
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient synthesis of epoxybutane from butanediol via a two-step process†
Xin Niuab,
Liguo Wang *bcd,
Junya Caoa,
Yan Caob,
Peng Heb,
Junya Zhouab and
Huiquan Li*bc
*bcd,
Junya Caoa,
Yan Caob,
Peng Heb,
Junya Zhouab and
Huiquan Li*bc
aChina University of Mining &Technology, Beijing, Beijing, 100083, P. R. China
bCAS Key Laboratory of Green Process and Engineering, National Engineering Laboratory for Hydrometallurgical Cleaner Production Technology, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: lgwang@ipe.ac.cn; hqli@ipe.ac.cn
cSino-Danish College University, Chinese Academy of Sciences, Beijing, 100049, P. R. China
dDalian National Laboratory for Clean Energy, Dalian, 116023, China
First published on 29th March 2019
Abstract
A novel approach for the synthesis of epoxybutane via decarboxylation of butenyl carbonate derived from butanediol was developed for the first time. For the carbonylation of butanediol with dimethyl carbonate, NaAlO2 has exhibited excellent catalytic activity under mild reaction conditions. The yield of butenyl carbonate reached as high as 96.2%. NaAlO2 provides suitable acid–base active sites to promote the transesterification reaction of butanediol and dimethyl carbonate. For the following step of decarboxylation of butenyl carbonate, ionic liquid 1-butyl-3-methylimidazolium bromide could effectively catalyze the decarboxylation process both in batch or continuous processes. Moreover, the catalytic mechanism for the crucial step of decarboxylation of butenyl carbonate over 1-butyl-3-methylimidazolium bromide was explored using DFT calculations. The results showed that the electrostatic and hydrogen-bond effects of 1-butyl-3-methylimidazolium bromide played a crucial role for the generation of epoxybutane. Briefly, the Br anion of the ionic liquid attacks the methylene of the ring and the H of the ionic liquid cation attacks the carbonyl oxygen, which facilitated the five-ring opening and subsequent decarboxylation to form BO. This study not only provided a new and green synthetic route for producing epoxybutane, but also contributed to the effective utilization of butanediol, which is inevitably produced as by-product in the process of coal to ethylene glycol, suggesting a promising application in the clean manufacture of epoxybutane with inexpensive cost.
Introduction
Epoxybutane (BO) is an important organic chemical raw material and mainly used to synthesize polyether polyol,1 which is being used in the manufacture of value-added polyurethane.2 Until now, the monomer of polyether polyol is mainly chosen from ethylene oxide (EO), propylene oxide (PO) and BO.3,4 Compared with EO and PO, BO not only possess a similar activity that can react with hydrogen containing compounds, such as water, alcohol, phenols, sulfur alcohol, ammine, acids and so on, but also has unique properties that can improve the durability of diphenylmethane diisocyanate (MDI)-based polyurethane. However, owing to the restrictions from the severe safety issues in the production technology and the expensive cost, the market share of polyether polyol derived from BO is smaller than that made from EO and PO. At present, BO is being synthesized by chlorohydrin process, direct oxidation method and indirect oxidation method.5 These conventional manufacturing processes of BO suffer from several drawbacks, such as environmental pollution, harsh operation conditions, and high cost of the raw materials. In the chlorohydrin route, butylene is reacted with water and Cl2 to produce chlorobutanol, which is then reacted with base to generate BO. In the production of BO by chlorohydrin process, salt waste and large amount of wastewater will be produced. In addition, the production of large amounts of hydrochloric acid will corrode the equipment. In the indirect oxidation method, the styrene process is the main process. Ethyl benzene is first oxidized with O2, where the peroxy compound is used to generate BO from butylene. The residual aromatic alcohol is then dehydrated to styrene. This method is energy-intensive and can produce sizeable waste streams. The direct oxidation method is the oxidation of butene to BO by hydrogen peroxide.6 Its disadvantages are harsh operation conditions and high costs of the raw materials. In this context, a new and environmentally friendly route for the synthesis of BO from butanediol via butenyl carbonate (BC) intermediate was investigated in this paper. The new route includes the synthesis of BC from butanediol and DMC, followed by decarboxylation of BC to BO, as shown in Scheme 1. | ||
| Scheme 1 (1) Synthesis of BC from butanediol and DMC, (2) BC decarboxylation to BO. | ||
In the previous reports, the intermediate of BC is generally synthesized from BO and CO2, which have the disadvantages of relatively high pressure and high cost.7–12 The development of a new route for the effective synthesis of BC via an inexpensive route is highly desired. In terms of this reason, a new approach based on the transesterification of butanediol with dimethyl carbonate (DMC) was developed in our group, where the crucial raw material of butanediol could be readily obtained from the by-product, which is inevitably produced in the well-established process of coal to ethylene glycol process.13 Owing to the high activity of alcohol hydroxyl group, ethylene glycol can be further hydrogenated to ethanol in the presence of hydrogen. Then the ethylene glycol can reacts with ethanol to produce butanediol due to the Golbert reaction.14,15 Thus, inexpensive butanediol is produced in large quantities in the coal to ethylene glycol process.
