
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterisation of α-carboxynitrobenzyl photocaged L-aspartates for applications in time-resolved structural biology†
Joanna I.
Zaitseva-Kinneberg
 a,
Anke
Puchert
b,
Yannik
Pfeifer
c,
Hao
Yan
d,
Briony A.
Yorke
e,
Henrike M.
Müller-Werkmeister
c,
Charlotte
Uetrecht
df,
Julia
Rehbein
g,
Nils
Huse
ab,
Arwen R.
Pearson
*a and
Marta
Sans
*a
a,
Anke
Puchert
b,
Yannik
Pfeifer
c,
Hao
Yan
d,
Briony A.
Yorke
e,
Henrike M.
Müller-Werkmeister
c,
Charlotte
Uetrecht
df,
Julia
Rehbein
g,
Nils
Huse
ab,
Arwen R.
Pearson
*a and
Marta
Sans
*a
aThe Hamburg Center for Ultrafast Imaging & Institute for Nanostructure and Solid State Physics, Luruper Chaussee 149, 22761 Hamburg, Germany. E-mail: arwen.pearson@cfel.de; marta.sans.valls@cfel.de
bDepartment of Physics and Centre for Hybrid Nanostructures, University of Hamburg, Luruper Chaussee 149, 22761 Hamburg, Germany
cUniversity of Potsdam, Institute of Chemistry, Physical Chemistry, Karl-Liebknecht-Str. 24-25, Golm, 14476 Potsdam, Germany
dHeinrich Pette Institute, Leibniz Institute for Experimental Virology, Martinistrasse 52, 20251 Hamburg, Germany
eSchool of Chemistry, University of Leeds, Leeds LS2 9JT, UK
fEuropean XFEL GmbH, Holzkoppel 4, 22869 Schenefeld, Germany
gFachbereich für Chemie und Pharmazie, Universität Regensburg, Universitätsstrasse 31, 93053 Regensburg, Germany
First published on 15th March 2019
Abstract
We report a new synthetic route to a series of α-carboxynitrobenzyl photocaged L-aspartates for application in time-resolved structural biology. The resulting compounds were characterised in terms of UV/Vis absorption properties, aqueous solubility and stability, and photocleavage rates (τ = μs to ms) and quantum yields (φ = 0.05 to 0.14).
In a time-resolved experiment the reactive species have to be produced instantaneously compared to the timescale of the reaction being studied. For time-resolved structural biology, time-resolutions spanning many decades in time are required in order to allow structural biologists to link the ultrafast (fs to ps) chemical steps with the slower (ns to ms) motions of the protein. These slower dynamics are of particular interest as they are thought to play a key role in modulating the selectivity control of enzymatic catalysis.1,2
For fast (sub ms) reactions most experiments are some variation of Porter's pump–probe3–6 approach where the reaction is initiated with a short laser pulse that either directly triggers the reaction of interest by photolysis or isomerisation,7–9 produces a T-jump,10 or releases a reactive moiety from a photocaged compound.6,11 The two limiting factors of such experiments are the time required for reaction initiation, which can range from a few fs (isomerisation/direct photolysis) to ms for photocaged compounds, and the degree of reaction initiation or quantum yield, which can often be only a small fraction of the sample. For state reversible systems stroboscopic illumination can result in high populations of particular intermediates.4 However, very few biochemical reactions are easily reversible, with the exception of photosensors such as rhodopsin8 and photoactive yellow protein,9 or the classical CO release and rebinding to heme groups.7
As only a small subset of proteins naturally contain photosensitive moieties, photocages12 (photoremovable protecting groups) have received increasing attention in recent years as a tool to study structure–function–dynamics relationships in a wide range of biomolecules both in vitro and in vivo,13,14 such as kinase activation15 and photoinduced gene expression in living cells.16 For time-resolved structural biology experiments both photocaged ligands that diffuse into or near the active site, and unnatural amino acids that can be site-specifically incorporated into the protein during translation can be used.6,17 An ideal photocage for these experiments should have a large absorption above approx. 320 nm, so that the enzyme is not excited directly, a high quantum yield with very rapid release of the substrate, be chemically stable in aqueous solution in the dark, and have good aqueous solubility.
