
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of Ni based mesoporous Al2O3 catalyst with enhanced CO2 methanation performance†
Jianghui Lin‡
 a,
Caiping Ma‡bc,
Jing Luod,
Xianghui Konga,
Yanfei Xua,
Guangyuan Maa,
Jie Wanga,
Chenghua Zhang*e,
Zhengfeng Lia and
Mingyue Ding*af
a,
Caiping Ma‡bc,
Jing Luod,
Xianghui Konga,
Yanfei Xua,
Guangyuan Maa,
Jie Wanga,
Chenghua Zhang*e,
Zhengfeng Lia and
Mingyue Ding*af
aSchool of Power and Mechanical Engineering, Hubei International Scientific and Technological Cooperation Base of Sustainable Resource and Energy, Wuhan University, Wuhan 430072, China. E-mail: dingmy@whu.edu.cn; zhangchh@sxicc.ac.cn
bState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dXiamen Tobacco Industrial Co., Ltd., Xiamen 361022, China
eSynfuels China Co. Ltd., Beijing 101407, China
fKey Laboratory of Advanced Energy Materials Chemistry, Ministry of Education, Nankai University, Tianjin 300071, China
First published on 15th March 2019
Abstract
A Ni based mesoporous γ-Al2O3 (MA) catalyst was prepared via partial hydrolysis without organic surfactants and employed in the carbon dioxide methanation reaction. The obtained catalysts were characterized by N2 adsorption–desorption, H2-TPR, XRD, XPS, TG, SEM and TEM-EDS techniques. CO2 methanation was performed in a fixed-bed reactor. A high surface area of MA with excellent hydrothermal stability was obtained, which promoted the dispersion of nickel species, producing a better catalytic performance. Incorporation of more NiO species into the Ni/MA catalyst increased the amount of active metallic Ni sties, further improving the catalytic activity and CH4 selectivity. Moreover, the monolithic skeleton of MA with fabric-like walls suppressed the aggregation of active metallic Ni sites and carbon deposition, enhancing the catalyst's stability, which provides a new insight for potential industrial applications.
1 Introduction
Synthetic natural gas (SNG), as an efficient and clean energy carrier, has the advantages of high combustion efficiency and using existing gas pipelines, attracting more and more attention with the depletion of natural gas and rapid rising of natural gas prices.1,2 The emission of carbon dioxide into the atmosphere is considered as the major cause of global warming. Many methods including CO2 capture, separation, storage and converting CO2 into useful compounds etc. have been used to reduce CO2 emissions, in which the CO2 methanation reaction (CO2 + H2 → CH4 + H2O, ΔG = −113 kJ mol−1) as an efficient approach for converting CO2 directly to SNG has drawn increasing interest.3In recent years, numerous studies on CO2 methanation have been carried out for metal-based catalysts with enhanced catalytic activity and stability.4–6 Many group VIIIB metals such as Fe, Ru, Rh, Ni, Co, etc. are used widely, in which Ru and Rh-based catalysts have been reported as the most effective catalysts for CO2 methanation, whereas higher cost and limited availability restrict their applications.7 Due to relatively high methanation activity and low price, Ni based catalysts are utilized extensively in the CO2 methanation process,8–10 but they are easily suffered from the deactivation due to carbon deposition and nickel sintering compared to noble metal catalysts.11,12 Low temperature is in favor of CO2 conversion and the formation of CH4 according to thermodynamic equilibrium that methanation reaction is an eight-electron process with kinetic barrier.13 However, complete conversion of CO2 into CH4 at low temperature for Ni based catalysts is very difficult from kinetics.14 It is necessary to modify the Ni-based catalyst for promoting its CO2 methanation performance.
Promotors (such as MgO, CaO, La2O3 and so on),15–17 supports (including SiO2, Al2O3, ZrO2 and CeO2) and reaction conditions (temperature, pressure, space velocity and so on) are extensively adopted to optimize Ni-based catalysts, which play an important role in improving the performance for CO2 methanation.18–21 Especially, the nature of the support has a significant influence on the dispersion of active metallic sites and interaction of metal–support.8 CeO2 (ref. 22) and/or TiO2 (ref. 23) supported Ni based catalysts display excellent CO2 methanation activity and CH4 selectivity by providing oxide vacancies and enhancing H2 adsorption, whereas active Ni nanoparticles undergo easily the thermal sintering due to their low Tammann temperature and “hot spots” existed inside the catalyst bed. In addition, mesoporous molecular sieves possessing large surface area and ordered mesoporous channel have been developed widely to optimize CO2 methanation performance.24–26 However, the silica based zeolites occur easily the collapse of mesoporous structures because of their intrinsic nature of the hydrothermal instability. As a result, designing mesoporous materials with high dispersion of Ni nanoparticles and excellent thermal stability is necessary for CO2 methanation.
Mesoporous alumina with large surface area and good thermal stability is used as a potential support.27 The sol–gel method is applied for the preparation of porous alumina, which results easily in the formation of inhomogeneous porous structure via the cracking of gels.28 Xu et al.29 developed a one-step evaporation induced self-assembly (EISA) strategy to synthesize the ordered mesoporous alumina material, which presented the strong metal–support interaction and outstanding thermal stability compared to the traditional non-mesoporous Al2O3 support, displaying better catalytic performance. However, low-temperature CO2 methanation performance is still needed to be improved. Developing a novel and economic stable mesoporous alumina with large surface area and narrow pore size distribution increasingly draws attention. Recently, Shang et al.30 prepared a new mesoporous γ-alumina (MA) material by partial hydrolysis without the adding of organic surfactants, which showed a high surface area and excellent thermal stability up to 900 °C. However, an application of this kind of MA supported Ni based catalyst to CO2 methanation has never been reported so far.
Herein, we prepared a Ni based catalyst supported on mesoporous γ-alumina synthesized by partial hydrolysis of a nontoxic and inexpensive inorganic aluminum salt. The nickel species loading, interaction of Ni–MA, and CO2 methanation performance were studied in details. The catalysts were characterized by N2 physisorption, scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), powder X-ray diffraction (XRD), hydrogen temperature-programmed reduction (H2-TPR) and thermogravimetric analysis (TG) for obtaining the structure–performance relationship.
2 Experimental
2.1 Catalyst synthesis
MA was prepared via a one-pot template-free partial hydrolysis path, as presented in the article.30 Typically, 0.1 mol of Al(NO3)3·9H2O was dissolved in 50 mL of deionized water at 75 °C, and 1 mol L−1 (NH4)2CO3 aqueous solution was dropped slowly under vigorous magnetic stirring until the formation of transparent gel. The gel was aged for 24 h, and then dispersed in a watch glass at 100 °C for 12 h. The as-prepared solid was treated at 200 °C in air for 10 h to remove ammonium nitrate, and further calcined at 500 °C for 10 h.The Ni/MA catalysts with different nickel contents were prepared by impregnation method. A certain amount of Ni(NO3)2·6H2O was dissolved in distilled water under the stirring and then the MA support was added into nickel nitrate solution under the stirring for 12 h at room temperature. Afterwards, the suspension was dried at 100 °C for 12 h and calcined at 550 °C for 5 h. In this process, the content (wt%) of Ni was changed from 10 to 25%. The prepared Ni-based catalysts were denoted by XNi/MA, where X denotes the content of Ni in weight percentage. In contrast, the commercial alumina (CA) modified Ni catalyst was prepared by the impregnation method. The content of Ni was 25% (wt) in the Ni/CA catalyst, which is denoted to 25Ni/CA.
