as well. These findings can thus serve as a guide for the direct industrial development of this new class of non-phthalate donors.Conflicts of interest
There are no conflicts to declare.Notes and references
- K. S. Thushara, E. S. Gnanakumar, R. Mathew, R. K. Jha, T. G. Ajithkumar, P. R. Rajamohanan, K. Sarma, S. Padmanabhan, S. Bhaduri and C. S. Gopinath, J. Phys. Chem. C, 2011, 115, 1952–1960 CrossRef CAS.
- B. Liu, R. Cheng, Z. Liu, P. Qiu, S. Zhang, T. Taniike, M. Terano, K. Tashino and T. Fujita, Macromol. Symp., 2007, 260, 42–48 CrossRef CAS.
- T. Sadashima, K. Katayama, T. Ota and H. Funabashi, in Current Achievements on Heterogeneous Olefin Polymerization Catalysts, ed. M. Terano, Sankeisha, Co. Ltd, Nagoya, Japan, 2004, pp. 228–233 Search PubMed.
- V. Busico, M. Causa, R. Cipullo, R. Credendino, F. Cutillo, N. Friederichs, R. Lamanna, A. Segre and V. V. Castellit, J. Phys. Chem. C, 2008, 112, 1081–1089 CrossRef CAS.
- B. Zhang, L. Zhang, Z. Fu and Z. Fan, Catal. Commun., 2015, 69, 147–149 CrossRef CAS.
- G. Monaco, M. Toto, G. Guerra, P. Corradini and L. Cavallo, Macromolecules, 2000, 33, 8953 CrossRef CAS.
- V. D. Noto, R. Zannetti, M. Viviani, C. Marega, A. Marigo and S. Bresadola, Makromol. Chem., 1992, 193, 1653 CrossRef.
- L. Pavanello and S. Bresadola, Stud. Surf. Sci. Catal., 1995, 91, 817 CrossRef CAS.
- L. Luciani, F. Milani and R. Zannetti, Macromol. Symp., 1995, 89, 139 CrossRef CAS.
- A. Marigo, C. Marega, R. Zannetti, G. Morini and G. Ferrara, Eur. Polym. J., 2000, 36, 1921 CAS.
- T. Wondimagegn and T. Ziegler, J. Phys. Chem., 2012, 116, 1027 CAS.
- H. X. Zhang, H. Zhang, C. Y. Zhang, C. X. Bai and X. Q. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4805 CrossRef CAS.
- E. Albizzati, G. Giannini, C. L. Norisiti and L. Resconi, in Polypropylene Handbook, ed. P. Edward and J. Moore, Hanser, New York, 1996, p. 12 Search PubMed.
- A. P. Van Wezel, P. van Vlaardingen and R. Posthumus, Ecotoxicol. Environ. Saf., 2000, 03, 305 CrossRef PubMed.
- K. Tully, D. Kupfer and A. M. Dopico, Toxicol. Appl. Pharmacol., 2000, 02, 183 CrossRef PubMed.
- S. Parodi, R. Nocci, U. Giannini, P. C. Barbe and U. Scata, JP S57-063310, Montedison, S.P.A., 1982.
- M. Kioka, N. Kashiwa and Y. Ushida, JP S58-138706, Mitsui Petrochemical Co. Ltd., 1983.
- G. H. Kim, B. H. Um, K. C. Son, K. Oh and H. L. Koh, J. Appl. Polym. Sci., 2014, 131, 40743 Search PubMed.
- M. Ratanasak and V. Parasuk, RSC Adv., 2016, 6, 112776–112783 RSC.
- A. K. Yaluma, J. C. Chadwick and P. J. T. Tait, Macromol. Symp., 2007, 260, 15–20 CrossRef CAS.
- M. V. Marques, R. Cardoso and M. Silva, Appl. Catal., A, 2010, 374, 65–70 CrossRef CAS.
- X. J. Wen, M. Ji, Q. F. Yi, H. Niu and J. Y. Dong, J. Appl. Polym. Sci., 2010, 118, 1853–1858 CAS.
