
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUranium speciation in weathered granitic waste rock piles: an XAFS investigation†
Akhil Tayal‡
 *a,
Steven D. Conradson§
b,
Aisha Kanzari¶
a,
Florian Lahroucha,
Michael Descostesc and
Martine Gerard*a
*a,
Steven D. Conradson§
b,
Aisha Kanzari¶
a,
Florian Lahroucha,
Michael Descostesc and
Martine Gerard*a
aInstitut de Minéralogie, de Physique des Matériaux, et de Cosmochimie (IMPMC), Sorbonne Université, UMR CNRS 7590, IRD UMR 206, MNHM, 4 Place Jussieu, F-75005 Paris, France. E-mail: akhil.tayal@desy.de; martine.gerard@sorbonne-universite.fr
bInstitut Jožef Stefan, Jamova cesta 39, 1000 Ljubljana, Slovenia
cORANO Mining, R & D Department., La Défense, Paris, France
First published on 15th April 2019
Abstract
Investigation of uranium migration in the waste piles of granite rock in the Limousin region of France is vital for developing strategies which address related environmental issues. Despite the fact that the concentration of uranium is far below the lower end of the cut off level in these piles, the large volume of rocks – which measure in the hundreds of metric tons – and their conditions of repository make this type of waste a source of concern for the international community. In this work, X-ray absorption spectroscopy techniques (XAFS) were employed in order to identify the speciation of uranium in the different categories of samples collected from various regions of the rock piles which had undergone 50 years of weathering. The samples, such as weathered granite, arena and technosoils, were studied in order to probe the transformation of the U bearing complex. XANES indicates U(VI) valence with uranyl species in all samples. Using a linear combination analysis and shell fitting approach, distinct speciation of uranium was observed in the different categories of samples. In the weathered rock and arena samples with relics of magmatic U minerals, uranyl phosphates comparable to autunite are shown to be dominantly linked with monodentate PO43−. However, the samples collected from technosoils are found to have a mixture of U-phosphate and U-clay minerals (phyllosilicates and silicates). Irrespective of the collection location, all the samples were found to contain U(VI)-oxo species The equatorial O ligands occur as two shells with an average separation of 0.14–0.21 Å. Moreover, all the samples have an Al/Si/P shell around 3.1 Å. A detailed EXAFS curve fit analysis shows that disorder afflicts the entire range of samples which can be attributed to either inhomogeneous binding sites on the disordered clay minerals or to the presence of a mixture of uranium-bearing minerals. XAFS investigations highlight the uranyl overriding forms of U (as U sorbed on clay minerals and secondary uranyl phosphates or silicates) contribute to the retention of U, even in oxidizing conditions known to enhance the mobility of U.
Introduction
Environmental contaminants can originate either through the release of waste from industrial sites, for example, or from natural sources that have been disrupted through processes such as mining. The significance of the latter is now recognized to such a degree that post-mining has now been established as part of the mining life-cycle.1 No matter the source of these contaminants, new conditions should induce reactions with other species in order for them to reach their new thermodynamic minima.2–5 Uranium post-mining in France is partly determined by the environmental fate of waste rock piles discarded in locations adjacent to former mines, most of which have undergone substantial revegetation. Uranium deposits in France are mostly vein-type granitic intrusions, coffinite, uraninite (UIV) paragenese, essentially located in the Massif Central (Limousin). These waste rocks come from overburden, non-mineralized material found on the surface which is removed in order to access geological features with more economic value between the years 1945 and 2001, 76![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of uranium (tU) at 0.2% average grade were produced concomitantly with 160 Mt of waste rocks.6–8 The typical U concentration is around 20 ppm depending on its geological background. A minor portion of the waste rocks corresponds to low-grade ore below the economic cut off value ranging from 100 to 300 ppm.9 The waste piles consist of decimetric to metric blocks submitted to weathering. Compared to massive granite, the waste rock piles have a much higher specific surface area, allowing them to break down more easily into granitic arena, leading to the formation of technosoils that may accumulate mobile U(VI).9–13 Weathered granitic material generally produces phyllosilicates, Fe oxyhydroxides or uranyl phosphates that are efficient traps for U sequestration with sorption or neoformation processes.14–19 The two major problems posed by these waste rocks lie in determining how oxidation related to weathering is affecting the piles based on the U valence of the minerals and, determining what the dominant speciation is. Geological timescales, however, are not necessarily sociological ones, and kinetic factors can cause the attainment of the new equilibrium to take a longer time than desired, during which intermediate species may be formed. The study of the mobilization of U is crucial to determining the intermediate species formation, especially during the incipient weathering period, which has been set at 50 years in the Limousin.
000 tons of uranium (tU) at 0.2% average grade were produced concomitantly with 160 Mt of waste rocks.6–8 The typical U concentration is around 20 ppm depending on its geological background. A minor portion of the waste rocks corresponds to low-grade ore below the economic cut off value ranging from 100 to 300 ppm.9 The waste piles consist of decimetric to metric blocks submitted to weathering. Compared to massive granite, the waste rock piles have a much higher specific surface area, allowing them to break down more easily into granitic arena, leading to the formation of technosoils that may accumulate mobile U(VI).9–13 Weathered granitic material generally produces phyllosilicates, Fe oxyhydroxides or uranyl phosphates that are efficient traps for U sequestration with sorption or neoformation processes.14–19 The two major problems posed by these waste rocks lie in determining how oxidation related to weathering is affecting the piles based on the U valence of the minerals and, determining what the dominant speciation is. Geological timescales, however, are not necessarily sociological ones, and kinetic factors can cause the attainment of the new equilibrium to take a longer time than desired, during which intermediate species may be formed. The study of the mobilization of U is crucial to determining the intermediate species formation, especially during the incipient weathering period, which has been set at 50 years in the Limousin.
Because of its sensitivity to low concentrations of the target element even in amorphous or crystalline form and to the parameters that determine the chemical speciation – valence and local geometry and the types of neighbor atoms and their distances from the absorber – EXAFS is acknowledged as one of the most incisive methods for obtaining such information.20–36 Here we apply it to study the U speciation in granitic rocks, arena and technosoils, related to the weathering of the waste rock piles, performing U L3 XAFS measurements on two categories of samples containing various concentration of U in phyllosilicates or uranyl phosphates paragenese. Reference samples of U bound to clay minerals (montmorillonite, kaolinite and illite) were prepared in the laboratory, and natural samples of autunite and soddyite were also measured. We demonstrate that all of the environmental samples are mononuclear uranyl species without amounts of U(IV) sufficiently large (<10%) to be detected in the XANES or EXAFS, in agreement with oxidizing conditions. In addition we find that this pattern continues in that the spectra divide into particular categories based on the underlying geometry of the equatorial O ligands and second and third shells. Furthermore, these sets correlate with categories of samples determined on field and confirmed by geochemical and mineralogical analyses.9,12 There are, however, also some peculiarities in these correlations pertaining to the extended geometries around the U. This disorder also reduces the precision of the metrical results from curve-fitting in describing the nuances of the U environments. The correlations were therefore often demonstrated by direct comparisons of components of the spectra analyzed to highlight the features that provided these results.
Materials and methods
A. Sampling
The geological background of the waste piles is the two-mica granite from the European Variscan belt.37 The granite paragenese is quartz, albite, orthoclase, biotite, muscovite. The ore is uraninite, pitchblende and coffinite, related to hydrothermal episodes. Pyrite, jarosite and clay minerals signed hydrothermal or supergene alterations processes.38–40 Samples were collected in the Limousin region of France, and specifically chosen from two sites, Fanay and Peny of the “La Crouzille” former mining district for a larger study (Fig. S1-ESI†). Both large waste rocks piles, have been revegetated. Technosoils on the foot of the Fanay piles board a dump. Arenisation and protosoils are the visible results of incipient weathering. For a better overview of the different compartments of the piles, the surface of the piles as well as soil pits were collected.9 The main U(IV) bearing magmatic or hydrothermal minerals are uraninite, zircon or monazite. Secondary neoformations of U(VI) rich minerals related to weathering are uranyl phosphates, and clay minerals and Fe oxyhydroxides with U sorbed.9,12Nine samples (Table 1) were selected for the determination of the U speciation by XAFS. They represent the three different categories among the two waste-rocks piles related to weathered material (weathered rocks, arena and technosoils). Sample AR is a weathered granite from a top pile pit with occurrence of uranyl phosphates and relics of magmatic U minerals. The arena chosen is sample A, from a top pile pit with large occurrence of uranyl phosphates, uranophane, clay minerals and Fe oxides. The four technosoil samples are from the top or the bottom part of the waste rock piles with a characteristic paragenesis of clay minerals (smectite/chlorite/kaolinite), Fe oxyhydroxides and various amounts of uranyl phosphates and disseminate uraninite, zircon and monazite. Three samples from the arena and technosoils (CFA, CFT1, CFT4) were chosen for <2 μm fraction extractions to remove the primary mineral from weathered products for a better signal/noise ratio.