2.2 Catalyst characterization
N2 adsorption and desorption isotherms were measured by an automatic physical adsorption analyzer (Micromeritics ASAP2020). The sample was degassed at 150 °C for 3 h to remove physical absorbed water and impurities on the surface before the measurement. The specific surface area was calculated by Brunauer–Emmett–Teller (BET) method and pore size distribution was analyzed by Barrett–Joyner–Halenda (BJH) method.Powder X-ray diffraction (XRD) was performed with a Bruker AXS-D8 Advance (Germany) diffractometer using Co Kα (γ = 1.78 Å) radiation at 35 kV and 40 mA. XPS spectra of the catalysts were checked by a VG system (MultiLab 2000), using an Al Kα X-ray source (1486.6 eV). The C 1s as a reference signal was adjusted to 284.6 eV.
H2-TPR measurements were performed in a quartz tube equipped with a thermal conductivity detector (TCD). The samples were pretreated with high purity N2 at 150 °C and a flow rate of 30 mL min−1 for 1.5 h to remove water and other contaminants. The samples were cooled down to room temperature, and then 5% H2/N2 was introduced into the system at a flow rate of 30 mL min−1. The TCD signal and sample temperature were recorded while the temperature being ramped to 900 °C at a heating rate of 10 °C min−1.
Scanning electron microscopy (SEM) images were obtained on a QUANTA F250 scanning electron microscope to characterize the surface morphology of the catalysts. Energy dispersive X-ray spectroscopy (EDX) was performed on a GENESIS EDX detector. High resolution transmission electron microscopy (HRTEM) experiments were performed in a Philips CM200 high resolution transmission electronic microscopy with 200 kV accelerating voltage.
The amounts of carbon deposition at the used catalysts were determined by a TGA/DSC analyzer (METTLER TOLEDO). After drying at room temperature, a certain amount of sample was heated to 900 °C in air with a heating rate of 10 °C min−1 on an Al2O3 crucible.
2.3 Catalyst testing
CO2 methanation was performed at atmospheric pressure in a vertical continuous-flow fixed-bed reactor (inner diameter of 12 mm and a length of 330 mm). The flow of reaction gases was controlled using mass flow controllers. The actual reaction temperature of the catalyst was monitored using a thermocouple placed in the middle of the tubular reactor. 500 mg of the catalyst mixed with 500 mg quartz sand (40–70 mesh) were placed between two layers of quartz wool. Before the reaction, the catalyst was reduced in situ at 500 °C with a heating rate of 2 °C min−1 under a flow of H2 at gas hourly space velocity (GHSV) of 2000 mL g−1 h−1 for 10 h. Subsequently, the temperature was cooled to 200 °C and the mixtures of H2 and CO2 (molar ratio H2/CO2 = 4/1) were introduced into the reactor at 6000 mL g−1 h−1 of GHSV. The catalytic tests were carried out at different temperatures, ranging from 200 to 500 °C.The gas products were analyzed using an on-line gas chromatogram (Fuli 9790II) equipped with a TCD detector using porapak Q and Molecular Sieve 5A packed column. The values of CO2 conversion (XCO2) and CH4 selectivity (SCH4) were calculated by the following equations:
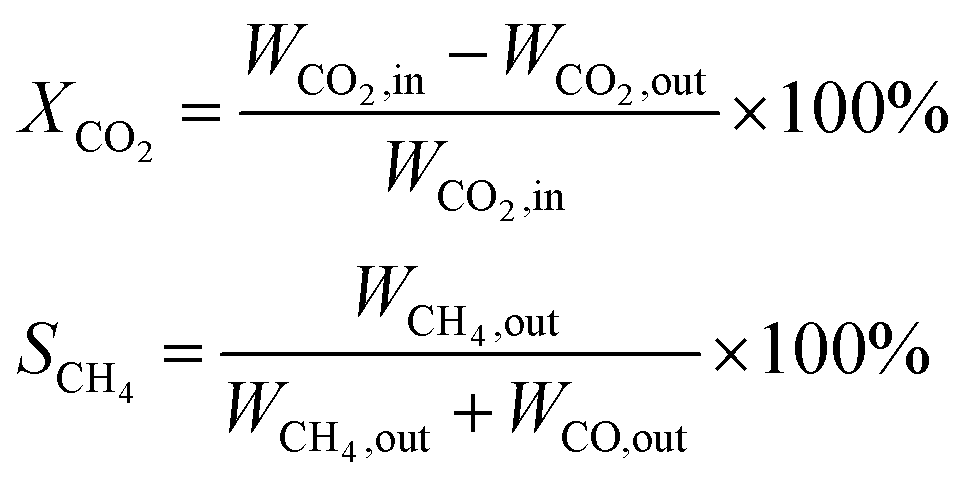
3 Results and discussion
3.1 Catalyst characterization
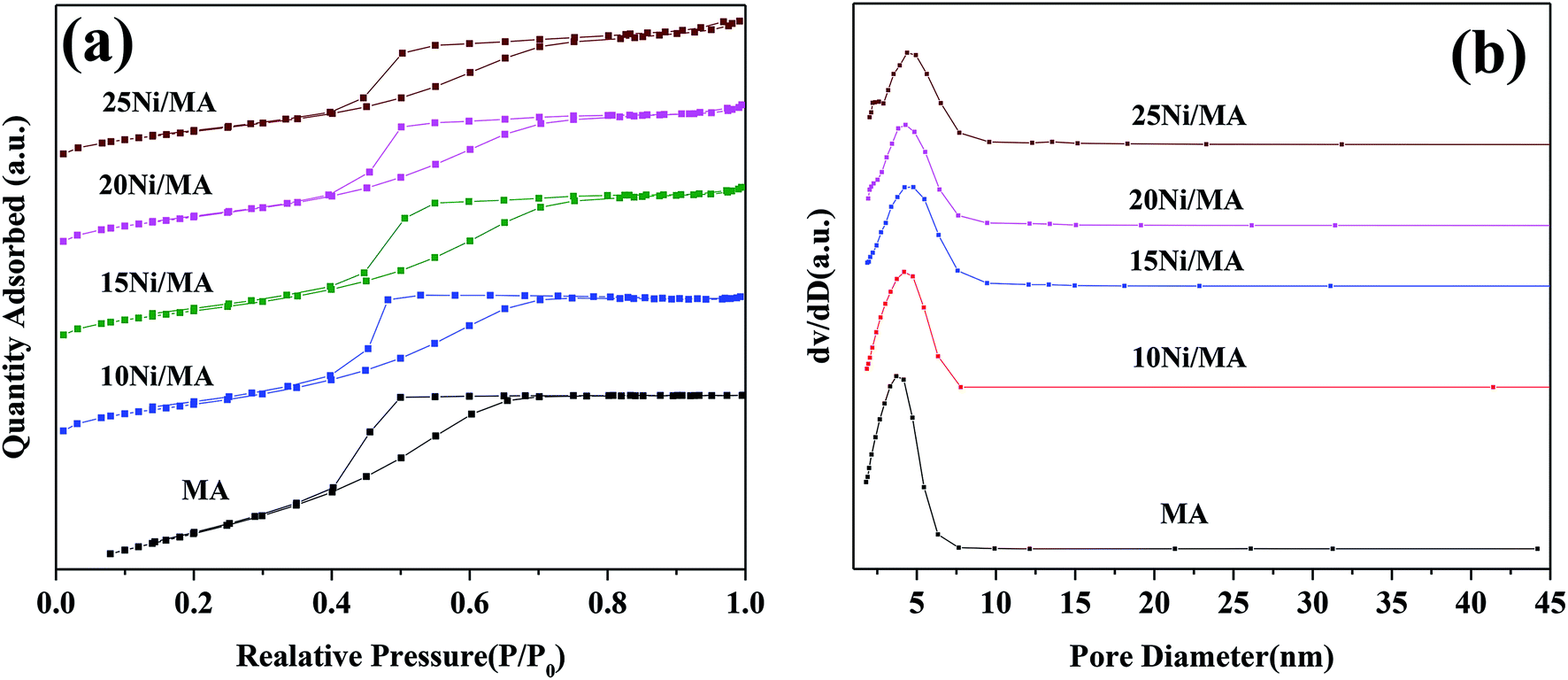 | ||
| Fig. 1 (a) N2 adsorption–desorption isotherms and (b) BJH pore size distributions of the support and Ni/MA catalysts. | ||