The development of effective catalyst system is crucial for the synthesis of BC from butanediol and DMC. Sodium aluminate (NaAlO2) is generally used as a cheaply available and highly basic catalyst.16–22 As it is insoluble in many organic solvents, NaAlO2 can be used as a truly heterogeneous catalyst.23,24 Some researchers reported that NaAlO2 have been used as an active catalyst for a number of base-catalyzed reactions (transesterification, isomerization,25,26 and condensation). Therefore, owing to its unique features in basicity, NaAlO2 was also employed in the synthesis of BC via carbonylation of butanediol with DMC. In addition, the synthesis of BO from decarboxylation of BC is worthy of special attention in terms of economic and environmental points of view. Imidazolium-based ionic liquid was demonstrated to be capable of catalyzing the decarboxylation of glycerol carbonate.27–32 In this work, imidazolium-based ionic liquids were tested and applied as catalyst in the decarboxylation of BC to produce BO.
In this paper, a novel approach for the synthesis of epoxybutane (BO) via decarboxylation of butenyl carbonate (BC) derived from butanediol were successfully developed. In this process, NaAlO2 was used as catalyst in the transesterification of butanediol with dimethyl carbonate (DMC) to produce BC, and subsequently 1-butyl-3-methylimidazolium bromide ([Bmim][Br]) was used as catalyst in the decarboxylation of BC to produce BO. The influence of reaction parameters, e.g. reaction temperature, reaction time and catalyst amount on the catalytic performance were optimized. Furthermore, the crucial catalytic mechanism of decarboxylation of BC to BO is deeply explored using DFT calculations.
Experimental
Materials
Dimethyl carbonate (99%) and decane were purchased from Aladdin Industrial Corporation. Butanediol (98%), 1,2-epoxybutane and butylene carbonate (98%) were purchased from Tokyo Chemical Industry Co., Ltd. Sodium aluminate and zinc bromide were purchased from Sinopharm Chemical Reagent Co., Ltd. 1-Butyl-3-methylimidazolium bromide ([Bmim][Br]), 1-butyl-3-methylimidazolium chloride ([Bmim][Cl]), 1-butyl-3-methylimidazolium iodide ([Bmim][I]) and 1-butyl-3-methylimidazolium tetrafluoroborate ([Bmim][BF4]) were purchased from Shanghai Cheng Jie Chemical Co. LTD.Catalytic activity test
While for continuous process, BC was loaded to the 250 mL three-necked flask from the sample bottle which was put on the balance. Thus, the amount of the BC introduced into the reactor can be directly calculated. Then BC was continuous fed into the flask by vacuum aspiration, and the feeding rate of BC was controlled by the valve under the vacuum degree of 0.005 MPa. During the reaction, the product was continuously collected in the bottom of flask and weighted. As a result, the yield of BO can be calculated.6 The device for the continuous process is shown in Fig. S1.†
Product analysis
After the reaction, the reaction mixture was analyzed using a gas chromatograph (GC-2010) using a RTX-WAX (30 m × 0.25 mm × 0.25 μm) capillary column and a flame ionization detector (FID). The decane was used as an internal standard for the quantitative analysis of butanediol, BC and BO. BC conversion, BC yield, BC selectivity, BO yield and formation rate of the decarboxylation of butenyl carbonate were calculated using the following equations:



where Cbutanediol is the conversion of butanediol; YBC is the yield of BC; SBC is the selectivity of BC; Mrt0 is the initial mole amount of butanediol; Mrtn is the mole amount of butanediol; Mitn is the mole amount of BC produced; YBO is the yield of BO; MBO is the mole amount of BO produced; MBC is the initial mole amount of BC; FRBO is the formation rate of the decarboxylation of butenyl carbonate; Mcatalyst is the mole amount of catalyst; h is the reaction time of decarboxylation.