| Sample | Description | U concentration (ppm) | Category |
|---|---|---|---|
| R1 | Soddyite | 712500 |
Natural reference |
| R2 | Autunite | 482700 |
Natural reference |
| R3 | Kaolinite | 500 | Synthetic |
| R4 | Montmorillonite (smectite) | 300 | Synthetic |
| R5 | Illite | 500 | Natural |
| AR | Yellow weathered granitic rock from the top of Fanay WRP | 997 | Altered rock |
| A | Yellow sandy arena from the top of PenyWRP | 1315 | Arena |
| T1 | Brown silty sandy horizon from the top of Fanay WRP | 431 | Technosoil |
| T2 | Ochre clayey-sandy technosoil horizon from the top of Peny WRP | 400 | Technosoil |
| T3 | Brown clayey silty horizon from the base of Fanay WRP | 2800 | Technosoil |
| T4 | Highly weathered rock in coarse sandy horizon from the base of Fanay WRP | 412 | Technosoil |
| CFA | Extracted clay fraction from sample A | 9932 | 2μ fraction |
| CFT4 | Extracted clay fraction from sample T4 | 1551 | 2μ fraction |
| CFT1 | Extracted clay fraction from sample T1 | 1661 | 2μ fraction |
B. Standards
Reference samples of U bound to natural phyllosilicates (montmorillonite, kaolinite, illite) from the IMPMC mineral collection were prepared in the laboratory by slow dropwise addition of uranyl nitrate to avoid precipitation ([U]sol = 4 mM, pH = 4.5) and centrifugation. Natural samples of autunite and soddyite, from the IMPMC collection were also measured.C. Experimental and analytical procedure
The amounts of concentrated samples were set to give a U concentration equivalent to ∼10 mg cm−2. Samples were pressed as uniform pellets and placed into 1 cm diameter holes in an aluminium holder designed for this experiment.At the light source the containers were fastened to the cold finger of a liquid nitrogen reservoir cryostat. The cryostat was evacuated and kept filled with nitrogen over the duration of the measurements. U L3 XAFS measurements were performed at the MARS beam line at Synchrotron Soleil in the fluorescence mode using 11 elements of a Ge detector. Each spectrum consists of one to as many as seven 90/120 minutes scans that are averaged together after pre-processing. The energy was calibrated by defining the first inflection point of an Y foil measured periodically as 17032.08 eV. The ionization energy, k = 0 Å−1, was defined as 17169.5 eV. Data was analyzed as described in ESI.† Analysis of the data was performed using a computer program written by Conradson et al.41 and Athena software package.42
Results
A. Identification of uranyl speciation
Inspection of the XANES (Fig. 1) shows that all of the spectra are very similar in the energies. The shoulder on the high energy side of the white line (∼17185 eV) originates from multiple scattering path along the trans-dioxo cations indicating presence of two widely separated U–O oxo and equatorial distances associated with U(VI)-oxo/uranyl speciation.28 The approximately 0.6 eV spread in the energies of the features is within the error of the measurements that were calibrated between rather than within the scans and on the MARS beamline are performed with only the transmitted and not the incident intensity. The variation in the white line amplitudes – the relatively low ones for autunite (R2) and soddyite (R1) – is not uncommon in actinide oxides and has been correlated with materials that had been heated and therefore were probably more ordered, as these two standard compounds would be relative to the other samples.
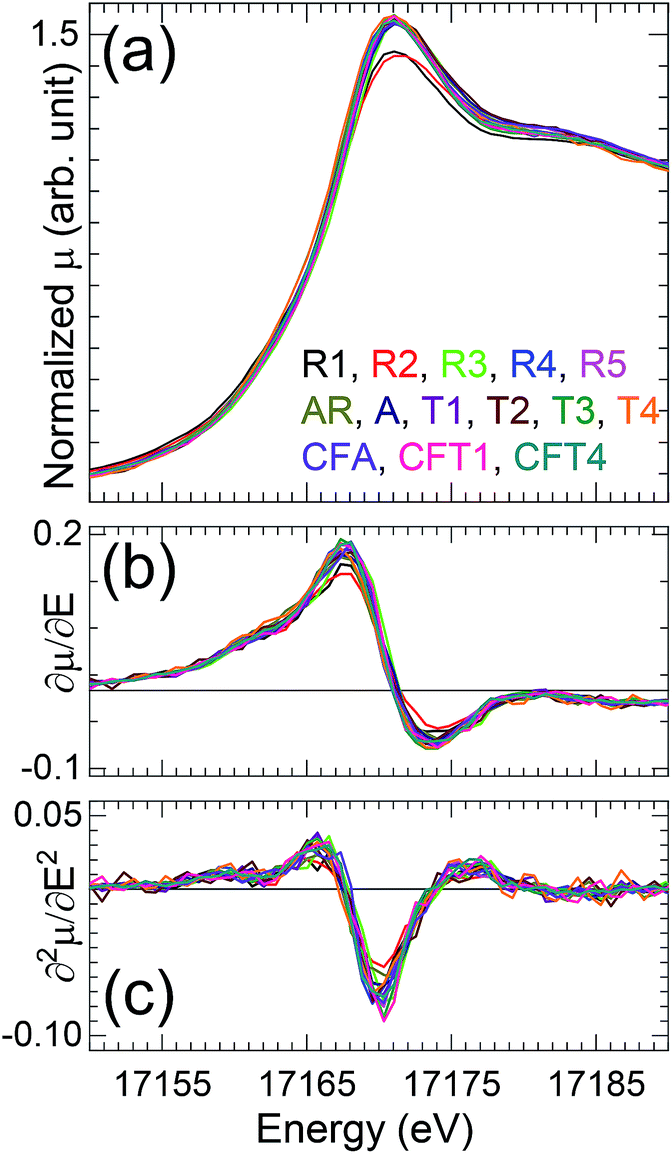 | ||
| Fig. 1 (a) U L3 XANES of all the samples and (b) first and (c) second derivatives associated. The absorption edge over all of the samples is within the uncertainty of the measurements as can be seen in the second derivative. The relatively low amplitude of the white line at 17171 eV and the shoulder at 17182 eV are characteristic of uranyl speciation. | ||
B. Linear combination analysis (LCA)
Beyond the expected demonstration that the speciation of the U is predominantly uranyl with (vide infra) O ligands in the equatorial plane, the principal motivation and challenge for the EXAFS is to determine the U speciation involving the second and more distant neighbors that will subsequently be used to postulate the mechanisms by which the U is concentrated in different sections of rock piles and as input into predictive models. A compilation of available EXAFS results describes four distinct uranyl species differentiated by the orientation of the second neighbor geometry that results in either monodentate or bidentate bridging of the equatorial ligands when, respectively, either a vertex or an edge of the cation-oxygen group faces the U.43 The common soil elements most likely to be constituents of the U speciation are Al, Si, and P as alumino-silicates (kaolinite, smectite, illite), phosphates (monazite, autunite) or silicates (soddyite). Using PO43− as the example, the issue is whether its tetrahedron is positioned end on to give a monodentate type of coordination with the U and a U–P distance around 3.6 Å, or if the tetrahedron is rotated so that two of its O ions are in position to bridge to the U to contract the U–P distance to around 3.2–3.4 Å. Autunite, one of the standards in this work, illustrates the former, with the PO43− group creating a three-dimensional network via single O bridges to multiple U sites. Example of the latter are the mineral Vanmeersscheite and ternary U–Ca–P oxyhydroxides where U–P distances as short as 3.0 Å have been proposed.44 The situation is analogous for Si and Al.The first problem that cannot be overcome solely by curve-fitting analysis of the one-dimensional U partial distribution function the difficulty in distinguishing between the expected Al, Si, and P neighbors because of their proximity in atomic number and resulting similarity of their EXAFS amplitude and phase. In addition, disorder is present within the individual U sites and also because the natural materials are most likely inhomogeneous, with more than one type of U species. This disorder will result in overlapping shells and prevent the analysis of the already low amplitude multiple scattering contributions that contain the three dimensional information on the site. At a minimum, identification of the speciation beyond the first neighbors requires complementary information from other methods. In the present study this includes bulk chemical and mineralogical analysis on the samples collected from different sections of the rock pile using X-ray Diffraction, Scanning Electron Microscopy coupled with microprobe, and Inductively Coupled Plasma-Mass Spectrometry. Clay minerals paragenesis in the order smectite, chlorite and kaolinite,9 is associated to secondary sub-micrometric uranyl-phosphates neoformations.12 Over time, U(VI) released from the alteration of pitchblende/uraninite-coffinite and other forms could therefore be expected to bind to clay minerals, iron oxyhydroxides and to precipitate as secondary phosphates.
The recurring theme of this report is that curve-fits alone are inadequate for providing unique and accurate solutions for the EXAFS of environmental uranyl samples with speciations that are complicated, disordered, and possibly heterogeneous.10,45–47 The three-dimensional geometry of the O-bridged moieties with Al, Si and P and their extended networks are not analyzable by single scattering curve-fits and too complicated for multiple-scattering analysis in these disordered systems. However a possible alternative is that subtle differences might provide information to distinguish these neighbors, Al, Si or P. Some degree of identification may result from their coupling to specific features in the spectra, with the caveat that these spectral “fingerprints” are necessary but not necessarily sufficient for making such assignments, with some degree of validation provided by additional corroborating data. The accuracy of this empirical, qualitative analysis will be enhanced when the species under investigation are closely related. This approach can be applied to the original spectra or, for lower amplitude spectral features, to the residuals after subtracting the principal elements of the structure found by curve fits. It was tested quantitatively for the first and second neighbors by adopting linear combination analysis48,49 using reference samples known to contain the relevant moieties. The basis for this empirical approach is shown in the k3χ EXAFS spectra of the standard samples, illite, smectite, kaolinite, soddyite, and autunite, specifically in the distinct different patterns displayed by two oscillations over the ranges k = 6.2–8.4 and 8.4–11.2 Å−1 (Fig. 2).