ortho-Nitrobenzyl (oNB) protecting groups are arguably the most used and best understood class of photocages.12 However, simple oNBs are not well suited for time-resolved structural biology, for example due to their absorbance maximum at 260 nm and slow photocleavage, which occurs on the millisecond timescale.18 Modified oNB cages, for example with a carboxyl substitution at the benzylic site to improve aqueous solubility and reduce photocleavage times to the microsecond timescale, have therefore been explored by a number of groups and have great potential for time-resolved structural biology.19–21
Here we chose 1-(αCNB)-L-aspartate (1a, Fig. 1) as a starting point.20 As inclusion of a methylenedioxy ring in oNB cages affords absorption at longer wavelengths,19 we combined 1a with an α-carboxy substitution and synthesised α-carboxynitropiperonyl L-aspartate (1-(αCNP)-L-aspartate, 1b). Concurrently, we also synthesised a p-bromo derivative (1c), as this has been previously reported to improve the rate of oNB photocleavage.22 We chose L-aspartate as the leaving group in this study as a photocaged amino acid exemplar system. α-Carboxylate caged amino acids are potential photocaged enzyme substrates, for example for amino acid decarboxylases. An analogous synthetic approach to that reported here can also be used to deliver γ-carboxylate caged amino acids that can serve as enzyme substrates or be incorporated into a peptide back-bone during translation or peptide synthesis. In our current work we are exploring the application of these photocages to studying the mechanism of the E. coli enzyme L-aspartate alpha-decarboxylase, which catalyses the irreversible conversion of L-aspartate to β-alanine and carbon dioxide, the first dedicated step in pantothenate and coenzyme A biosynthesis.23
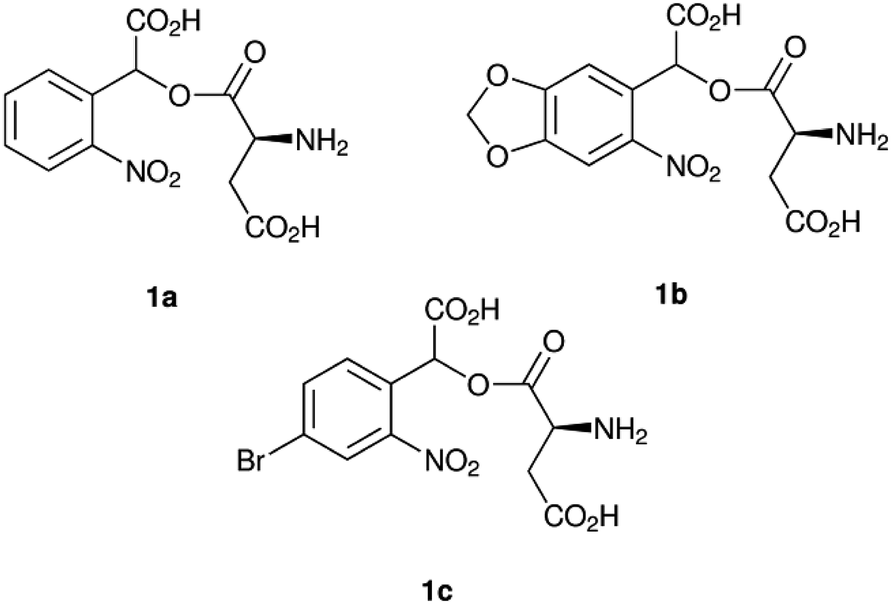 | ||
| Fig. 1 α-Carboxynitrobenzyl photocaged L-aspartate compounds synthesised and investigated in this work: α-carboxynitrobenzyl (αCNB, 1a), α-carboxynitropiperonyl (αCNP, 1b) and p-bromo-α-carboxynitrobenzyl (BrαCNB, 1c) L-aspartate. | ||
The synthesis of α-carboxynitrobenzyl L-aspartate is summarised in Scheme 1. In comparison with the synthesis of Grewer and co-workers,‡,20 this improved route avoids the use of stoichiometric in situ generated HCN. Following a literature procedure,24 trimethylsilyl cyanide was added to commercially available o-nitrobenzaldehydes (2) to afford the corresponding racemic trimethylsilyl (TMS) protected cyanohydrins (3). These were hydrolysed with concentrated HCl to afford o-nitromandelic acids (4), which were then acetylated to give 5. Next, the carboxyl groups were protected with tert-butyl trichloroacetimidate to give 6 and treatment with catalytic Cs2CO3 in methanol afforded the alcohols (7).25 These were coupled to a commercially available amino acid derivative (N-Boc 4-tBu L-aspartate) using EDC and DMAP to give the esters (8), as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture, which were deprotected in the final step with TFA/CH2Cl2 to afford the photocaged L-aspartates (1). Conveniently, steps (i)–(iv) can be telescoped to afford 6 with only a single purification step and the final compounds were obtained in good to very good yields (48–74% over 7 steps).