Surface area (SBET), pore volume (VP) and pore diameter (DP) of the MA support and Ni/MA catalysts with different nickel loadings are presented in Table 1. BET surface area, average pore volume and pore diameter of MA is 237.0 m2 g−1, 0.32 cm3 g−1 and 3.1 nm, respectively. In contrast, BET surface area of CA is 166.2 m2 g−1, which is lower than that of MA (237.0 m2 g−1), demonstrating that preparation of MA by partial hydrolysis of inorganic aluminum salt facilitates the increase of BET surface area compared to the traditional preparation method of CA. As Ni species is added into the MA support, the BET surface area presents a slight decreasing trend. The BET surface area decreases slowly from 158.8 m2 g−1 of 10Ni/MA to 152.4 m2 g−1 of 25Ni/MA. The average pore diameter of Ni/MA catalysts is in the range of 3.3–4.0 nm (Table 1), while the average NiO crystalline diameter calculated by Scherrer equation from XRD is over 10 nm, indicating that Ni species are dispersed mainly on the MA surface without entering into the mesoporous structures of MA. Therefore, it is possible that partial blockage of NiO particles on the surface of MA results in the decrease of BET surface area and pore volume.32 In addition, with the gradual increasing of Ni species content, the NiO nanoparticles size presents an increasing trend. The crystalline size of NiO increases from 13.6 to 22.7 nm as the Ni content increases from 10 to 25 wt%, implying that the adding of more Ni species facilitates the formation of larger crystallites. On the other hand, the 25Ni/CA catalyst presents 100.3 m2 g−1 of BET surface area, which is only two-thirds of the 25Ni/MA catalyst (152.4 m2 g−1), whereas the crystallites size (31.7 nm) of Ni species over the 25Ni/CA is 1.4 times bigger than that of 25Ni/MA, revealing better dispersion ability of MA compared to CA.
| Sample | SBETa/(m2 g−1) | Dpb/(nm) | Vpc/(cm3 g−1) | Crystalline sized/(nm) |
|---|---|---|---|---|
| a Calculated by the BET equation.b BJH desorption average pore diameter.c BJH desorption pore volume.d Average NiO crystalline diameter calculated by Scherrer equation from XRD. | ||||
| CA | 166.2 | 4.69 | 0.32 | — |
| MA | 237.0 | 3.10 | 0.25 | — |
| 10Ni/MA | 158.8 | 3.36 | 0.18 | 13.6 |
| 15Ni/MA | 156.7 | 3.81 | 0.20 | 15.8 |
| 20Ni/MA | 156.6 | 3.74 | 0.19 | 17.6 |
| 25Ni/MA | 152.4 | 4.00 | 0.22 | 22.7 |
| 25Ni/CA | 100.3 | 4.97 | 0.25 | 31.7 |
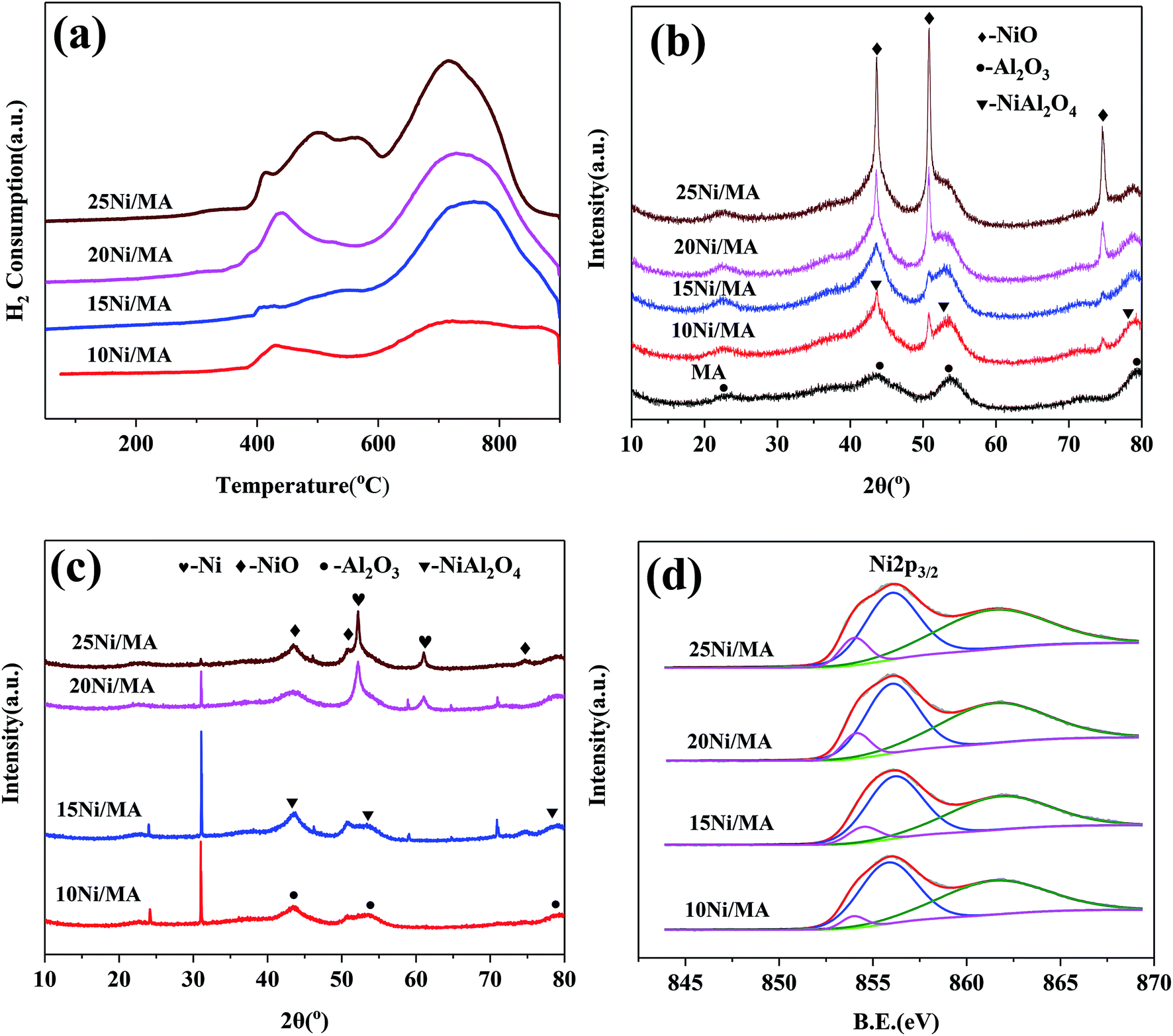 | ||
| Fig. 2 (a) H2-TPR profiles of the fresh Ni/MA catalysts, (b) XRD patterns of the fresh Ni/MA catalysts, (c) XRD patterns of the spent Ni/MA catalysts and (d) XPS spectra in Ni 2p of the fresh Ni/MA catalysts. | ||
After reaction, new diffraction peaks at 2θ = 52.2°, 61.0° appeared in the XRD patterns (shown in Fig. 2c) are assigned to Ni phase (JCPDS no. 87-0712). It is found that the intensity of Ni diffraction peaks presents an increasing trend with the increasing in nickel species content. The highest fraction of metallic Ni phase is observed over the 25Ni/MA catalyst, which confirms the formation of more metallic Ni sites for the Ni/MA catalyst with higher Ni loading. It is accepted that metallic Ni sites play a crucial role in promoting the CO2 methanation reaction.
| Sample | SNiO/ST (%) |
|---|---|
| 10Ni/MA | 8.1 |
| 15Ni/MA | 12.7 |
| 20Ni/MA | 19.1 |
| 25Ni/MA | 20.3 |
 | ||