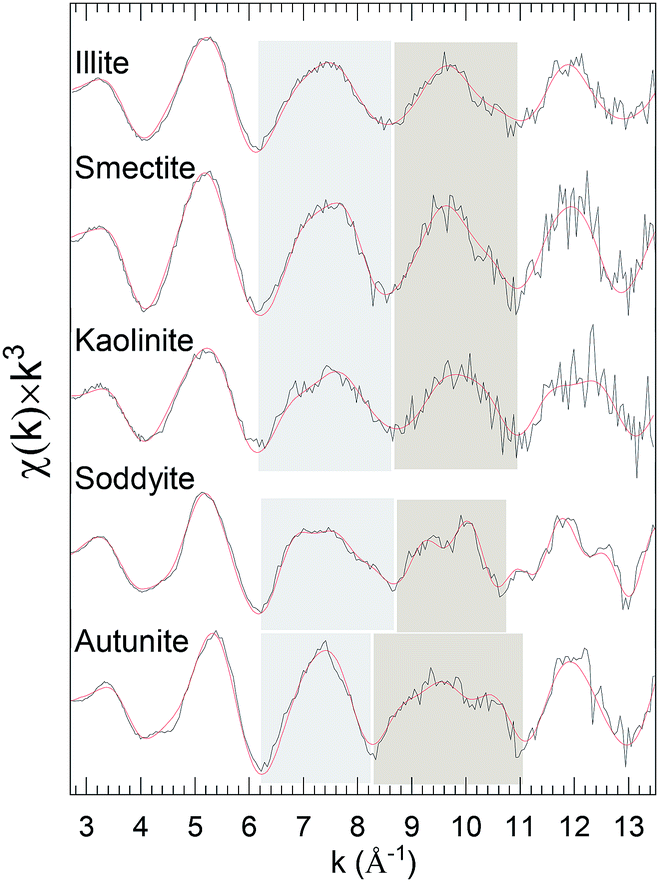 | ||
| Fig. 2 U L3χ(k) × k3 EXAFS of clay standards. The highlighted regions delineate the features used as spectral “fingerprints” that correlate with particular species. A sample description is provided in Table 1. Solid red line represents EXAFS fit to the data. | ||
Curve-fits demonstrate that these patterns can only be fully duplicated by including the second neighbor cations, which means that they correlate with the extended geometries around the U through 3–3.5 Å (cf. EXAFS fit parameters: Table S1 and autunite: Fig. S2†). The uranyl phosphate, autunite, requires both of its P neighbor shells at 3.10 and 3.27 Å. The other standards contain different arrangements of SiO42− tetrahedral and AlO63− octahedral. In soddyite the U resides on one of the crystallographic sites with edge sharing with neighboring SiO42− groups to give bidentate type (Fig. S3†) coordination in its equatorial oxygen shell (Oeq). Illite and smectite differ in that in illite the AlO63− groups are located on the edges of the crystal and in smectite they are internal between the layers. The edge positions give them a high affinity for sorbing uranyl via inner sphere complexes, whereas in the latter the sorption via outer sphere complexes is much weaker. SiO42− and AlO63− constitute the primary structural motif in kaolinite, promoting direct bridging with uranyl in all of the four possible geometries.
The four different types of k-space spectra that encompass several of the common P and Al–Si containing species can be used as the basis for LCA that will augment visual comparisons. Using this approach the spectra of the soil samples divide into three sets: (A) dominant end on, monodentate PO43− (Fig. S3†), (B) a mixture of clay (smectite/illite/kaolinite) and uranyl phosphate, (C) a similar mixture of clay minerals and U–P complex that could not be uniquely determine alone by comparing chi spectra and are distinguished using combination of geochemistry and mineralogy.
The LCA analysis of the soils samples was performed analogous to individual shell or wave fits but using a sum of the standard spectra, minimizing the error by adjusting their amplitudes. This process found three types of behavior. As could be expected from the similarity of their spectra in the two variable regions with those of autunite, the curve-fits of the EXAFS from samples A and AR had significantly lower errors when the autunite spectrum was added to those of the smectite, illite, kaolinite, and soddyite (Fig. 3a, Table 2, Fig. S4†). Sample A was collected from an arena and sample AR from the weathered granitic rock. Fig. S4† shows representative plots of fitting results obtained on T2 and AR samples by performing the LCA. It can be seen that for the AR sample inclusion of autunite component shows a significant improvement in the fits around the shaded and lower k region (5 Å) with a marginal improvement observed for the T2 sample. The obtained ponderation factors after the inclusion of autunite component are listed in Table 2. These results (Table 2) indicate that the uranyl phosphates constitute a substantial fraction of the U species: ∼59% for A and AR, and around 30% for CFA and T2. When this analysis is performed on the spectra from sample T2, collected from a top WRP technosoil, and sample CFA, the extracted clay fraction (<2 μm) from sample A (arena), the errors (Table 2) are appreciably lower with the inclusion of the autunite spectrum. The improvement is even better for samples A and AR (Fig. 3b, Table 2). These results imply that the uranyl phosphate could still constitute a substantial fraction of the U. This is consistent with mineralogical and geochemical characteristics showing a smectite, chlorite kaolinite, and micas paragenese with μm-size particles of uranyl phosphate observed by EDS and SEM (Fig. S5†).
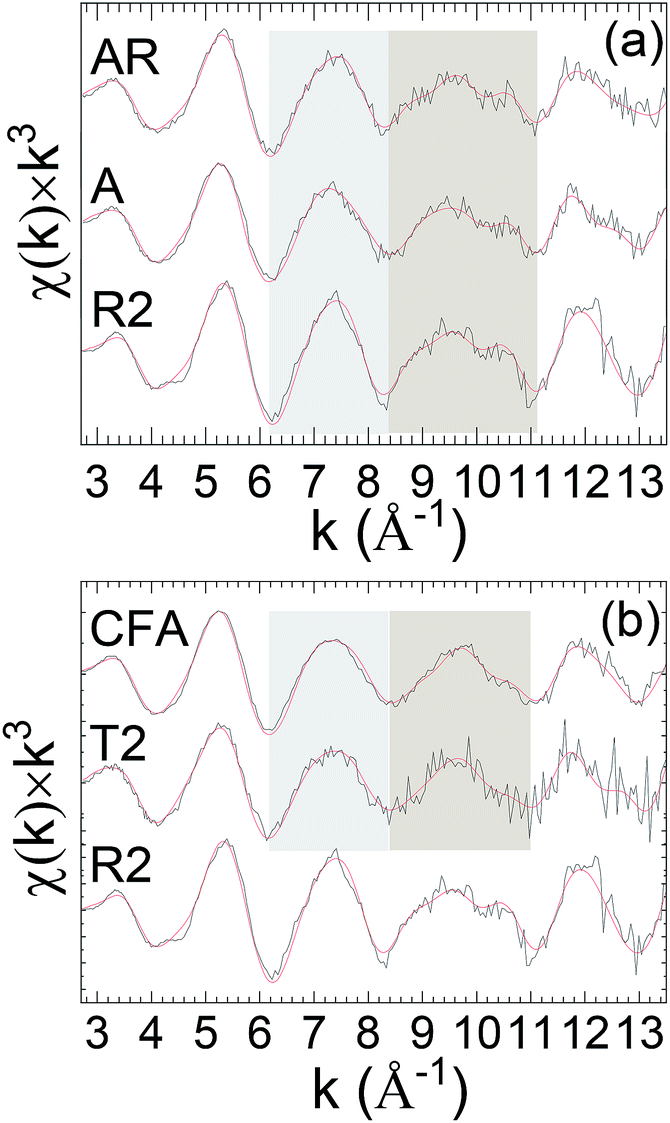 | ||
| Fig. 3 (a) U L3k3χ(k) of autunite (R2) and indicated samples. The shaded areas delineate the spectral regions that contain the signatures assigned to the monodentate Oeq coordination of the PO43− tetrahedron to the U(VI). (b) U L3χ(k) × k3 EXAFS of autunite and indicated samples. Fitting using LCA shows a significant improvement in the fits around the shaded region with the addition of the autunite component. Solid red line represents EXAFS fit to the data. | ||
| Sample | Si/Al minerals | Autunite and Si/Al minerals |
|---|---|---|
| A | R-factor = 0.0799, chir2 = 0.0119 | R-factor = 0.0218, chir2 = 0.0032, weight: 0.59 ± 0.05 (R2) + 0.41 ± 0.05 (R3/R5) |
| AR | R-factor = 0.1134, chir2 = 0.0162 | R-factor = 0.0242, chir2 = 0.0034, weight: 0.66 ± 0.05 (R2) + 0.34 ± 0.05 (R5) |
| CFA | R-factor = 0.0216, chir2 = 0.0034 | R-factor = 0.0111, chir2 = 0.0017, weight: 0.29 ± 0.05 (R2) + 0.71 ± 0.05 (R5) |
| T2 | R-factor = 0.0575, chir2 = 0.0075 | R-factor = 0.0426, chir2 = 0.0056, weight: 0.28 ± 0.05 (R2) + 0.72 ± 0.05 (R5) |
Finally, LCA failed to give curve-fits of sufficiently quality to conclude that the spectra from the remaining last set of samples could be interpreted as combinations of the standards (Fig. 4). These results indicate other types of uranyl speciation.
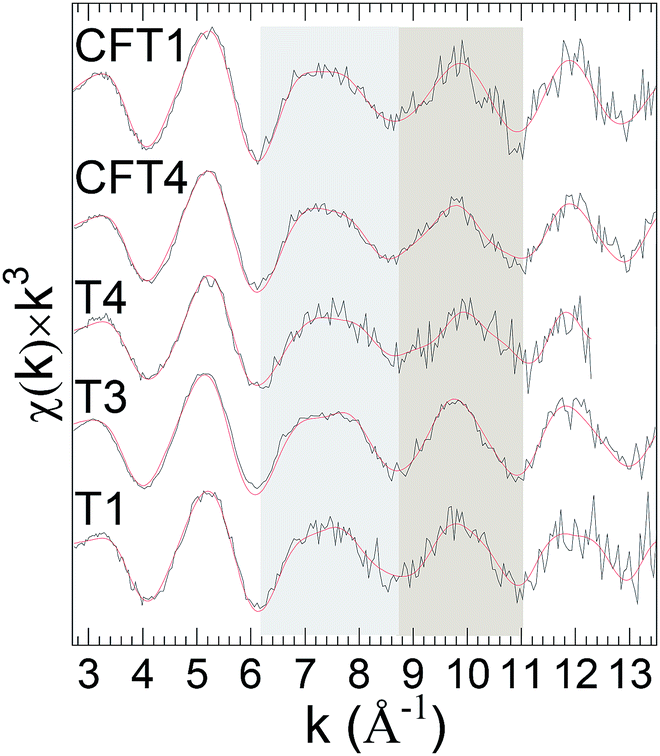 | ||
| Fig. 4 U L3χ(k) × k3 EXAFS of indicated samples. Solid red line represents EXAFS fit to the data. | ||
C. Comparison of χ(R) EXAFS spectra
The samples of C group (Fig. 4) could not be distinguished by performing LCA hence for further classification Fourier transform representation, χ(R), was performed because of its relationship to the local structure that allows for some interpretation of the specific changes in speciation. Caveats are: (1) the peak positions are lower than the actual pairwise distances because of the phase shift that decreases with increasing Z of the neighbor shell; (2) strong multiple scattering of the photoelectron can produce extra features; and (3) this correlation between spectrum and structure is lost in complicated and especially disordered structures because of interference over the relatively short range of the spectra between waves with similar distances. Previous publications of uranyl speciation with clays minerals or phosphates often individually show only relatively small differences because they focus on sets of closely related samples, either from the field or prepared in the laboratory.50,51 In contrast, the spectra from the eleven samples from numerous sources that we report here display significant diversity after accounting for the spectral signature of the U(VI) trans dioxo moiety. This neighbor shell contributes to the peak around R-ϕ = 1.4 Å that dominates all the spectra; its consistent position and amplitude corroborate the interpretation of the XANES that the U in all of the samples is >85–90% uranyl.Closer inspection shows that these spectra can be divided into two groups based on the shapes of the Fourier transform modulus and real (or imaginary) component over R-ϕ = 1.6–2.6 Å that is the contribution from the Oeq neighbors, although this classification is approximate and some of the spectra display intermediate characteristics. The obtained spectral, group 2, have strong correlation with those obtained from LCA (set A and B) suggesting uranyl phosphate environment, reflects as well in FT of χ(k) spectra.