:1 diastereomeric mixture, which were deprotected in the final step with TFA/CH2Cl2 to afford the photocaged L-aspartates (1). Conveniently, steps (i)–(iv) can be telescoped to afford 6 with only a single purification step and the final compounds were obtained in good to very good yields (48–74% over 7 steps).
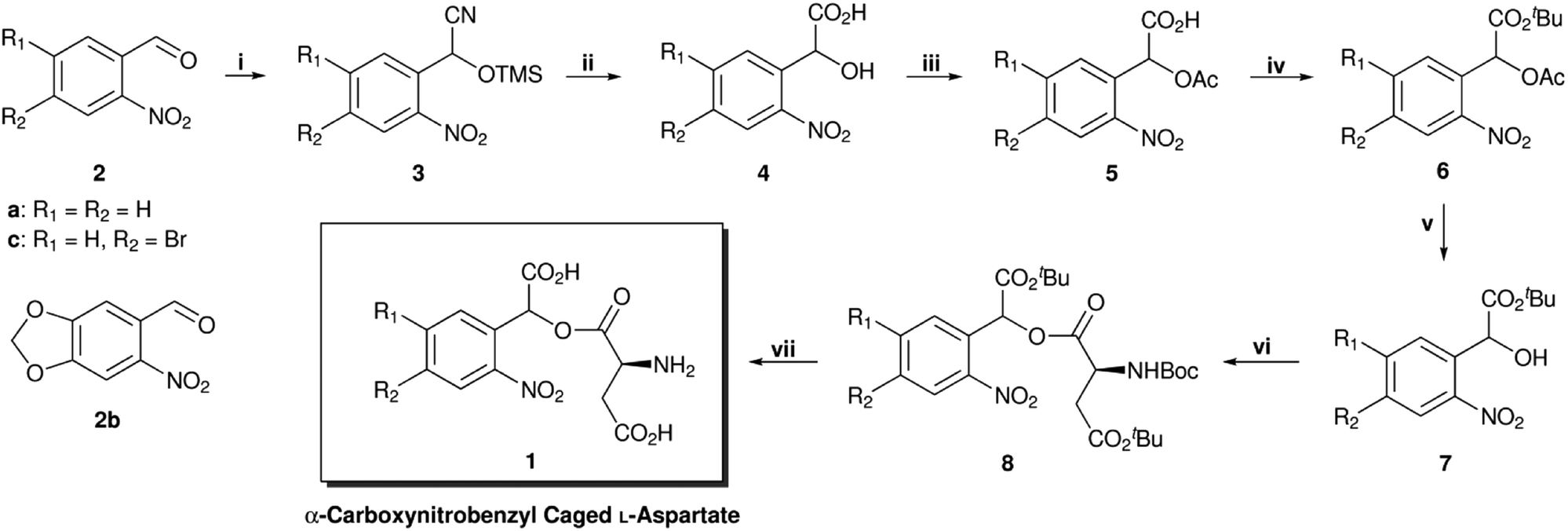 | ||
| Scheme 1 Synthesis of photocaged L-aspartates 1a–1c (structures of final products shown in Fig. 1). (i) TMSCN, MeCN, 18 h, r.t. (ii) HCl, 24 h, 0–100 °C. (iii) Ac2O, 18 h, r.t. or 4 h, 60 °C. (iv) TBTA, benzene, 2 d, r.t., (6a, 72%, 6b, 85%, 6c, 66%; 4 steps). (v) Cs2CO3, MeOH, 1 h, r.t. (vi) N-Boc 4-tBu L-aspartate, DMAP, EDC·HCl, CH2Cl2, 24 h, 0 °C – r.t., (8a, 81%, 8b, 88%, 8c, 73%; 2 steps). (vii) TFA, CH2Cl2, 24 h, r.t., quant. TMSCN = trimethylsilyl cyanide, TBTA = tert-butyl trichloroacetimidate, DMAP = 4-(dimethylamino)pyridine, EDC = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, TFA = trifluoroacetic acid. | ||
For time-resolved applications, photocages must be stable under dark conditions in either solid state, or in organic or aqueous solution, depending on the application. To test aqueous stability, hydrolysis was followed by 1H NMR spectroscopy, with a water suppression pulse sequence. The methylenedioxy substituted photocage (1b) was much more stable than the parent α-carboxynitrobenzyl (1a) (Table S1 in the ESI†) and the hydrolysis was found to be retarded with decreasing pH. The decreased stability at high pH is due to a neighbouring group participation mechanism: the α-carboxylate attacks the ester carbonyl intramolecularly, forming a mixed anhydride which is readily cleaved. When protonated at lower pH, the α-carboxylate can no longer attack in this way.25
Photocaged compound 1b displayed the expected additional absorption band at λmax = 365 nm, with a relatively high extinction coefficient (~4000 M−1 cm−1 – approx. 10-fold that of 1a, 1c or other oNB photocages without either methylenedioxy or dimethoxy substitution patterns at that wavelength). In addition, the photoreaction (Scheme 2) was investigated by laser flash photolysis spectroscopy. Excitation at 355 nm (in aerated phosphate buffer at pH 7) of compound 1b resulted in a transient absorption at 460 nm which decayed with a time constant τ = 199 ± 6 ns (Fig. S8 in the ESI†). We believe this to be the initially formed aci-nitro species,§ which is deprotonated on this timescale (Il'ichev and co-workers reported a similar time