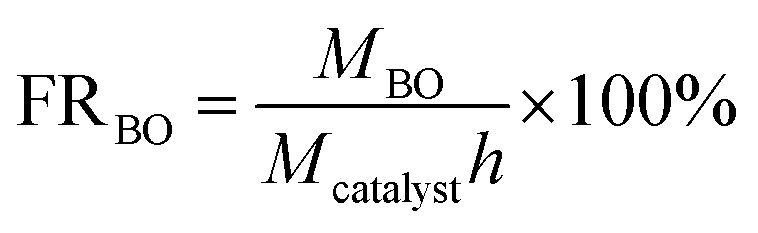
Characterization
XRD: phase structure of catalyst was determined with X-ray diffractometer using Cu Kα radiation. The working voltage and current were 40 kV and 40 mA, respectively. The 2θ range used was from 5° to 90°.Infrared (IR) analysis of the catalyst samples (diluted by KBr at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200) were characterized using a TENSOR-27 spectrometer (Bruker) with the scanning range is 400–4000 cm−1.
:200) were characterized using a TENSOR-27 spectrometer (Bruker) with the scanning range is 400–4000 cm−1.
1H NMR spectra were recorded on a Bruker Avance III 600 MHz spectrometer in CDCl3 with tetramethylsilane (TMS) as internal standard.
CO2-TPD was carried out to determine the basicity of samples using an AutoChem II 2920 equipped with a thermal conductivity detector (TCD). About 100 mg of sample was heated in situ in 25 mL min−1 of helium gas at 220 °C for 0.5 h in order to remove adsorbed impurities. After that, the sample was cooled to 50 °C and saturated with 25 mL min−1 of 10% vol% CO2/He for 1 h. Weakly adsorbed CO2 was eliminated by flushing with a 25 mL min−1 of helium gas at 50 °C for 0.5 h, and increased to 800 °C with a ramping rate of 10 °C min−1
N2 sorption measurement was performed using a Quantachrome Autosorb-1 at 77 K and the samples were degassed under vacuum at 573 K for 3 h before measurement. The specific surface areas were calculated by the Brunauer–Emmett–Teller (BET) method using N2 adsorption data ranging from P/P0 = 0.05 to 0.30.
Computational calculations
All the Density Functional Theory (DFT) calculations were performed using the Dmol3 program available in Materials studio 6.1 package. The generalized gradient approximation (GGA) with Perdew–Wang 1991 function was used.33,34 Geometry optimizations for minima were carried out using the standard Berny algorithm in redundant internal coordinates up to the neighborhood of the solution. If not converged, the optimization was further continued using analytical second derivatives. Optimizations for transition states were carried out with an initial guess for the transition state being generated from manual manipulation of the geometry using MOLDEN. Complete linear synchronous transit and quadratic synchronous transit (LST/QST) calculations were performed to obtain the structures of transition states (TS).35,36Results and discussion
Transesterification of butanediol with DMC to synthesize BC
The effects of reaction conditions, such as catalyst amount, reaction temperature, DMC/butanediol molar ratio, as well as reaction time on the transesterification performance were investigated and discussed below.
The effect of catalyst amount ranging from 0.1–5.0 wt% (based on the butanediol amount) on the reaction was investigated in Fig. 1. It is observed that, without catalyst, the reaction efficiency is so low that, the butanediol conversion is only 6.7%. However, the introduction of 0.1 mol% catalyst led to the rapid increase of butanediol conversion to 95.5%, then hardly changed when continue increasing the catalyst amount to 3.0 wt%. Thus, the 0.1 mol% catalyst was good enough for the transesterification of butanediol with DMC.
 | ||
| Fig. 1 Effect of catalyst amount on BC synthesis. (Reaction conditions: butanediol: 0.05 mol; n(DMC)/n(butanediol) = 4; temperature: 120 °C; reaction time: 60 min.) | ||
Fig. 2 showed the influence of temperature on the BC synthesis with NaAlO2 as catalyst. It can be observed that the BC is effectively synthesized with excellent selectivity within the temperature range investigated. Meanwhile, the butanediol conversion increased sharply from 52.2% to 94.8% at first with the temperature increased from 50 to 105 °C. When the temperature increased to higher temperatures, i.e. 120 °C and 150 °C, butanediol conversion gradually increased to 96.9%. The above results indicated that temperature had a great effect on butanediol conversion, and the conversion rate became very fast and nearly completely converted at 120 °C.