The first group (Fig. 5a) consisting of the spectra from samples T3, T4, T1 and CFT1 resembles many of the previously reported measurements. These spectra could not be distinguished using LCA (set C). The largest peak from the uranyl contribution is at R-ϕ = 1.4 Å, a well separated smaller one at R-ϕ = 1.9 Å, a distinct shoulder around R-ϕ = 2.4 Å, and a peak around R-ϕ = 3.0 Å that may be small (sample T3), shifted (samples T1), overlapping with others (sample T4), or not present presumably because of interference with the waves from other shells (sample CFT1). The real component (Fig. 5a2) corroborates this pattern, showing two local maxima between R-ϕ = 1.5 and 1.9 Å with the second higher than the first.
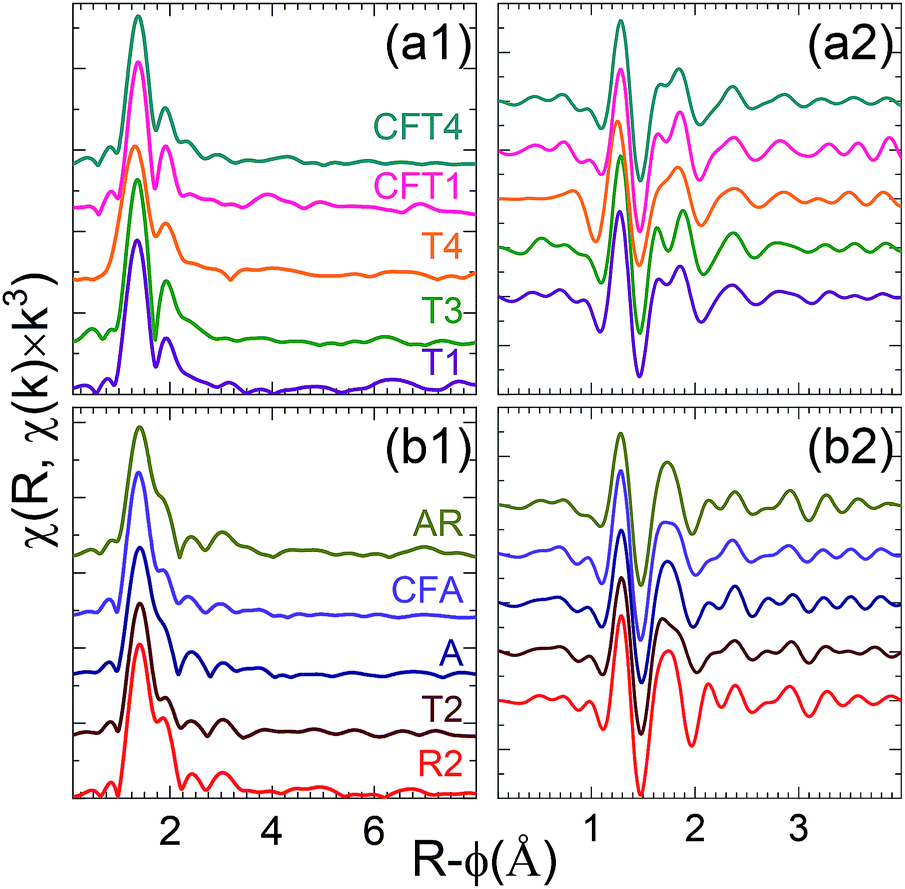 | ||
| Fig. 5 Fourier transforms moduli [(a1) and (b1)] and real components [(a2) and (b2)] of EXAFS spectra of indicated samples for the first [(a1) and (a2)] and second [(b1) and (b2)] groups of spectra. | ||
A, AR, CFA, T2 consist of a phosphate rich sample and/or mixture of phosphate and Si/Al minerals (set A and B from the LCA). Interestingly, comparing χ(R) for those samples (T2, A, AR, CFA) shows they can be grouped together (Fig. 5b1). They differ from the first group in that the second peak is shifted to lower R and not well separated from the first, with only a small or no dip between them. This causes a better resolution of the feature at R-ϕ = 2.4 Å, it appears as a distinct peak. Their real component (Fig. 5b2) between R-ϕ = 1.6 and 1.9 Å consists of a broad local maximum.
This classification is corroborated by examining the real component of the Fourier transform after subtraction of the U-oxo wave as determined by curve-fits (Fig. 6). This procedure enhances the sensitivity of the resulting spectrum to the Oeq and other components. Group 1 spectra display a real component peak at R-ϕ>1.8 Å, a simple minimum around R-ϕ = 2.03 Å, and a simple relative maximum around R-ϕ = 2.35 Å. For group 2 the first maximum is located at R-ϕ < 1.8 Å, the region around R-ϕ = 2.03 Å exhibits a narrow minimum followed by a narrow maximum and another dip or shoulder before it also shows a relative maximum at or slightly above R-ϕ = 2.35 Å.
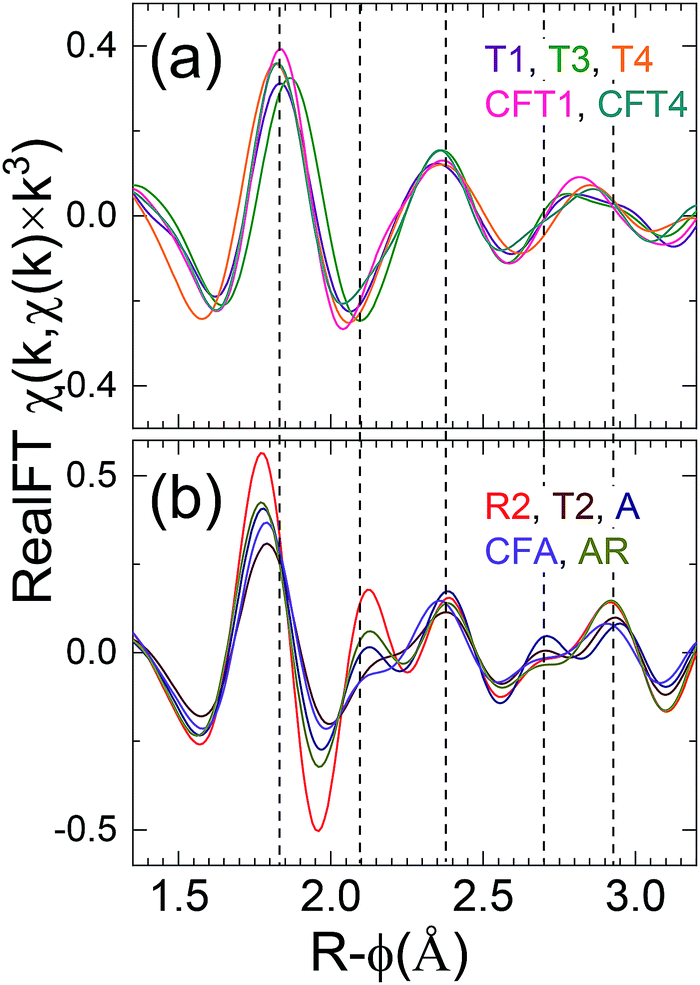 | ||
| Fig. 6 Real component of Fourier transform of EXAFS spectra after subtraction of the U-oxo contribution. These are dominated by the components of the equatorial O contribution. (a), and (b) are, respectively, groups 1 and 2. The listed numbers for each sample show the shorter U–Oeq distance, the separation between this one and the next longest one that can be compared with the 0.12 Å resolution limit, and the ratio of their two numbers that does not account for differences in the Debye–Waller factors. The dashed lines are guides to the positions of particular features discussed in the text. | ||
One notable result is the variability in the χ(R) spectra around R-ϕ = 1.95 Å that occurs not only as the differences between the groups but also between the spectra within the second group, where it often includes diminished amplitudes. Since this is the region where the Oeq neighbors make their contributions it would be caused by interference between the waves from two or more U–Oeq neighbor shells that would have different frequencies. It indicates that among this group, different samples possess disorder in the form of mixtures of U species or in the U–O pair distributions within the Oeq neighbor shells that could be not only wider but also anharmonic.
This disorder afflicts the second and third neighbor shells as well. The first group of spectra show more similarity amongst themselves in the pattern of features around R-ϕ = 2.03 Å than the second, but exhibit the opposite trend around R-ϕ = 2.8 Å. These results thus corroborate the contradiction of the theme of the previous reports of the local structure being homogeneous and therefore deriving from only a single U species in the samples. This inference is further addressed by curve-fits of the EXAFS. The very low amplitudes of any features at higher R indicates no U–U pair suggesting the absence of a U(VI) precipitate or other polynuclear material such as uranyl hydroxides.