constant for the deprotonation of the aci-nitro formed after excitation of o-nitrobenzyl methanol).18 The aci-nitro/nitronate species are understood to be key intermediates in oNB photoreactions.12,18 Formation and deprotonation of the initial aci-nitro is followed by decay of the nitronate species, observed at 430 nm, which fits to an exponential decay function with a time constant of 0.87 ms (see Fig. 2). In the case of compounds 1a and 1c, however, the nitronate decay kinetics were much faster and followed a bi-exponential decay (see Table 1). The two components may be due to the formation of both isomers about the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. The decay of the nitronate is often assumed to be the rate determining step, and Jayaraman showed this to be the case with γ-CNB-L-glutamate (in aq. phosphate buffer at pH 7) using both laser flash photolysis and time-resolved IR spectroscopy.26 However, Il'ichev and co-workers demonstrated, for o-nitrobenzyl methanol, that the rate-determining step is pH-dependent (the nitronate decay is acid-catalysed and the benzisoxazolol ring-opening is base-catalysed – at pH ≤ 4, the ring-opening is the slower step) and a hemiacetal intermediate can persist when the leaving group is poor.18 Thus, the measured time constants give only an indication of the release rate of photocages investigated in this work. Nevertheless, carboxylate is a good leaving group and our experiments were carried out in buffer at neutral pH. Therefore, it is likely that product release coincides with nitronate decay in this case.
C double bond. The decay of the nitronate is often assumed to be the rate determining step, and Jayaraman showed this to be the case with γ-CNB-L-glutamate (in aq. phosphate buffer at pH 7) using both laser flash photolysis and time-resolved IR spectroscopy.26 However, Il'ichev and co-workers demonstrated, for o-nitrobenzyl methanol, that the rate-determining step is pH-dependent (the nitronate decay is acid-catalysed and the benzisoxazolol ring-opening is base-catalysed – at pH ≤ 4, the ring-opening is the slower step) and a hemiacetal intermediate can persist when the leaving group is poor.18 Thus, the measured time constants give only an indication of the release rate of photocages investigated in this work. Nevertheless, carboxylate is a good leaving group and our experiments were carried out in buffer at neutral pH. Therefore, it is likely that product release coincides with nitronate decay in this case.