 | ||
| Fig. 2 Effect of temperature on BC synthesis. (Reaction conditions: butanediol: 0.05 mol; n(DMC)/n(butanediol) = 5; catalyst: 10 wt% of NaAlO2 based on butanediol; reaction time: 60 min.) | ||
The effect of molar ratio of DMC and butanediol was studied, as shown in Fig. 3. The conversion of butanediol increased from 91.5% to 96.6% with increasing the molar ratio from 2 to 4, and further increase the molar ratio did not show any change. Thus DMC/butanediol molar ratio of 4:1 was chosen as the prefered value, in which case butanediol was completely converted into the decarboxylation precursor of BC.
 | ||
| Fig. 3 Effect of molar ratio between DMC and butanediol on BC synthesis. (Reaction conditions: butanediol: 0.05 mol; catalyst: 10 wt% of NaAlO2 based on butanediol; temperature: 120 °C; reaction time: 60 min.) | ||
At last, the effect of reaction time was also studied, and the result is shown in Fig. 4. As can be seen from the figure, the reaction is very fast that the butanediol conversion reached to 78.9% only after 15 min, and then quickly reached to 93.8% and kept unchanged anymore at 30 min, which means 30 min is the equilibrium and optimum time for this reaction.
 | ||
| Fig. 4 Effect of reaction time on BC synthesis. (Reaction conditions: butanediol: 0.05 mol; n(DMC)/n(butanediol) = 4; catalyst: 10 wt% of NaAlO2 based on butanediol; temperature: 120 °C.) | ||
Recyclability of the catalysts
Additionally, the recyclability of the catalyst was also studied. After the reaction, the catalyst was separated by centrifugation, then washed by DMC and reused again. The result was shown in Fig. 5, it can be seen that, although BC selectivity was almost remained at 99.7%, the butanediol conversion increased from 92.2% to 96.8% when fresh and reused catalyst were applied, respectively. From this result, it can be deduced that the catalytic activity was further enhanced during the first run, and exhibiting higher catalytic activity in the subsequent recycling experiments. | ||
| Fig. 5 Reuse of catalyst NaAlO2. (Reaction conditions: butanediol: 0.05 mol; n(DMC)/n(butanediol) = 4; catalyst: 10 wt% of NaAlO2 based on butanediol; temperature: 120 °C; reaction time: 30 min.) | ||
To further explore the structure change of the catalyst during the reusability, the fresh and reused catalyst were investigated by XRD. As can be seen in Fig. S3,† the characteristic peaks of sodium aluminate at 2θ = 20.8, 30.2, 33.6, 35.0, 45.8, 48.4, 51.8, 58.1, 61.1, 63.0 (JCPDS 00-019-1179 and 01-083-0316) were clearly observed in both fresh and reused catalyst, and the peak strength is basically the same. These results suggested that crystalline structure and catalyst activity primarily remained during the recycle process, the results of which is consistent with the experimental data.
From the spectra in Fig. S4,† we can see that the peaks at 3300 to 3700 cm−1 and 1650 cm−1 are attributed to the bending mode of the –OH group of physically adsorbed water and the peaks at 1455 cm−1 corresponding to the carbonate after it was exposed to the air. The peaks at 810, 610, 560 and 490 cm−1 had no obvious changes after the sample was used for the reaction, indicating that the composition of NaAlO2 is stable under the reaction conditions used.
The basicity of the NaAlO2 (both before and after reaction) was evaluated by using CO2 temperature-programmed desorption (TPD) measurements as shown in Fig. S5.† The used NaAlO2 was separated by centrifugation and washed by ethanol. Then dried it at 100 °C for 4 h. To evaluate the amount of different types of basic sites, the desorption curve can be arbitrarily differentiated into two regions corresponding to weak and strong basic sites (below or above 400 °C, respectively). The desorption peaks at 256 °C for fresh NaAlO2 indicated basic sites of weak strength. The desorption peaks at 460 °C and 740 °C of used NaAlO2 can be attributed to the interaction of CO2 with strong basic sites because the higher temperature is needed to desorb the CO2 adsorbed on the more strongly basic sites.
The surface area is also tested using BET characterization. As shown in Table S1,† it can be observed that the BET surface area of the used NaAlO2 (13.6 m2 g−1) was higher than that of fresh NaAlO2 (1.7 m2 g−1). The enlarged surface area could be presumably attributed to the leaching of sodium element. Thus, more basic sites could be exposed.