| Fig. 3 SEM images of (a) MA support, (b–e) fresh catalysts: 10/15/20/25 Ni/MA and (f–i) spent catalysts: 10/15/20/25 Ni/MA. | ||
As nickel species are added into MA, the fabric-like walls with inside diameter about 100 nm are displayed on the surface layers (Fig. 3b–e). These porous frameworks are well cross-linked together to form the fabric-like walls. It is possible that the combination of Ni species with MA during the impregnation and calcination processes changes the surface morphologic structures of Ni/MA. NiO spherical particles are observed inside the fabricate walls, and the nanoparticles size increases with the increasing nickel content in the Ni/MA catalysts. After reaction, the fabric-like structures remain almost unchanged (Fig. 3f–i), validating the excellent hydrothermal stability of Ni/MA. There is no particle aggregation of Ni species is during reaction, suggesting that the MA support played an important role in restraining the sintering of Ni particles. In addition, no obvious carbon deposition is observed for the Ni/MA catalyst in the SEM patterns, indicating that the distinct monolithic skeleton of MA facilitates the transfer of reaction and product molecules, and suppresses the carbon deposition on the surface of MA.
TEM measurements are performed to characterize the structure and distribution of Ni species over the 25Ni/MA catalyst (Fig. 4). Two kinds of lattice spacing about 0.24 nm and 0.21 nm are observed on the surface of MA (Fig. 4a and b), corresponding to NiO (111) and (200) crystals, respectively.38,39 The high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) imaging and elemental mapping from Fig. 4c–f show the distribution of Ni, Al and O elements, respectively. To the marked nanoparticle (red circle marked in Fig. 4c) anchored outside the MA surface, only Ni and O are present while no Al is observed in the mapping shown in Fig. 4d–f, demonstrating the only presence of NiO nanoparticles on the MA surface. This further validates that Ni species added in the Ni/MA catalysts is mainly covered on the surface layers without entering into the mesoporous structures of MA. The NiO nanoparticle size is about 30 nm, which is comparative with the value calculated by XRD. The combination of TEM, SEM and XRD provides an unambiguous illustration on the NiO particle size distribution of Ni/MA.
 | ||
| Fig. 4 (a, b) TEM, (c) STEM-HAADF, (d) Al map, (e) Ni map and (f) O map images of the fresh 25Ni/MA catalyst. | ||
TEM images and elemental mapping of the used 25Ni/MA catalyst are shown in Fig. 5. As shown in Fig. 5a, the mesoporous structures of 25Ni/MA remain unchanged during reaction, indicating that the reaction of CO2 methanation does not destroy the mesoporous structures. The elemental mapping shown in Fig. 5b–e presents the existing of Ni, Al and O, respectively. Only metallic Ni element (Fig. 5d) is observed for the marked nanoparticle (red circle in Fig. 5b) on the surface layers, whereas no Al or O exists (Fig. 5c and e), indicating that the NiO phase on the surface of 25Ni/MA has been converted completely to metallic Ni in the CO2 methanation reaction. The spent Ni nanoparticles size is about 25 nm, indicating that the Ni particles remain a stable state, and no sintering of Ni nanoparticles appeared during reaction. This might be attributed to the fabric-like walls of MA, suppressing the aggregation of Ni nanoparticles during the reaction.