D. Curve-fit results, axial and equatorial O neighbor shells
The metrical results from the curve-fits (Table 2) corroborate this interpretation of exclusively U(VI)-oxo/uranyl speciation a representative fits for each of the three samples is shown in Fig. 7. The first U–O distance is at the 1.78–1.82 Å, which is expected for uranyl, and the numbers for all nine samples are within <±10% of each other. All of the fits find an equatorial shell split between a shorter (2.28–2.36 Å) and a longer (2.43–2.55 Å) distance that bracket an average of ∼2.4 Å. The spectra from samples T2, AR and T1 require a third O shell at 2.57–2.80 Å to fit the data exactly. The separations between the principal first two equatorial shells range from 0.12–0.21 Å (Table 2). Interference between the two U–Oeq waves would thus account for the relatively low amplitude and variations found in the χ(R) spectra. Comparing these distances with the resolution limit of 0.12 Å and the 0.15 Å = π value that gives the maximum destructive interference, correlation between the waves from these two (or three) shells will reduce the accuracy of the metrical parameters found by the fits even while the correspondence between the data and fit are excellent. This will be especially true when the numbers of atoms in two principal O neighbor shells are unequal or if their distributions are disordered and broadened. In terms of the separation into the groups, inspection of the real component around R-ϕ = 1.5–2.3 Å (Fig. 6) shows that the first group where the real component maximum is slightly above R-ϕ = 1.8 Å give longer distances to the first O (with the one exception T4), group two gives the converse. However, there is no apparent correlation between the separations or relative numbers of the two principal O shells that determine the interference condition and thus the shape of the EXAFS. Therefore the structures are disordered, implying that the curve-fit results should not necessarily be interpreted as defining distinct neighbor shells but instead describing, in combination properties of the overall distribution, averaged over all of the U atoms in the sample.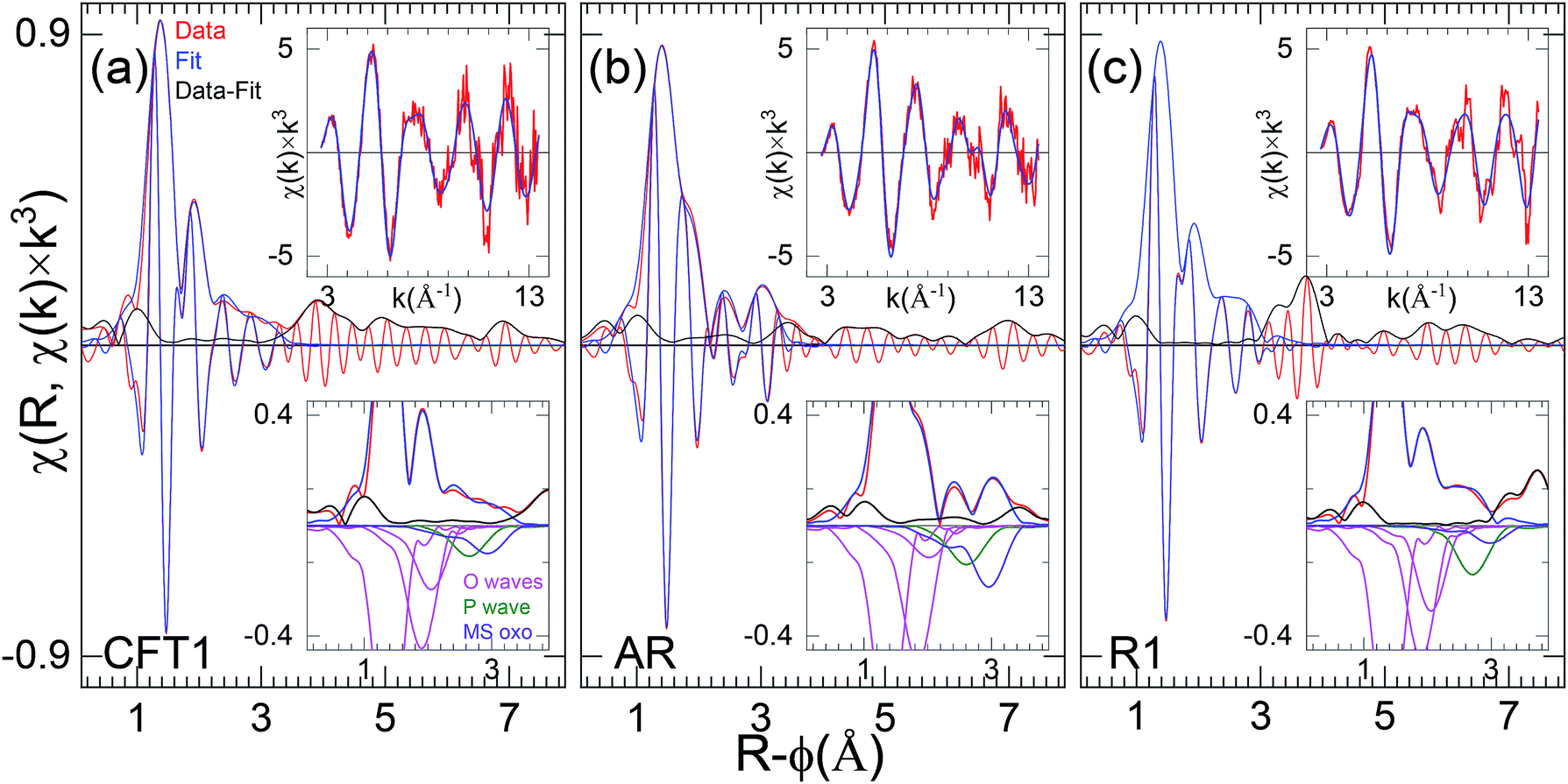 | ||
| Fig. 7 Fourier transforms of EXAFS spectra and curve-fits of representative samples. The upper inset is the original χ(k) data, the lower are the transform moduli of the data, fit, and difference, and (inverted) the contributions of the individual neighbor shells. The curve-fits included the oxo, two equatorial O, a P/Si/Al at 3.1 Å (P wave), and the multiple scattering oxocontribution (MS oxo). | ||
E. Characterization of second near neighbor region, direct comparison of spectra
The second near neighbor shells are arguably even more important than the first because they better delineate whether the U occurs as a separate phase or, if not, the specific mode by which it binds to the Si/Al minerals or other. As with the equatorial O neighbors, the ability to extract information on the more distant neighbors depends on the size of the signal being analyzed, which in turn depends on how well ordered the structure is. Thus, the soddyite standard spectrum (R1, Table S1†) contains large features out to R-ϕ = 4 Å that curve-fits identify as components of the crystal structure (Fig. 7). In contrast, spectral features in the other spectra past R-ϕ = 3.2 Å are much smaller or non-existent for the spectra with low levels of high frequency noise and those that are not as small reflect the overall noise level indicated by the amplitudes of features for R-ϕ ≥ 4 Å. For this reason our analysis and interpretation only extends to features below R-ϕ = 3.6 Å in χ(R), corresponding to neighbor shells with distances < ∼3.8 Å.It is useful to perform the same direct examination of the real component of χ(R) of the second neighbor contributions, as was performed on the first. The spectra of the first group all display (Fig. 7) a feature at R-ϕ = 2.4 Å that, although quite variable in the modulus, gives very similar real components. However, as already described, the R-ϕ = 3.0 Å feature in the group one, spectra undergoes substantial shifts. For group two, the features at R-ϕ = 2.4 and 3.0 Å are relatively consistent in the moduli but less so in the real component, where some show more features of comparable amplitude.
F. Curve-fit results, second neighbor shells
Using LCA it was observed that in some samples uranyl is dominantly linked to PO43− (samples A, AR),whereas in samples T2 and CFA uranyl is bonded to mixtures of Al/Si minerals and phosphates. However, in the other samples LCA was not efficient in discerning U speciation. Although the EXAFS curve fit did not lead to distinguish between Al, Si or P neighbor it helped to reveal the nature of second shell coordination as the association Al/Si/P. In this regards, in the subsequent EXAFS fits second neighbor shell is index as Al/Si/P for samples in which U is bound to phosphate, illite, soddyite, smectite, or kaolinite. Curve fits results show that the region between R-ϕ = 2.3–2.4 Å originates primarily in the first Al/Si/P neighbor shell at around 3.1 Å. A, or rather “the,” pivotal issue in extracting information on the second and third neighbors by curve fits of uranyl species is the multiple scattering oxo (Oaxms) contribution consisting of one three and two four leg paths. Its overall contribution is therefore a composite or sum of these three, making the actual wave complicated. Its χ(R) spectrum is comparably complicated, with a significant component extending to lower R than the main peak that originates in the relatively simple highest amplitude four leg path. Calculating this wave accurately is difficult because the relative, k-dependent amplitudes of its three constituents depend on the Debye–Waller factors for the motions both parallel and perpendicular to the O–U–O axis and currently these cannot be estimated with sufficient accuracy. However, since the U-oxo (or U–Oax) bond is quite rigid and isolated in molecular complexes, except those with OH– and other strong π donor ligands that are not present in these samples, it is reasonable to assume that its pair distribution and consequently the overall shape of the amplitude of its EXAFS are identical in all of the spectra. This is corroborated by the range of U–Oax distances being only 0.03 Å total for all eleven samples, within the error of the measurement. The constraint that the U–Oaxms distance be within ±0.01 Å of twice the single scattering one was easily adhered to in the curve-fits (Table 2).The U–Oax multi scattering problem is crucial because of its connections with not only the generic second neighbor question but more specifically in the case of uranyl-Fe complexation. Indeed, numbers of these samples contain chlorite, which is a Fe-, Mg-rich phyllosilicates, and are suspected to contain Fe oxides in the clay fraction. It has been observed that the phase of a U–Fe shell around 3.45 Å is almost perfectly correlated with that of the U–Oaxms wave with U–O = 1.8 Å (Table 3).