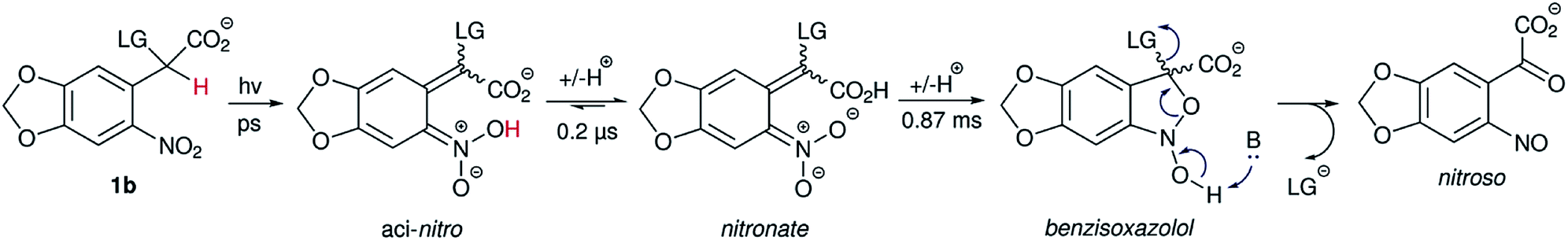 | ||
| Scheme 2 Proposed mechanistic pathway (compound 1b is shown as an example) of the nitrobenzyl photocleavage reaction. Photoinduced HAT leads to the aci-nitro/nitronate intermediates, which subsequently cyclise before releasing the leaving group (LG). Refer to the text for a detailed discussion. | ||
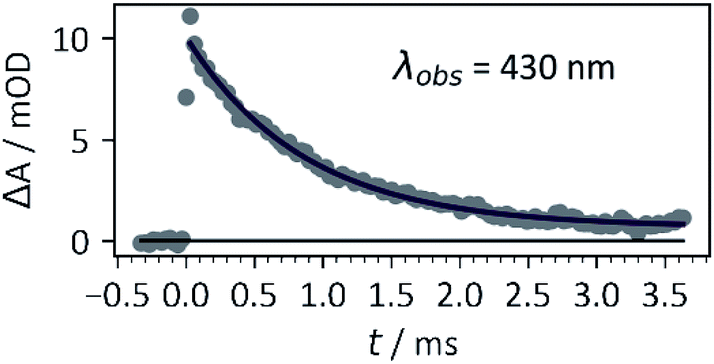 | ||
| Fig. 2 Kinetic trace of transient species (assigned to the nitronate) formed by excitation of 1b with a 355 nm (third harmonic) pulse from a Nd:YAG laser, 30 mJ pulse energy, 1 cm × 1 cm cuvette, 0.4 mM 1b in 5× PBS buffer, pH 7. Data were fitted with the exponential decay function ΔA = ae−t/τ. The time constant obtained was τ = 0.87 ± 0.04 ms. See ESI† for more information. | ||
| Photocaged compound | τ 1/μs | τ 2/μs | φ 365 | ε 355/M−1 cm−1 | t 1/2/h (hydrolysis)b |
|---|---|---|---|---|---|
| a Time constants were derived by exponential fitting of kinetic traces from laser flash photolysis data and represent the two biexponential components of nitronate decay (suspected to coincide with the release of L-aspartate). Quantum yield determinations were carried out with a 365 nm UV LED, and were measured using phenylglyoxylic acid actinometry and HPLC. Thermal half-lives were obtained by fitting 1H NMR data recorded over 24 h with a water-suppression pulse sequence. See ESI for more details. b Half-life of the less stable diastereoisomer at pH 7. | |||||
| 1a | 1.5 ± 0.1 | 10.4 ± 0.4 | 0.105 ± 0.005 | 430 | 13.9 ± 0.2 |
| 1b | 870 ± 40 | n/a | 0.048 ± 0.007 | 4060 | 12.6 ± 0.1 |
| 1c | 1.48 ± 0.08 | 38 ± 1 | 0.14 ± 0.02 | 520 | 10.2 ± 0.1 |
Contrary to our expectation, the pBr-αCNB (1c) caged compound did not display faster nitronate decay kinetics compared to αCNB (1a). Both compounds 1a and 1c absorb about an order of magnitude less than 1b, however their kinetics appear to be a hundred fold faster, with quantum yields of about 10% and 14%, respectively. Conversely, the effective quantum yield of 1b was only 5%, half that of 1a. The αCNP cage would therefore be more suited to applications where longer wavelength UV light is required, and high downstream time-resolution is not required, or applications involving continuous irradiation. The low quantum yields are also non-ideal for time-resolved crystallographic applications where the resulting electron density is an average of all molecules in the X-ray illuminated volume (i.e. both caged and uncaged), and improving them is therefore a future goal. Especially interesting would be a photocage with fast kinetics, high quantum yield and a high extinction coefficient at ca. 350 nm.
In summary, in the experiments described here we have presented a new and improved synthetic route to α-carboxynitrobenzyl photocages. These have been fully characterised, showing good aqueous stability in buffer. The α-carboxynitropiperonyl group shows a new absorbance maximum at 360 nm, which is advantageous for time-resolved structural studies of biomolecules. Work towards confirming the nitronate decay as the rate determining step is ongoing, and we are also working towards an asymmetric synthesis of 2-nitromandelic acid derivatives (4) to address the issue of the racemic and diastereomeric compounds presented here.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Cluster of Excellence ‘The Hamburg Centre for Ultrafast’ of the Deutsche Forschungsgemeinschaft (DFG) – EXC 1074 – project ID 194651731. B. A. Y. thanks the Wellcome Trust (Grant code 110296/Z/15/Z) for support. J. R. thanks the DFG (Emmy-Noether, grant code 251211948) for generous funding. The Heinrich Pette Institute, Leibniz Institute for Experimental Virology is supported by the Free and Hanseatic City of Hamburg and the Federal Ministry of Health. We thank Prof Malte Brasholz for the use of laboratory equipment and helpful discussions. We thank Dr Diana C. F. Monteiro for helpful discussions. The authors acknowledge the Scientific Service of Hamburg University's Chemistry Department for compound characterisation and we are grateful to Claudia Wontorra in particular for NMR (hydrolysis studies). J. I. Z.-K. wishes to thank Dana Komadina, Dr Iosiphina Sarrou and Prof Henry N. Chapman for the use of their HPLC equipment. The authors wish to thank Prof Godfrey Beddard for reading the manuscript and providing helpful comments. J. I. Z.-K. thanks Owen Tuck for help preparing buffers and standards, and Bastian Wulff for assistance with the operation of the Applied Photophysics LKS80 instrument used for laser flash photolysis experiments.Notes and references
- P. K. Agarwal, S. R. Billeter, P. T. R. Rajagopalan, S. J. Benkovic and S. Hammes-Schiffer, Proc. Natl. Acad. Sci., 2002, 99, 2794–2799 CrossRef CAS PubMed.
- S. Hay, M. J. Sutcliffe and N. S. Scrutton, Proc. Natl. Acad. Sci., 2007, 104, 507–512 CrossRef CAS PubMed.
- G. Porter, Proc. R. Soc. A, 1950, 200, 284–300 CAS.
- P. Coppens, M. Pitak, M. Gembicky, M. Messerschmidt, S. Scheins, J. Benedict, S. Adachi, T. Sato, S. Nozawa, K. Ichiyanagi, M. Chollet and S. Koshihara, J. Synchrotron Radiat., 2009, 16, 226–230 CrossRef CAS PubMed.
- B. A. Yorke, G. S. Beddard, R. L. Owen and A. R. Pearson, Nat. Methods, 2014, 11, 1131–1134 CrossRef CAS PubMed.
- E. C. Schulz, P. Mehrabi, H. M. Müller-Werkmeister, F. Tellkamp, A. Jha, W. Stuart, E. Persch, R. De Gasparo, F. Diederich, E. F. Pai and R. J. D. Miller, Nat. Methods, 2018, 15, 901–904 CrossRef CAS PubMed.
- F. Schotte, M. Lim, T. A. Jackson, A. V. Smirnov, J. Soman, J. S. Olson, G. N. Phillips, M. Wulff and P. A. Anfinrud, Science, 2003, 300, 1944–1947 CrossRef CAS PubMed.
- M. Andersson, E. Malmerberg, S. Westenhoff, G. Katona, M. Cammarata, A. B. Wöhri, L. C. Johansson, F. Ewald, M. Eklund, M. Wulff, J. Davidsson and R. Neutze, Structure, 2009, 17, 1265–1275 CrossRef CAS PubMed.
- K. Pande, C. D. M. Hutchison, G. Groenhof, A. Aquila, J. S. Robinson, J. Tenboer, S. Basu, S. Boutet, D. P. DePonte, M. Liang, T. A. White, N. A. Zatsepin, O. Yefanov, D. Morozov, D. Oberthuer, C. Gati, G. Subramanian, D. James, Y. Zhao, J. Koralek, J. Brayshaw, C. Kupitz, C. Conrad, S. Roy-Chowdhury, J. D. Coe, M. Metz, P. L. Xavier, T. D. Grant, J. E. Koglin, G. Ketawala, R. Fromme, V. Šrajer, R. Henning, J. C. H. Spence, A. Ourmazd, P. Schwander, U. Weierstall, M. Frank, P. Fromme, A. Barty, H. N. Chapman, K. Moffat, J. J. van Thor and M. Schmidt, Science, 2016, 352, 725–729 CrossRef CAS PubMed.