 | ||
| Fig. 5 (a) TEM, (b) STEM-HAADF, (c) Al map, (d) Ni map, (e) O map images of the spent 25Ni/MA catalyst. | ||
3.2 Catalyst testing
CO2 methanation performances of the Ni/MA catalysts are shown in Fig. 6. As shown in Fig. 6a, the catalytic activity of all of the catalysts increases firstly with the increasing reaction temperature, and then reaches the maximum value at a specified temperature. Subsequently, the catalytic activity begins to decrease with further increasing temperature. A similar changing trend is observed for CH4 selectivity (Fig. 6b). The maximum value (77.2% of CO2 conversion and 99.9% of CH4 selectivity) is displayed at 360 °C over the 25Ni/MA catalyst. When the temperature is higher than 360 °C, CO2 conversion and CH4 selectivity begin to drop. As we known, CO2 methanation is an exothermic reaction, which is favorable at low temperature based on its thermodynamic equilibrium.40 Raising the reaction temperature facilitates the reverse water-gas shift reaction, increasing the amount of CO vice-product.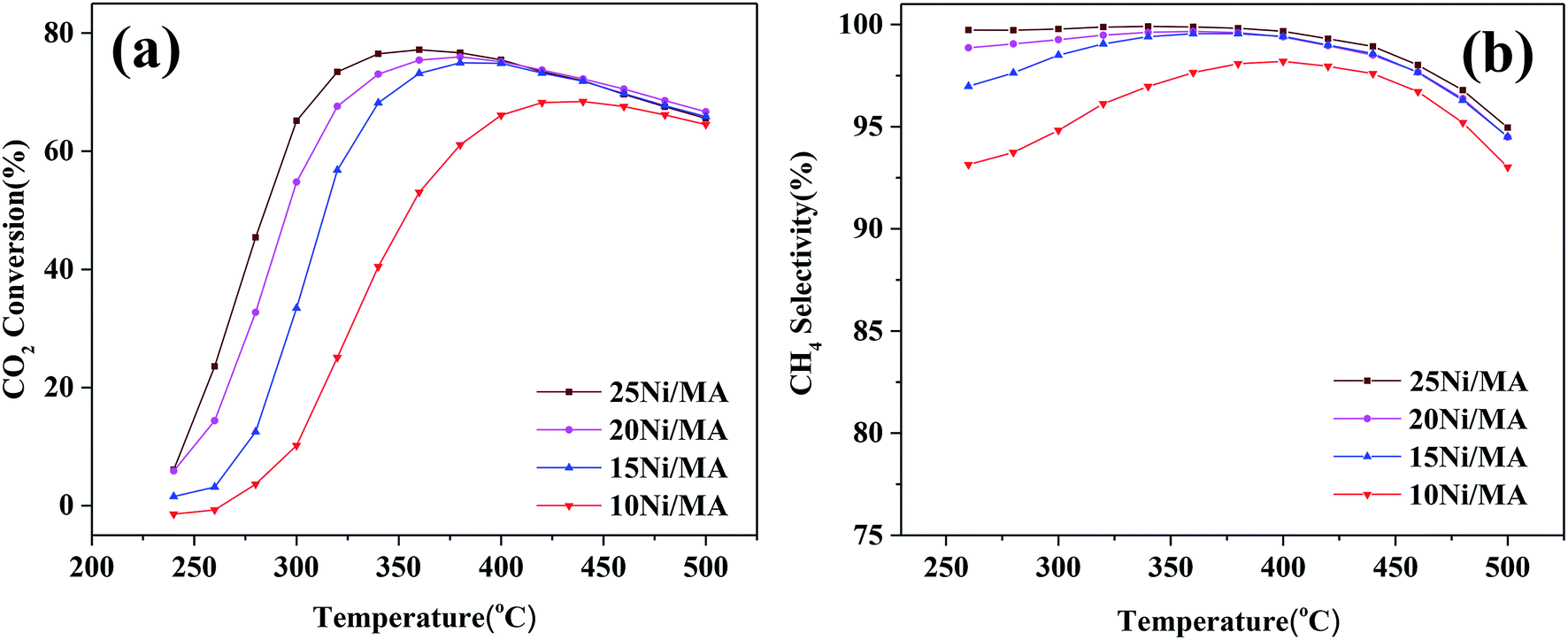 | ||
| Fig. 6 (a) CO2 conversion and (b) CH4 selectivity of Ni/MA catalysts, GHSV = 6000 mL g−1 h−1, H2/CO2 = 4, 1 atm. | ||
The effect of nickel loadings on the catalytic performances is further studied. From Fig. 6a it can be found that CO2 conversion presents an increasing trend with the increasing Ni content. The optimal catalytic activity increases from 62 to 77% as the Ni content increases from 10 to 25%, accompanied with the obvious decrease of suitable reaction temperature from 420 to 360 °C. It is generally accepted that metallic Ni plays a unique role in providing active sites for CO2 methanation. Rahmani et al.35 prepared the Ni/Al2O3 catalysts with different Ni loadings, and found that CO2 conversion increased gradually with the increasing nickel loading in CO2 methanation. Du et al.26 investigated CO2 methanation performance of Ni-MCM-41 catalysts containing 1–3 wt% Ni species, and found that higher selectivity and space-time yield were obtained for the Ni-MCM-41 catalyst with higher Ni loading. In the present study, Ni species added into the MA support is dispersed well on the MA support without entering into the mesoporous structures, confirmed by the BET, SEM and TEM results. In addition, the H2-TPR results indicate that the amount of α-type and β-type Ni species increases with the Ni loading increasing, implying that incorporation of more Ni species into MA weakens the metal–support interaction, promoting the reduction of NiO and increasing the amount of active metallic Ni sites, which plays a critical role in improving the CO2 methanation performance.7 The used Ni/MA catalyst with higher Ni loading presents more active metallic Ni sites confirmed also by both the XRD and XPS results, which further provides more active sites for CO2 methanation. Especially, the adding of more Ni loading increases the particles size of NiO, as confirmed by XRD, which facilitates the weakened interaction of Ni–MA, promoting the formation of active metallic Ni. It is also considered that the particle size of NiO plays an indirect role in promoting the catalytic activity.
On the other hand, it is seen that the CH4 selectivity in the range of 250–400 °C increases gradually with the increasing in nickel loading (shown in Fig. 6b). As the reaction temperature is over 400 °C, CH4 selectivity for all of the catalysts begins to decrease with the increasing temperature on stream, which may be attributed to the reverse water gas shift reaction,26 resulting in the conversion of CO2 to CO, decreasing the CH4 selectivity.
In addition, comparison of CO2 methanation performance for MA modified and CA modified Ni catalysts are shown in Fig. 7. It can be found that the MA modified Ni catalyst presents higher catalytic activity and CH4 selectivity compared to the traditional CA modified Ni catalyst. The optimal CO2 conversion of 25Ni/CA is 73.6% at 380 °C, whereas that of 25Ni/MA is 77.2% at 360 °C, revealing better low-temperature CO2 methanation activity by the modification of MA (Fig. 7a). According to the N2 adsorption–desorption analysis and XRD results mentioned above, the MA support prepared by partial hydrolysis method displays BET surface area of 237 m2 g−1 and average pore size of 3.1 nm. The commercial CA presents a lower BET surface area about 1.4 times (166 m2 g−1) and a higher pore size about 1.5 times (4.7 nm). Therefore, larger surface area and narrower pore size of MA facilitate the dispersion of active nickel species on the surface, promoting the catalytic activity at lower temperature.