| O – oxo | O – eq. 1 | O – eq. 2 | O – eq. 3 | Al/Si/P [1] | Al/Si/P [2] | MS oxo | Extra | ||
|---|---|---|---|---|---|---|---|---|---|
| a N is the coordination number and R the distance (Å). σ2 is the Debye–Waller factor (mÅ) of the considered scattering path. ΔE0 is the energy of the threshold (eV). Fitting range was k = 2.7–13.5 Å−1, except T4 k = 2.7–12.3 Å−1. If no error is listed the parameter was either fixed at the listed value or at the value of that parameter for a related neighbor shell. Constraints: (numbers refer to the column number in the table of the wave). ΔE0(2) = ΔE0(1) ± 1; ΔE0(3) = ΔE0(2); ΔE0(5) = ΔE0(1) ± 1.5; ΔE0(6) = ΔE0(5); ΔE0(7) = ΔE0(1) ± 1.0. σ(2) = σ(1) + 0.02 ± 0.02; σ(3) = σ(2) + 0.01 ± 0.02; σ(5) = σ(2) ± 0.03; σ(6) = σ(5); σ(7) = σ(1) + 0.01 ± 0.01. N(2) + N(3) = 4.5 ± 1.5; 2R(7) = 2 × R(1) ± 0.01; N(7)αN(1) ± 10%. | |||||||||
| Fe | |||||||||
| T3 | R | 1.79 ± 0.01 | 2.34 ± 0.02 | 2.46 ± 0.02 | 3.09 ± 0.02 | 3.30 ± 0.02 | 3.59 ± 0.03 | 3.75 ± 0.02 | |
| N | 2.4 ± 0.5 | 2.0 ± 0.6 | 4.1 ± 1.2 | 3.1 ± 0.9 | 0.7 ± 0.2 | 2.2 ± 1.0 | 0.3 ± 0.1 | ||
| σ2 | 34 ± 30 | 74 ± 20 | 103 ± 21 | 102 ± 16 | 70 | 54 ± 75 | 70 | ||
| ΔE0 | −2.3 ± 3.8 | −3.3 ± 3.1 | −3.3 ± 2.5 | −3.8 ± 2.6 | −3.8 | −3.3 ± 3.1 | −1.4 | ||
| P | |||||||||
| T2 | R | 1.82 ± 0.02 | 2.32 ± 0.02 | 2.52 ± 0.03 | 2.75 ± 0.02 | 3.13 ± 0.02 | 3.31 ± 0.02 | 3.65 ± 0.06 | 3.54 ± 0.02 |
| N | 2.2 ± 0.6 | 3.0 ± 0.9 | 3.5 ± 1.0 | 1.0 ± 0.3 | 1.8 ± 0.5 | 2.1 ± 0.6 | 2.0 ± 1.2 | 0.7 ± 0.2 | |
| σ | 44 ± 24 | 67 ± 21 | 86 ± 24 | 40 | 37 ± 40 | 40 | 64 ± 80 | 40 | |
| ΔE0 | 2.6 ± 4.2 | 2.7 ± 3.2 | 2.7 ± 2.5 | 3.6 | 4.1 ± 3.4 | 4.1 | 1.6 ± 4.4 | 3.5 | |
| Fe | |||||||||
| A | R | 1.81 ± 0.02 | 2.31 ± 0.02 | 2.52 ± 0.03 | 3.12 ± 0.02 | 3.33 ± 0.02 | 3.64 ± 0.05 | 3.74 ± 0.01 | |
| N | 2.5 ± 0.6 | 3.9 ± 1.0 | 2.6 ± 0.8 | 3.2 ± 1.0 | 1.4 ± 0.4 | 2.2 ± 1.4 | 0.4 ± 0.1 | ||
| σ | 56 ± 17 | 68 ± 18 | 84 ± 21 | 92 ± 17 | 50 | 76 ± 13 | 30 | ||
| ΔE0 | 1.3 ± 3.5 | 0.3 ± 2.9 | 0.3 ± 2.6 | 2.8 ± 2.8 | 2.8 | 0.3 ± 5.0 | 1.7 | ||
| Fe | |||||||||
| T4 | R | 1.78 ± 0.02 | 2.33 ± 0.02 | 2.48 ± 0.03 | 3.10 ± 0.02 | 3.29 ± 0.02 | 3.54 ± 0.04 | 3.68 ± 0.02 | |
| N | 2.4 ± 0.6 | 3.5 ± 1.1 | 3.0 ± 1.0 | 1.4 ± 0.4 | 1.2 ± 0.4 | 2.1 ± 1.0 | 0.5 ± 0.2 | ||
| σ | 60 ± 18 | 86 ± 22 | 115 ± 31 | 56 ± 19 | 40 | 60 ± 38 | 40 | ||
| ΔE0 | −2.7 ± 4.2 | −1.7 ± 2.9 | −1.7 ± 2.6 | −1.2 ± 3.5 | −1.2 | −3.7 ± 4.5 | −1.7 ± 3.3 | ||
| Fe | |||||||||
| AR | R | 1.80 ± 0.02 | 2.28 ± 0.02 | 2.44 ± 0.02 | 2.62 ± 0.2 | 3.08 ± 0.02 | 3.26 ± 0.2 | 3.62 ± 0.06 | 3.80 ± 0.02 |
| N | 2.3 ± 0.6 | 3.5 ± 0.9 | 3.0 ± 0.8 | 1.9 ± 0.6 | 2.5 ± 0.7 | 2.5 ± 0.7 | 2.4 ± 0.8 | 0.3 ± 0.1 | |
| σ | 51 ± 17 | 51 ± 18 | 59 ± 19 | 65 | 67 ± 16 | 60 | 71 ± 45 | 40 | |
| ΔE0 | −0.5 ± 3.8 | 0.1 ± 3.0 | 0.1 ± 2.7 | −1.3 | 1.0 ± 3.0 | 1.0 | −1.5 ± 4.0 | 0.5 | |
| P | |||||||||
| T1 | R | 1.79 ± 0.02 | 2.36 ± 0.02 | 2.55 ± 0.03 | 2.80 ± 0.02 | 3.16 ± 0.02 | 3.38 ± 0.02 | 3.59 ± 0.06 | 3.62 ± 0.02 |
| N | 2.6 ± 0.7 | 4.4 ± 1.3 | 2.1 ± 0.7 | 0.9 ± 0.3 | 1.0 ± 0.3 | 0.8 ± 0.2 | 2.3 ± 0.1 | 0.7 ± 0.2 | |
| σ | 51 ± 19 | 86 ± 23 | 80 ± 25 | 60 | 56 ± 17 | 56 | 71 ± 11 | 30 | |
| ΔE0 | −1.2 ± 4.3 | −0.6 ± 2.9 | −0.6 ± 2.9 | −0.6 | 0.0 ± 3.7 | 0.0 | −2.2 ± 3.5 | 0.9 | |
| Fe | |||||||||
| CFA | R | 1.80 ± 0.01 | 2.30 ± 0.02 | 2.49 ± 0.02 | 3.10 ± 0.03 | 3.28 ± 0.02 | 3.61 ± 0.05 | 3.73 ± 0.01 | |
| N | 2.4 ± 0.5 | 4.5 ± 1.0 | 2.0 ± 0.6 | 2.7 ± 0.8 | 0.6 ± 0.2 | 2.1 ± 0.8 | 0.3 ± 0.1 | ||
| σ | 45 ± 17 | 79 ± 17 | 69 ± 20 | 109 ± 21 | 50 | 45 ± 132 | 30 | ||
| ΔE0 | −1.6 ± 3.7 | −2.6 ± 2.7 | −2.6 ± 2.9 | −0.1 ± 2.6 | −0.1 | −2.6 ± 4.5 | 2.0 | ||
| Fe | |||||||||
| CFT4 | R | 1.80 ± 0.01 | 2.34 ± 0.02 | 2.51 ± 0.02 | 3.03 ± 0.02 | 3.19 ± 0.03 | 3.62 ± 0.04 | 3.71 ± 0.03 | |
| N | 2.4 ± 0.6 | 3.4 ± 0.9 | 2.2 ± 0.6 | 0.9 ± 0.3 | 2.2 ± 0.7 | 2.2 ± 1.3 | 0.2 ± 0.1 | ||
| σ | 47 ± 18 | 62 ± 18 | 53 ± 21 | 93 ± 29 | 109 ± 20 | 67 ± 16 | 42 ± 13 | ||
| ΔE0 | 0.1 ± 3.7 | −0.9 ± 2.9 | −0.9 ± 3.2 | 1.6 ± 2.8 | 1.6 ± 2.4 | −0.9 ± 3.4 | −0.9 ± 6.0 | ||
| CFT1 | R | 1.80 ± 0.02 | 2.35 ± 0.02 | 2.53 ± 0.02 | 3.10 ± 0.03 | 3.25 ± 0.02 | 3.60 ± 0.03 | ||
| N | 2.2 ± 0.6 | 4.4 ± 1.2 | 1.9 ± 0.6 | 2.8 ± 0.9 | 0.5 ± 0.2 | 1.9 ± 0.9 | |||
| σ | 36 ± 20 | 76 ± 21 | 66 ± 22 | 106 ± 21 | 60 | 56 ± 44 | |||
| ΔE0 | −0.4 ± 4.4 | −1.4 ± 2.8 | −1.4 ± 3.2 | 1.1 ± 2.6 | 1.1 | −1.4 ± 2.9 | |||
This U–Fe/U–Oaxms correlation, the complexity of the structures that contain additional neighbor shells overlapping the U–Oaxms and, if present, U–Fe waves with U–Fe <3.6 Å, has resulted in the absence of consensus on the definitive approach to this problem. EXAFS spectra have been analyzed based on 3 different statements: (1) the U–Oaxms wave was negligible – in which case this entire spectral feature originated in an Fe; (2) the U–Oaxms wave was the source of the entire spectral feature – in which case there was no Fe at this distance but only at a much longer one; or (3) this feature included contributions from both types of wave – which gave a smaller number of Fe atoms at this distance. Although uranyl can bind to an Fe-oxide to give a U–Fe distance around 3.45 Å, a result also obtained in calculations, the correlation of this distance with the Oaxms wave makes this finding almost too coincidental. Although, as discussed above, correlating the spectra with the overall sample composition implies that Fe is part of the U speciation in several of the samples, we are unable to quantify this. Four sets of Oaxms amplitudes and phases were tested on these spectra, one calculated from the crystal structure of autunite, one calculated from a contrived totally symmetric UO2O4 structure derived from and tested against the spectrum of Cs2UO2Cl4, an empirical one based on an average of all of the spectra after subtracting the contributions from the other structure components, and an empirical one based on an average of the two spectra with the lowest amplitude of the R = 3.0 Å feature under the assumption that the higher amplitudes derived from the contributions of other shells. As could be expected, the empirical parameters corresponded better to the Oaxms EXAFS, particularly in its low R region. But because they were derived from the spectra after subtraction of the other components they exhibited a distinct tendency to return those same fits, or closely related ones with Fe. The calculated ones did not give as good results, and we found almost complete correlation with the Fe, when the Oaxms amplitude was allowed to float Fe around 3.45 Å could always be included. However, when the Oaxms amplitude was fixed at the value obtained from our Cs2UO2Cl4 spectrum, which was relatively low compared with the size of this feature from the spectra of our eleven samples here, a Fe added to the fit gave an unrealistically short distance of ∼3.1 Å.
Discussion
This EXAFS study, focused on the uranium speciation related to the waste rock weathering, permitted to characterize non-modified environmental samples. The sample set composed of altered granitic rock (AR), arena (A and CFA) and technosoils (T1, T2, T3, T4, CFT1 and CFT4) representing the weathering trend of the waste rock. The XANES observations show clearly that the U valence in all of the samples is U(VI) with uranyl species. Thereby the mobility of uranium is fully governed by its speciation through sorption and precipitation mechanisms.A combination of different analytical approaches identifies distinct U speciation in heterogeneous sample environment. LCA has helped to discern samples into three sets based on χ(k) spectral features. Uranyl species linked to phosphate tetrahedral in monodentate fashion are found to dominate in sample A and AR (set a). In sample T2 and CFA (set b) presence of uranyl phosphate could not be ruled out, however it remains as secondary component, the dominant phase being silicates. This distinction between the two sets is particularly highlighted by XRD data obtained from sample A and its clay fraction, CFA. Autunite, uranophane occur within a dominant chlorite, smectite, kaolinite clay fraction (CFA). Interestingly, the four samples in set a, b show similar χ(R) features and could be well compared to autunite, thus further confirming the presence of U–P speciation. LCA on third set (set c) of samples could not provide any distinction on possible U speciation and are further investigated by comparing χ(R) features.
The equatorial O ligands occur primarily as two overlapping shells with an average distance of 2.35–2.40 Å, a separation of 0.14–0.21 Å, and a total of 4–5 atoms, although some samples require a third O neighbor at a longer distance to completely fit the data. Within these common attributes of the speciation there are three distinct types of equatorial O geometries, and therefore three species that differ in the overall width of the U–Oeq distribution, the degree of continuity or separation of the two principal subshells, and the relative numbers of atoms in the two principal subshells, albeit with some mixing that indicates that some of these samples contain two of these types of U(VI) species.
All samples can be adjusted with a second Al/Si/P shell at longer distance whose contribution overlaps with this 3.1 Å one, so that overall Al/Si/P distribution duplicates the Oeq one in being described by two wide, overlapping shells. The finding of disorder, is linked to inhomogeneous U binding sites because of disorder in the minerals and/or mixtures of distinct U species on different binding sites. Whether intentional or not, the impression left by prior reports is that the EXAFS curve-fit results signify distinct, discrete, well separated neighbor shells centered at the single distance found by the fit and described by Gaussian with narrow peaks. We propose a somewhat different or at least more complete interpretation, acknowledging that those in the field would probably not disagree. First, at least some of the samples contain two binding sites, either on the same mineral or possibly located on different ones. This is not surprising but does demonstrate an important characteristic of natural samples, as opposed to laboratory substrates. In addition, the binding sites possess substantial disorder. Defects and departures from long range order in the minerals result in static disorder in the U–Oeq bond lengths in terms of their not being single valued but spanning a range whose distribution is not necessarily Gaussian so that the actual distributions are semi-continuous as the broadened neighbor shells overlap. This disorder subsequently propagates into the second and more distant neighbor shells. The curve-fits approximate the EXAFS of these quasi-continuous distributions via a small number of waves from discrete but overlapping shells, which can be done quite accurately when the range of the data is limited. The U–O distributions, however, are not amorphous despite the disorder and continuity of U–O distances. They are sufficiently well defined to give the three types of spectra and associated structures that we have observed. It has been demonstrated through this XAFS study that uranyl phosphates or silicates, as well as phyllosilicates are the dominant phases controlling U speciation in the weathered waste rock piles. It must be emphasized that the examination of the disordered U-complex can be performed using the high-resolution (HR) XANES technique which can provide better geometric information due to its insensitivity to structural disorder and superior elemental sensitivity.52 The technique has been utilized for the structural characterization of model U(VI) minerals with the combination of ab initio multiple scattering theory. However, it was noted that this technique is complimentary and must be coupled with ab initio quantum chemical calculations.53–55 In the present work, the observed wide distribution of axial and equatorial U–O distance in all the samples indicates the presence of disorder in the heterogeneous soil mixture. Even in the samples displaying similar U-speciation a wide range of U–O distances occurs. Therefore, it is challenging to define precisely all the theoretical parameters for the simulation in the heterogeneous soil mixture. In the absence of well-defined minimum energy species allowed by the soil chemistry, a combined approach with EXAFS providing essential information can be useful for structural modeling.
There is also the question of the reconstruction of the binding sites and speciation via the identification of the second and third neighbor cations. In addition to the similarities in the behavior of the Oeq and Al/Si/P neighbor shells, the EXAFS indicates the presence of additional neighbors <3.6 Å from the U. Unfortunately, as described it was not possible to devise a method for evaluating these via curve-fits. However, absence of evidence is not evidence of absence. We defer to the discussion of the results in Fig. 7 for the presence of Fe or other metals in the 3.2–3.6 Å range which could be attributed to the occurrence of chlorite. This type of difference in the appearance of χ(R), and in particular its real component, has been identified before in these systems as a means of discriminating between species with Fe and ones with Al. Verification will require a more complete study of the behavior of the Oaxms wave, which is beyond the scope of the current work.
Conclusions
In conclusion, uranium speciation in the waste rock piles submitted to weathering is investigated using the XAFS technique. Samples collected from granitic rock, arena and technosoil display distinct local environment around U. In all the samples U is found to be in U(VI)-oxo/uranyl species with equatorial shells have two different type of U–O distance. Linear combination analysis helps to categorizes the samples into three sets (I) U is mainly linked to monodentate PO43−, (II) samples with a mixture of uranyl phosphate and U-clay minerals, (III) and samples that could not be distinguishable just by comparing chi(k) spectra. EXAFS curve fits show that the second shell is invariably composed of Al/Si/P whose contribution overlaps at 3.1 Å shell distance. Moreover, EXAFS analysis helps to identify the disorder in all the samples which can be attributed to either inhomogeneous U binding sites or presence of a mixture of distinct U species on different binding sites. The retention of U even in oxidizing conditions is facilitated by the occurrence of uranyl phosphates and silicates as well as U sorption on phyllosilicates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the SOLEIL Synchrotron Facility for radiation facilities and thank Dr Pier Lorenzo Solari MARS beamline for assistance. We are grateful to Dr Catherine Beaucaire CEA for her assistance and expertise in the U sorption process. This work was supported by ORANO Mining, R & D Department.Notes and references
- H. E. Jamieson, S. R. Walker and M. B. Parsons, Appl. Geochem., 2015, 57, 85–105 CrossRef CAS.
- E. C. Buck, D. J. Wronkiewicz, P. Finn and J. K. A. Bates, J. Nucl. Mater., 1997, 249, 70 CrossRef CAS.
- A. P. Deditius, S. Utsunomiya and R. C. Ewing, J. Alloys Compd., 2007, 444, 584 CrossRef.
- D. J. Wronkiewicz, J. K. Bates, T. J. Gerding, E. Veleckis and B. S. Tani, J. Nucl. Mater., 1992, 190, 107 CrossRef CAS.
- D. J. Wronkiewicz, J. K. Bates, S. F. Wolf and E. C. Buck, J. Nucl. Mater., 1996, 238, 78 CrossRef CAS.
- AREVA-NC, Expertise globale du bilan décennal environnemental d’AREVA NC 1ère artie : stockage de Bellezane et impact environnemental à l’échelle du bassin versant du Ritord, France, 2007 Search PubMed.
- AREVA-NC, Global expertise of the ten-year environmental situation of AREVA NC2 part: environmental impact at the level of catchment basins and surveillance evaluation, France, 2007 Search PubMed.
- AREVA-NC, Bilan Décennal Environnemental 1994–2003, Etablissement de Bessines, France, 2004 Search PubMed.
- A. Kanzari, M. Gérard, F. Boekhout, L. Galoisy, G. Calas and M. Descostes, J. Geochem. Explor., 2017, 183, 114–126 CrossRef CAS.
- T. Allard, P. Ildefonse, C. Beaucaire and G. Calas, Chem. Geol., 1999, 158, 81–103 CrossRef CAS.
- Y. Wang, M. Frutschi, E. Suvorova, V. Phrommavanh, M. Descostes, A. A. A. Osman, G. Geipel and R. Bernier-Latmani, Nat. Commun., 2013, 4, 2942 CrossRef PubMed.
- F. Boekhout, M. Gérard, A. Kanzari, A. Michel, A. Déjeant, L. Galoisy, G. Calas and M. Descostes, Appl. Geochem., 2015, 58, 123–135 CrossRef CAS.
- M. Leermakers, V. Phrommavanh, J. Drozdzak, Y. Gao, J. Nos and M. Descostes, Chemosphere, 2016, 155, 142–151 CrossRef CAS PubMed.
- M. A. Sequeira Braga, H. Paquet and A. Begonha, Catena, 2002, 49, 41–56 CrossRef CAS.
- P. S. Munasinghe, M. E. Elwood Madden, S. C. Brooks and A. S. Elwood Madden, Appl. Geochem., 2015, 58, 147–160 CrossRef CAS.
- M. Gérard, S. Caquineau, J. Pinheiro and G. Stoops, Eur. J. Soil Sci., 2007, 58, 496–515 CrossRef.
- J.-K. Lee, M.-H. Baik, J.-W. Choi and M.-S. Seo, Nucl. Eng. Des., 2011, 241, 5316–5324 CrossRef CAS.
- Q. Jin, L. Su, G. Montavon, Y. Sun, Z. Chen, Z. Guo and W. Wu, Chem. Geol., 2016, 433, 81–91 CrossRef CAS.
- F. Cretaz, S. Szenknect, N. Clavier, P. Vitorge, A. Mesbah, M. Descostes, C. Poinssot and N. Dacheux, J. Nucl. Mater., 2013, 442, 195–207 CrossRef CAS.
- R. J. Baker, Coord. Chem. Rev., 2014, 266, 123 CrossRef.
- J. Plasil, J. Geosci., 2014, 59, 99 CrossRef CAS.
- P. C. Burns, R. C. Ewing and A. Navrotsky, Science, 2012, 335, 1184 CrossRef CAS PubMed.
- O. N. Batuk, S. D. Conradson, O. N. Aleksandrova, H. Boukhalfa, B. E. Burakov, D. L. Clark, K. R. Czerwinski, A. R. Felmy, J. S. Lezama-Pacheco, S. N. Kalmykov, D. A. Moore, B. F. Myasoedov, D. T. Reed, D. D. Reilly, R. C. Roback, I. E. Vlasova, S. M. Webb and M. P. Wilkerson, Environ. Sci. Technol., 2015, 49, 6474–6484 CrossRef CAS PubMed.
- C. Chisholm-Brause, S. D. Conradson, C. T. Buscher, P. G. Eller and D. E. Morris, Geochim. Cosmochim. Acta, 1994, 58, 3625–3631 CrossRef CAS.
- S. D. Conradson, Appl. Spectrosc., 1998, 52, 252A–279A CrossRef CAS.
- S. D. Conradson, D. A. Andersson, K. S. Boland, J. A. Bradley, D. D. Byler, T. Durakiewicz, S. M. Gilbertson, S. A. Kozimor, K. O. Kvashnina, D. Nordlund, G. Rodriguez, G. T. Seidler, P. S. Bagus, S. M. Butorin, D. R. Conradson, F. J. Espinosa-Faller, N. J. Hess, J. J. Kas, J. S. Lezama-Pacheco, P. Martin, M. B. Martucci, J. J. Rehr, J. A. Valdez, A. R. Bishop, G. Baldinozzi, D. L. Clark and A. Tayal, Phys. Rev. B, 2017, 96, 125114 CrossRef.
- C. H. Delegard, V. F. Peretrukhin, V. P. Shilov and A. K. Pikaev, Alkaline chemistry of transuranium elements and technetium and the treatment of alkaline radioactive wastes, United States, 1995 Search PubMed.
- C. Den Auwer, E. Simoni, S. Conradson and C. Madic, Eur. J. Inorg. Chem., 2003, 3843–3859 CrossRef CAS.
- C. Fillaux, J.-C. Berthet, S. D. Conradson, P. Guilbaud, D. Guillaumont, C. Hennig, P. Moisy, J. Roques, E. Simoni, D. K. Shuh, T. Tyliszczak, I. Castro-Rodriguez and C. Den Auwer, C. R. Chim., 2007, 10, 859–871 CrossRef CAS.
- J. J. Katz, The Chemistry of the Actinide and Transactinide Elements, Springer Science & Business Media, vol. 1–5, 2007 Search PubMed.
- S. D. Kelly, Dev. Soil Sci., 2010, 34, 411–466 CAS.
- D. E. Morris, P. G. Allen, J. M. Berg, C. J. Chisholm-Brause, S. D. Conradson, R. J. Donohoe, N. J. Hess, J. A. Musgrave and C. D. Tait, Environ. Sci. Technol., 1996, 30, 2322–2331 CrossRef CAS.
- D. E. Morris, C. J. Chisholm-Brause, M. E. Barr, S. D. Conradson and P. G. Eller, Geochim. Cosmochim. Acta, 1994, 58, 3613–3623 CrossRef CAS.
- R. T. Pabalan, D. R. Turner, F. Paul Bertetti and J. D. Prikryl, in Adsorption of Metals by Geomedia, Academic Press, San Diego, 1998, pp. 99–130, DOI:10.1016/B978-012384245-9/50004-9.
- T. E. Payne, G. R. Lumpkin and T. D. Waite, in Adsorption of Metals by Geomedia, Academic Press, San Diego, 1998, pp. 75–97, DOI:10.1016/B978-012384245-9/50003-7.
- M. L. Schlegel and M. Descostes, Environ. Sci. Technol., 2009, 43, 8593–8598 CrossRef CAS PubMed.
- IAEA, World Distribution of Uranium Deposits (UDEPO) with Uranium Deposit Classification, International Atomic Energy Agency, Vienna, 2009 Search PubMed.
- F. J. Dahlkamp, in Uranium Ore Deposits, Springer-Verlag Berlin Heidelberg, 1993, vol. XII, p. 460, DOI:10.1007/978-3-662-02892-6.
- J. Leroy, Econ. Geol., 1978, 73, 1611–1634 CrossRef CAS.
- W. Salama, I. González-Álvarez and R. R. Anand, Ore Geol. Rev., 2016, 73, 500–521 CrossRef.
- S. D. Conradson, T. Durakiewicz, F. J. Espinosa-Faller, Y. Q. An, D. A. Andersson, A. R. Bishop, K. S. Boland, J. A. Bradley, D. D. Byler, D. L. Clark, D. R. Conradson, L. L. Conradson, A. L. Costello, N. J. Hess, G. H. Lander, A. Llobet, M. B. Martucci, J. Mustre de Leon, D. Nordlund, J. S. Lezama-Pacheco, T. E. Proffen, G. Rodriguez, D. E. Schwarz, G. T. Seidler, A. J. Taylor, S. A. Trugman, T. A. Tyson and J. A. Valdez, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 115135 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- E. R. Sylwester, E. A. Hudson and P. G. Allen, Geochim. Cosmochim. Acta, 2000, 64, 2431–2438 CrossRef CAS.
- C. C. Fuller, J. R. Bargar, J. A. Davis and M. J. Piana, Environ. Sci. Technol., 2002, 36, 158–165 CrossRef CAS PubMed.
- G. Catalano Jeffrey and E. Brown Gordon, Am. Mineral., 2004, 89, 1004 CrossRef.
- T. Reich, H. Moll, T. Arnold, M. A. Denecke, C. Hennig, G. Geipel, G. Bernhard, H. Nitsche, P. G. Allen, J. J. Bucher, N. M. Edelstein and D. K. Shuh, J. Electron Spectrosc. Relat. Phenom., 1998, 96, 237–243 CrossRef CAS.
- H. A. Thompson, G. E. Brown Jr and G. A. Parks, Am. Mineral., 1997, 82, 483–496 CAS.
- M. S. Massey, J. S. Lezama-Pacheco, F. M. Michel and S. Fendorf, Environ. Sci.: Processes Impacts, 2014, 16, 2137–2144 RSC.
- X. Rui, M. J. Kwon, E. J. O'Loughlin, S. Dunham-Cheatham, J. B. Fein, B. Bunker, K. M. Kemner and M. I. Boyanov, Environ. Sci. Technol., 2013, 47, 5668–5678 CrossRef CAS PubMed.
- N. Clavier, F. Crétaz, S. Szenknect, A. Mesbah, C. Poinssot, M. Descostes and N. Dacheux, Spectrochim. Acta, Part A, 2016, 156, 143–150 CrossRef CAS PubMed.
- S. Lantenois, R. Champallier, J. M. Bény and F. Muller, Appl. Clay Sci., 2008, 38, 165–178 CrossRef CAS.
- A. Manceau, P. Bustamante, A. Haouz, J. P. Bourdineaud, M. Gonzalez-Rey, C. Lemouchi, I. Gautier-Luneau, V. Geertsen, E. Barruet, M. Rovezzi, P. Glatzel and S. Pin, Chem.–Eur. J., 2019, 25, 997–1009 CrossRef CAS PubMed.
- T. Vitova, M. A. Denecke, J. Göttlicher, K. Jorissen, J. J. Kas, K. Kvashnina, T. Prüßmann, J. J. Rehr and J. Rothe, J. Phys.: Conf. Ser., 2013, 430, 012117 CrossRef.
- T. Vitova, K. O. Kvashnina, G. Nocton, G. Sukharina, M. A. Denecke, S. M. Butorin, M. Mazzanti, R. Caciuffo, A. Soldatov, T. Behrends and H. Geckeis, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 235118 CrossRef.
- K. O. Kvashnina and F. M. F. de Groot, J. Electron Spectrosc. Relat. Phenom., 2014, 194, 88–93 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00961b |
| ‡ Deutsches Elektronen-Synchrotron, Notkestraße 85, 22607 Hamburg, Germany. |
| § Washington State University, USA. |
| ¶ ERAMET Research, 1, Avenue Albert Einstein, 78193 Trappes Cedex, France. |
| This journal is © The Royal Society of Chemistry 2019 |