- J. Kubelka, Photochem. Photobiol. Sci., 2009, 8, 499–512 CrossRef CAS PubMed.
- I. Schlichting, S. C. Almo, G. Rapp, K. Wilson, K. Petratos, A. Lentfer, A. Wittinghofer, W. Kabsch, E. F. Pai, G. A. Petsko and R. S. Goody, Nature, 1990, 345, 309–315 CrossRef CAS PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- D. P. Nguyen, M. Mahesh, S. J. Elsässer, S. M. Hancock, C. Uttamapinant and J. W. Chin, J. Am. Chem. Soc., 2014, 136, 2240–2243 CrossRef CAS PubMed.
- E. R. Ballister, C. Aonbangkhen, A. M. Mayo, M. A. Lampson and D. M. Chenoweth, Nat. Commun., 2014, 5, 5475 CrossRef PubMed.
- H.-D. Gao, P. Thanasekaran, C.-W. Chiang, J.-L. Hong, Y.-C. Liu, Y.-H. Chang and H.-M. Lee, ACS Nano, 2015, 9, 7041–7051 CrossRef CAS PubMed.
- C. Chou, D. D. Young and A. Deiters, ChemBioChem, 2010, 11, 972–977 CrossRef CAS PubMed.
- I. Josts, S. Niebling, Y. Gao, M. Levantino, H. Tidow and D. Monteiro, IUCrJ, 2018, 5, 667–672 CrossRef CAS PubMed.
- Y. V. Il'ichev, M. A. Schwörer and J. Wirz, J. Am. Chem. Soc., 2004, 126, 4581–4595 CrossRef PubMed.
- D. Binder, C. Bier, A. Grünberger, D. Drobietz, J. Hage-Hülsmann, G. Wandrey, J. Büchs, D. Kohlheyer, A. Loeschcke, W. Wiechert, K.-E. Jaeger, J. Pietruszka and T. Drepper, ChemBioChem, 2016, 17, 296–299 CrossRef CAS PubMed.
- C. Grewer, J. Jäger, B. K. Carpenter and G. P. Hess, Biochemistry, 2000, 39, 2063–2070 CrossRef CAS PubMed.
- A. G. Russell, M. J. Sadler, H. J. Laidlaw, A. Gutiérrez-Loriente, C. W. Wharton, D. Carteau, D. M. Bassani and J. S. Snaith, Photochem. Photobiol. Sci., 2012, 11, 556–563 CrossRef CAS PubMed.
- C. G. Bochet, Tetrahedron Lett., 2000, 41, 6341–6346 CrossRef CAS.
- M. E. Webb, A. G. Smith and C. Abell, Nat. Prod. Rep., 2004, 21, 695–721 RSC.
- K. Manju and S. Trehan, J. Chem. Soc., Perkin Trans. 1, 1995, 2383–2384 RSC.
- F. M. Rossi, M. Margulis, C.-M. Tang and J. P. Y. Kao, J. Biol. Chem., 1997, 272, 32933–32939 CrossRef CAS PubMed.
- Q. Cheng, M. G. Steinmetz and V. Jayaraman, J. Am. Chem. Soc., 2002, 124, 7676–7677 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR stability studies, laser flash photolysis data, quantum yield determination, and compound synthesis and characterisation. See DOI: 10.1039/c9ra00968j |
| ‡ Regarding Grewer's synthesis,20 we found that cyanohydrin formation was not complete within 1.5 h (overnight is sufficient) and 1 h at reflux (100 °C bath temp.) – rather than 8 h at 80 °C – was enough for hydrolysis of the amide to afford 4a. |
| § Il'ichev and co-workers argue that, though the Z-isomer about the aci-nitro group would form first, both E- and Z-forms would rapidly equilibrate in aqueous solution via solvent caged H3O+ and would not be spectroscopically distinguishable (except perhaps using ultrafast techniques).18 |
| This journal is © The Royal Society of Chemistry 2019 |