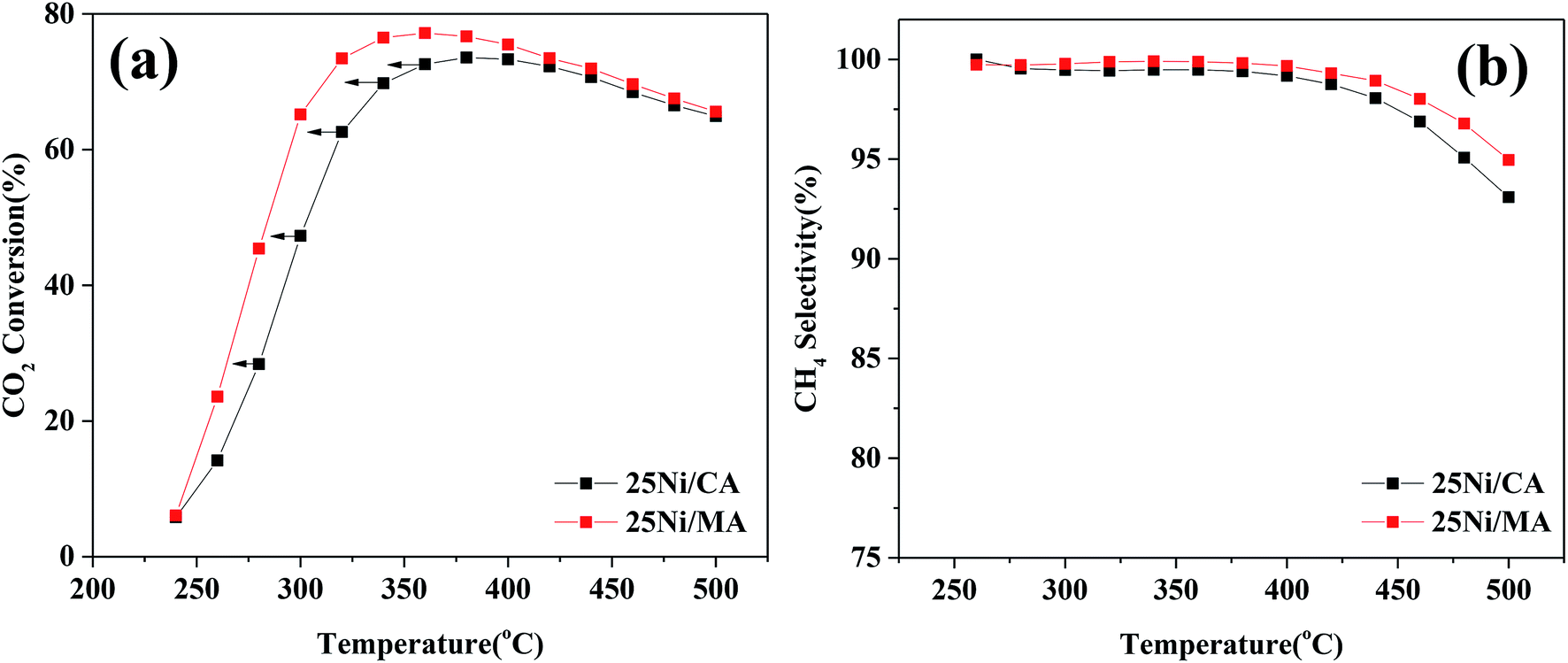 | ||
| Fig. 7 (a) CO2 conversion and (b) CH4 selectivity of 25Ni/CA and 25Ni/MA catalysts, GHSV = 6000 mL g−1 h−1, H2/CO2 = 4, 1 atm. | ||
In order to further analyze the effect of other Al2O3 support with different pore size on the CO2 methanation performance, the results of Rahmani et al.41 have been selected herein. Comparative data are listed in Table 1S (in the ESI†). From Table 1S† it can be found that the Al2O3 support is prepared by sol–gel method, which exhibits higher pore size of 8.1 nm compared to the MA support in the present study. However, the optimized CO2 conversion for 25Ni/Al2O3 reported by Rahmani et al.41 is 73% at 350 °C, which is lower than the value of 25Ni/MA in this study, suggesting that the MA support with narrower pore size by partial hydrolysis preparation method displays an outstanding CO2 methanation performance. In addition, some typical Ni-based catalysts for CO2 methanation are selected. From Table 2S† it is seen that the Ni/MA catalyst presents the highest CO2 conversion at a low temperature, further verifying excellent CO2 methanation performance of the MA support modified Ni catalyst.
The optimized 25Ni/MA catalyst is carried out for the stability test. As shown in Fig. 8, the 25Ni/MA catalyst presents 77% of catalytic activity with almost 100% of CH4 selectivity, which remains unchanged during the reaction for 100 h, indicating that modification by the MA support improves obviously the catalyst stability. Furthermore, higher temperature methanation (400 °C) of MA and CA supported Ni catalysts is tested (Fig. 1S†). From Fig. 1Sa† it can be seen that the Ni/MA catalyst displays the excellent stability in 400 °C, and there is no deactivation appeared during reaction. In contrast, the catalytic activity of the Ni/CA catalyst decreases gradually in the initial stage and then remained unchanged basically with time on stream. Especially, the CH4 selectivity begins to decrease after reaction for 27 h (Fig. 1Sb†). Besides, the reusability of 25Ni/MA at 400 °C is further tested (Fig. 2S†), and no deactivation of the catalyst is observed during reaction for 80 h. All of these results suggest that the CA supported Ni catalyst takes place easily the deactivation in the CO2 methanation reaction, while the MA support plays a critical role in improving the stability of the catalyst.
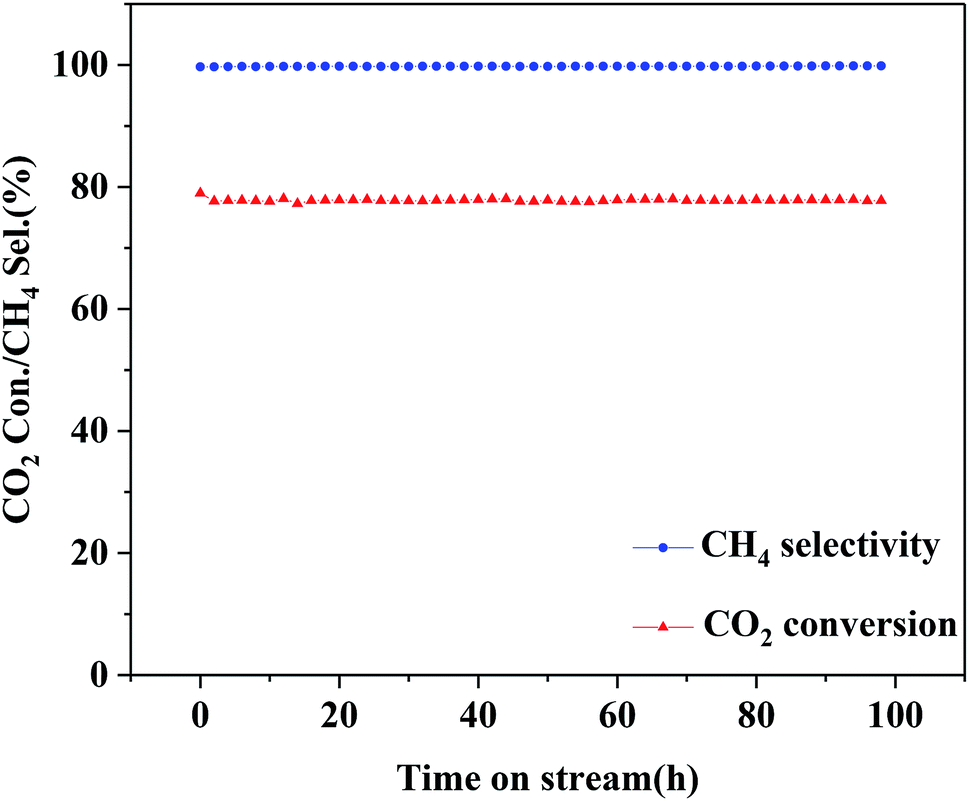 | ||
| Fig. 8 Stability test of the 25Ni/MA catalyst at 340 °C, GHSV = 6000 mL g−1 h−1, H2/CO2 = 4, 1 atm. | ||
The TEM results indicate that metallic Ni is formed on the surface of MA. No aggregation of Ni particles and no formation of carbon deposition are exhibited. It is possible that the MA support with cross-linked fabricate walls restrains the aggregation of metallic Ni particles, improving the catalyst stability. The micro-structures of the spent catalyst are further analyzed. The spent 25Ni/MA catalyst after reaction for 100 h is characterized by XRD. From Fig. 9a it is seen that phase composition of the spent 25Ni/MA is composed mainly of metallic Ni. The Ni particle size calculated using Scherrer equation is about 21.5 nm, which is close to the value in initial reaction, further validating the stability of Ni active nanoparticles during the CO2 methanation process.