- Q. Zhou, T. Zheng, H. Li, Z. Luo, A. Wang, L. Zhang and Y. Hu, RSC Adv., 2016, 6, 75023–75031 RSC.
- A. Matta, P. Chammingkwan, B. K. Singh, M. Terano, T. Kaneko and T. Taniike, Appl. Catal., A, 2018, 554, 80–87 CrossRef CAS.
- L. Cavallo, S. Del Piero, J.-M. Ducere, R. Fedele, A. Melchior, G. Morini, F. Piemontesi and M. Tolazzi, J. Phys. Chem. C, 2007, 111, 4412–4419 CrossRef CAS.
- Q. Zhou, H. Xu, A. Wang, Z. Ma, H. Li, L. Zhang and Y. Hu, J. Appl. Polym. Sci., 2017, 134, 44704 Search PubMed.
- H. Matsuoka, B. Liu, H. Nakatani and M. Terano, Macromol. Rapid Commun., 2001, 22, 326 CrossRef CAS.
- H. Ikeuchi, T. Yano, S. Ikai, H. Sato and J. Yamashita, J. Mol. Catal. A: Chem., 2003, 193, 207–215 CrossRef CAS.
- M. C. Sacchi, F. Forlini, I. Tritto, P. Locatelli, G. Morini, L. Noristi and E. Albizzati, Macromolecules, 1996, 29, 3341 CrossRef CAS.
- X. Bai, M. Gao and H. Li, Petrochem. Technol., 2004, 34, 617–620 Search PubMed.
- Toho Catalysts Co., Ltd., WO Patent 2,006,129,773, 2006.
- Dow Global Technologies LLC, US8088872B2, 2012.
- Dow Global Technologies LLC, WO2009/085649A1, 2009.
- M. Giampiero and A. Enrico, EP0728769B1, 1998.
- G. Morini and A. Cristofori, EP 0728724A1, 1996.
- X. Wen, J. Appl. Polym. Sci., 2010, 118, 1853–1858 CAS.
- K. Soga, T. Shiono and Y. Doi, Makromol. Chem., 1988, 189, 1531 CrossRef CAS.
- J. Kumawat, V. K. Gupta and K. Vanka, Organometallics, 2014, 33, 4357–4367 CrossRef CAS.
- M. Chang, X. Liu and P. J. Nelson, et al., J. Catal., 2006, 239(2), 347–353 CrossRef CAS.
- N. Pasquini, Polypropylene Handbook, Munich: Carl Hanser Verlag, 2nd edn, 1996 Search PubMed.
- R. A. Hutchinson, C. M. Chen and W. H. Ray, J. Appl. Polym. Sci., 1992, 44(8), 1389–1414 CrossRef CAS.
- D. Fregonese, S. Mortara and S. Bresadola, J. Mol. Catal. A: Chem., 2001, 172, 89–95 CrossRef CAS.
- N. M. Ostrovskii and F. Kenig, Chem. Eng. J., 2005, 107, 73–77 CrossRef CAS.
- M. C. Sacchi, F. Forlini, I. Tritto, R. Mendichi, G. Zannoni and L. Noristi, Macromolecules, 1992, 25(22), 5914–5918 CrossRef CAS.
- A. Correa, F. Piemontesi, G. Morini and L. Cavallo, Macromolecules, 2007, 40, 9181–9189 CrossRef CAS.
- E. Albizzati, U. Giannini, G. Morini, A. C. Smith and R. C. Ziegler, Ziegler Catalysts, ed. G. Fink, R. Mulhaupt and H. H. Britzinger, Springer-Verlag, Berlin, 1995, pp. 413–425 Search PubMed.
- X. Dang, Q. Li, H. Li, Y. Yang, Y. Hu and L. Zhang, J. Polym. Res., 2014, 21, 619 CrossRef.
- H. Zhang, Y. Lee, J. Park, D. Lee and K. Yoon, Macromol. Res., 2011, 19, 622–628 CrossRef CAS.
- Y. V. Kissin, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1745–1758 CrossRef CAS.
- B. Liu, T. Nitta, H. Nakatani and M. Terano, Macromol. Chem. Phys., 2003, 204, 395–402 CrossRef CAS.
- J. C. Chadwick, F. P. T. J. van der Burgt and S. Rastogi, Macromolecules, 2004, 37, 9722–9727 CrossRef CAS.
- J. C. Chadwick, G. Morini, E. Albizzati, G. Balbontin, I. Mingozzi, A. Cristofori, O. Sudmeijer and G. M. M. Van Kessel, Macromol. Chem. Phys., 1996, 197, 2501–2510 CrossRef CAS.
- J. B. P. Soares, Macromol. Symp., 2007, 257, 1–12 CrossRef CAS.
- M. Terano, T. Kataoka and T. Keii, in Catalytic Polymerization of Olefins, ed. T. Keii and K. Soga, Kodansha-Elsevier, Amsterdam, 1986, p. 407 Search PubMed.
- V. Busico, P. Corradini, L. DeMartino, A. Proto, V. Savino and E. Albizzati, Makromol. Chem., 1985, 186, 1279 CrossRef CAS.
- V. Busico, P. Corradini, A. Ferraro and A. Proto, Makromol. Chem., 1986, 187, 1125 CrossRef CAS.
- A. G. Potapov, G. D. Bukatov and V. A. Zakharov, J. Mol. Catal. A: Chem., 2006, 246, 248 CrossRef CAS.
- R. Credendino, J. T. M. Pater, D. Liguori, G. Morini and L. Cavallo, J. Phys. Chem. C, 2012, 116, 22980–22986 CrossRef CAS.
- R. Credendino, D. Liguori, G. Morini and L. Cavallo, J. Phys. Chem. C, 2014, 118, 8050–8058 CrossRef CAS.
- A. Piovano, M. D'Amore, K. S. Thushara and E. Groppo, J. Phys. Chem. C, 2018, 122(10), 5615–5626 CrossRef CAS.
- C. B. Yang, C. C. Hsu, Y. S. Park and H. F. Shurvell, Eur. Polym. J., 1994, 30, 205 CrossRef CAS.
- P. Sobota, J. Utko and T. Lis, J. Organomet. Chem., 1990, 393, 349 CrossRef CAS.
- M. Ystenes and E. Rytter, Spectrochim. Acta, Part A, 1992, 48, 543–555 CrossRef.
- M. Ystenes and E. Rytter, Spectrochim. Acta, Part A, 1989, 45, 1127–1135 CrossRef.
- E. Rytter, S. Kvisle, O. Niriswin, M. Ystenes and H. A. Oya, in Transition Metal Catalyzed Polymerizations. Ziegler–Natta and Metathesis Polymerizations, ed. R. P. Quirk, Cambridge Univ. Press, New York, 1988, p. 292 Search PubMed.
- M. Ystenes and E. Rytter, Spectrosc. Lett., 1987, 20, 519 CrossRef CAS.
- M. Vittadello, P. E. Stallworth, F. M. Alamgir, S. Suarez, S. Abbrent, C. M. Drain, V. D. Noto and S. G. Greenbaum, Inorg. Chim. Acta, 2006, 359, 2513–2518 CrossRef CAS.
- Y. Hu and J. C. W. Chien, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 2003 CrossRef CAS.
- G. Singh, S. Kaur, U. Makwana, R. B. Patankar and V. K. Gupta, Macromol. Chem. Phys., 2009, 210, 69–76 CrossRef CAS.
- V. Di Noto and S. Bresadola, Macromol. Chem. Phys., 1996, 197, 3827–3835 CrossRef CAS.
- V. N. Panchenko, L. V. Vorontsova and V. A. Zakharov, Polyolefins J., 2017, 4(1), 87–97 CAS.
- X.-r. Shen, Z.-s. Fu, J. Hu, Q. Wang and Z.-q. Fan, J. Phys. Chem. C, 2013, 117, 15174–15182 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00715f |
| This journal is © The Royal Society of Chemistry 2019 |