
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNIR-to-visible upconversion in quantum dots via a ligand induced charge transfer state†
Noga Meir a,
Iddo Pinkasb and
Dan Oron*a
a,
Iddo Pinkasb and
Dan Oron*a
aDepartment of Physics of Complex Systems, Weizmann Institute of Science, Rehovot 7610001, Israel. E-mail: dan.oron@weizmann.ac.il
bDepartment of Chemical Research Support, Weizmann Institute of Science, Rehovot 7610001, Israel
First published on 17th April 2019
Abstract
Nanomaterials that possess the ability to upconvert two low-energy photons into a single high-energy photon are of great potential to be useful in a variety of applications. Recent attempts to realize upconversion (UC) in semiconducting quantum dot (QD) systems focused mainly on fabrication of heterostructured colloidal double QDs, or by using colloidal QDs as sensitizers for triplet–triplet annihilation in organic molecules. Here we propose a simplified approach, in which colloidal QDs are coupled to organic thiol ligands and UC is achieved via a charge-transfer state at the molecule–dot interface. We synthesized core/shell CdSe/CdS QDs and replaced their native ligands with thiophenol molecules. The alignment of the molecular HOMO with respect to the QD conduction band resulted in the formation of a new charge-transfer transition from which UC can be promoted. We performed a series of pump–probe experiments and proved the non-linear emission exhibited by these QDs is the result of UC by sequential photon absorption, and further characterized the QD–ligand energy landscape by transient absorption. Finally, we demonstrate that this scheme can also be applied in a QD solid.
Introduction
Photon upconversion (UC) is a non-linear process in which two or more low-energy photons are converted to one high-energy photon. In the context of nanomaterials, realization of UC holds great promise for the use of these nanoparticles in a variety of applications. One example for that is the potential to overcome the Shockley–Queisser limit in photovoltaic devices,1,2 by upconverting low-energy photons in the near-infrared (NIR) range of the solar spectrum, whose energies are below the band gap of the absorber material. Upconverting nanoparticles can also be beneficial for bioimaging, as tags for two-photon excitation fluorescence, which enables the use of NIR radiation for excitation, thus allowing deeper penetration to scattering biological samples and providing a higher signal-to-noise ratio and reduced autofluorescence.3 In contrast to conventional two-photon dyes, UC potentially has an additional advantage since it requires a lower photon fluence, consequently reducing the damage caused to the specimen by the excitation beam. UC nanoparticles have also recently been suggested for controlled optogenetic stimulation deep inside scattering tissue.4,5Unlike other non-linear phenomena such as two-photon absorption or sum-frequency generation (SFG), UC is an incoherent process whose mechanism is based on the sequential absorption of photons by long-lived, metastable states. As such, the requirements for efficient UC include a ladder-like arrangement of the energy states, a mechanism which inhibits cooling of hot charge carriers and relatively long-lived excited states (from which UC can be promoted).6 UC by sequential photon absorption in nanocrystals can take place via several mechanisms, such as excited-state absorption (ESA), energy-transfer UC (ETU) and photon avalanche (PA). In all three mechanisms the absorption leads to population of highly excited states, however they differ by the number of fluorophores involved in the process.7,8 Another mechanism, more typical to the use of organic molecules, is triplet–triplet annihilation (TTA),2 in which the energy from an excited triplet state of a sensitizer molecule is transferred to an excited triplet state of an emitter (also known as activator) molecule, followed by interaction between two emitter molecules, resulting in one emitter in the ground state and another in the excited singlet state (from which UC emission occurs). UC systems based on this mechanism can exhibit relatively high UC efficiencies, however it has several drawbacks such as photobleaching and limited availability of NIR fluorophores due to the use of organic molecules.
Although the majority of attempts to realize UC in nanomaterials were based on the use of host materials doped with rare-earth metals or transition metal ions,8–10 in recent years a different approach was implemented, employing semiconducting colloidal QD heterostructures (CQDs). The advantage of using CQDs for UC lies in their higher color tunability and absorption cross section. UC is facilitated by recent developments in the field of QDs synthesis, allowing the fabrication of a great variety of QDs in different compositions and architectures. The ability to achieve UC in such systems depends mostly on rational design of the energy landscape in the quantum dot heterostructure, allowing coupling of two quantum emitters, as well as confinement of at least one of the charge carriers (electron or hole) in different areas of the crystal. This can be accomplished by careful synthesis of the QD heterostructure in specific architectures, such that the two quantum emitters are spatially separated by a potential barrier. This was previously demonstrated for UC both in the vis-to-vis11,12 and the NIR-to-vis13,14 regime. In both of these realizations, a low band-gap dot is first excited, followed by further excitation of the hole either by intraband absorption or by an Auger mechanism. The excited hole can relax into the higher band gap dot, which then recombines with the spatially delocalized electron. Other recently introduced methods for achieving UC by the use of colloidal QDs are to couple them to organic molecules exhibiting TTA, where the QDs function as sensitizers for UC by TTA,15–17 or relying on hot carrier injection from a plasmonic nanoparticle.18 Yet, the range of accessible emission energies in the QD–TTA scheme is still limited by the energy gap (S0 → T1) of the emitter molecule, which typically does not go below ∼1.1 eV. Alternatively, NIR-to-vis UC was achieved by cooperative sensitization of organic molecules by PbS QDs,19 leading to UC efficiency which significantly exceeds previous reports. Notably, the latter requires coupling between neighboring QDs and was thus only realized in a QD film architecture.
In this work, we suggest a different approach and demonstrate how UC can be realized in a coupled QD–ligand system, in which the upconverted luminescence is emitted from the QD. This particular system is composed of a II–VI core/shell QD and thiol ligands attached to the surface of the crystal. The attachment of these specific ligands gives rise to a new electronic transition, occurring at the interface between the molecule and the QD surface. Thus, the first transition in this UC scheme involves excitation of a charge-transfer state between the HOMO of the ligand and the conduction band of the QD. From a synthetic standpoint, this ligand-to-dot UC system is much easier to fabricate than previous dot-to-dot UC design, since it requires fewer synthetic steps and has an overall higher synthetic yield. Moreover, relying on a charge-transfer transition, this poses no requirements on the HOMO–LUMO gap of the organic molecule. We use a pump–probe experimental setup to discern the nature of the non-linear emission exhibited by these QDs and prove it is the result of UC by sequential photon absorption. Finally, we observed this type of upconversion both in the case of isolated QDs dispersed in an organic solvent, and in a close-packed array of the QDs, which was deposited on a solid substrate.
Experimental
Synthesis
Ligand exchange to TP
CdSe/CdS core/shell dots were purified by centrifugation with acetone and methanol and re-dispersed in toluene. 2 ml of 1 μM of purified QDs solution was mixed with 20 μl of 0.1 M TP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene solution and stirred for 10 min.
:toluene solution and stirred for 10 min.
QD film self-assembly and ligand exchange to 1,4-BDT
QD film preparation was performed in a Teflon well, based on a known procedure with some modifications and additions.22–24 A solution of purified QDs in toluene was gently dropped on the surface of diethylene glycol, and the solvent was allowed to evaporate for ∼2 h. Then, the film was separated from the DEG surface by slow injection of acetonitrile. The film was collected by removal of the DEG using a syringe pump and transferred to an ITO-coated glass substrate that was submerged in the DEG at a 30° angle. The substrate was kept under vacuum overnight to remove residual acetonitrile. For ligand-exchanged film preparation, 0.03 M solution of 1,4-BDT in acetonitrile was used instead of clean acetonitrile.25 An additional step of ligand exchange was performed after 1 day, in which the films were dipped in 0.1 M solution of 1,4-BDT in acetonitrile for 10 min and then washed with clean acetonitrile.EM and optical characterization
TEM images were taken at 120 kV, using a Philips CM-120 transmission electron microscope. HRTEM images were taken at 200 kV, using a Themis-Z ultra high-resolution, double aberration-corrected transmission electron microscope. SEM images were taken at 2 kV by an in-lens SE detector, using a Zeiss Ultra 55 scanning electron microscope. UV-vis absorption spectra were measured using a UV-vis/NIR spectrophotometer (V-670, JASCO). Absolute O.D. was measured using an absolute quantum yield characterization system (Hamamatsu Quantaurus QY).Time-resolved fluorescence spectroscopy and pump–probe experiment
Excitation pulses were generated by a frequency tripled Nd:YAG Q-switched laser oscillator pumping an optical parametric oscillator (OPO) (NT342/C/3/UVE, EKSPLA) with pulse durations of ∼5 ns at a repetition rate of 10 Hz. Excitation pulses at 480–680 nm were obtained from the OPO and focused into a 10 × 10 mm rectangular quartz cuvette (Starna Cells), containing a solution of a low concentration of QDs dispersed in toluene, and stirred with a magnetic stirrer. For the pump–probe experiment, the 1064 nm probe pulse was transferred through a delay line and combined with the pump pulse by a dichroic mirror. Fluorescence signals were collected at a right-angle by a 0.4 NA objective. After spectral filtering by a dielectric filter, the signal was further passed through a monochromator (SpectraPro 2150i, Acton) for complete filtering of the laser pulses from the fluorescence. Time-resolved signals were measured by a photo-multiplier tube (R10699, Hamamatsu Photonics, Hamamatsu City, Japan). Data was collected by a 600 MHz oscilloscope (WaveSurfer 62Xs, Teledyne LeCroy). Pulse energies were measured by a pyroelectric sensor (PE9-C, Ophir Optronics).Transient absorption
Transient absorption (TA) of the core/shell QDs before and after LE was measured in toluene. The excitation (pump) pulse was obtained from the amplified 120 fs Ti:sapphire system (Spitfire ACE) using a computer controlled OPA (TOPAS) at either 550 nm or 670 nm, operating at 1 kHz and chopped at 500 Hz. The pump was followed at different delay times by a continuum white-light pulse, generated by focusing a part of the 800 nm fundamental beam onto a 6 mm thick CaF2 plate, creating a supercontinuum. The CaF2 plate was translated continuously in a circular motion to avoid thermal damage, which would affect the CaF2 white-light spectrum. For each time delay, the signal was accumulated over 5000 pulses for optimal signal-to-noise ratio (SNR).
Results and discussion
For our QD–ligand system we used CdSe/CdS core/shell QDs and thiophenol (TP) ligands, connected to the surface of the CdS shell through the S atom (Fig. 1a). CdSe QDs with a 2.8 nm diameter were synthesized by hot-injection method and later coated with CdS shell by slow continuous injection of Cd and S precursors at an elevated temperature. The final average diameter of the QDs was 7.4 nm. Following the synthesis, the original ligands attached to the QDs surface (mainly oleic acid) were replaced with TP. Further description of the QDs synthesis and the ligand exchange (LE) process can be found in the experimental section. Due to the location of the TP HOMO level relative to the CdS band gap, the exchange of the QDs native ligands with TP resulted in the formation of a trap state located above the valence band of the CdS shell. Upon the formation of an exciton in the charge-transfer transition between the molecule HOMO and the CdS, the electron tends to be localized in the QD core (see Fig. S1†), while the hole is trapped in the ligand HOMO. This trapped hole can be further excited by additional absorption of a low-energy photon, which can lead to its recombination with the electron in the CdSe core, and emission of an upconverted photon. A schematic description of the UC process in this system is shown in Fig. 1c.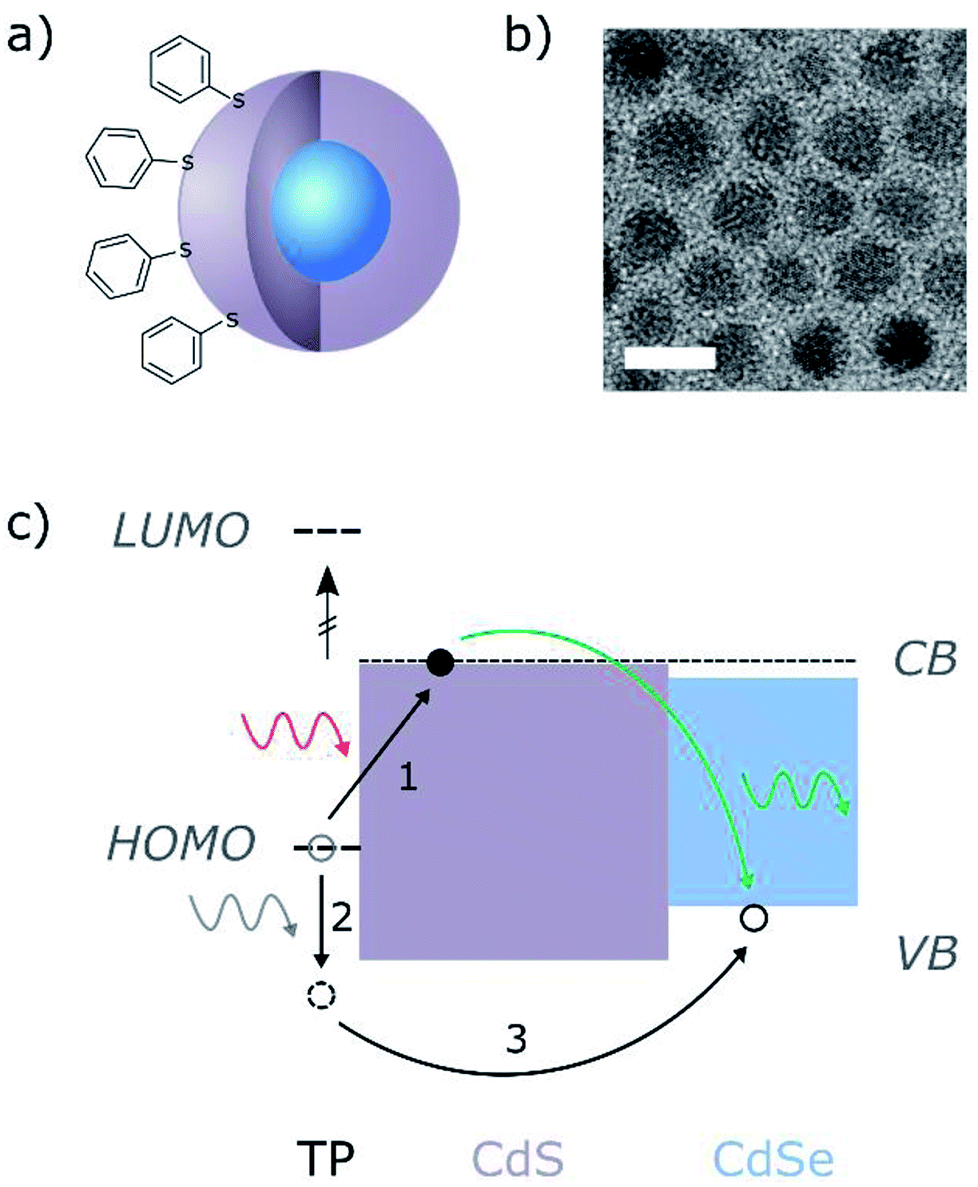 | ||
| Fig. 1 (a) Schematic illustration of the CdSe/CdS core/shell structure of the QDs and the TP ligands connected to the surface of the particle. (b) TEM image of the as-synthesized core/shell QDs. Scale bar is 10 nm. (c) Schematic illustration of the QD–ligand band alignment and the process of upconversion by sequential photon absorption. The electron is excited to the CdS band edge and relaxes such that it is mostly localized in the CdSe core (blue), but is also slightly extended into the CdS shell (purple). After the absorption of a second photon, the hole trapped in the TP HOMO is further excited and relaxes to the CdSe, where it recombines with the electron and emits an upconverted photon. The TP HOMO and LUMO levels are labeled with short dashed lines. The TP LUMO is well above all other energy levels (HOMO–LUMO gap is ∼4.4 eV (ref. 26)) and is not involved in the UC process. The TP HOMO level is ∼0.4 eV above the VB of the CdSe. The long dashed line represents the calculated 1S(e) level of the CdSe/CdS QD (see Fig. S1†). | ||
We first examine the effect of the LE on the linear optical properties of the QDs. As can be seen in Fig. 2a, the absorption spectrum of the QDs before and after the LE is mostly unaltered. Close examination of the lowest-energy absorption band by absolute O.D. measurements reveals a very small red-shift of about 3 nm following the ligand exchange (inset of Fig. 2a), while the spectral shape of the absorption band is preserved. This can be attributed to slight exciton delocalization caused by the thiophenol ligand shell.27,28 No additional features were observed in the absorption spectrum of the ligand-exchanged dots compared to the as-synthesized ones.
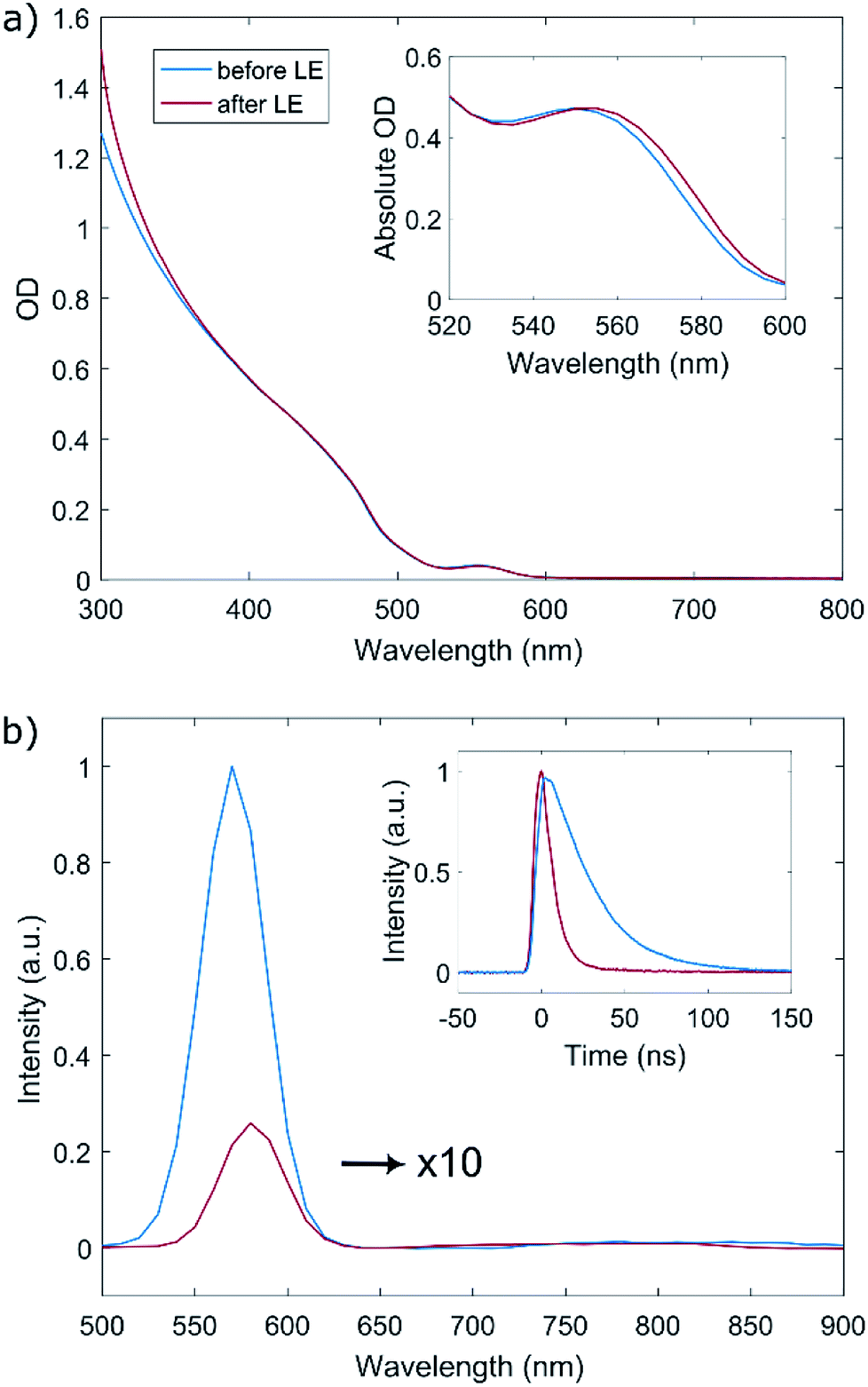 | ||
| Fig. 2 (a) Absorption spectra of the core/shell QDs, before and after the ligand exchange to thiophenol. The inset depicts the optical density around the first exciton absorption peak, showing a ∼3 nm red-shift following the ligand exchange. (b) Emission spectra of the core/shell QDs, before and after ligand exchange. The emission spectrum of the post-LE dots (red line) was scaled up by a factor of 10 for clarity. The inset shows time-resolved fluorescence signal of the QD taken at 570 nm (PL peak), before and after LE. | ||
Fig. 2b shows the photoluminescence spectrum of the QDs before and after ligand exchange (blue and red lines, respectively) and time-dependent PL traces measured at the peak of the emission (Fig. 2b inset). The PL spectrum and lifetime measurements were performed by exciting a dilute sample of the QDs with a 5 ns pulsed laser at 480 nm, filtering the time-resolved fluorescence signal through a monochromator and collecting it with a photomultiplier tube. Following the attachment of TP ligands, the band-edge emission of the QDs is considerably quenched to ∼3% of its original value and is red-shifted by ∼10 nm. This finding supports the claim regarding the location of the TP HOMO level, causing the TP molecule to be an efficient hole trap when attached to this type of QDs, consistent with the work of Meijerink and coworkers.29 Additionally, we notice the lifetime measured at the peak of the PL (570 nm), which is comprised of both the radiative and non-radiative decay routes of the excited state, is significantly shortened, indicating as well quenching of the radiative recombination pathway in the QD band gap. The measured lifetime is fit to a biexponential decay 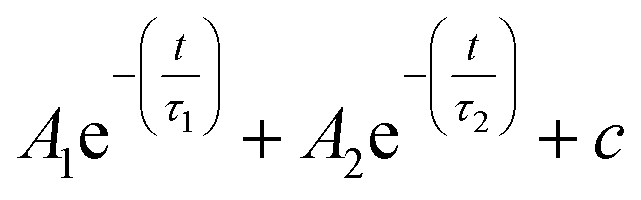 , convolved with the instrument response function (∼8 ns, see Fig. S3†). In both as-synthesized and ligand-exchanged dots the PL trace exhibited a very fast component (well below 5 ns) which corresponds to a fast decay to non-radiative trap states, and a slower component dominated by radiative recombination in the CdSe core. This longer lifetime component decreased from 30 ns to 16 ns as a result of the ligand exchange, and the amplitude ratio between the non-radiative and radiative lifetime was decreased by a factor of 20, in agreement with the decrease of the PL intensity.
, convolved with the instrument response function (∼8 ns, see Fig. S3†). In both as-synthesized and ligand-exchanged dots the PL trace exhibited a very fast component (well below 5 ns) which corresponds to a fast decay to non-radiative trap states, and a slower component dominated by radiative recombination in the CdSe core. This longer lifetime component decreased from 30 ns to 16 ns as a result of the ligand exchange, and the amplitude ratio between the non-radiative and radiative lifetime was decreased by a factor of 20, in agreement with the decrease of the PL intensity.
We then turned to examine the non-linear photophysical properties of our QD–TP system. Excitation with a 680 nm beam, which is well below the energy of the QD first exciton absorption band leads to a non-linear photoluminescence signal whose spectral shape is identical to that of the linear PL spectrum of the QDs (see Fig. S4 in the ESI†). However, this could also be the result of simultaneous two-photon absorption in the CdSe core, and indeed we observed a similar PL spectral shape for the original QDs prior to ligand exchange. Thus, in order to gain more compelling evidence for the occurrence of UC, and to differentiate between this process and two-photon absorption, we performed a series of PL pump–probe experiments. The setup for the pump–probe measurements is schematically presented in Fig. 3a. After the initial excitation of the QDs solution by a 5 ns pulse ranging from 650 nm to 680 nm, the sample is excited again by a second pulse at a longer wavelength of 1064 nm which is delayed by approximately 15 ns. This delay was chosen in order to prevent overlap of the two pulses.
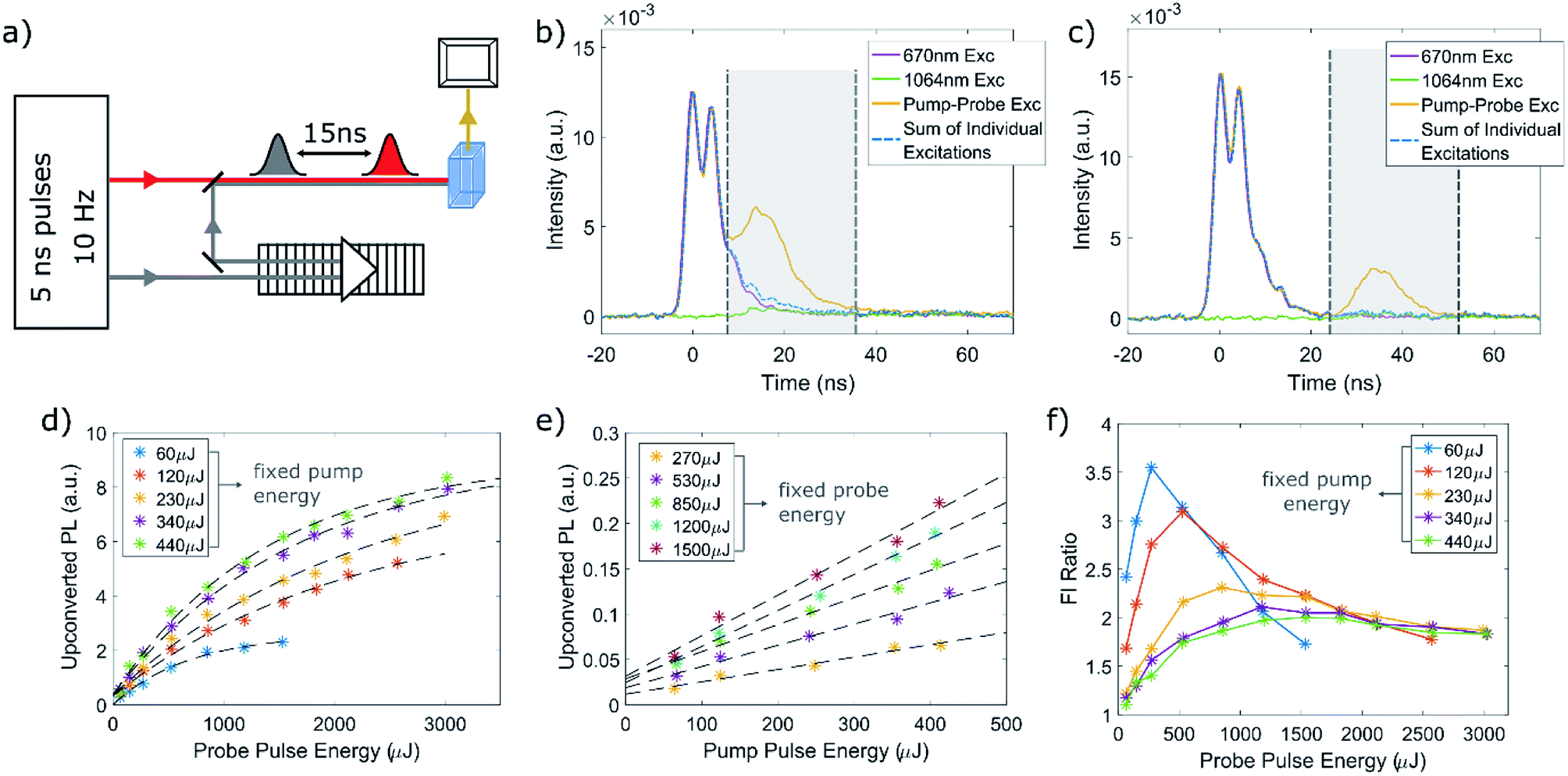 | ||
| Fig. 3 (a) Schematic illustration of the optical setup used for the pump–probe measurements. (b) Typical result of a pump–probe measurement of the QD–TP system, excited by a 670 nm pump with a pulse energy of 100 μJ and a 1064 nm probe at 600 μJ, delayed by 15 ns. The PL traces depicted are detected at 580 nm, and correspond to excitation of the QDs with either the pump (purple line), the probe (green line), or the pump and delayed probe (yellow line). The ratio between the fluorescence signal of the combined excitation (pump + probe) and the sum of the individual excitations (labeled with a blue dashed line) for this measurement was ∼3. (c) Typical result of a pump–probe measurement performed on the same sample presented in (b), with a 32 ns delay. The sample was excited with a 100 μJ pump. The probe intensity was adjusted such that the energy density at the sample will be the same for both delays. (d) Dependence of the upconverted PL on the probe pulse energy, for fixed pump intensities. The data was fit to a linear saturation assuming Poissonian distribution for the excitation probability (black dashed lines). (e) Dependence of the upconverted PL on the pump pulse energy, for fixed probe intensities. Black dashed lines depict linear fits of the data. (f) Dependence of the Fl ratio with the probe pulse energy, for fixed pump excitation intensity, summarizing all the pump–probe measurements performed on this sample with 670 nm pump and 1064 nm probe at increasing intensities. All the results presented in (d)–(f) relate to the measurements taken with a 15 ns delay. | ||
A typical pump–probe experiment performed on the QD–TP system, in which the QDs are excited using a 670 nm beam is presented in Fig. 3b. The photoluminescence transient from the QD sample, collected at the peak of the PL spectrum (580 nm), was measured three times: once only with the pump pulse, another only with the probe pulse and a third time with both the pump and probe pulses. As can be seen in the figure, both of the individual excitations – either the red pump or IR probe – can lead to upconverted PL (purple and green time traces, correspondingly), likely by two-photon absorption (certainly for the IR probe) or by consecutive absorption of two photons with similar energies. However, when the QDs are excited by both the pump and probe, we notice a significant increase in the PL signal that corresponds to the arrival of the probe pulse. This behavior was consistent for all the measurements performed on the QD–TP system, and was not observed in any of the pre-LE QDs. As an additional control, we tested for UC PL in QDs which underwent ligand exchange to octanethiol, an aliphatic thiol, but observed none (see Fig. S6 in the ESI†). When integrating over 30 ns from the arrival of the probe pulse, we find that the fluorescence in the case of the dual excitation is about three times higher than the combination of the two individual excitations (for the measurement depicted in Fig. 3b). Therefore, it is clear that a considerable contribution to the non-linear PL in this system comes from UC by sequential absorption of two photons where the intermediate state lifetime is at least several tens of nanoseconds. This is further corroborated by increasing the delay time between the pump and probe to 32 ns (Fig. 3c). The intensity of the upconverted PL decreased on average by ∼35%, indicating a significant portion of the QDs population remained in the excited state. As a side note, it is important to clarify the appearance of what seems to look like a second emission peak resulting from the 670 nm excitation. This is an artifact caused by double-humped temporal shape of the pulse generated by the OPO close to its degeneracy point. Therefore, the two peaks correspond to the same electronic transition in the QD–TP system. This artifact becomes detectable due to the shortening of the emission lifetime upon ligand exchange, which becomes comparable with the instrument response time.
We repeated the pump–probe measurement for a series of increasing pump and probe excitation intensities. Fig. 3d and e plots the upconverted luminescence extracted from the measurements taken with a 15 ns delay against either pump or probe intensity, after subtraction of the background emission resulting from two-photon absorption of either the pump or probe. As expected, the upconverted PL scales linearly with both the pump and the probe energy for low excitation intensities. Further increasing the probe intensity results in linear saturation of the upconverted PL (Fig. 3d) at approximately 1000–1500 μJ (corresponds to ∼1 J cm−2). Similarly to an above-band gap excitation in QDs, this can be modeled using Poissonian distribution 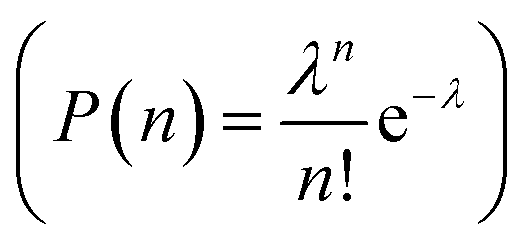 , in which the probability to excite n excitons (or in our case, generate n trials to transfer an excited hole by intraband absorption) is given by the distribution parameter
, in which the probability to excite n excitons (or in our case, generate n trials to transfer an excited hole by intraband absorption) is given by the distribution parameter 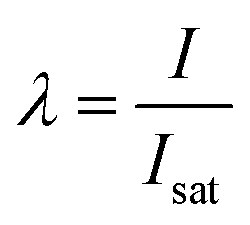 , when Isat is the saturation energy. Thus, the probability to transfer the excited hole above the potential barrier will depend on
, when Isat is the saturation energy. Thus, the probability to transfer the excited hole above the potential barrier will depend on 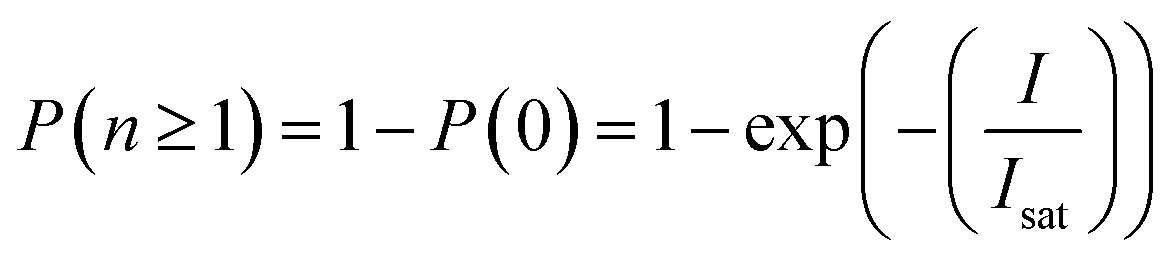 . Additionally, since the upconverted PL as a function of the pump intensity does not reach saturation in the region of our measurement, and based on the maximal pump energy density (which corresponds to ∼0.18 J cm−2), we can give an upper bound for the 670 nm absorption cross section of ∼6 × 10−17 cm2, almost two orders of magnitude lower than the characteristic cross section of the band edge transition in these QDs.
. Additionally, since the upconverted PL as a function of the pump intensity does not reach saturation in the region of our measurement, and based on the maximal pump energy density (which corresponds to ∼0.18 J cm−2), we can give an upper bound for the 670 nm absorption cross section of ∼6 × 10−17 cm2, almost two orders of magnitude lower than the characteristic cross section of the band edge transition in these QDs.
Fig. 3f plots the ratio of the total luminescence intensity in the 15 ns pump–probe experiment to the sum of the luminescence upon excitation with the pump pulse alone or the probe pulse alone. The result of a series of such pump–probe experiments using different excitation intensities for the pump beam at 670 nm is presented. Since the upconverted PL signal contains contributions from both simultaneous and sequential absorptions of photon pairs from the pump and probe beam, it will be proportional to the following expression:
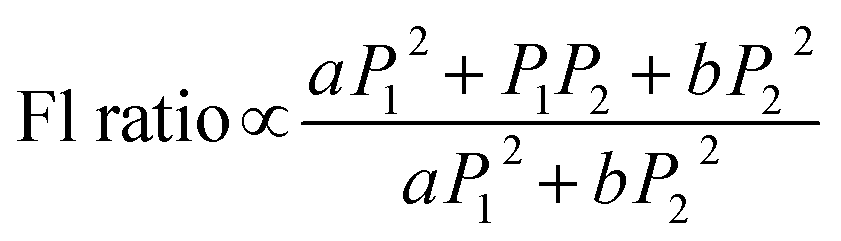
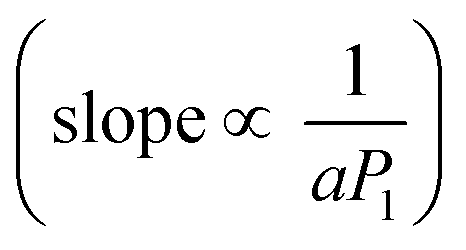 . Additionally, the maximal Fl ratio will be obtained under the condition of
. Additionally, the maximal Fl ratio will be obtained under the condition of 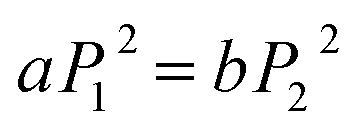 . This expectation is also corroborated by our experimental findings, as can be seen in Fig. 3e, where the maximal Fl ratio appears at higher probe intensities with the increase of the pump intensity. Further details regarding this analysis are included in the ESI.† Overall, the results presented in Fig. 3d–f corroborate our claim regarding the feasibility of UC by sequential absorption of two photons in the QD–TP system.
. This expectation is also corroborated by our experimental findings, as can be seen in Fig. 3e, where the maximal Fl ratio appears at higher probe intensities with the increase of the pump intensity. Further details regarding this analysis are included in the ESI.† Overall, the results presented in Fig. 3d–f corroborate our claim regarding the feasibility of UC by sequential absorption of two photons in the QD–TP system.
Although the results of the pump–probe experiments presented so far in this manuscript provide proof for the existence of UC in our QD–TP system, it does not give clear indication for the spectral position of the meta-stable state from which UC takes place. Time-resolved PL transients of both types of QDs (before and after the LE) did not clearly reveal any new radiative transitions created due to the attachment of the new ligands. Additionally, no new absorption features appeared in the QDs spectrum following the LE process (Fig. 2a) that can be attributed to a direct absorption of the pump wavelength. This can be explained by the weak oscillator strength of the charge-transfer transition, which is below the detection limit of our instrumentation, yet still requires a closer inspection. Thus, an alternative spectroscopic method is needed in order to gain insight regarding the electronic transitions in the two types of QD systems. We therefore performed two sets of transient absorption (TA) measurements using a 120 fs pump pulse. In the first set we used a 670 nm pump (same as in our PL pump–probe experiment) at increasing excitation intensities, and examined the bleach evolution of the first excitonic transition in the CdSe core. The second set of measurements presented here used a 550 nm pump. This allowed us to probe the optically excited state of the system close to the band-edge and with ps resolution.
We first plot the difference of the particles' absorption with and without the 670 nm pump (ΔO.D.) against the wavelength of the white-light probe (Fig. 4a and b), focusing on the region of the two lowest-energy transitions in the CdSe core, located around 500 and 560 nm. This spectrum was constructed by summing all the TA spectra taken with time delays between 25 to 50 ps from the arrival of the pump pulse. In both cases (before and after LE) excitation with the 670 nm pump results in a bleach of these two transitions due to the excitation of an electron in the CdSe core. However, a more careful examination of the bleach properties reveals a significantly different dependence of the bleach intensity on the excitation power for the two different QDs samples. Fig. 4c presents the dependence of bleach magnitude of the 1st exciton (at ∼560 nm) with the pump power, before and after LE. Both curves showed a good fit to a power law y = axb, where the as-synthesized QDs showed a nearly quadratic power dependence (b = 1.9), typical of two-photon absorption, while the ligand-exchanged QDs power dependence was close to a linear one (b = 1.3), indicating a significant component of linear absorption. These findings support our claim regarding the origin of the non-linear PL exhibited by the CdSe core in both types of QD systems. As the energy of the pump is lower than the first absorption band of the CdSe core, state filling of the CdSe in the pre-LE QDs will only be allowed by absorption of photon pairs. However, in the QDs coated with TP it can be achieved either by absorption of photon pairs, or by absorption of single photons via the charge-transfer state, leading to a much lower exponent, and proving that single 670 nm photons can be absorbed by the QD–TP complex.
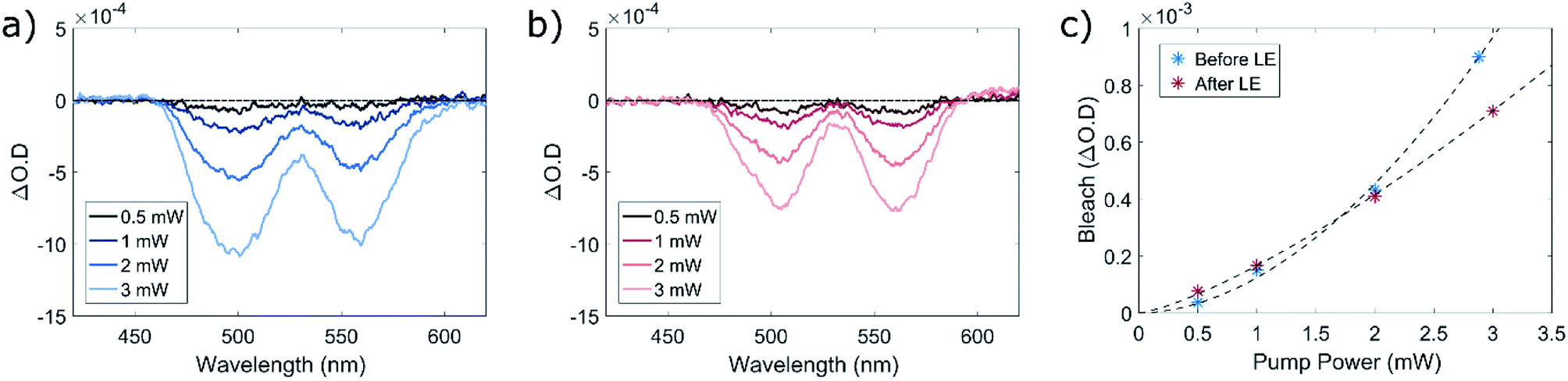 | ||
| Fig. 4 Power-dependent transient absorption measurements with a 670 nm pump. TA spectra of the QDs, (a) before and (b) after the LE, taken between 25 and 50 ps after the arrival of the pump pulse, with increasing pump power. (c) Bleach intensity of the 1st excitonic transition (∼560 nm) in the QDs before and after LE, plotted against the 670 nm pump power. The black dashed lines depict the power law fit (y = axb) of either one of the QDs samples, giving a power dependence of b = 1.9 for the pre-LE QDs and b = 1.3 for the QDs coated with TP. | ||
Due to the weak absorption induced by 670 nm excitation we were not able to detect new electronic transitions in the ligand-exchanged QDs compared to the original ones. However, using a 550 nm pump, which matches the band edge absorption of the CdSe core, proved to be more beneficial in this context. Since the TA measurements already clearly showed the presence of linear absorption of the charge transfer state, we expect to observe a bleach feature at a wavelength corresponding to this transition. Notably, this is difficult, since the white light continuum we use is generated with an 800 nm pump, limiting the accessible spectral range. Considering this, the most distinguishable difference between the TA spectrum of the QDs before and after the LE (Fig. 5a) is a new bleach band found around 875 nm. As can be seen in the figure, following the LE to TP we detect a bleach in the absorption, which did not exist prior to the LE process. This indicates a new transition in the QD–TP system induced by the LE process, whose absorption decreases upon excitation of a band edge exciton in the QD. As such, it most likely corresponds to the charge transfer absorption occurring at the interface between the QD and the molecules (schematically drawn in Fig. 1c). Examining the kinetics probed around 875 nm for the QDs before and after the LE (Fig. S7 in the ESI†), we notice both types of QD systems show induced absorption, followed by a quick relaxation upon carrier cooling into a surface trap state. Fitting this relaxation trace to a biexponential decay dynamics, we find the time constant for trapping becomes shorter due to the LE. This dynamic witnessed in both QD systems is typical of hole trapping,30 although in this case the time constants for relaxation are longer than for an intra-dot trap, most likely due to slower relaxation dynamics to the trap state, affected by the molecule–QD interface. Following the trapping, in the case of the QDs before the LE, we witness excited state absorption (ESA) which does not recover within the timeframe of this measurement (∼2 ns). However, for the QD–TP system we observe a clear bleach which partially recovers after ∼500 ps but does not reach the induced absorption value observed prior to LE, even on the time scale of 2 ns (Fig. 5b and c, correspondingly). This bleach indicates the existence of a transient filling of an electronic transition arising from the connection of the TP ligands to the QD surface. Therefore, the observed feature at 875 nm is likely implicated with the UC process. Notably, this is in agreement with the results of Meijerink and coworkers on the position of aromatic thiol traps.29
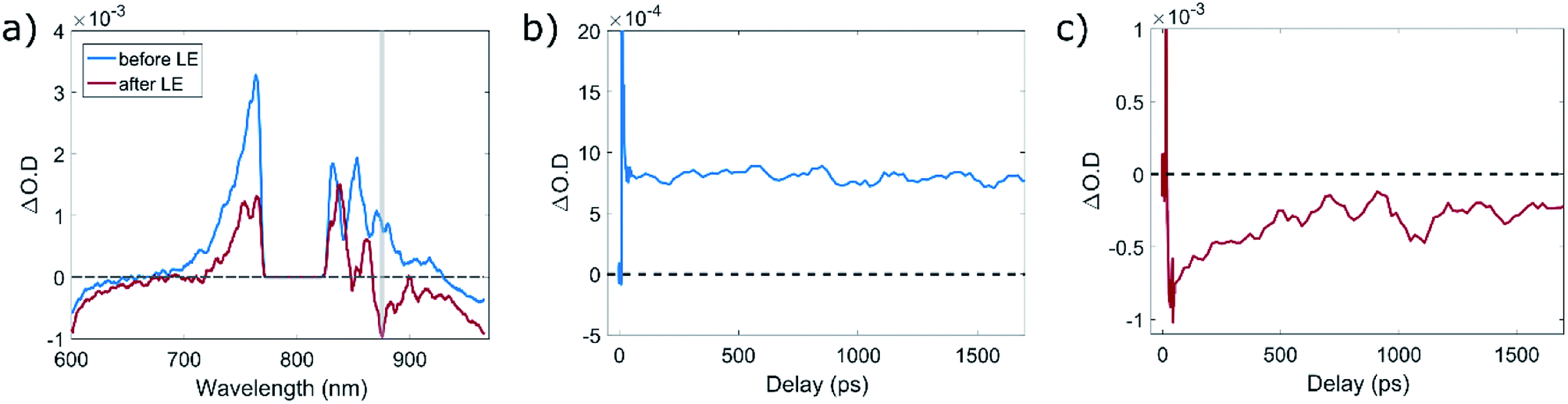 | ||
| Fig. 5 Transient absorption measurements with a 550 nm pump. (a) TA spectra of the QDs, before and after ligand exchange, taken between 25 and 50 ps after the arrival of the pump pulse. The area around 800 nm was filtered out before reaching the sample since it contained the fundamental 800 nm beam which was used to create the white-light probe. The grey rectangle around 875 nm (±2 nm) corresponds to the ΔO.D. time traces depicted in (b) and (c) for the quantum dots before and after the LE, correspondingly. The y axis in (b) and (c) is cropped in order to gain clearer image of the kinetics throughout the whole timeframe of the measurement. A zoomed-in figure of the shorter time delays (up to 50 ps), presenting the full magnitude of the induced absorption can be found in the ESI.† | ||
So far we discussed the dynamics of UC in isolated QDs, suspended in an organic medium. These results show that UC in the QD–TP system is an intrinsic property; in the following we show that this type of ligand-to-dot UC can be also realized in a QD solid. Demonstrating the feasibility of UC in dense, closely-packed QD structures is an important step towards utilizing these non-linear processes in light harvesting applications. QD films were formed by self-assembly on an immiscible liquid surface in a Teflon well, using a previously published procedure, with some adaptations.22–24 Briefly, a purified QD solution in toluene was gently drop-casted on the surface of diethylene glycol (DEG), and the toluene was allowed to evaporate completely. Then the formed QD film was separated from the DEG by slow injection of acetonitrile and transferred to an ITO-coated glass substrate which was submerged in the DEG, by slow removal of the DEG with a syringe pump. This method of QD film preparation results in close-packed structure of the QDs (see SEM image, Fig. 6a), which extends for tens and even hundreds of micrometers and is composed of up to 10 monolayers. For ligand-exchanged QD films, we used 1,4-benzenedithiol (1,4-BDT) molecules, thus achieving maximal similarity to our QD–TP system. The LE process was performed after the formation of the film and before the transfer to the substrate. Performing LE in this manner helps maintaining the close-packed arrangement of the QDs and prevents cracks and damage to the film.25 A more detailed description of the QD films formation and LE is given in the experimental section.
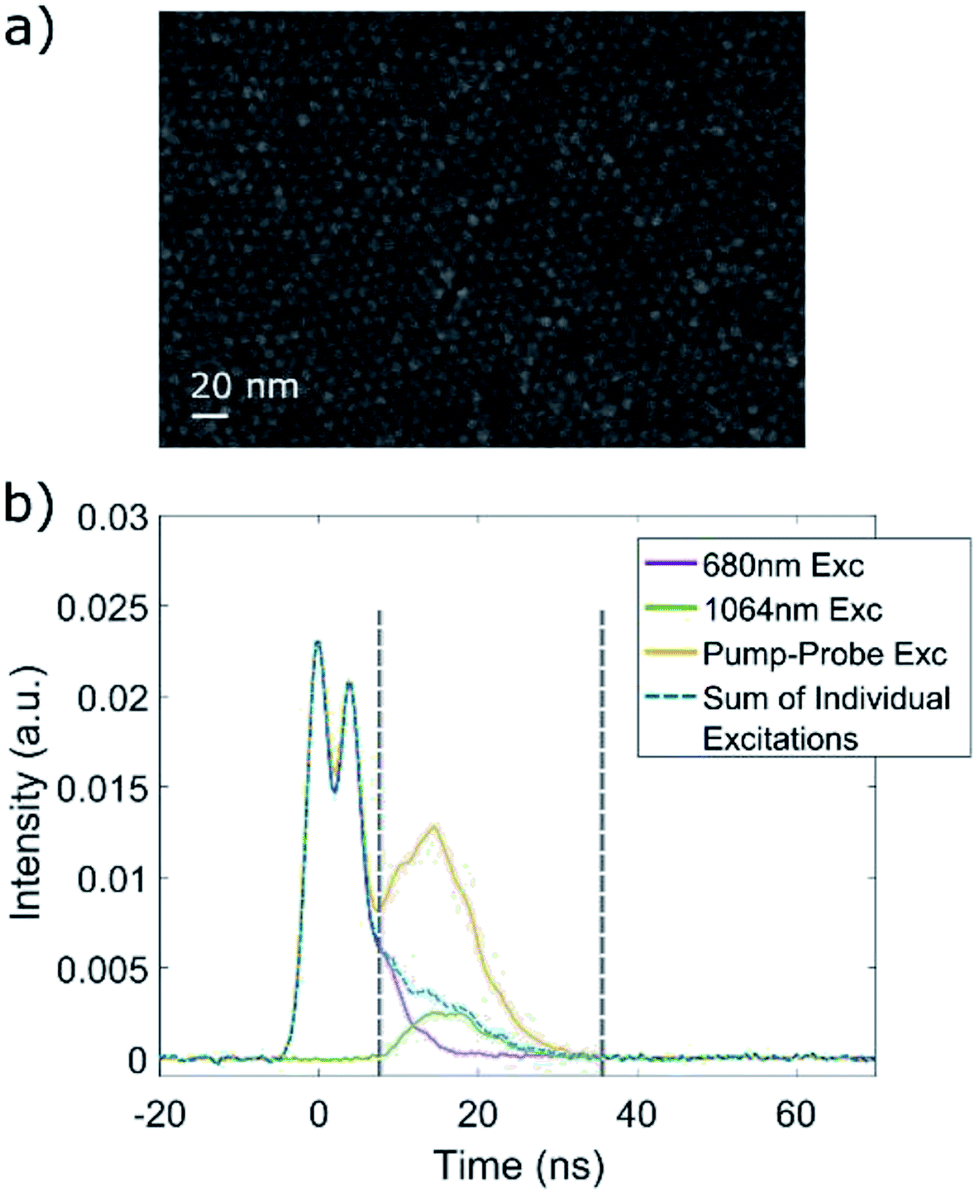 | ||
| Fig. 6 UC in QD solids. (a) SEM image of self-assembled QD film. (b) PL traces of a typical pump–probe experiment, performed on a QD film after LE to 1,4-BDT, using a 680 nm pump at 120 μJ and a 1064 nm probe at 1200 μJ. The Fl ratio in this case was 2.8. | ||
In a similar manner to the solution suspended QDs, the QD films also exhibited non-linear PL when subjected to excitation with lower energy than the first electronic transition, and have also shown a decrease in the measured lifetime of the linear PL upon LE to 1,4-BDT. Here too we performed a series of pump–probe experiments, using the same optical setup described earlier, and a typical pump–probe measurement is presented in Fig. 6b. Generally, the results were consistent with the measurements performed on the solution-suspended QD – an increase of the PL signal was detected with the arrival of the probe pulse, and the overall PL in this time frame was higher than the sum of the emission caused by 2-photon absorption of either the 680 or 1064 nm excitation, for all of the ligand-exchanged QD films measured. Due to the similar effect of the LE on both the solution-suspended QDs and the QD films, and the consistent results of the pump–probe experiments, we can safely assume that ligand-to-dot UC takes place by the same mechanism in the QD films as well as in the QD–TP solution.
Conclusions
In conclusion, we demonstrated how UC can be realized in a coupled QD–ligand system via a mechanism which was not previously considered. The attachment of specific thiol ligands to the surface of a core/shell CdSe/CdS QDs resulted in the formation of a new charge transfer transition, absorbing in the NIR spectral region. Upon excitation with energy below the band gap of the CdSe core, the exciton formed in the QD–ligand complex can be further promoted by intraband absorption of an additional photon and recombine in the CdSe core, thus emitting an upconverted photon. We performed a series of pump–probe experiments, in which the probe pulse could only generate intraband absorption. These measurements, and particularly the linear scaling of the upconverted emission with both pump and probe intensities, provided proof for the occurrence of UC by sequential photon absorption. Additionally, transient absorption measurements helped us build a more complete picture of the UC process by providing further insight regarding the spectral position of the meta-stable state from which UC was promoted, whose energetic position was found to be consistent with past studies on aromatic thiols. Finally, we showed that this system can also be constructed as a QD solid and exhibit the same optical properties as the solution-suspended QDs, demonstrating how this UC scheme can transpire both in single QDs and in an ensemble of coupled QDs.Due to the dramatic quenching of the band edge luminescence by the TP ligands, the efficiency of the UC process in this particular coupled QD–ligand system is very low, making quantitative assessment of the yield very difficult. Indeed, it is well known that thiol-based ligands can dramatically quench photoluminescence from QDs. Nevertheless, and unlike the inherently low absorption cross section of the charge-transfer transition, this quenching is not due to an intrinsic flaw of this upconversion scheme. We believe that with proper choice of the ligand, enabling better control of the energy gap between the molecular HOMO and the QD conduction band, the efficiency of the ligand-mediated upconversion process can be significantly enhanced, although the saturation intensity would likely remain high due to the weak absorption. Moreover, and in contrast with alternative hybrid organic–inorganic upconversion nanoparticles based on triplet–triplet annihilation15–17 or cooperative sensitization,19 the spectral range of the absorption in ligand-mediated upconversion is decoupled from the HOMO–LUMO gap of the ligand molecule and the band gap of the QD, readily allowing for upconversion even deeper into the infrared spectral range. Finally, this scheme presents a dramatic simplification over upconversion schemes based on double quantum dots.11,13,14 All in all, and particularly when considering the simplicity of the synthetic procedure, we believe these results provide an alternative route for the formation of broadband upconversion nanocrystals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank L. Houben for performing HRTEM of the QDs, T. Udayabhaskararao for his help with the fabrication of the QD solids and A. Teitelboim for her assistance with the data analysis and for helpful discussions throughout this project. Financial support by the Israeli Science Foundation (grant 270/16) is gratefully acknowledged.References
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510 CrossRef CAS.
- J. de Wild, A. Meijerink, J. K. Rath, W. G. J. H. M. van Sark and R. E. I. Schropp, Energy Environ. Sci., 2011, 4, 4835 RSC.
- J. Zhou, Z. Liu and F. Li, Chem. Soc. Rev., 2012, 41, 1323–1349 RSC.
- S. Hososhima, H. Yuasa, T. Ishizuka, M. R. Hoque, T. Yamashita, A. Yamanaka, E. Sugano, H. Tomita and H. Yawo, Sci. Rep., 2015, 5, 1–10 Search PubMed.
- B. Zheng, H. Wang, H. Pan, C. Liang, W. Ji, L. Zhao, H. Chen, X. Gong, X. Wu and J. Chang, ACS Nano, 2017, 11, 11898–11907 CrossRef CAS PubMed.
- T. Trupke, M. A. Green and P. Würfel, J. Appl. Phys., 2002, 92, 4117–4122 CrossRef CAS.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed. Engl., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- S. Wu, G. Han, D. J. Milliron, S. Aloni, V. Altoe, D. V Talapin and B. E. Cohen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1–5 CrossRef.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed.
- Z. Deutsch, L. Neeman and D. Oron, Nat. Nanotechnol., 2013, 8, 649–653 CrossRef CAS PubMed.
- C. C. Milleville, E. Y. Chen, K. R. Lennon, J. M. Cleveland, A. Kumar, J. Zhang, J. A. Bork, A. Tessier, J. M. Lebeau, D. B. Chase, M. O. Joshua and M. F. Doty, ACS Nano, 2019, 13, 489–497 CrossRef CAS PubMed.
- A. Teitelboim and D. Oron, ACS Nano, 2016, 10, 446–452 CrossRef CAS PubMed.
- N. S. Makarov, Q. Lin, J. M. Pietryga, I. Robel and V. I. Klimov, ACS Nano, 2016, 10, 10829–10841 CrossRef CAS PubMed.
- M. Wu, D. N. Congreve, M. W. B. Wilson, J. Jean, N. Geva, M. Welborn, T. van Voorhis, V. Bulović, M. G. Bawendi and M. A. Baldo, Nat. Photonics, 2015, 10, 31–34 CrossRef.
- Z. Huang, X. Li, M. Mahboub, K. M. Hanson, V. M. Nichols, H. Le, M. L. Tang and C. J. Bardeen, Nano Lett., 2015, 15, 5552–5557 CrossRef CAS PubMed.
- M. Mahboub, Z. Huang and M. L. Tang, Nano Lett., 2016, 16, 7169–7175 CrossRef CAS PubMed.
- G. V. Naik, A. J. Welch, J. A. Briggs, M. L. Solomon and J. A. Dionne, Nano Lett., 2017, 17, 4583–4587 CrossRef CAS PubMed.
- J. Lauth, G. Grimaldi, S. Kinge, A. J. Houtepen, L. D. A. Siebbeles and M. Scheele, Angew. Chem., Int. Ed., 2017, 56, 14061–14065 CrossRef CAS PubMed.
- L. Carbone, C. Nobile, M. De Giorgi, F. Della Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- S. Christodoulou, G. Vaccaro, V. Pinchetti, F. De Donato, J. Q. Grim, a. Casu, a. Genovese, G. Vicidomini, a. Diaspro, S. Brovelli, L. Manna and I. Moreels, J. Mater. Chem. C, 2014, 2, 3439 RSC.
- A. Dong, J. Chen, P. M. Vora, J. M. Kikkawa and C. B. Murray, Nature, 2010, 466, 474–477 CrossRef CAS PubMed.
- M. P. Boneschanscher, W. H. Evers, J. J. Geuchies, T. Altantzis, B. Goris, F. T. Rabouw, S. A. P. van Rossum, H. S. J. van der Zant, L. D. a Siebbeles, G. Van Tendeloo, I. Swart, J. Hilhorst, A. V. Petukhov, S. Bals and D. Vanmaekelbergh, Science, 2014, 344, 1377–1380 CrossRef CAS PubMed.
- A. Dong, J. Chen, S. J. Oh, W. K. Koh, F. Xiu, X. Ye, D. K. Ko, K. L. Wang, C. R. Kagan and C. B. Murray, Nano Lett., 2011, 11, 841–846 CrossRef CAS PubMed.
- A. Dong, Y. Jiao and D. J. Milliron, ACS Nano, 2013, 7, 10978–10984 CrossRef CAS PubMed.
- R. H. Abu-Eittah and R. H. Hilal, Appl. Spectrosc., 1972, 26, 270 CrossRef CAS.
- K. O. Aruda, V. A. Amin, C. M. Thompson, B. Lau, A. B. Nepomnyashchii and E. A. Weiss, Langmuir, 2016, 32, 3354–3364 CrossRef CAS PubMed.
- M. S. Azzaro, A. Dodin, D. Y. Zhang, A. P. Willard and S. T. Roberts, Nano Lett., 2018, 18, 3259–3270 CrossRef CAS PubMed.
- S. F. Wuister, C. de Mello Donegá and A. Meijerink, J. Phys. Chem. B, 2004, 108, 17393–17397 CrossRef CAS.
- A. Avidan, I. Pinkas and D. Oron, ACS Nano, 2012, 6, 3063–3069 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A calculation for the electron and hole energy levels, HRTEM image of the CdSe/CdS QDs, a fit of the band-edge lifetime for the QD–TP system, the upconverted spectrum of the QD–TP, examples of the instrument response transient, pump–probe control experiment, additional TA results and a more detailed explanation regarding the trends observed in the pump–probe results. See DOI: 10.1039/c9ra01273g |
| This journal is © The Royal Society of Chemistry 2019 |