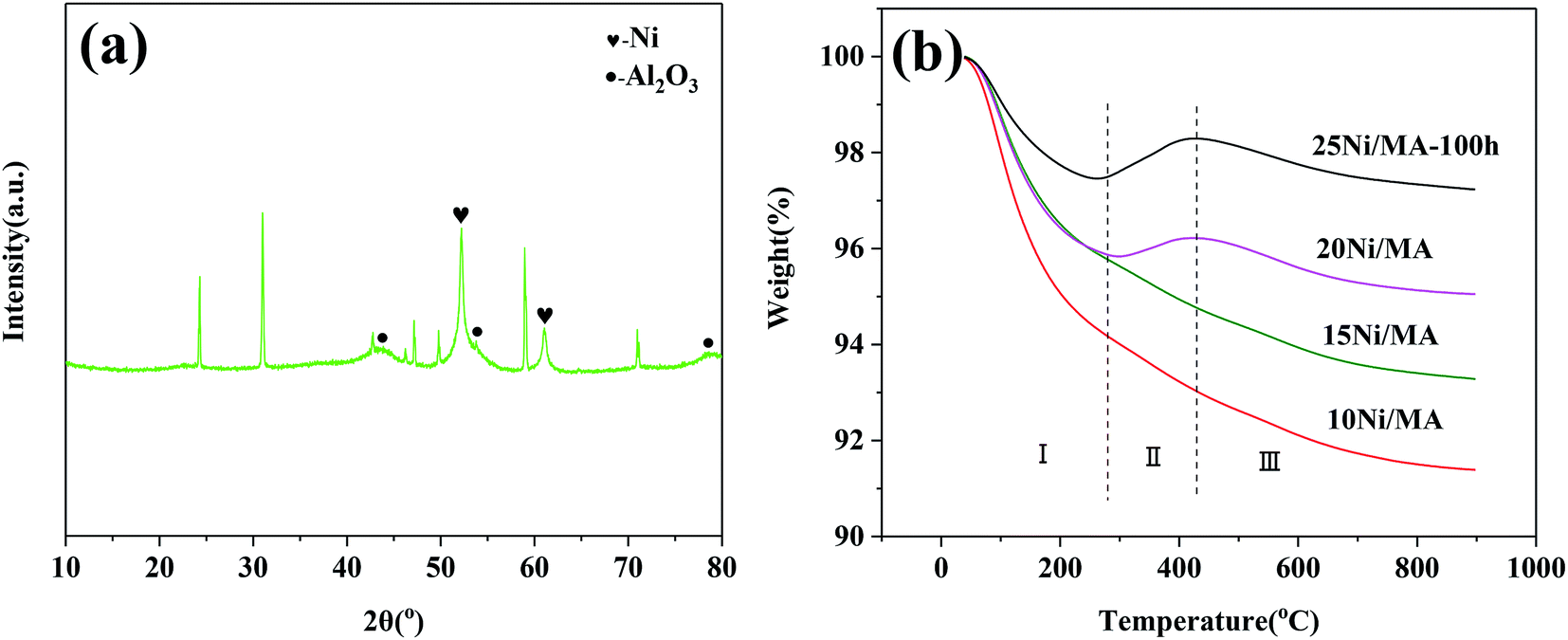 | ||
| Fig. 9 (a) XRD pattern of the 25Ni/MA catalyst after 100 h reaction and (b) TG curves of the spent Ni/MA catalysts. | ||
The behavior of carbon deposition over the Ni/MA catalysts is analyzed by TG. As shown in Fig. 9b, two weight loss steps can be seen for all of the spent catalysts. The first weight loss in zone I (below 290 °C) is attributed to thermal desorption of physical adsorbed H2O, and the second weight loss in zone III (above 420 °C) is related to the removal of graphite coke that led to the catalyst deactivation.42,43 Besides, there is a weight gain in zone II (290–420 °C) for the 20Ni/MA and 25Ni/MA catalysts, which is ascribed to the oxidation of reduced Ni species.44 It is possible that more metallic Ni sites formed over the spent Ni/MA catalyst with higher Ni loading facilitate the oxidation of metallic Ni. From Fig. 9b, it is seen that all catalysts display excellent carbon resistance with only 2.8% carbon deposition amount at most. The weight loss of the used 25Ni/MA catalyst (100 h) is only 1.1%, revealing excellent anti-coking ability. This is consistent with the TEM results mentioned above.
4 Conclusions
In summary, the Ni/MA catalysts with different Ni loadings were facilely prepared using partial hydrolysis using inorganic aluminum salt and employed for CO2 methanation. The MA modified Ni catalyst displayed higher catalytic activity and CH4 selectivity as well as excellent stability compared to the traditional Ni/CA catalyst. The characterization results indicated that the MA support was synthesized successfully by partial hydrolysis method, and displayed excellent hydrothermal stability. Modification of MA decreased the metal–support interaction, promoting the reduction of NiO species and formation of metallic Ni active sites over the Ni/MA catalysts. The monolithic skeleton of MA with fabric-like walls stabilized the nano-nickel particle sizes, which restrained the sintering of metallic Ni particles and carbon deposition on the catalyst, promoting obviously the catalyst stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from International cooperation and exchange program of the National Natural Science Foundation of China (51861145102), International Science and Technology cooperation project of Xinjiang Production and Construction Corps (2017BC008), 111 project (B12015) and Fundamental Research Fund for the Central Universities (2042017kf0173).References
- M. Streibel, N. Nakaten, T. Kempka and M. Kühn, Energy Procedia, 2013, 40, 202–211 CrossRef CAS.