Synthesis of BO from decarboxylation of BC
As aforementioned, imidazolium-based ionic liquids have been shown to be capable of catalyzing the decarboxylation of alkylene carbonates. In this work, [Bmim][Br] ionic liquid was tested and applied as the catalyst for the decarboxylation of BC. With using ionic liquids [Bmim][Br] as catalyst and ZnBr2 as promoter. The decarboxylation of BC can be effectively carried out, and the effect of reaction conditions, such as promoter/catalyst mass ratio, catalyst amount, were investigated and discussed below.Fig. 6 showed the influence of promoter/catalyst mass ratio on the BO synthesis with ionic liquids [Bmim][Br] as catalyst and ZnBr2 as promoter. It can be observed that the selectivity of BO reduced from 97.4% to 80.2% with increasing the mass ratio from 0 to 2. Therefore, the catalytic system without promoter is more superior choice in the selective synthesis of BO.
 | ||
| Fig. 6 Effect of promoter/catalyst mass ratio on BO synthesis. (Reaction conditions: BC: 40 g; catalyst: 0.83 g; reaction time: 90 min; vacuum degree: 0.005 MPa.) | ||
The effect of catalyst amount on the reaction was investigated, and the result is shown in Fig. 7. It is seen that the reaction could not occur without any catalyst. However, with the increase of ionic liquids [Bmim][Br] from 0.83 g to 2.00 g, the selectivity of BO slightly increased from 97.4% to 98.2%. After that, the selectivity of BO reduced with the further increase of catalyst amount.
 | ||
| Fig. 7 Effect of catalyst amount on BO synthesis. (Reaction conditions: BC: 40 g; reaction time: 90 min; vacuum degree: 0.005 MPa.) | ||
However, this result obtained is from the commercial BC rather than BC from the carbonylation of butanediol with DMC. Thus, in order to realize the two-step process, the synthesis of BO was furtherly carried out by using BC generated from butanediol. In this process, BC was firstly synthesized by the transesterification reaction of butanediol with DMC over NaAlO2 catalyst under the optimized reaction conditions. After the reaction, BC was separated from the product mixture and purified by vacuum distillation. The obtained BC was further used for the synthesis of BO over [Bmim][Br] catalyst. As a result, 94.0% of separated BO yield could be obtained. In this sense, a novel approach for BO synthesis is realized by a two-step process of carbonylation and decarboxylation method for the first time.
The decarboxylation of butenyl carbonate was undertaken in the presence of several [Bmim]-based ionic liquids with different anions (Cl−, I−, BF4−), and the effect of different IL on formation rate of the decarboxylation of butenyl carbonate was shown in Fig. S6.† It can be observed that IL with halide anions [Bmim][Cl], [Bmim][Br] and [Bmim][I] can catalyze the decarboxylation of BC. The FRBO values are 8.7, 3.9, 1.2, respectively, which are in the sequence of [Bmim][Cl] > [Bmim][Br] > [Bmim][I]. While the [Bmim][BF4] can not catalyze the decarboxylation of BC. The selectivity of BO is 96.4% when the catalyst is [Bmim][Cl], while the higher selectivity of 98.2% can be obtained when the catalyst is [Bmim][Br]. Thus, it is clear that anion of ILs have great impact on the decarboxylation of BC.
Mechanism of the decarboxylation
As it is shown in Fig. 7, the involvement of [Bmim][Br] catalyst resulted in the BO product, while the yield of BO is negligible when there is no introduction of any catalyst. Thus, the DFT calculations were performed to reveal the interaction between [Bmim][Br] and BC.As showed in Fig. 8, it was found that the carbonyl oxygen O1 of the BC was the main nucleophilic attack area and the carbon C1 of the BC were the main electropositivity attack area. The nitrogen atoms of the [Bmim][Br] were the main nucleophilic attack area. The Br− anions, which formed the hydrogen bonds of C–H⋯Br, was the main electropositivity attack area of [Bmim][Br]. Accordingly, six initial configurations between BC and [Bmim][Br] was assumed and shown in Fig. 9.
 | ||
| Fig. 8 Optimized structure of the BC and the [Bmim][Br]. | ||
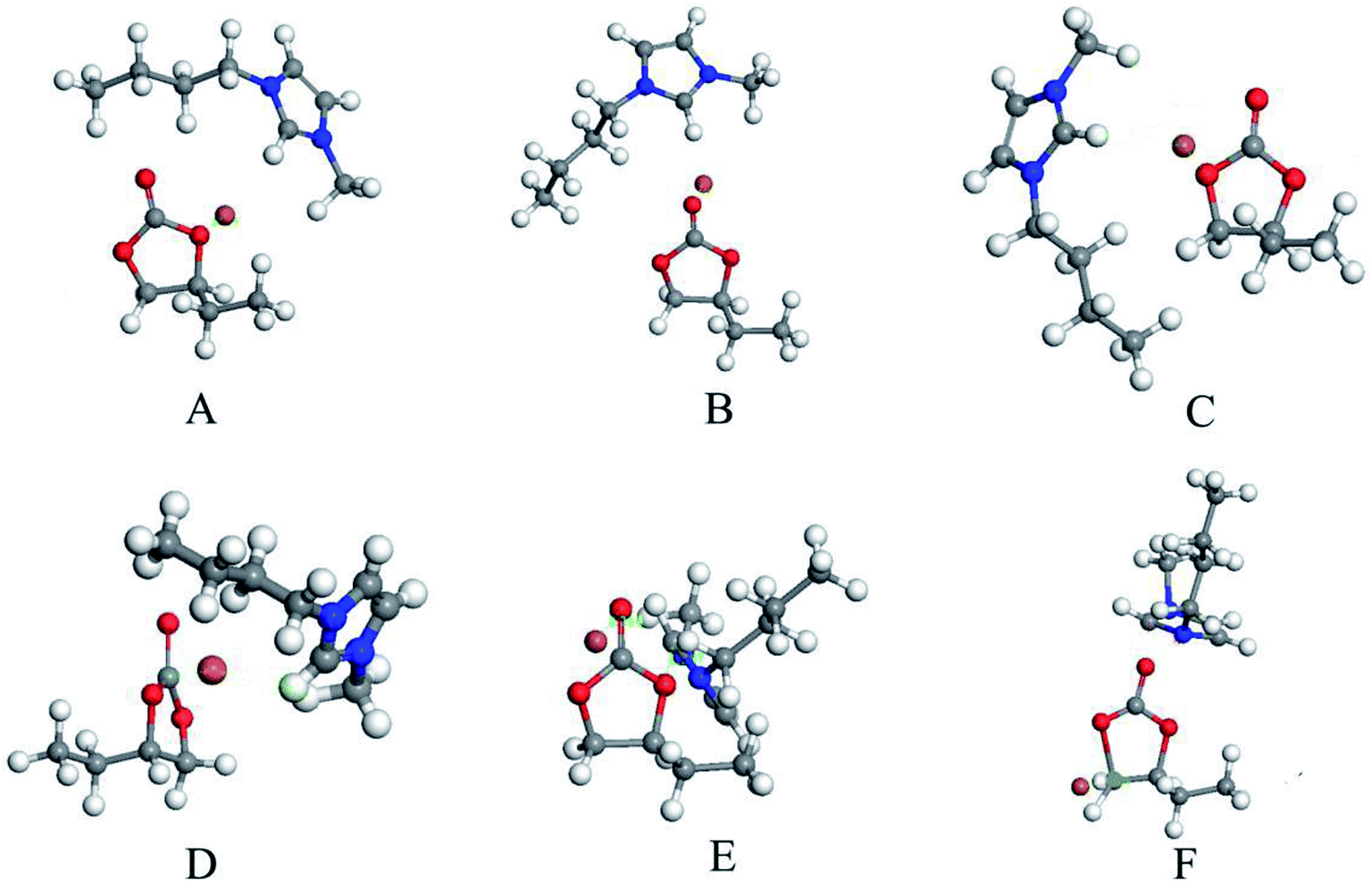 | ||
| Fig. 9 Six initial configurations in DFT calculation. | ||
According to the reports and DFT calculation results, a possible mechanism for decarboxylation was illustrated in Scheme 2.37,38 The optimized geometries for the intermediates and transition states were shown in Fig. S7†. The vibrational mode analysis shows that the reaction mechanism is reliable. When using vibrational mode analysis to elucidate the mechanism, we find that the only imaginary frequency of a transition state is always related to the breaking or forming bond. The potential energy surface profiles of the [Bmim][Br]-catalyzed process was shown in Fig. 10.
 | ||
| Scheme 2 The possible decarboxylation reaction mechanism. | ||
 | ||
| Fig. 10 Potential energy surface profiles of the [Bmim][Br]-catalyzed process and the optimized geometries for the intermediates and transition states. | ||
As shown in Scheme 2 and Fig. 10, first, the five-ring-opening step was the rate-limiting step with the highest energy barrier of 31.1 kcal mol−1. It was observed that the nucleophilic attack of Br anions on the least-hindered carbon C2 of BC played a very important role to break the covalent bond between the C2 and O2. Under the interaction with Br anions, BC was activated with the C2–O2 bond length enlarged from 1.45 Å and 2.74 Å. However, a small probability was observed that the C3 of BC was nucleophilically attacked by Br anions because of the steric effect. Besides, the hydrogen bond between H1 and the carbonyl oxygen (O1) made the ring-opening much easier. Thus, the five-ring-opening step made A convert into intermediate B via transition state TS1. Then, a new complex C was formed when CO2 was released into the reaction system via the TS2 with an energy barrier of 4.6 kcal mol−1. The third step is the proton transfer from O3⋯H to the nitrogen atom of the imidazolium ring along with the simultaneous loss of Br anion via transition state TS3 with an energy barrier of 11.4 kcal mol−1. Finally, the epoxy ring was obtained via an intramolecular nucleophilic attack, and the catalyst was regenerated (TS4) with an energy barrier of 29.1 kcal mol−1 (TS3). Besides, there was some negative effect of the hydrogen bonding between the ·C⋯H on the ring-closing event.
Conclusions
In conclusion, this paper firstly reported a novel approach for the synthesis of BO via decarboxylation of BC derived from butanediol. By using a cheap and easily available raw material NaAlO2 as catalyst, 99.7% BC selectivity and 96.2% BC yield can be obtained from the transesterification of butanediol with DMC, and the catalyst can be easily recovered and reused. Furthermore, BC can be effectively decarboxylated to BO when using [Bmim][Br] as catalyst. Finally, BO was obtained by a two-step carbonylation and decarboxylation method for the first time by using butanediol as raw material. This new process is very significant for the production of epoxy compounds and the utilization of butanediol by-product inevitably generated in the process of coal to ethylene glycol.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
“Transformational Technologies for Clean Energy and Demonstration”, Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDA 21030600; National Natural Science Foundation of China (No. 21576272 and 21476244) and Youth Innovation Promotion Association CAS (2016046).References
- Y. Zuo, M. Liu, M. T. Ma, C. S. Song and X. W. Guo, Ind. Eng. Chem. Res., 2017, 56, 7462–7467 CrossRef CAS.
- S. Sang, US Pat., US2018271191-A1, 2018.
- J. Hilf, P. Schulze, J. Seiwert and H. Frey, Macromol. Rapid Commun., 2014, 35, 198–203 CrossRef CAS PubMed.
- I. Kim, J. T. Ahn, S. H. Chang, C. S. Yang and I. Park, Polymer, 2003, 44, 3417–3428 CrossRef CAS.
- L. I. Hua, W. Wang, R. Lin and X. She, CN Patent, CN101085763, 2010.
- H. Fang, J. Ge, J. Li, Y. Xue and K. Zhang, CN Patent, CN104098531-B, 2016.
- M. Hong, Y. Kim, H. Kim, H. J. Cho, M. H. Baik and Y. Kim, J. Org. Chem., 2018, 83, 9370–9380 CrossRef CAS PubMed.
- V. Onyenkeadi, S. Kellici and B. Saha, Energy, 2018, 165, 867–876 CrossRef CAS.
- A. I. Adeleye, D. Patel, D. Niyogi and B. Saha, Ind. Eng. Chem. Res., 2014, 53, 18647–18657 CrossRef CAS.
- M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651–2684 RSC.
- W. L. Dai, W. Y. Yang, Y. Zhang, D. Wang, X. B. Luo and X. M. Tu, J. CO2 Util., 2017, 17, 256–262 CrossRef CAS.
- M. Ulusoy, E. Cetinkaya and B. Cetinkaya, Appl. Organomet. Chem., 2009, 23, 68–74 CrossRef CAS.
- R. H. Shi, W. W. Dong, Z. Han, T. Zhang and L. H. Zhang, Yingyong Huagong, 2013, 42, 2056–2060 CAS.
- H. Li, W. Huang, X. Li and X. Gao, Ind. Eng. Chem. Res., 2016, 55, 9994–10003 CrossRef CAS.
- A. Yin, X. Guo, W.-L. Dai, H. Li and K. Fan, Appl. Catal., A, 2008, 349, 91–99 CrossRef CAS.
- S. Ramesh, F. Devred, V. D. B. Ludivine and D. P. Debecker, ChemCatChem, 2017, 10, 1398–1405 CrossRef.
- Y. T. Algoufi, G. Kabir and B. H. Hameed, J. Taiwan Inst. Chem. Eng., 2016, 70, 179–187 CrossRef.
- S. Wang, P. F. Hao, S. X. Li, A. L. Zhang, Y. Y. Guan and L. N. Zhang, Appl. Catal., A, 2017, 542, 174–181 CrossRef CAS.
- Y. K. Endah, S. K. Min, J. Choi, J. Jae, D. L. Sang and H. Lee, Catal. Today, 2016, 293, 136–141 Search PubMed.
- P. Zhang, L. Liu, M. Fan, Y. Dong and P. Jiang, RSC Adv., 2016, 6, 76223–76230 RSC.
- V. P. Indran, N. A. S. Zuhaimi, M. A. Deraman, G. P. Maniam, M. M. Yusoff, T. Y. Y. Hin and M. H. A. Rahim, RSC Adv., 2014, 4, 25257–25267 RSC.
- A. Axelsson, A. Antoine-Michard and H. Sunden, Green Chem., 2017, 19, 2477–2481 RSC.
- T. Wan, P. Yu, S. Wang and Y. Luo, Energy Fuels, 2009, 23, 1089–1092 CrossRef CAS.
- R. Bai, P. Liu, J. Yang, C. Liu and Y. Gu, ACS Sustainable Chem. Eng., 2015, 3, 1292–1297 CrossRef CAS.
- M. M. Wang, M. A. A. Gasmalla, H. A. Tessema, X. Hua and R. J. Yang, Food Chem., 2017, 233, 151–158 CrossRef CAS PubMed.
- A. A. Marianou, C. M. Michailof, A. Pineda, E. F. Iliopoulou, K. S. Triantafyllidis and A. A. Lappas, ChemCatChem, 2016, 8, 1100–1110 CrossRef CAS.
- J. Kadokawa, Y. Iwasaki and H. Tagaya, Macromol. Rapid Commun., 2002, 23, 758–760 CrossRef.
- R. Abdul-Karim, A. Hameed and M. I. Malik, RSC Adv., 2017, 7, 11786–11795 RSC.
- J. S. Choi, F. S. H. Simanjuntak, J. Y. Oh, K. I. Lee, S. D. Lee, M. Cheong, H. S. Kim and H. Lee, J. Catal., 2013, 297, 248–255 CrossRef CAS.
- S. M. Gade, M. K. Munshi, B. M. Chherawalla, V. H. Rane and A. A. Kelkar, Catal. Commun., 2012, 27, 184–188 CrossRef CAS.
- D. J. Darensbourg and A. D. Yeung, Green Chem., 2014, 16, 247–252 RSC.
- X. L. N. Su, W. W. Lin, H. Y. Cheng, C. Zhang, Y. Wang, X. J. Yu, Z. J. Wu and F. Y. Zhao, Green Chem., 2017, 19, 1775–1781 RSC.
- L. Zhao, P. He, L. Wang, M. Ammar, Y. Cao and H. Li, Catal. Today, 2016, 281, 392–401 CrossRef.
- L. Wang, M. Ammar, P. He, Y. Li, Y. Cao, F. Li, X. Han and H. Li, Catal. Today, 2016, 281, 360–370 CrossRef.
- S. Fischer and M. Karplus, Chem. Phys. Lett., 1992, 194, 252–261 CrossRef CAS.
- Y. Gao, W. Peng, Z. Ning, W. Wei and Y. Sun, J. Mol. Catal. A: Chem., 2011, 351, 29–40 CrossRef CAS.
- X. Meng, H. Y. He, Y. Nie, X. Zhang, S. J. Zhang and J. Wang, ACS Sustainable Chem. Eng., 2017, 5, 3081–3086 CrossRef CAS.
- L. Wang, H. Li, S. Xin, P. He, Y. Cao, F. Li and X. Hou, Appl. Catal., A, 2014, 471, 19–27 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01220f |
| This journal is © The Royal Society of Chemistry 2019 |