- J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Fuel, 2010, 89, 1763–1783 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 28, 2975–2992 RSC.
- T. Abe, M. Tanizawa, K. Watanabe and A. Taguchi, Energy Environ. Sci., 2009, 2, 315–321 RSC.
- J. Liu, W. Bing, X. Xue, F. Wang, B. Wang, S. He, Y. Zhang and M. Wei, Catal. Sci. Technol., 2016, 6, 3976–3983 RSC.
- A. H. Zamani, R. Ali and W. A. W. A. Bakar, J. Taiwan Inst. Chem. Eng., 2014, 45, 143–152 CrossRef CAS.
- M. Younas, L. Loong Kong, M. J. K. Bashir, H. Nadeem, A. Shehzad and S. Sethupathi, Energy Fuels, 2016, 30, 8815–8831 CrossRef CAS.
- T. A. Le, M. S. Kim, S. H. Lee, T. W. Kim and E. D. Park, Catal. Today, 2017, 293–294, 89–96 CrossRef CAS.
- G. Zhou, H. Liu, K. Cui, H. Xie, Z. Jiao, G. Zhang, K. Xiong and X. Zheng, Int. J. Hydrogen Energy, 2017, 42, 16108–16117 CrossRef CAS.
- Y. Yan, Y. Dai, H. He, Y. Yu and Y. Yang, Appl. Catal., B, 2016, 196, 108–116 CrossRef CAS.
- I. Ganesh, Renewable Sustainable Energy Rev., 2014, 31, 221–257 CrossRef CAS.
- S. Rönsch, J. Schneider, S. Matthischke, M. Schlüter, M. Götz, J. Lefebvre, P. Prabhakaran and S. Bajohr, Fuel, 2016, 166, 276–296 CrossRef.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- J. Gao, Q. Liu, F. Gu, B. Liu, Z. Zhong and F. Su, RSC Adv., 2015, 5, 22759–22776 RSC.
- J. N. Park and E. W. McFarland, J. Catal., 2009, 266, 92–97 CrossRef CAS.
- Y. Feng, W. Yang and W. Chu, Integr. Ferroelectr., 2016, 172, 40–48 CrossRef CAS.
- G. Zhi, X. Guo, X. Guo, Y. Wang and G. Jin, Catal. Commun., 2011, 16, 56–59 CrossRef CAS.
- H. C. Wu, Y. C. Chang, J. H. Wu, J. H. Lin, I. K. Lin and C. S. Chen, Catal. Sci. Technol., 2015, 5, 4154–4163 RSC.
- F. Song, Q. Zhong, Y. Yu, M. Shi, Y. Wu, J. Hu and Y. Song, Int. J. Hydrogen Energy, 2017, 42, 4174–4183 CrossRef CAS.
- K. Zhao, W. Wang and Z. Li, J. CO2 Util., 2016, 16, 236–244 CrossRef CAS.
- G. Zhou, H. Liu, K. Cui, A. Jia, G. Hu, Z. Jiao, Y. Liu and X. Zhang, Appl. Surf. Sci., 2016, 383, 248–252 CrossRef CAS.
- S. Tada, T. Shimizu, H. Kameyama, T. Haneda and R. Kikuchi, Int. J. Hydrogen Energy, 2012, 37, 5527–5531 CrossRef CAS.
- S. Tada, D. Minori, F. Otsuka, R. Kikuchi, K. Osada, K. Akiyama and S. Satokawa, Fuel, 2014, 129, 219–224 CrossRef CAS.
- M. C. Bacariza, I. Graça, S. S. Bebiano, J. M. Lopes and C. Henriques, Chem. Eng. Sci., 2018, 175, 72–83 CrossRef CAS.
- R. Merkache, I. Fechete, M. Maamache, M. Bernard, P. Turek, K. Al-Dalama and F. Garin, Appl. Catal., A, 2015, 504, 672–681 CrossRef CAS.
- G. Du, S. Lim, Y. Yang, C. Wang, L. Pfefferle and G. L. Haller, J. Catal., 2007, 249, 370–379 CrossRef CAS.
- S. A. Hassanzadeh-Tabrizi and E. Taheri-Nassaj, Mater. Lett., 2009, 63, 2274–2276 CrossRef CAS.
- A. Khaleel and B. Dellinger, Environ. Sci. Technol., 2002, 36, 1620–1624 CrossRef CAS PubMed.
- L. Xu, F. Wang, M. Chen, H. Yang, D. Nie, L. Qi and X. Lian, RSC Adv., 2017, 7, 18199–18210 RSC.
- X. Shang, X. Wang, W. Nie, X. Guo, X. Zou, W. Ding and X. Lu, J. Mater. Chem., 2012, 22, 23806–23814 RSC.
- L. Xu, F. Wang, M. Chen, J. Zhang, K. Yuan, L. Wang, K. Wu, G. Xu and W. Chen, RSC Adv., 2016, 6, 28489–28499 RSC.
- A. Zhao, W. Ying, H. Zhang, H. Ma and D. Fang, Catal. Commun., 2012, 17, 34–38 CrossRef CAS.
- J. Zhang, H. Xu, X. Jin, Q. Ge and W. Li, Appl. Catal., A, 2005, 290, 87–96 CrossRef CAS.
- M. Cai, J. Wen, W. Chu, X. Cheng and Z. Li, J. Nat. Gas Chem., 2011, 20, 318–324 CrossRef CAS.
- S. Rahmani, M. Rezaei and F. Meshkani, J. Ind. Eng. Chem., 2014, 20, 4176–4182 CrossRef CAS.
- M. Lenglet, A. D'huysser, J. P. Bonelle, J. Dürr and C. K. Jørgensen, Chem. Phys. Lett., 1987, 136, 478–482 CrossRef CAS.
- N. S. Mcintyre and M. G. Cook, Anal. Chem., 1975, 47, 2208–2213 CrossRef CAS.
- J. Liu, C. Li, F. Wang, S. He, H. Chen, Y. Zhao, M. Wei, D. G. Evans and X. Duan, Catal. Sci. Technol., 2013, 3, 2627–2633 RSC.
- Y. Li, G. Lu and J. Ma, RSC Adv., 2014, 4, 17420–17428 RSC.
- L. Zhou, Q. Wang, L. Ma, J. Chen, J. Ma and Z. Zi, Catal. Lett., 2015, 145, 612–619 CrossRef CAS.
- S. Rahmani, M. Rezaei and F. Meshkani, J. Ind. Eng. Chem., 2014, 20, 1346–1352 CrossRef CAS.
- A. I. Tsyganok, T. Tsunoda, S. Hamakawa, K. Suzuki, K. Takehira and T. Hayakawa, J. Catal., 2003, 213, 191–203 CrossRef CAS.
- Z. Xu, M. Zhen, Y. Bi and K. Zhen, Appl. Catal., A, 2000, 198, 267–273 CrossRef CAS.
- D. Liu, R. Lau, A. Borgna and Y. Yang, Appl. Catal., A, 2009, 358, 110–118 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10348h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |