
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReview of α-nucleosides: from discovery, synthesis to properties and potential applications
Guangcheng Ni†
,
Yuqi Du†,
Fan Tang,
Jiang Liu*,
Hang Zhao * and
Qianming Chen
* and
Qianming Chen
State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, Chinese Academy of Medical Sciences Research Unit of Oral Carcinogenesis and Management, West China Hospital of Stomatology, Sichuan University, Chengdu, Sichuan 610041, P. R. China. E-mail: zhaohangahy@scu.edu.cn; liujiang1220@hotmail.com
First published on 7th May 2019
Abstract
Nucleic acids play an important role in the genetic process of organisms; nucleosides, the building block of nucleic acids, typically exist in nature in a β configuration. As an anomer of β-nucleoside, α-nucleoside is extremely rare in nature. Because of their unique and interesting properties such as high stability, specific parallel double-stranded structure and some other biochemical properties, α-nucleosides have attracted wide attention. Various methods including but not limited to the mercuri procedure, fusion reaction and Vorbrüggen glycosylation have been used to synthesize α-nucleosides and their derivatives. However, to the best of our knowledge, there is no review that has summarized these works. Therefore, we systematically review the discovery, synthesis, properties, and potential applications of α-nucleosides in this article and look to provide a reference for subsequent studies in the coming years.
1. Introduction
As genetic molecules, nucleic acids are widely present in living organisms. According to their different chemical components, they can be divided into ribonucleic acid (RNA) and deoxyribonucleic acid (DNA). These molecules play important roles in the replication, transmission, and transcription of genetic information in living organisms. As building blocks of nucleic acids, nucleotides are composed of a nucleobase, ribose or deoxyribose sugar, and phosphate. A nucleoside lacks a phosphate at the C5′ position. Nucleosides can be classified into five types: adenosine, guanosine, cytidine, thymidine, and uridine (Fig. 1).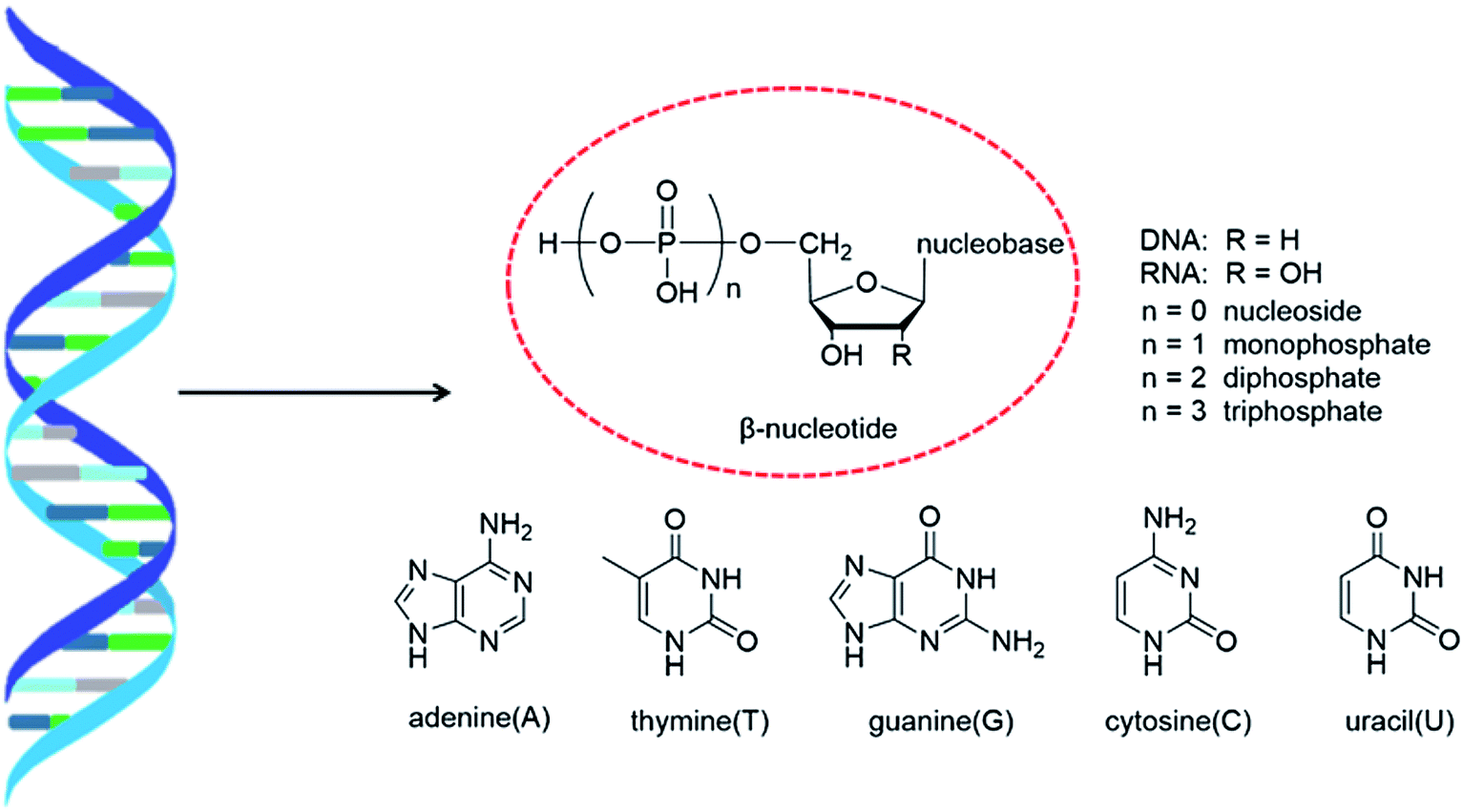 | ||
| Fig. 1 Double helix structure of DNA or RNA and its components: nucleotides. | ||
In general, in naturally occurring nucleosides, the ribose or deoxyribose is linked to nucleobases through β-glycosidic bonds, which means that the nucleobase at C1 is cis with respect to the hydroxymethyl group at C4, known as the β-configuration; thus, these molecules are referred to as β-nucleosides. In an α-nucleoside, the nucleobase and hydroxymethyl group in the ribose or deoxyribose are in a trans relationship1 (Fig. 2). As an anomer of β-nucleoside, α-nucleoside is extremely rare in nature. Compared with nucleosides, due to the removal of the hydroxyl group at the C2′ position, deoxynucleosides have reduced steric hindrance, which making the α-anomer more easily formed. Because of their subtle differences in configuration, the properties exhibited by α-nucleosides differ from those of β-nucleosides. For example, the α-oligonucleotide can form a parallel double-stranded structure with the complementary β-oligonucleotide, show tolerance to or even inhibit some enzymes, and inhibit certain bacteria and tumors. Since their discovery, numerous studies of α-nucleosides have been carried out and the characteristics of α-nucleosides have gradually emerged. Here, we describe the discovery, extraction, synthesis, and separation of α-nucleosides and their unique properties that differ from those of β-nucleosides.
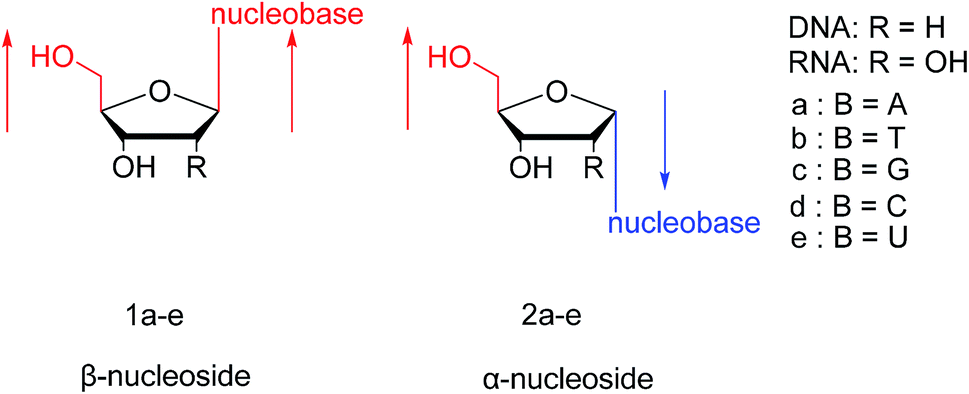 | ||
| Fig. 2 Configuration of β-nucleosides and α-nucleosides. | ||
2. α-Nucleosides and their derivatives found in nature
As shown in Table 1, α-nucleosides are extremely rare in nature, with only a few α-anomers of nucleoside or nucleoside derivatives having been reported. In 1955, an α-nucleoside derivative was first discovered in diphosphopyridine nucleotide (DPN), now known as nicotinamide adenine dinucleotide (NAD). Kaplan2 et al. found that although DPN activity was destroyed by DPNase treatment, there was still a reaction with cyanide. Thus, they separated the cyanide-reacting material by column chromatography followed by acidic acetone precipitation and named this fraction as the “DPN isomer”. Optical rotation tests showed that the DPN isomer differed from “normal” DPN in its nicotinamide glycosidic linkage; the DPN isomer showed a positive rotation, while “normal” DPN showed a negative rotation. They first proposed the concept of the α-anomer. After that, in 1965, Suzuki3 et al. detected α-NAD, α-NADP, α-nicotinic acid mononucleotide, and α-nicotinic acid adenine dinucleotide in Azotobacter vinelandii. Besides, in 1963, Bonnett4 et al. found that 5,6-dimethyl-1-α-D-ribofuranosyl benzimidazole-3′-phosphate was a structural unit in vitamin B12. Additionally, in 1965, Gassen5 et al. used formic acid gradient elution to obtain a mixture of mononucleotides, which included α-cytidine, from a hydrolysate of yeast RNA on a Dowex-1 X2 column. Furthermore, after hydrolysis of the corrinoid factor Cx from Propionibacterium shermanii with cerous hydroxide, Dinglinger6 et al. isolated α-adenosine by column chromatography in 1971. These are the first reports of α-anomers in naturally occurring nucleosides.| Time | Author | α-Nucleosides or α-nucleoside derivatives | Source |
|---|---|---|---|
| 1955 | Kaplan et al. | α-DPN | DPN |
| 1963 | Bonnett et al. | 5,6-Dimethyl-1-α-D-ribofuranosyl benzimidazole 3′-phosphate | Vitamin B12 |
| 1965 | Suzuki et al. | α-NAD, α-NADP, α-nicotinic acid mononucleotide | Azotobacter vinelandii |
| α-Nicotinic acid adenine dinucleotide | |||
| 1965 | Gassen et al. | α-Cytidine | Hydrolysate of yeast RNA |
| 1971 | Dinglinger et al. | α-Adenosine | Corrinoid factor Cx from Propionibacterium shermanii |
3. Synthesis of various α-nucleosides
Although α-nucleosides and their derivatives are rare in nature, they have recently received attention from researchers. To study their properties in detail and evaluate their application potential, various methods have been developed to chemically synthesize α-nucleosides. Here, we mainly summarize the first methods developed for synthesizing various α-nucleosides, such as α-A/dA, α-rT/T, α-U/dU, α-G/dG, and α-C/dC. Additionally, interesting synthetic methods will be described.3.1. α-A & α-dA
As shown in Fig. 3, Wright7 et al. first proposed the chemical synthesis method for α-adenosine in 1958, which was also the first synthesis of α-nucleosides. Adopting the classic mercuri procedure, they used 5-O-benzoyl-D-ribofuranosyl bromide 2,3-cyclic carbonate to condense with chloromercuri-6-benzamidopurine, followed by removal of the protecting group to obtain β-adenosine and α-adenosine at yields of 15% and 24%, respectively. After that, in 1967, Schramm8 et al. reported a new method for synthesizing α-adenosine. After heating a mixture of ribose, adenine, and phenyl polyphosphate at 100 °C for 3 min in the presence of protons provided by concentrated HCl, they directly obtained a mixed product of adenosine, including common β-adenosine and its α-anomer at 20% yield. Under similar conditions, they obtained 40% yield of β- and α-deoxyadenosine. Using unprotected ribose and nucleobase avoids the need to prepare intermediates, making this new method simpler and faster than traditional methods. The new method also avoids the stereochemical limitations of acylated sugars and makes it easier to prepare α-anomers.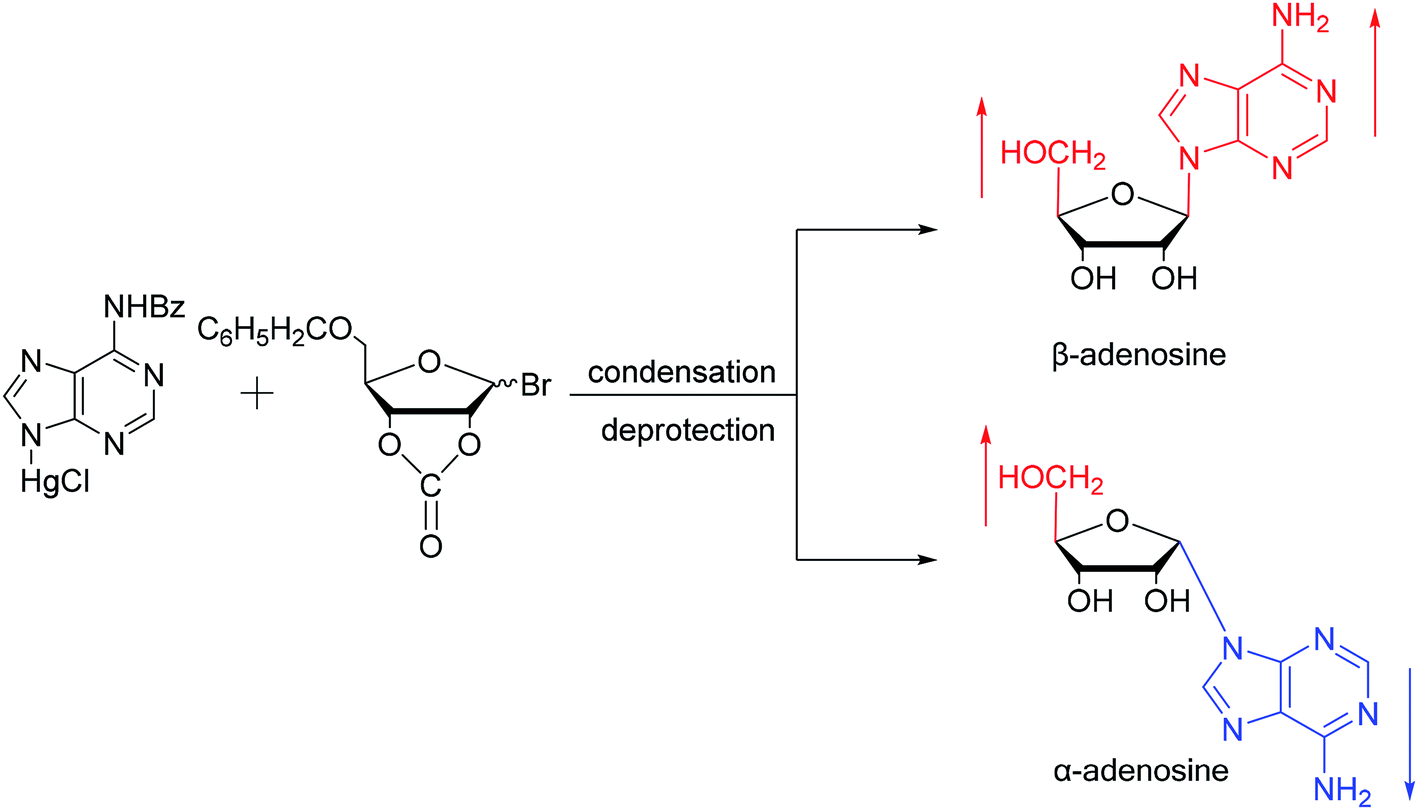 | ||
| Fig. 3 First synthesis of α-adenosine. | ||
Similarly, using the mercuri procedure, Ness9 et al. first proposed the synthesis of α-deoxyadenosine in 1960 (Fig. 4). They used 2-deoxy-5-O-p-nitrobenzoyl-D-ribose diisobutyl dithioacetal as a starting material, which was converted to 2-deoxy-3,5-di-O-p-nitrobenzoyl-D-ribosyl chloride through a series of reactions, and then condensed with chloromercuri-6-benzamidopurine in dimethyl sulfoxide solution. After deprotection, β-deoxyadenosine and its α-anomer were obtained at 10% and 19% yields, respectively. However, when mercuri procedure is used to synthesize nucleosides, contamination with mercury ions affect the biological functions of nucleosides,10 and thus Robins11 et al. attempted to synthesize nucleosides using a new method to avoid using mercury. The main starting material is 1,3,5-tri-O-acetyl-2-deoxy-D-ribofuranose, which was obtained by treating 6-acetamido-9-(3′5′-di-O-acetyl-2′-deoxy-β-D-ribofuranosyl)purine with acetic acid and acetic anhydride at 100 °C, the obtained nucleoside derivative was mainly in the α-configuration. Then fused with 2,6-dichloropurine, and after treatment with methanolic ammonia and catalytic dehalogenation, α-dA is synthesized. In addition, various reported α-adenosine derivatives are shown in Table 2.
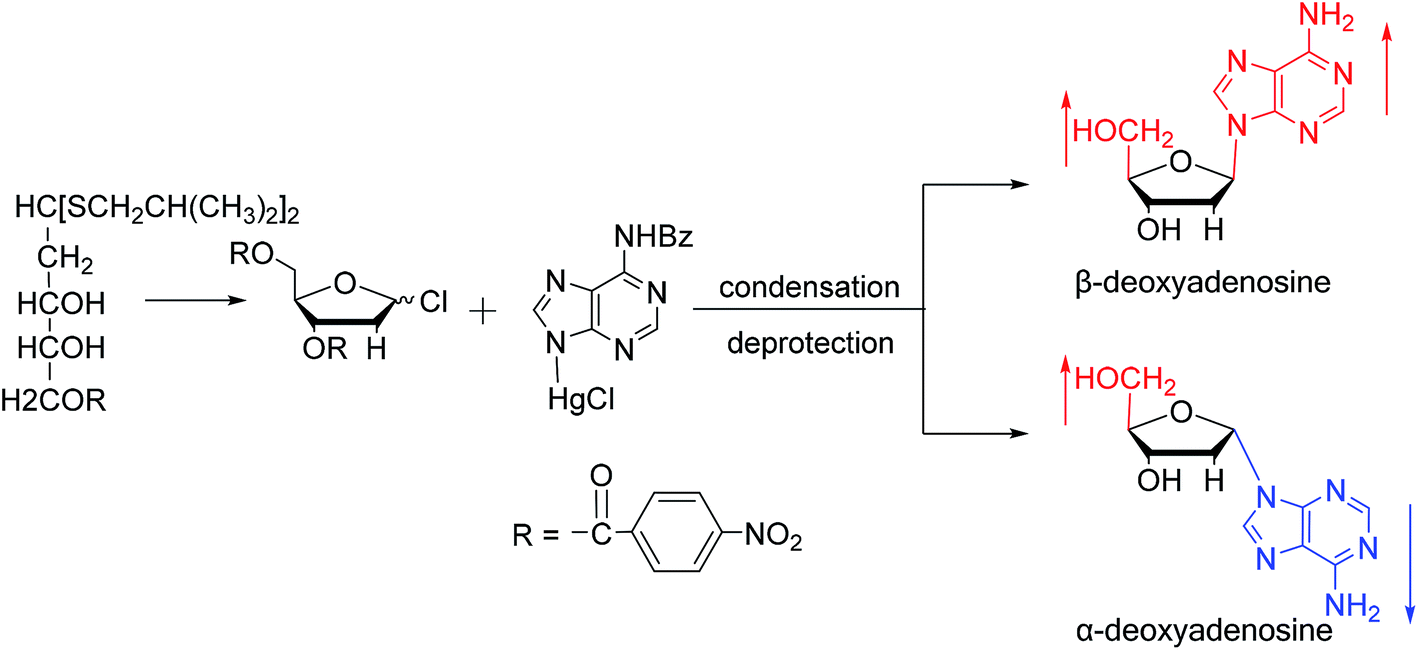 | ||
| Fig. 4 First synthesis of α-deoxyadenosine. | ||
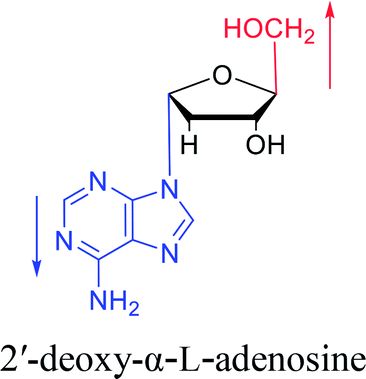
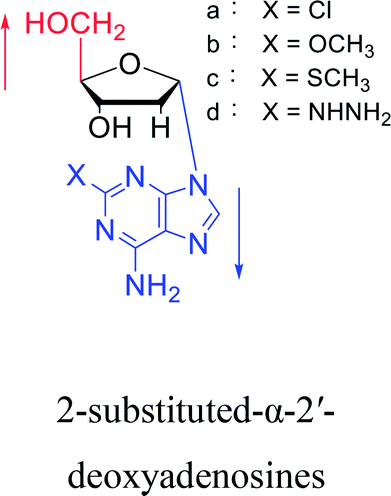
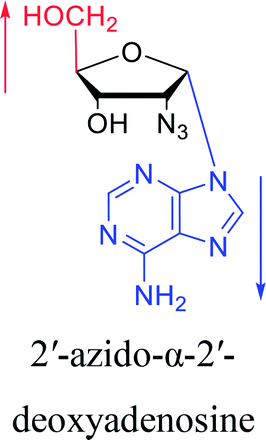
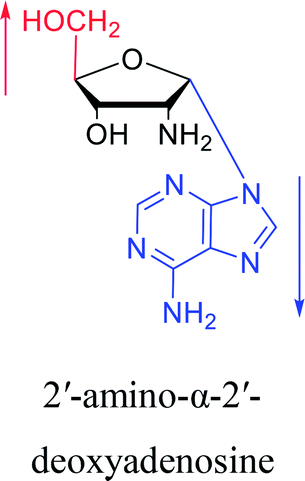
3.2. α-rT/U & α-T/dU
In 1964, Nishimura19 et al. used trimethylsilyl groups to protect nucleobases, followed by condensation with tribenzoylribofuranosyl chloride, an acyl halo sugar. After deacylation, they obtained nucleosides corresponding to the nucleobases. They found that when this method was used to synthesize uridine and thymidine, the corresponding α-anomer could be separated from the product (Fig. 5). That is the first available report of artificial synthesis of α-rT/U.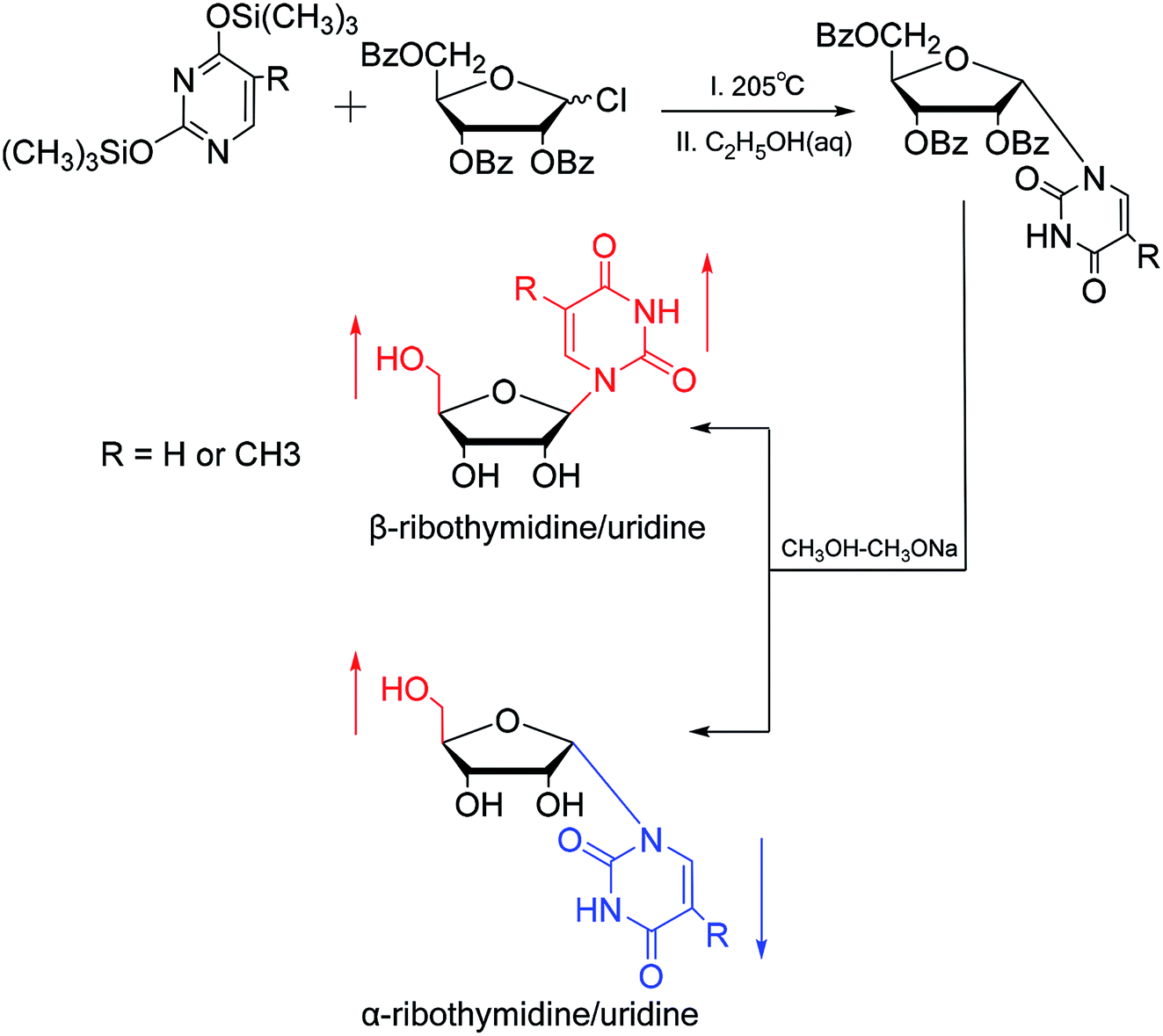 | ||
| Fig. 5 First synthesis of α-ribothymidine/uridine. | ||
While exploring the stereoselective synthesis of α-nucleosides, Aoyama20 proposed a method for synthesizing deoxyuridine and deoxythymidine in a stereoselective manner in 1987, including the β- and α-anomers, as illustrated in Fig. 6. In the condensation reaction of 5-substituted-2,4-di(trimethylsilyloxy)pyrimidines and 3,5-di(O-p-chlorobenzoyl)-2-deoxy-α-D-ribofuranosyl chloride, they found that when Brønsted acid (p-nitrophenol) was present, β-T/dU was stereoselectively synthesized in 96% yield; and on this basis, when organic bases (pyridine) were added, α-T/dU was formed in approximately 70% yield.
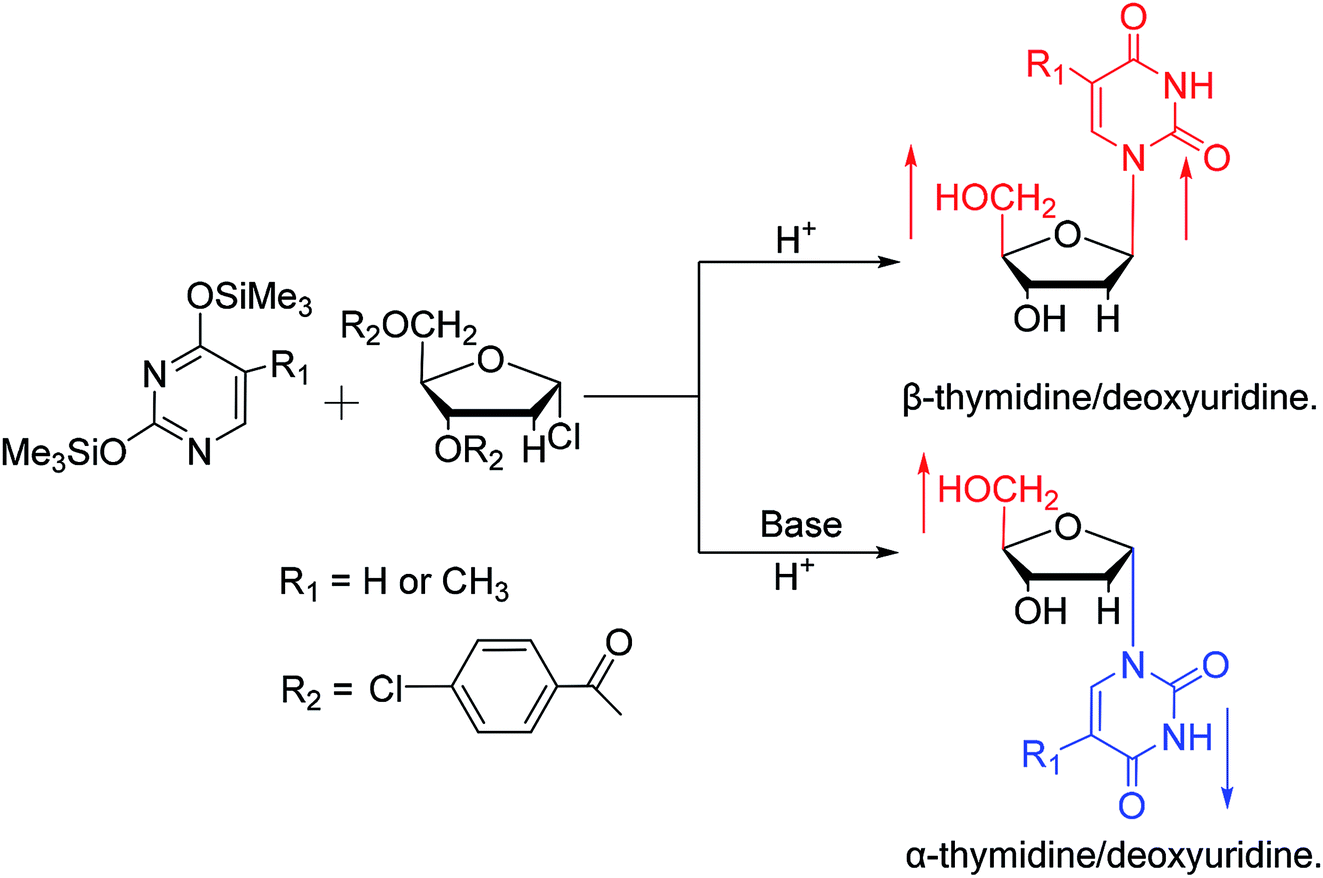 | ||
| Fig. 6 First synthesis of α-thymidine/deoxyuridine. | ||
Using a similar method, Shimizu21 et al. used trimethylsilyl and benzoyl as protective groups, in a series of reactions, they first synthesized 5′-phosphate-α-D-cytidine, and with similar conditions, 5′-phosphate-α-L-uridine and 5′-phosphate-α-L-thymidine was also synthesized at a ratio of α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β of approximately 2:1. Besides, we summarized the α-uridine/thymidine derivatives in Table 3.
:β of approximately 2:1. Besides, we summarized the α-uridine/thymidine derivatives in Table 3.
| The structure and name of α-uridine/thymidine derivatives | Time | Author | The structure and name of α-uridine/thymidine derivatives | Time | Author |
|---|---|---|---|---|---|
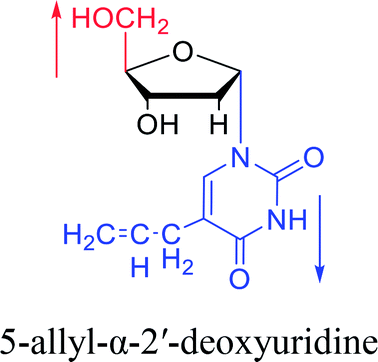 |
1964 | Minnemeyer22 et al. | 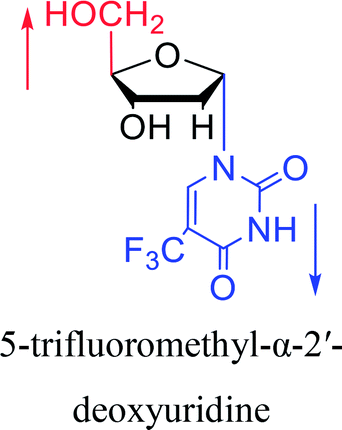 |
1966 | Ryan23 et al. |
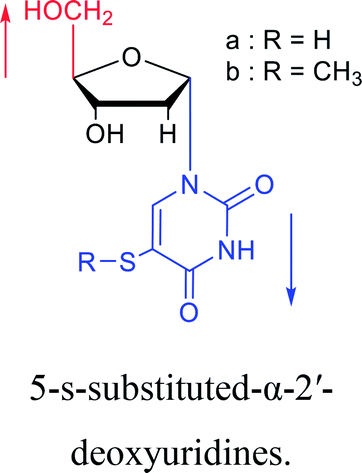 |
1969 | Kotick24 et al. | 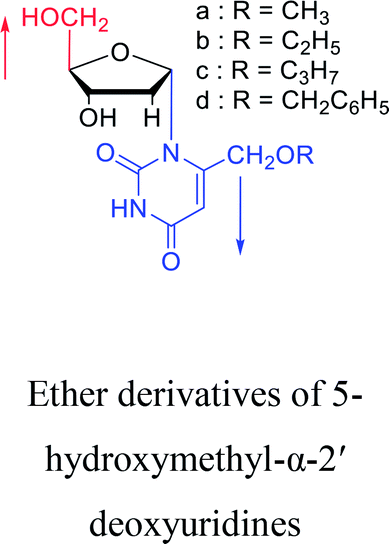 |
1970 | Bubbar25 et al. |
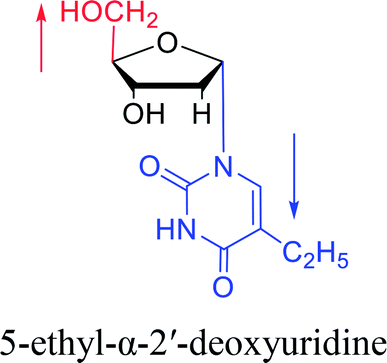 |
1974 | Kulikowski26et al. | 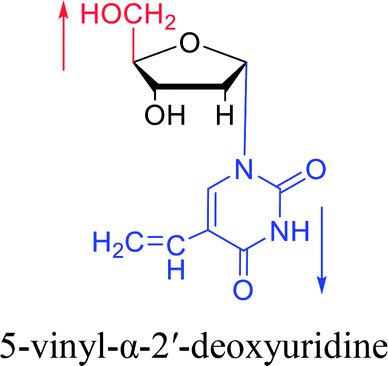 |
1975 | Sharma27 et al. |
 |
1976 | Armstrong28 et al. | 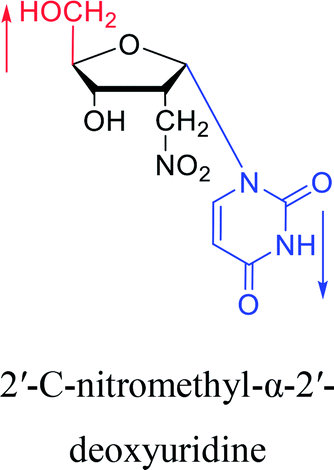 |
1977 | Brink29 et al. |
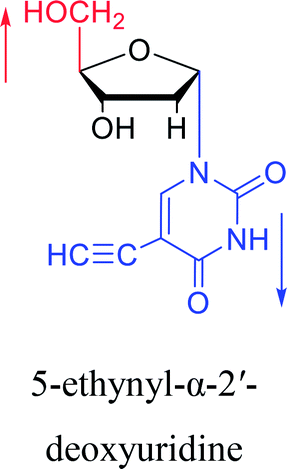 |
1978 | Barr30 et al. | 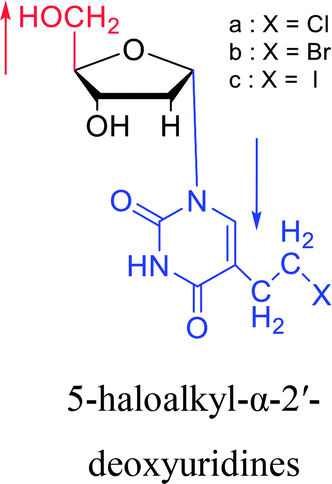 |
1985 | Griengl31 et al. |
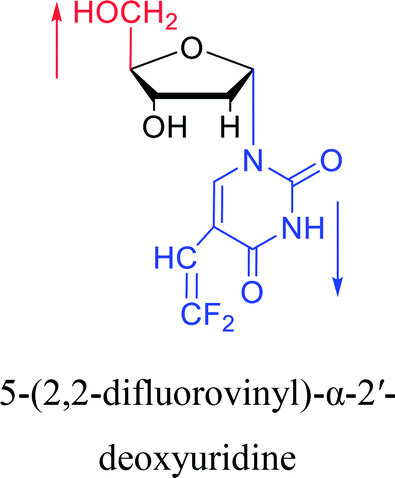 |
1987 | Bobek32 et al. | 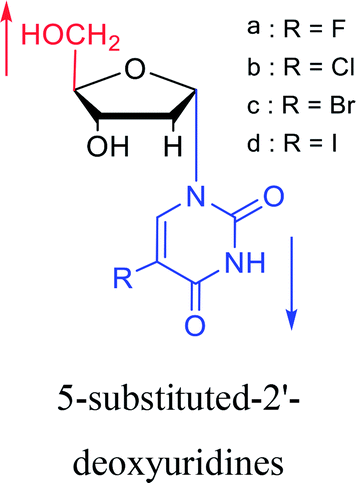 |
1987 | Aoyama20 |
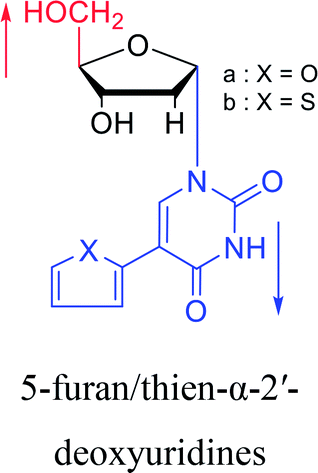 |
1991 | Wigerinck33 et al. | 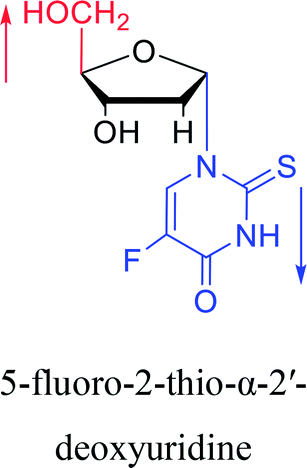 |
1993 | Bretner34 et al. |
 |
1994 | Elbarbary35 et al. | 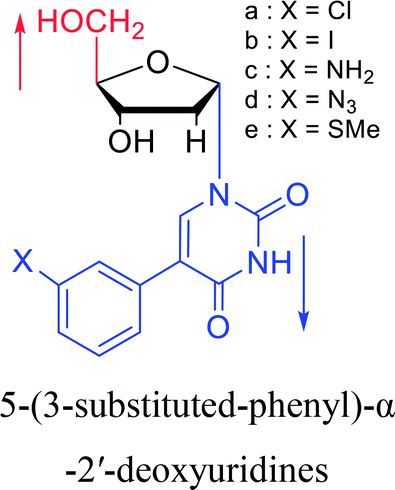 |
1996 | Wellmar36 et al. |
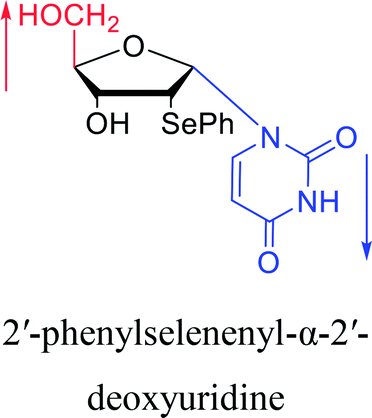 |
1997 | Diaz37 et al. | 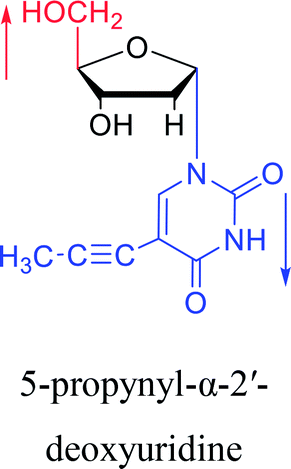 |
1998 | Morvan38 et al. |
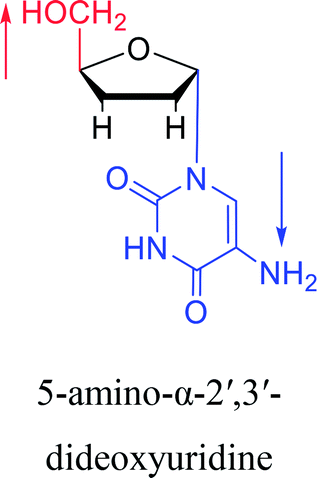 |
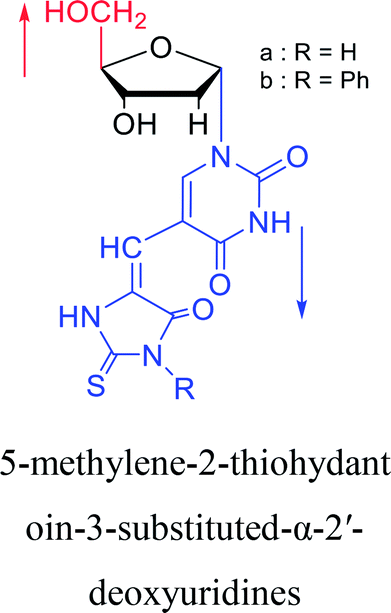
2003 | Colacino39 et al. | 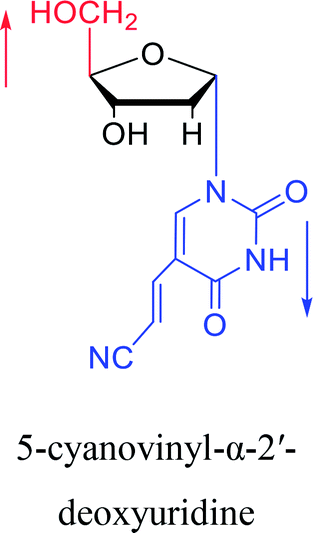 |
2005 | Ogino40 et al. |
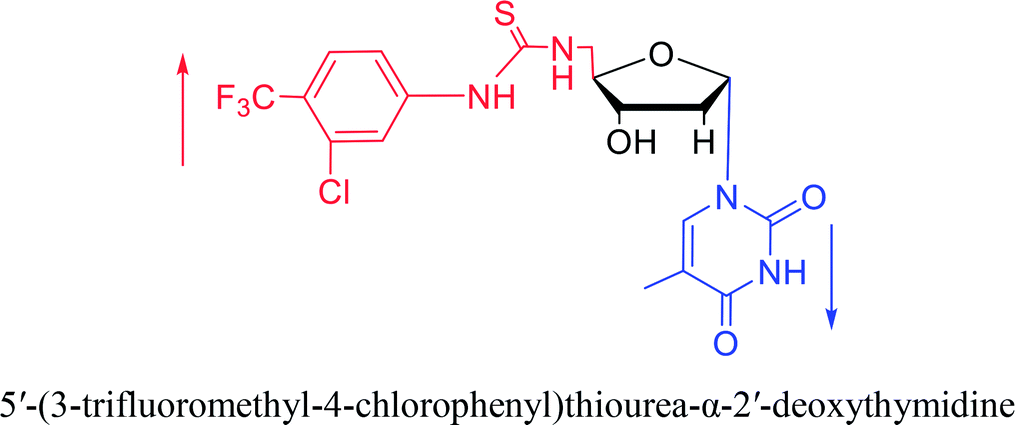 |
2007 | Van Daele41 et al. | |||
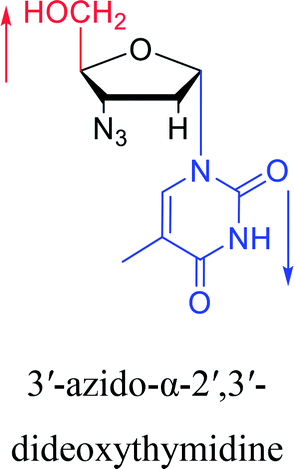 |
2010 | Cui42 et al. | 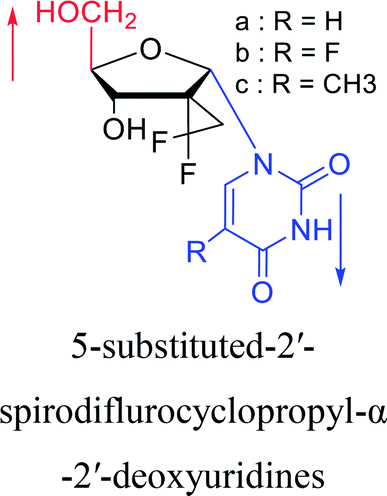 |
2016 | Liu43 et al. |
 |
2012 | Cui44 et al. | |||
3.3. α-G & α-dG
A method for stereoselective synthesis of α-guanosine was first proposed by Gosselin45 et al. in 1990. They used α-D-ribofuranothioxooxazolidine as a starting material, after four-step reaction involving tert-butyldimethylsilyl chloride (TBDMSCl), RANEY® Ni, NH2CH(CN)CONH2 and tetrabutylammonium fluoride (TBAF) respectively, they transformed it to 1-α-D-ribofuranosyl-4-carbamoyl-5-aminoimidazole, which was treated with sodium methylxanthate at 180 °C. After oxidized with hydrogen peroxide and amination with ammonia, they obtained α-guanosine in a 6.6% yield (Fig. 7). Although there have been several reports on the synthesis of α-guanosine,46,47 it is typically produced only as a by-product of other reactions. Therefore, we consider that this was the first synthesis of α-guanosine.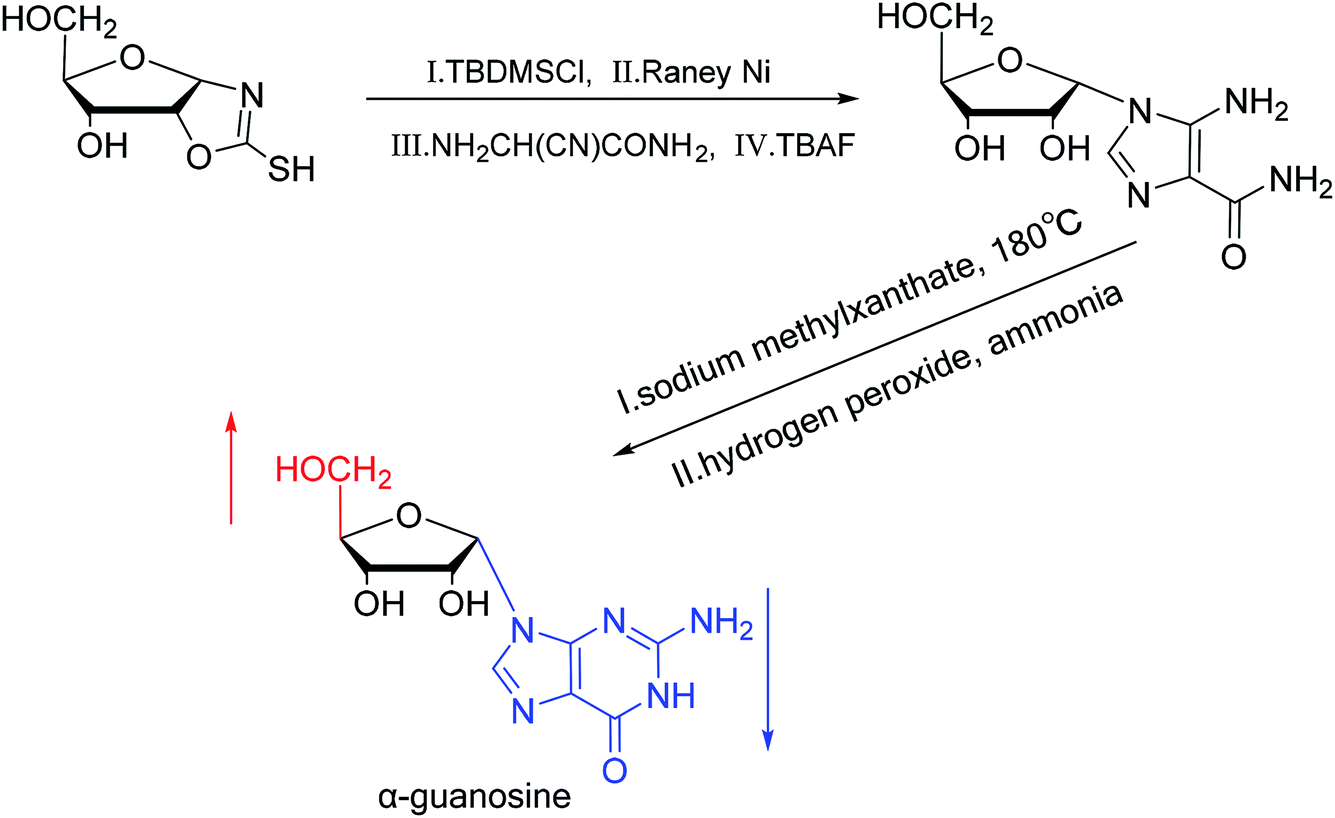 | ||
| Fig. 7 First synthesis of α-guanosine. | ||
Believing that some of the previous methods are somewhat unsuited for the preparation of deoxyguanosines, in 1969, Robins48 et al. first reported a fusion procedure to synthesize α-deoxyguanosine, which shown in Fig. 8. They used 2-fluoro-6-benzyloxypurine and 1,3,5-tri-O-acetyl-2-deoxyribose as raw materials for acid-catalyzed fusion reactions, and the obtained product was treated with methanol ammonia at 80 °C. After catalytic hydrogenation by palladium, they obtained β-deoxyguanosine and α-deoxyguanosine in 14% and 16% yields, respectively. In addition, in 1993, Morvan49 et al. proposed two new methods for the synthesis of α-deoxyguanosine derivatives. One method used 2-N-palmitoyl-guanine and 3′5′-di-O-acetyl-4-N-benzoyl-2′-deoxycytidine as starting materials to carry out transglycosylation reaction in the presence of Lewis acid and silylating agent, and after deprotection, 2-N-palmitoyl-α-deoxyguanosine is obtained. In the second method, silylated guanine and 1-O-acetyl-3,5-di-O-p-nitrobenzoyl-2-deoxyribose were glycosylated under phase transfer conditions. And with deprotection after acylation of guanine, they finally obtained 2-N-isobutyryl-α-deoxyguanosine.
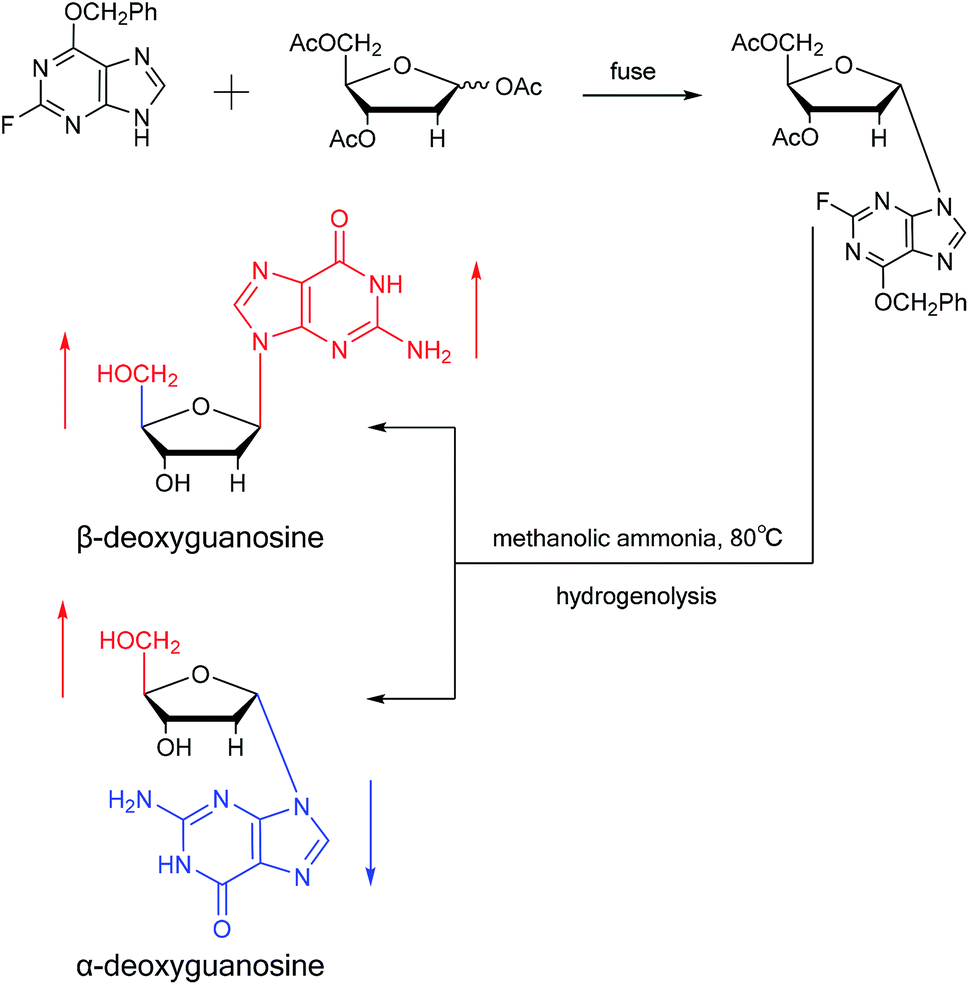 | ||
| Fig. 8 First synthesis of α-deoxyguanosine. | ||
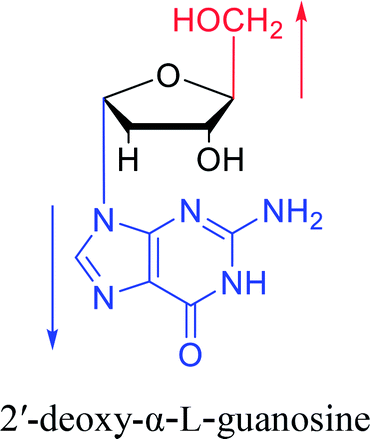
Furthermore, due to steric hindrance, as illustrated in Table 4, α-guanosine derivatives are rarer than other nucleosides.
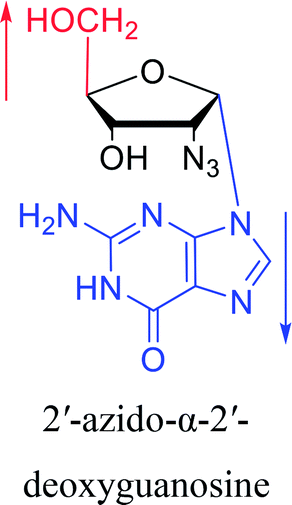
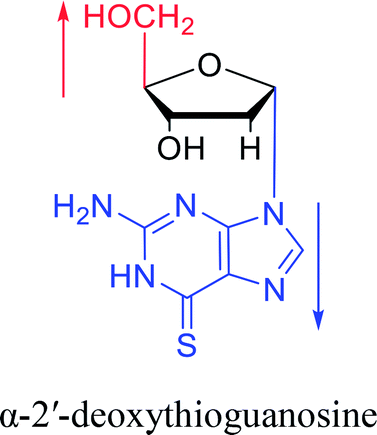
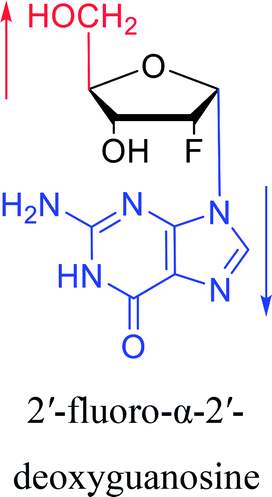
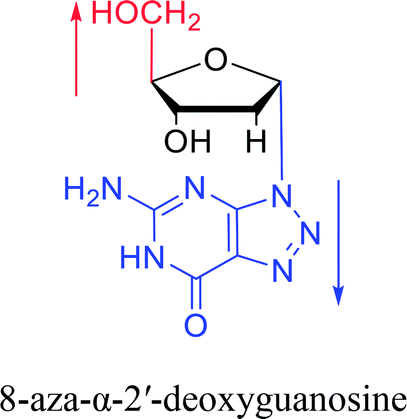
3.4. α-C & α-dC
Sanchez52 et al. first synthesized α-cytidine in 1970 (Fig. 9). They reacted D-ribose and cyanamide in aqueous solution to give the corresponding aminooxazoline derivative. The derivative was reacted with cyanoacetylene to synthesize α-cytidine for the first time. Due to found that the previous synthesis method of α-nucleoside had the problem of stereo selection, in 1994, Sawai53 et al. improved this method to stereoselectively synthesize α-nucleosides based on Sanchez et al.'s research. They not only synthesized α-cytidine and α-deoxycytidine, but also obtained α-thymidine in 10% yield. In this process, no β-nucleosides were synthesized.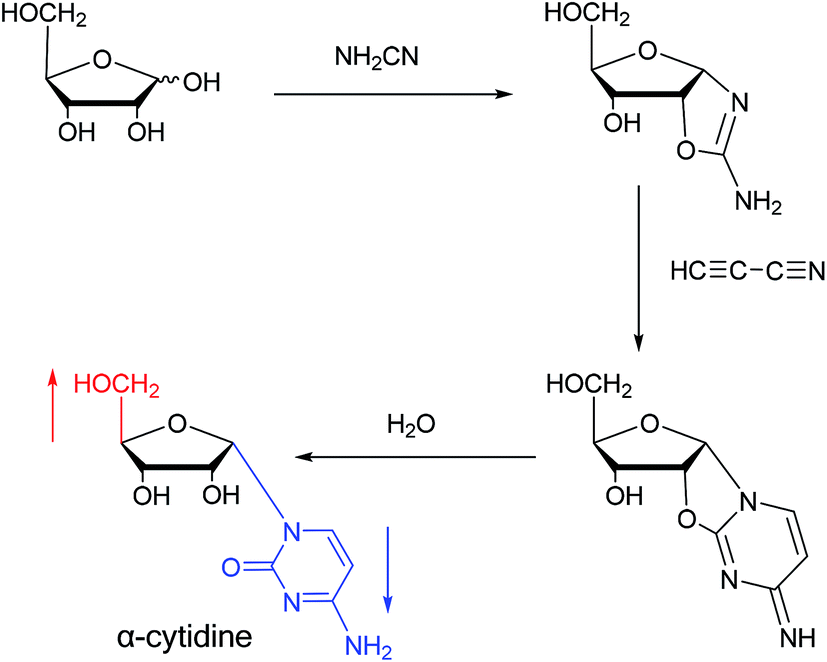 | ||
| Fig. 9 First synthesis of α-cytidine. | ||
Additionally, in 1961, as illustrated in Fig. 10, Fox54 et al. reported the synthesis of α-deoxycytidine using the mercuri procedure. They condensed 3,5-di-O-(p-chlorobenzoyl)-2-deoxy-D-ribosyl chloride with mercuri-N-acetylcytosine to obtain deoxycytidine derivatives. After deacylation with alcoholicammonia, the α- and β-deoxynucleosides were synthesized, which was the first synthetic α-deoxycytidine. The α-cytidine derivatives are summarized in Table 5.
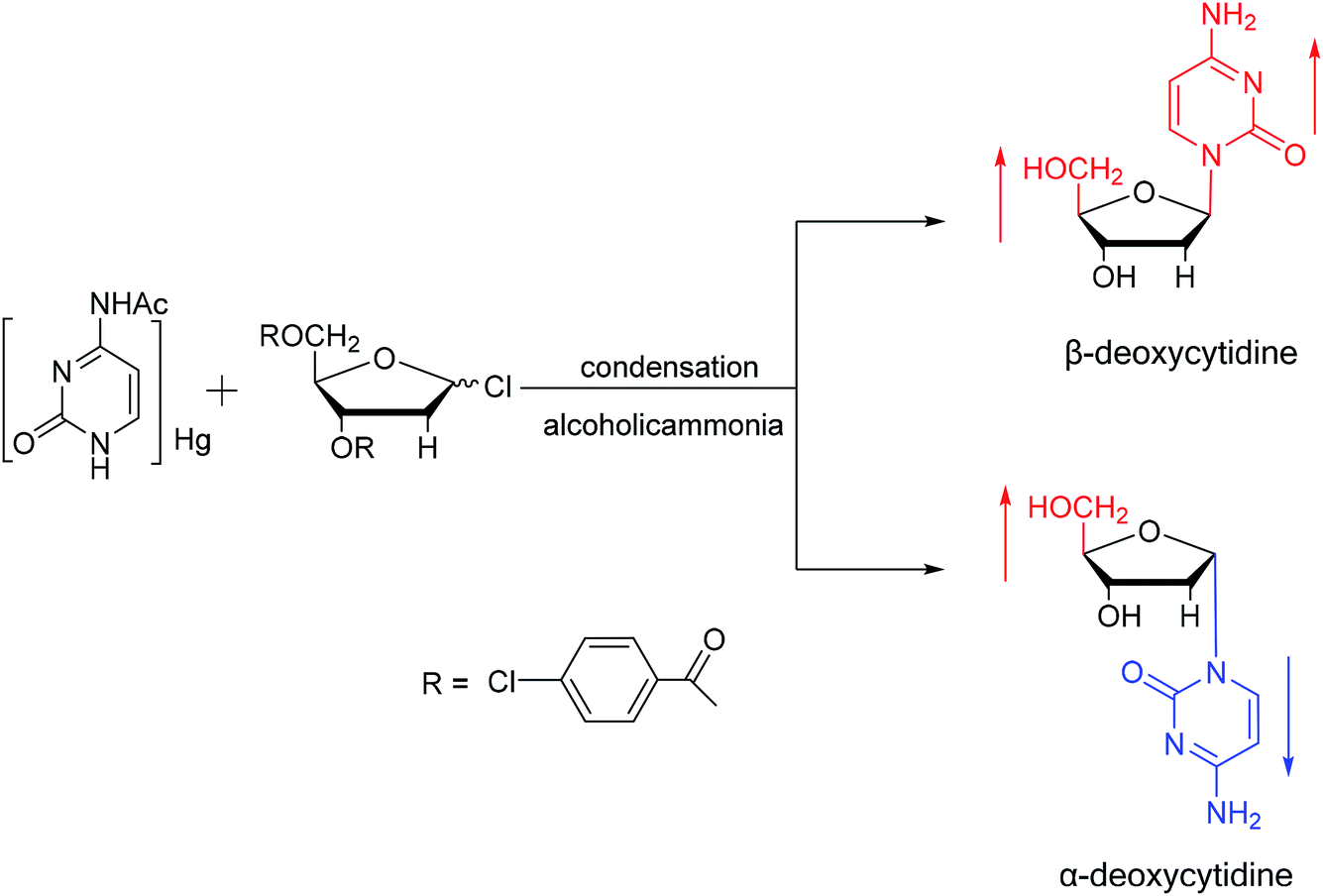 | ||
| Fig. 10 First synthesis of α-deoxycytidine. | ||
| The structure and name of α-cytidine derivatives | Time | Author | The structure and name of α-cytidine derivatives | Time | Author |
|---|---|---|---|---|---|
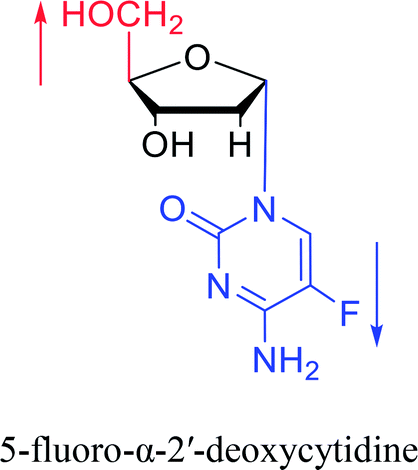 |
1966 | Duschinsky55 et al. | 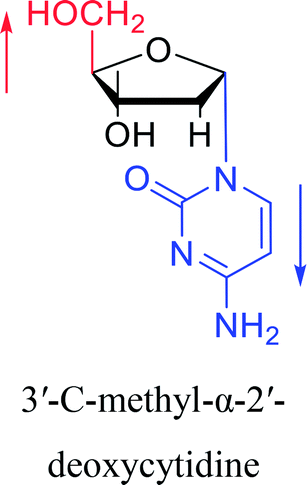 |
1969 | Walton56 et al. |
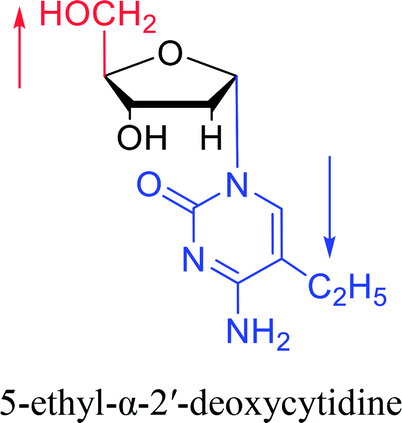 |
1974 | Kulikowski26 et al. | 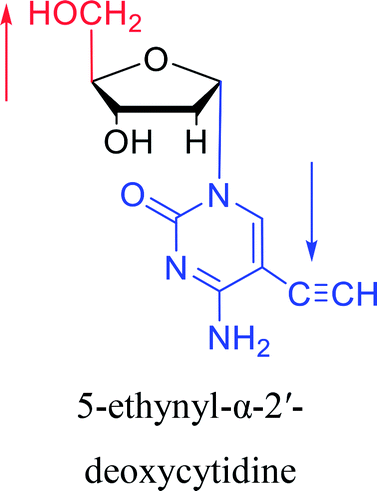 |
1978 | Barr30 et al. |
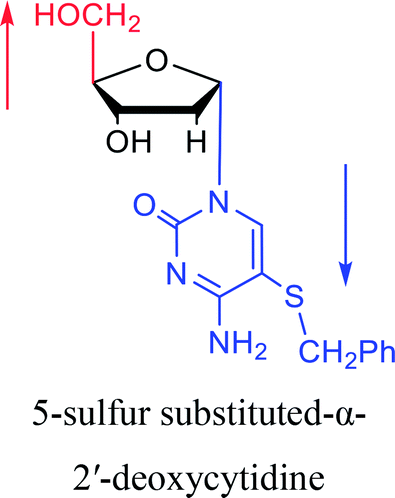 |
1983 | Solan57 et al. | 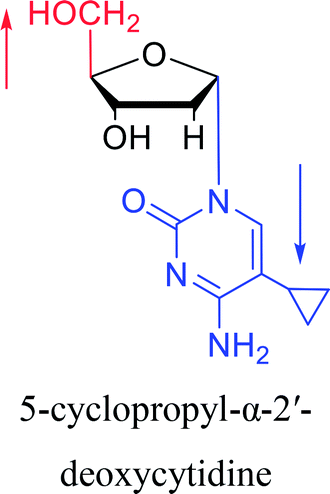 |
1992 | Peters58 et al. |
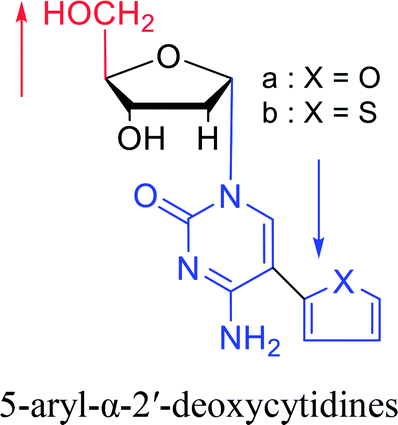 |
1992 | Peters58 et al. | 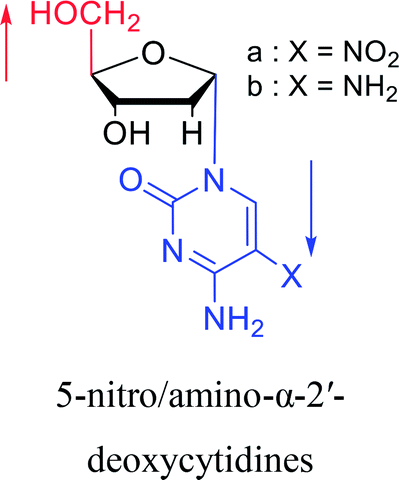 |
2003 | Colacino39 et al. |
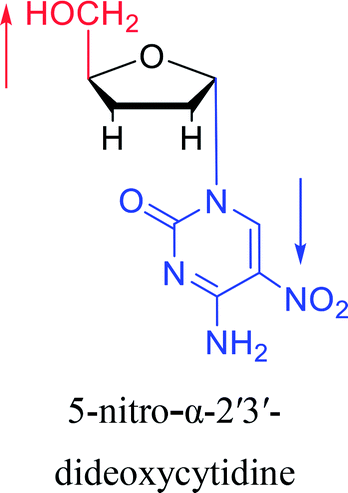 |
2003 | Colacino39et al. | 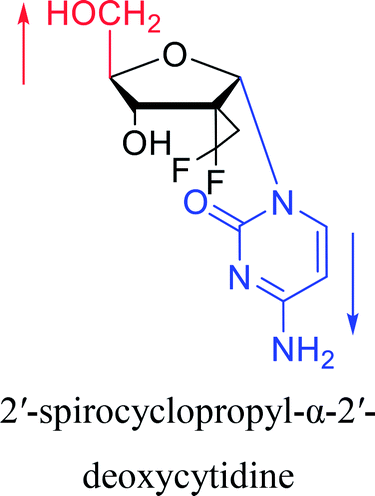 |
2016 | Liu43 et al. |
Interestingly, in 1978, Imazawa59 et al. proposed a novel method for the synthesis of α-nucleoside derivatives. They used trimethylsilyl trifluoromethanesul-fonate (TMS-triflate) and bis(trimethylsilyl)-acetamide (BSA) as catalysts and reacted 3′-azido-3′-deoxy-5′-O-acetylthymidine with silylated N6-octanoyladenine to obtain α-3′-azido-2′,3′-dideoxyadenosine. Through a similar reaction, they also obtained the α-anomer of 9-(3′-azido-2′,3′-dideoxy-D-ribofuranosyl)guanine. Referring to this transglycosylation reaction, Yamaguchi60 et al. reported a new method for the synthesis of α-nucleosides in 1984 (Fig. 11). They used TMS-triflate and BSA to induce the self-anomerization of β-nucleosides to α-nucleosides. Specifically, they reacted 3′,5′-di-O-acetyl-N4-benzoyl-2′-deoxycytidine with TMS-triflate and BSA in dry acetonitrile at 70 °C. After saponification, α-deoxycytidine was synthesized in 41% yield. Using a similar synthetic pathway, they also synthesized α-deoxyadenosine and α-deoxythymidine at yields of 33% and 28%, respectively. Referring to this reaction, in 2002, Sato61 et al. used TMS-triflate to treat β-thymidine derivatives in acetonitrile solution, after deprotection, they achieved epimerization of β-thymidine and obtained α-thymidine in an overall yield of 50% from β-thymidine.
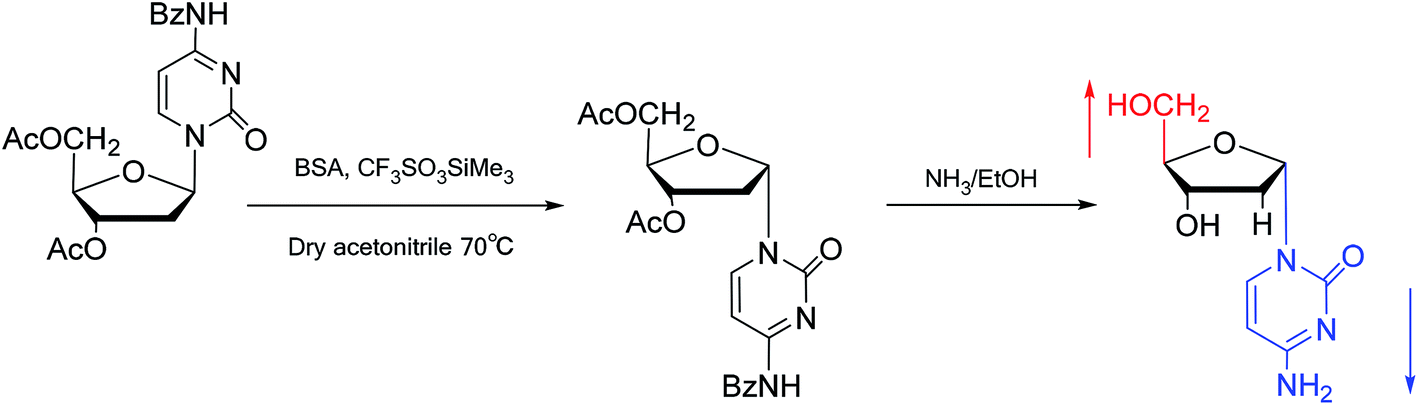 | ||
| Fig. 11 Self-anomerization of β-nucleosides to α-nucleosides. | ||
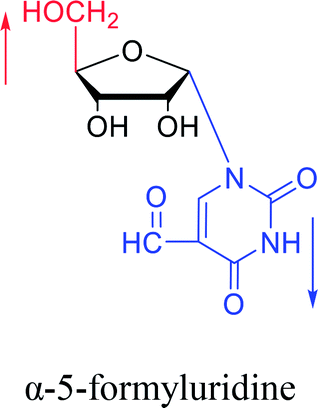
In fact, self-anomerization occurring in nucleoside derivatives are not rare. For example, in 1976, Armstrong28 et al. found that after treatment with strong base, part of the β-5-formyluridines were anomeric to α-5-formyluridine. And in 1986, Seela62 et al. accidentally found that after treatment with 1 M aqueous hydrochloric acid, β-2-deoxy-2-methoxy tubercidin was isomerized by a transglycosylation reaction to obtain its α-anomer in a yield of 13%. Similarly, in 1993, Ward63 et al. pointed out that when a mixture of sulfuric acid and acetic anhydride was added to a solution of β-3′,5′-di-O-acetylthymidine in acetonitrile, rapid isomerization occurred to establish a balance of α:β = 2:1 in a few minutes. They speculated that this catalytic effect was caused by the CH3CO– group.
Furthermore, classic Vorbrüggen glycosylation can also be used to synthesize α-nucleosides. In 1994, Janardhanam64 et al. protected the nucleobase with trimethylsilyl and protected deoxyribose with p-methylbenzoyl. They adopted Vorbrüggen glycosylation to synthesize all five α-nucleoside or α-nucleoside derivatives by the catalysis of SnCl4. At the same time, they found that carrying the reaction out in the presence of ten equivalents of SnCl4 resulted in the highest production of α-nucleoside (α:β = 75:25). Additionally, in 1997, as shown in Fig. 12, Wang65 et al. showed that Vorbrüggen glycosylation was guided by benzoate at the C2 position of arabinose to achieve stereoselective synthesis of α-nucleosides derivatives. After photoinduced electron-transfer (PET) deoxygenation and deprotection, they synthesized α-dA, T, dG and dC.
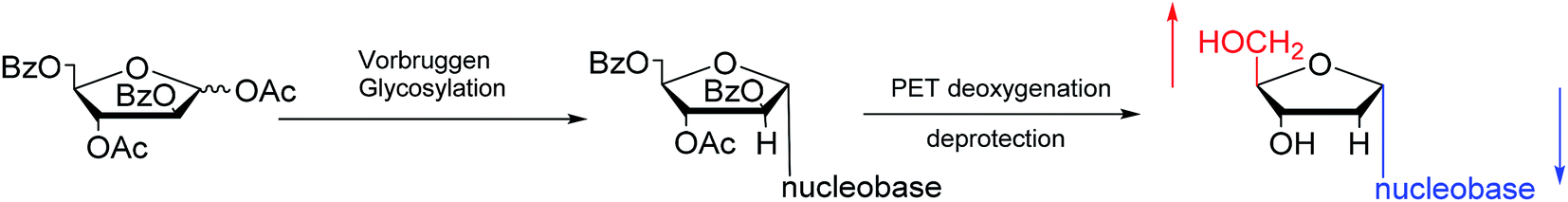 | ||
| Fig. 12 Stereoselective synthesis of α-nucleosides by Vorbrüggen glycosylation. | ||
As we talk about stereoselective synthesis, it should be noted that in 1969, when Kotick24 et al. synthesized 5-s-substituted-α-2′-deoxyuridines, they found that when trimethylsilyl chloride was present during the condensation process, it was more favorable for the synthesis of α-uridine derivatives, while under the condition of removing trimethylsilyl chloride, the main product was the β-configuration. And they suspected that this might be caused by the exchange of chlorides.
Notably, Garcia66 et al. found that Pseudomonas cepacia lipase (PSL-C) is highly chemo- and regio-selective for the 3′ position of the β-2′-deoxynucleoside derivative, displaying opposite selectivity for the 5′-position of the corresponding α-anomer. They considered this to be an effective method for separating α and β-2′-deoxynucleosides. In fact, they had separated an α and β-mixture of thymidine derivatives from industrial waste streams in 2006.
In summary, as shown in Fig. 13, we mainly compiled the first reports of the syntheses of various α-nucleosides, it was clear that α-nucleosides were originally only by-products of the nucleoside synthesis process, but with the discovery of various unique properties of α-nucleosides, which will be discussed in the next section, the synthesis of α-nucleosides has also begun to attract the attention of researchers. In the early stage of α-nucleoside synthesis research, as the above-mentioned first synthesis of α-A/dA, the main method is condensation reaction under mercury protection. After realizing that the presence of mercury affects the biological properties of nucleosides, researchers have begun to replace mercury with other protecting groups such as acetyl, benzoyl, halogen, and so on. With the deepening of the study, the researchers found that under the function of certain chemicals such as TMS-triflate and BSA, the self-anomerization of β-nucleosides can be achieved to obtain the corresponding α-nucleosides. Later, since the previous synthetic methods of α-nucleoside were found to generally obtain a large number of β-nucleosides as by-products while synthesizing α-nucleosides, the researchers proposed some methods like oxazoline method and Vorbrüggen glycosylation to stereoselectively synthesize α-nucleosides. Unfortunately, to date, there is no systematic method for synthesizing all α-nucleosides, which we believe should be one of the directions for future research.
 | ||
| Fig. 13 First synthesis of various α-nucleosides. | ||
4. Property and application of α-nucleosides
While synthesizing α-nucleosides, researchers also investigated their properties. Because of the unique structure of α-nucleosides, some of their properties differ from those of β-nucleoside apart from their optical rotation. Although nearly all studies are in the laboratory stage, it is undeniable that α-nucleosides have potential application value in some fields. Here, we summarize the main unique properties of α-nucleosides, for example, their higher stability compared to the β-anomer, specific parallel double-stranded structure of α-β duplex, inhibition of tumors, bacteria and malaria parasites, and other biological properties.4.1. Enzymatic stability of α nucleosides and α-oligonucleotides
It is generally thought that α-nucleosides are more stable than natural beta nucleosides, as indicated by their resistance to various enzymes. In 1961, when synthesizing 2′-deoxycytidine and its α-anomer, Fox54 et al. found that when 2′-deoxycytidine was deaminated and enzymatically hydrolyzed into uracil by nucleoside deaminases and nucleosidases in resting cell suspensions of Escherichia coli B, its α-anomer was inert to these reactions. Similarly, the α-anomer of thymidine was resistant to glycosyl cleavage by E. coli B nucleosidases. In 2008, Hatano67 et al. found that α-thymidine was not destroyed when β-thymidine and its derivatives were converted to the 1-phosphate form and a free thymine nucleobase under the function of thymidine phosphorylase (TP). They suggested that this was because the nucleobase of the α form did not fit into the pocket of TP. And in 1974, Sequin68 et al. used four dinucleoside monophosphates βT-βT, αT-βT, βT-αT and αT-αT as substrates for snake venom phosphodiesterase and spleen phosphodiesterase. The results showed that although all four compounds are substrates for both enzymes, if enzymic attack occurred in the α-nucleoside portion of the dinucleoside monophosphate, the rate of the hydrolysis was quite slow. In 1987, Morvan69 et al. used d(CATGCG) as a substrate for endonucleases and exonucleases. The α-hexamer remained nearly intact when the natural β-hexamer was completely degradated by nuclease S1 and calf spleen phosphodiesterase. Also in 1987, Thuong70 et al. showed that α-oligothymidylates are much more resistant to endonucleases than their β analogs, while additional protection against the corresponding exonuclease was provided by acridine incorporation. They suggested that this was because insertion of the acridine ring formed a miniduplex structure and provided additional binding energy, which strongly stabilized the double strand with a complementary sequence. In addition, since β-oligodeoxynucleotides (fully β-modified oligonucleotides) were rapidly destroyed by serum enzymes and degraded in cells, in 1988, Bacon71 et al. synthesized and studied the degradation process of α-oligodeoxynucleotides (fully α-modified oligonucleotides) in different media such as rabbit reticulocyte lysates. The results suggested that, in contrast to β-oligodeoxynucleotides, the degradation of α-oligodeoxynucleotides in various media was quite slow, even without degradation, which indicated the strong enzyme stability of α-oligodeoxynucleotides. Because of the resistance of α-nucleosides and α-oligonucleotides to these enzymes, they may be useful as gene control agents.4.2. Parallel double helix
In general, the naturally occurring duplexes DNA or RNA follow the Watson–Crick model, and the two strands of the double helix are in an antiparallel relationship with each other, and this rule also applies to α-duplexes. In fact, in 1987, Morvan72 et al. have pointed out that α-[d(CATGCG)] can form an antiparallel double-stranded structure with complementary α-oligodeoxynucleotide. However, differences are observed when the hybridization occurs between the α-oligonucleotide and the β-oligonucleotide. Also in 1987, Sun73 et al. synthesized an α-oligodeoxyribonucleotide with the sequence 5′d(TCTAAACTC)3′, and used a pentamethylene linker to link a 9-amino acridine derivative with its 5′-phosphate. They also synthesized two β-oligodeoxyribonucleotides, containing complementary sequences in the 5′ → 3′ or 3′ → 5′ orientation, respectively. As illustrated in Fig. 14, the results indicate that an α-oligodeoxyribonucleotide can form a double helix structure with an β-oligodeoxyribonucleotide, and interestingly, they adopted a parallel 5′ → 3′ orientation. And in the same year, Morvan74 et al. also confirmed that the α-oligodeoxynucleotides, α-d[CATGCG] can form parallel double-stranded structure with complementary β-oligodeoxynucleotides. Similarly, in 1988, Praseuth75 et al. also demonstrated that α-octathymidylate can form a parallel double-stranded structure with complementary β-oligodeoxynucleotide. In addition, they found that α-octathymidylate can be incorporated into complementary DNA duplexes to form a local triple helix structure which parallel to the complementary adenine strand. And in 1991, because homopyrimidine oligonucleotides can recognize the base pair sequence of double helix DNA and bind to the homopurine, Sun76 et al. synthesized 11-mer α- and β-oligodeoxynucleotides with the sequence 5′-d(TCTCCTCCTTT)-3′ and bound them to the corresponding DNA duplex to form a local triple helix. Here, the α-oligodeoxynucleotide has an anti-parallel structure with the homopurine strand, and the β-oligodeoxynucleotide adopted a parallel orientation. They believe that the formation of Hoogsteen hydrogen bonds in the triple helix is responsible for the anti-parallel structure of the α-oligonucleotides to the homologous strand. Furthermore, In 1992, Debart77 et al. synthesized an α-oligoribonucleotide, α-[r(UCUUAACCCACA)], consisting entirely of α-ribonucleotides, this oligoribonucleotide with highly nuclease-resistant can form a 2 α-RNA:1 β-DNA triple helix structure with complementary β-DNA sequence, wherein both α-RNAs are parallel to β-DNA. In addition, this α-RNA also exhibits inhibition of the de novo HIV-1 infection in cells without sequence specific.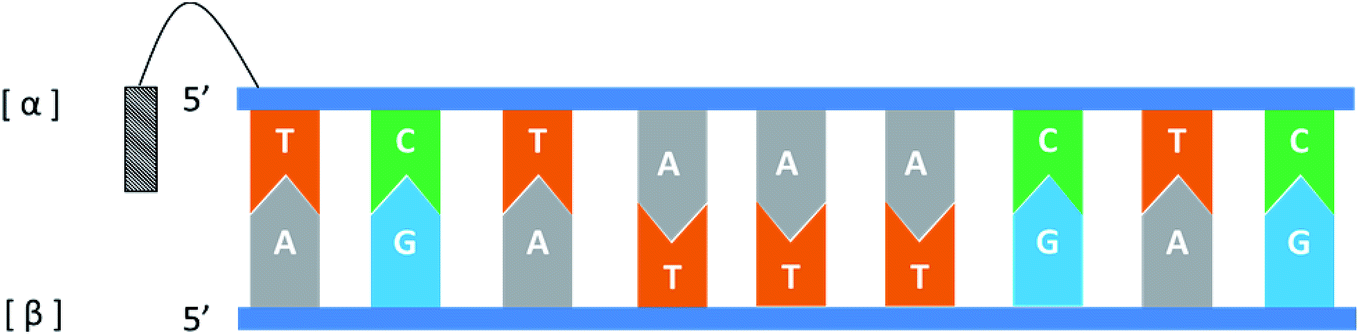 | ||
| Fig. 14 An α-oligodeoxyribonucleotide forms a parallel double strand with the β-oligodeoxyribonucleotide. | ||
Interestingly, in 1995, Koga78 et al. used synthetic 5′-phosphoramidite derivatives of α-deoxynucleoside and commercial 3′-phosphoramidite derivatives of β-deoxynucleoside as raw materials, and adopted standard solid-phase synthesis methods to construct α-containing oligodeoxynucleotides with alternating α-deoxynucleosides and β-deoxynucleosides, which have strong resistance to phosphatase. In recognition of the complementary native β-oligodeoxynucleotide, the sequence specificity of hybrid strand was similar to that of the native oligonucleotide, even though the thermal stability of the duplex which containing the hybrid strand is lower than the native DNA duplex. Furthermore, compared to the both strands are hybrid strands, CD spectrum confirmed that the helicity of a duplex containing only one hybrid strand is more similar to the native DNA. It is worth mentioning that this hybrid strand can also form a duplex with a complementary β-oligoribonucleotide, but the thermal stability is significantly lower than that of the hybrid strand: β-oligodeoxynucleotide complex.
It should be clear that this parallel double helix structure was limited to regions containing α-nucleotides and unusual phosphodiester linkages; in fact, the α-containing duplexes could form an approximately antiparallel double-stranded structure except for the position of the α-nucleoside,79 which shown in Fig. 15.
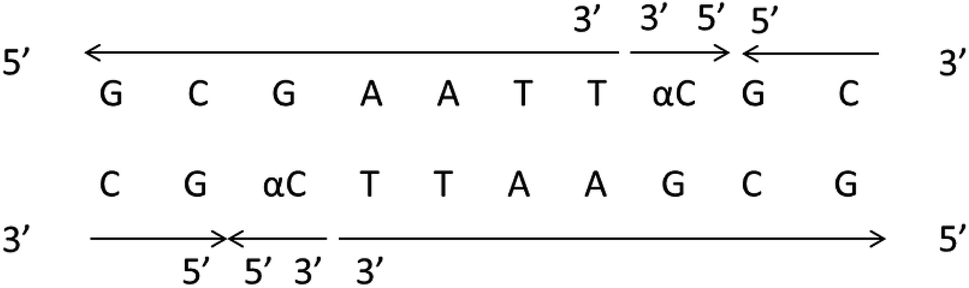 | ||
| Fig. 15 α-Containing duplexes can form an overall anti-parallel β-DNA structure, the interference limited to the regions containing α nucleotides and unusual phosphodiester linkages. | ||
Furthermore, in 1987, Gagnor80 et al. hybridized α and β-anomer d(G2T12G2) oligodeoxyribonucleotides to rA12 and compared their properties. They observed melting temperatures of 27.4 °C for β-oligodeoxyribonucleotide/RNA hybrid and 52.8 °C for α-oligodeoxyribonucleotide/RNA, indicating that the double strand containing the α-nucleoside has a higher melting temperature. Interestingly, in 1989, Paoletti81 et al. studied the melting temperatures of three sequences, α-d(CCTTCC): β-d(GGAAGG), β-d(CCTTCC): β-d(GGAAGG) and α-d(GGAAGG): β-d(CCTTCC), and found values of 28.1 °C, 20.2 °C, and 13.8 °C, respectively. These results suggest that the properties of the α sequence greatly impact the stability of the double strand.
4.3. Biological properties
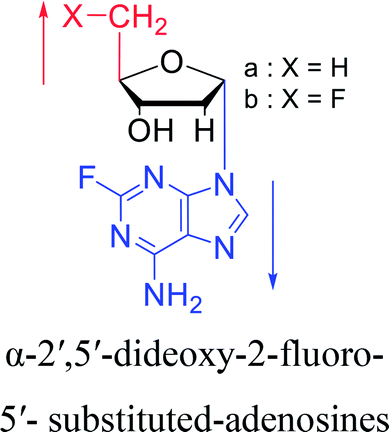
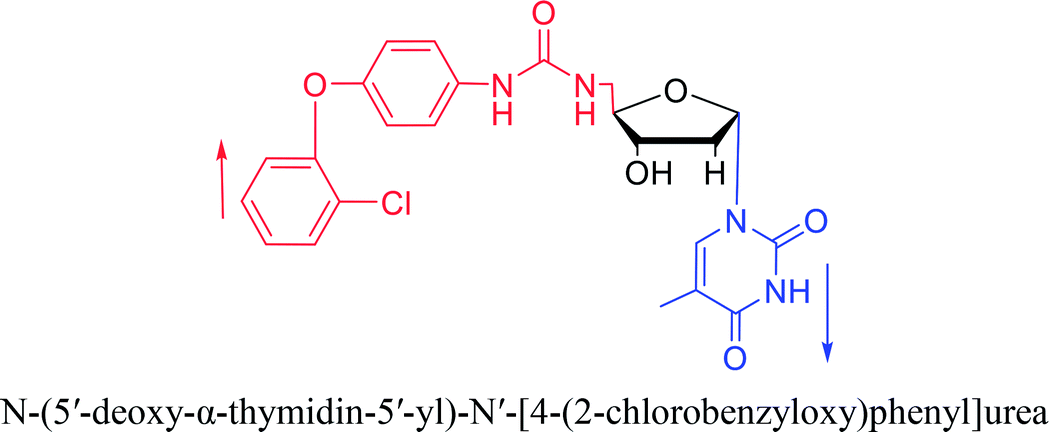
Interestingly, α-nucleosides and their derivatives also have a variety of specific biological properties, such as the anti-tumor, anti-bacterial, anti-malarial effects, inhibition of RNase H and potential for antisense therapy.In 1972, Christensen13 et al. pointed out that although the inhibitory activity is lower than the corresponding β-nucleoside derivative, 2-chloro-α-2′-deoxyadenosine and 2-methoxy-α-2′-deoxyadenosine significantly inhibits leukemia L1210 cells, in addition, 2-hydrazino-α-2′-deoxyadenosine and 2-chloro-α-2′-deoxyadenosine also showed growth inhibition of Escherichia coli and Streptococcus faecium, respectively. And In 1975, Bobek82 et al. discovered that α anomers of 4′-thio-5-fluorouridine has similar properties. Moreover, in 1979, Acton50 et al. proposed that α-2′-deoxythioguanosine has antitumor (lymphosarcoma) activity. Importantly, this compound is less toxic to the bone marrow than the corresponding β anomer, and thus higher concentrations can be used to achieve higher efficacy. Furthermore, in 1993, when studying the inhibitory effect of nucleoside derivatives on tumors, Bretner34 et al. found that although the α-anomer was less active than the corresponding β-anomer, it showed higher selectivity for tumor cells. Additionally, in 1998, Townsend83 et al. synthesized a series of 2-substituted derivatives of 5,6-dichloro-1-(α-L/D-lyxofuranosyl)benzimidazole and 1-(5-deoxy-α-L/D-lyxofuranosyl)-5,6-dichloro-benzimidazole and studied their inhibition of human cytomegalovirus (HCMV) and herpes simplex virus type 1 (HSV-1). The results indicated that all of these derivatives had no or only weak inhibition on HSV-1, while for HCMV, most derivatives such as 2-isopropylamino or benzylthio derivatives had only weak inhibition, however, 2-halogen derivatives have significant inhibitory effects on the Towne strain of HCMV, especially 5-deoxy α-L-analogues exhibit the strongest inhibitory effect. And in 2007, Van Daele41 et al. synthesized a series of 5′-thiourea-substituted α-thymidine derivatives and found that the derivative with a (3-trifluoromethyl-4-chlorophenyl)thiourea moiety significantly inhibited thymidine monophosphate kinase, and thus strongly inhibited growing Mycobacterium bovis (MIC99 = 20 μg mL−1) and Mycobacterium tuberculosis (MIC50 = 6.25 μg mL−1) strains. Based on these results, Cui42 et al. synthesized the α-anomer of 3′-azido-2′,3′-dideoxythymidine in 2010, which significantly inhibited the growth of Plasmodium species with EC50 values in the micromolar range. Later, in 2012,44 they synthesized a series of thymidine analogues that showed anti-malarial effects by inhibiting Plasmodium falciparum thymidylate kinase (PfTMPK). Among them, N-(5′-deoxy-α-thymidin-5′-yl)-N′-[4-(2-chlorobenzyloxy)phenyl]urea was reported to be the most effective inhibitor of P. falciparum growth, with an EC50 value of 28 nM and CC50 of 29 μM. Therefore, α-nucleosides may have development potential as anti-tumor, anti-bacterial, and anti-parasitic drugs.
Furthermore, α-nucleosides strongly inhibit RNase H. In 1988, Bloch84 et al. showed that RNase H, which uses a DNA:RNA duplex as a natural substrate, was inhibited when it bound to a non-physiological α-DNA:β-RNA hybrid. And In 1996, Shinozuka85 et al. conducted a more in-depth study of the inhibitory effects of α-dC12, β-dC12, α-S-dC12, β-S-dC12, α-T15, β-T15, α-S-T15, and β-S-T15 on RNase H(“-S-” stands for a phosphorothioate linkage). The results showed that the α-anomeric-2-deoxycytidylate phosphorothioate was the most potent inhibitor (>40%). In fact, RNase H catalyzes the degradation of the RNA portion of DNA–RNA hybrids, which is an inevitable step in the reverse transcription of retroviruses and thus essential for the replication of retroviruses such as HIV. Therefore, α-nucleosides also show potential as antiretroviral drugs.
To date, there are no reports of α-anomeric lesions in mammals. However, tumor cells can grow and proliferate in hypoxic environments. Radiation treatments create conditions that promote α-anomeric damage. For example, when DNA is subjected to γ-irradiation under anoxic conditions, the α-anomer of 2′-deoxyadenosine (α-dA) is the major product of DNA damage,86 as illustrated in Fig. 16.
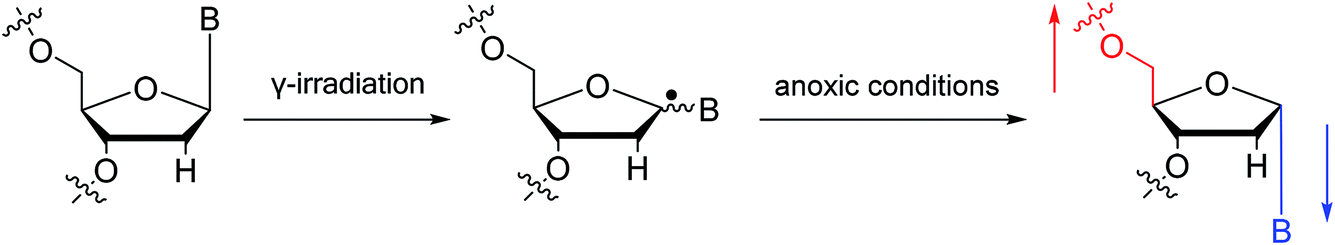 | ||
| Fig. 16 Formation of α-2′-deoxynucleosides when DNA is subjected to γ-irradiation under anoxic conditions. And ‘B’ indicates nucleobase. | ||
In fact, when α-dA is present in a DNA duplex, it is mutagenic and directs the incorporation of dC, dA, or T during replication in vitro, and in vitro. Under in vivo conditions, it will result in the deletions of single nucleotides.87 Even so, α-dA is “gentler” than other α-anomeric lesions. In 2015, Amato88 et al. found that without SOS induction, α-T, α-dG and α-dC nearly completely blocked DNA replication with only 1–3% bypass efficiency, while α-dA only caused medium blockade of replication with a bypass efficiency of 24%. Interestingly, unlike many other damaged DNA nucleobases, the αdA lesion was not repaired by DNA glycosylases and AP lyases, Instead, E. coli endonuclease IV (Nfo) directly incised the phosphodiester bond 5′ to the lesion in DNA. And in 2004, Ishchenko89 et al. pointed out that not only Nfo, but also Saccharomyces cerevisiae Apn1 protein, a homologue of Nfo and human major AP endonuclease 1, are involved in the alternative nucleotide incision repair pathway. However, in 2016, Timofeyeva90 et al. showed that the efficiency of α-dA lesion conversion by APE1 is very low. They suggested that this is because although the α-dA structure promotes enzyme recognition, the formation of the catalytically active complex and hydrolysis of the 5′-phosphodiester bond are greatly hindered. However, the identification and efficacy of enzymes is affected by the flanking sequences of α-dA, as suggested by Johnson87 et al. in 2012. They found that the 5′CαdAG3′ sequence was unique because its KM value was approximately four-fold that of other substrates (5′GαdAC3′, 5′GαdAG3′, 5′CαdAC3′), suggesting a lower affinity with Nfo for the 5′CαdAG3′ duplex compared to that of the other substrates. Furthermore, in 2017, Williams91 et al. pointed out that human polymerase (Pol) η can bypass all α-dN damage and extend primers to generate full-length replication products.
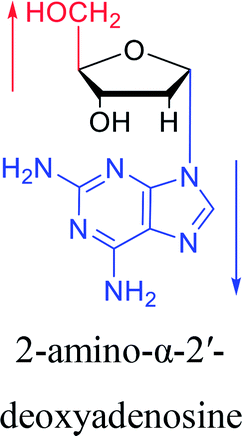
However, although α-anomeric lesions cause genetic damage, they show potential for use in antisense therapy. Antisense nucleotides refer to RNA or DNA molecules capable of precisely complementary to a specific mRNA and specifically blocks the expression of the target gene, resulting in low or no expression. As a gene regulatory factor, it plays an important role in inhibiting the expression of some harmful genes and overexpression of uncontrolled genes. As we mentioned above, α-anomeric lesions can precisely pair with the target sequence and significantly block DNA replication. In fact, in 1995, Boiziau92 et al. have constructed 17-mer oligodeoxynucleotides containing 2, 7, or 12 α-nucleotides, respectively, and studied the inhibitory effects of these hybrids on the reverse transcription of Moloney Murine Leukemia Virus (MMLV). The results showed that these α-containing oligodeoxynucleotides with only 5′ end can inhibit reverse transcription, and the hybrid strand with a longer β region has a stronger inhibitory effect. This suggests that oligonucleotides containing α-nucleotides have the potential to be retroviral antisense therapeutics. It should be noted that purely α-oligonucleotide (the entire strand is composed of α-nucleotides) is not suitable for the field of antisense therapy. Because in 1996, Aramini93 et al. pointed out that although purely α-oligonucleotide can form stable structures with their targets and are not degraded by nucleases, these complexes are RNase H resistant. Unlike purely α-anomeric, the oligodeoxynucleotides incorporating a single α-nucleoside have RNase H activity. Thus, these oligodeoxynucleotides show ideal properties of antisense nucleotides as proposed by Germann79 et al. in 1997, including (1) highly nuclease resistant; (2) capable of forming a stable complex with its messenger RNA target; and (3) sensitive to RNase H. Thus, α-nucleosides have potential value for use in antisense therapy. And in 1997, Aramini94 et al. constructed four self-complementary DNA decamer duplexes [GCGAATTCGC] and inserted αA, αT, αG and αC into different locations in the strand in the opposite direction through 3′ → 3′ and 5′ → 5′ phosphodiester linkages. They demonstrated that all of these DNA sequences formed stable duplexes, and the structure of the four double-strands was similar to the control β–β double strand. The double-strands containing αA, αT, and αG showed similar thermal stability values as the control, and αC insertion had the most deleterious effects on thermodynamic and structural properties. Therefore, the type, content and location of α-nucleosides are important for the rational design of antisense molecules. It is worth mentioning that in 1998, Morvan38 et al. synthesized 5-propynyl-α-oligodeoxynucleotides mainly containing deoxyuridine and deoxycytidine, and they confirmed that these oligodeoxynucleotides can form more stable duplexes with the corresponding DNA or RNA. Similarly, in 2002, Naval16 et al. synthesized 2-amino-α-2′-deoxyadenosine and incorporated it into the methoxyethylphosphoramidate α-oligodeoxynucleotides, which made the α-oligodeoxynucleotides to form more stable complexes with the target RNA. And in 2010, Morvan95 et al. also concluded that modified, especially cationically modified α-oligonucleotides can form more stable double-strands or three-stranded structure with target RNA or DNA than modified β-oligonucleotides. And these backbone-modified α-oligonucleotides are also considered to have potential for application in the field of antisense therapy due to their good selectivity to targets, high nuclease resistance and other properties.
4.4. Other properties
In addition to the various properties mentioned above, some other properties such as higher affinity for protons, unique fluorescent properties and reasons for selection of β-RNA need to be mentioned.In 2005, Müller96 et al. found that some nucleoside derivatives such as imidazole nucleosides can form stable complexes with metal ions like silver ions. On this basis, they conducted a similar study on the α-anomers of these nucleoside derivatives in 2007.97 The results showed that the α-nucleoside derivatives had significantly higher affinity for protons than their β-anomers. Similarly, the metal-ion complexes of the α-anomers were more stable than their β-anomers. Thus, α-nucleosides have the potential to be developed into nucleoside-based nanostructures and devices. And in 2017, Seela98 et al. constructed a silver-mediated base pair by using α-dC and β-dC. They found that compared to the β–β duplex (Tm = 34 °C), a 12-mer duplex with α-dC exhibited better thermal stability (Tm = 43 °C). They also found that α-dC has an excellent ability to identify DNA single-nucleotide polymorphism mismatches.
And in 2018, Seela51 et al. prepared a series of 12-mer oligodeoxyribonucleotides incorporating α-anomers of 8-aza-2′-deoxyguanosine 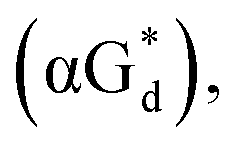 αGd, β-anomers of 8-aza-2′-deoxyguanosine
αGd, β-anomers of 8-aza-2′-deoxyguanosine 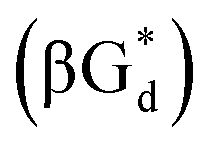 and βGd. They found that oligodeoxyribonucleotides containing
and βGd. They found that oligodeoxyribonucleotides containing 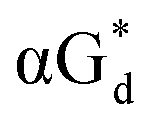 showed high fluorescence at pH (8.0). However, the fluorescence changed when a duplex DNA was formed with A, T, G, and C, and the duplex containing the
showed high fluorescence at pH (8.0). However, the fluorescence changed when a duplex DNA was formed with A, T, G, and C, and the duplex containing the 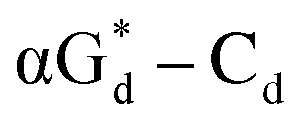 base pair showed the strongest fluorescence decrease. They also found that decreased fluorescence corresponded to a higher Tm value, and the duplex containing
base pair showed the strongest fluorescence decrease. They also found that decreased fluorescence corresponded to a higher Tm value, and the duplex containing 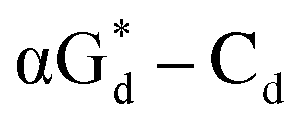 showed better thermal stability than αGd–Cd. Additionally, the results showed that the α-anomers were more efficient than β-nucleosides for mismatch distinction.
showed better thermal stability than αGd–Cd. Additionally, the results showed that the α-anomers were more efficient than β-nucleosides for mismatch distinction.
Interestingly, in 1997, Sawai99 et al. discussed the selective advantages of β-RNA compared to α-RNA. They suggested that during the process of chemical evolution, both α- and β-glycosidic nucleosides could be formed. However, they found that under the same conditions, the yield and chain length of the β-oligomer were much higher than those of the corresponding α-oligomer. Therefore, they hypothesized that a possible explanation for the selectivity advantage of β-RNA is that the formation of β-oligomers is easier than that of α-oligomers. Additionally, depending on changes in temperature, forming a ternary structure of single-stranded oligoribonucleotides more easily and better conformational flexibility may be important factors in the selection of β-RNA over α-RNA.
5. Conclusion
In summary, since Kaplan proposed the concept of the α-configuration in 1955, only five α-nucleosides or derivatives present in nature have been reported. A series of α-nucleosides and derivatives were artificially synthesized by methods such as the mercuri procedure, fusion reactions, self-anomerization or Vorbrüggen glycosylation. Additionally, some unique properties of α-nucleosides, including the stability or inhibition of enzymes, parallel double-stranded structure of α–β duplex, fluorescent properties, and inhibition of tumors, bacteria and Plasmodium, suggest their application prospects. These molecules may be useful in gene regulation, antiretroviral drugs, antitumor drugs, and genetic mismatch identification, among others. It is conceivable that more applications of α-nucleosides have not yet been discovered. For example, because of the special spatial conformation of α-nucleosides, whether they achieve base pairing in a new manner that differs from the traditional Watson–Crick principle requires further evaluation. Additionally, studies are needed to determine whether α-nucleosides can form structures such as G-quartets or C-pentamers like their β-anomers. Although the antitumor and antiretroviral effects of α-nucleosides have been confirmed, they have not been used in clinical treatment. Therefore, as a drug or drug carrier, α-nucleosides may have broad applications in the future. Furthermore, the use of β-guanosine in the field of supramolecular hydrogels has made considerable progress,100 but no studies have reported the properties of hydrogels constructed from α-guanosine. Considering the better stability of α-nucleosides against external conditions such as enzymes and temperature, if α-nucleosides can be used to form supramolecular hydrogels, they can be used in more extreme environments. In conclusion, through multidisciplinary integration, including the fields of biochemistry, materials science, and medicine, additional properties of α-nucleosides will be identified to broaden their application prospects. Finally, if we inadvertently miss some important research by any scholars, we sincerely apologize for this, and we hope that readers will point these omissions out, which will help us further summarize the α-nucleosides in the future.Conflicts of interest
There are no conflicts to declare.Funding
National Natural Science Foundation of China (81621062).Acknowledgements
The authors gratefully acknowledge support from the National Natural Science Foundation of China (81621062, 81500860, 81700988) and Outstanding Young Scholar Fund (20826041A4403) from the Sichuan University for financial support.References
- U. Sequin, Experientia, 1973, 29, 1059–1062 CrossRef CAS PubMed.
- N. O. Kaplan, M. M. Ciotti, F. E. Stolzenbach and N. R. Bachur, J. Am. Chem. Soc., 1955, 77, 815–816 CrossRef CAS.
- S. Suzuki, K. Suzuki, T. Imai, N. Suzuki and S. Okuda, J. Biol. Chem., 1965, 240, P554–P556 Search PubMed.
- R. Bonnett, Chem. Rev., 1963, 63, P573–P605 CrossRef.
- H. G. Gassen and H. Witzel, Biochim. Biophys. Acta, 1965, 95, 244–250 CrossRef CAS.
- F. Dinglinger and P. Renz, Hoppe-Seyler's Z. Physiol. Chem., 1971, 352, 1157–1161 CrossRef CAS PubMed.
- R. S. Wright, G. M. Tener and H. G. Khorana, J. Am. Chem. Soc., 1958, 80, 2004–2006 CrossRef CAS.
- G. Schramm, G. Lunzmann and F. Bechmann, Biochim. Biophys. Acta, 1967, 145, 221–227 CrossRef CAS.
- R. K. Ness and H. G. Fletcher, J. Am. Chem. Soc., 1960, 82, 3434–3437 CrossRef CAS.
- J. J. K. Novak and F. Sorm, Collect. Czech. Chem. Commun., 1962, 27, 902–905 CrossRef CAS.
- M. J. Robins and R. K. Robins, J. Am. Chem. Soc., 1965, 87, 4934–4940 CrossRef CAS PubMed.
- M. J. Robins, T. A. Khwaja and R. K. Robins, J. Org. Chem., 1970, 35, 636–639 CrossRef CAS PubMed.
- L. F. Christensen, A. D. Broom, M. J. Robins and A. Bloch, J. Med. Chem., 1972, 15, 735–739 CrossRef CAS PubMed.
- J. B. Hobbs and F. Eckstein, J. Org. Chem., 1977, 42, 714–719 CrossRef CAS PubMed.
- M. Imazawa and F. Eckstein, J. Org. Chem., 1979, 44, 2039–2041 CrossRef CAS.
- M. Naval, T. Michel, J. J. Vasseur and F. Debart, Bioorg. Med. Chem. Lett., 2002, 12, 1435–1438 CrossRef CAS PubMed.
- S. Ye, M. M. Rezende, W. P. Deng, B. Herbert, J. W. Daly, R. A. Johnson and K. L. Kirk, J. Med. Chem., 2004, 47, 1207–1213 CrossRef CAS PubMed.
- G. G. Sivets, E. N. Kalinichenko and I. A. Mikhailopulo, Helv. Chim. Acta, 2007, 90, 1818–1836 CrossRef CAS.
- T. Nishimura, B. Shimizu and I. Iwai, Chem. Pharm. Bull., 1964, 12, 1471–1478 CrossRef CAS PubMed.
- H. Aoyama, Bull. Chem. Soc. Jpn., 1987, 60, 2073–2077 CrossRef CAS.
- B. Shimizu, A. Saito, T. Nishimura and M. Miyaki, Chem. Pharm. Bull., 1967, 15, 2011–2014 CrossRef CAS PubMed.
- H. J. Minnemeyer, P. B. Clarke and H. Tieckelmann, J. Med. Chem., 1964, 7, 567–569 CrossRef CAS PubMed.
- K. J. Ryan, E. M. Acton and L. Goodman, J. Org. Chem., 1966, 31, 1181–1184 CrossRef CAS PubMed.
- M. P. Kotick, C. Szantay and T. J. Bardos, J. Org. Chem., 1969, 34, 3806–3813 CrossRef CAS PubMed.
- G. L. Bubbar and V. S. Gupta, Can. J. Biochem., 1970, 48, 3147–3153 CrossRef CAS.
- T. Kulikowski and D. Shugar, J. Med. Chem., 1974, 17, 269–273 CrossRef CAS PubMed.
- R. A. Sharma and M. Bobek, J. Org. Chem., 1975, 40, 2377–2379 CrossRef CAS PubMed.
- V. W. Armstrong, J. K. Dattagupta, F. Eckstein and W. Saenger, Nucleic Acids Res., 1976, 3, 1791–1810 CrossRef CAS PubMed.
- A. J. Brink, O. G. de Villiers and A. Jordaan, J. Chem. Soc., Perkin Trans. 1, 1977, 1608–1612 RSC.
- P. J. Barr, A. S. Jones, P. Serafinowski and R. Walker, J. Chem. Soc., Perkin Trans. 1, 1978, 1263–1267 RSC.
- H. Griengl, M. Bodenteich, W. Hayden, E. Wanek, W. Streicher, P. Stutz, H. Bachmayer, I. Ghazzouli and B. Rosenwirth, J. Med. Chem., 1985, 28, 1679–1684 CrossRef CAS PubMed.
- M. Bobek, I. Kavai and E. De Clercq, J. Med. Chem., 1987, 30, 1494–1497 CrossRef CAS PubMed.
- P. Wigerinck, C. Pannecouque, R. Snoeck, P. Claes, E. De Clercq and P. Herdewijn, J. Med. Chem., 1991, 34, 2383–2389 CrossRef CAS PubMed.
- M. Bretner, T. Kulikowski, J. M. Dzik, M. Balinska, W. Rode and D. Shugar, J. Med. Chem., 1993, 36, 3611–3617 CrossRef CAS PubMed.
- A. A. Elbarbary, A. I. Khodair, E. B. Pedersen and C. Nielsen, Monatsh. Chem., 1994, 125, 593–598 CrossRef CAS.
- U. Wellmar, A. B. Hornfeldt, S. Gronowitz and N. G. Johansson, Chem. Heterocycl. Compd., 1996, 32, 1312–1318 CrossRef.
- Y. Diaz, A. ElLaghdach and S. Castillon, Tetrahedron, 1997, 53, 10921–10938 CrossRef CAS.
- F. Morvan, J. Zeidler and B. Rayner, Tetrahedron, 1998, 54, 71–82 CrossRef CAS.
- E. Colacino, G. Sindona, G. Gosselin and C. Mathe, Nucleosides, Nucleotides Nucleic Acids, 2003, 22, 2013–2026 CrossRef CAS PubMed.
- M. Ogino, Y. Yoshimura, A. Nakazawa, I. Saito and K. Fujimoto, Org. Lett., 2005, 7, 2853–2856 CrossRef CAS PubMed.
- I. Van Daele, H. Munier-Lehmann, M. Froeyen, J. Balzarini and S. Van Calenbergh, J. Med. Chem., 2007, 50, 5281–5292 CrossRef CAS PubMed.
- H. Q. Cui, L. M. Ruiz-Perez, D. Gonzalez-Pacanowska and I. H. Gilbert, Bioorg. Med. Chem., 2010, 18, 7302–7309 CrossRef CAS PubMed.
- X. Liu, X. L. Xia, C. H. Sun, C. Lin, Y. Q. Zhou, M. Hussain, F. Tang, L. Liu, X. Li and J. C. Zhang, Nucleosides, Nucleotides Nucleic Acids, 2016, 35, 479–494 CrossRef CAS PubMed.
- H. Q. Cui, J. Carrero-Lerida, A. P. G. Silva, J. L. Whittingham, J. A. Brannigan, L. M. Ruiz-Perez, K. D. Read, K. S. Wilson, D. Gonzalez-Pacanowska and I. H. Gilbert, J. Med. Chem., 2012, 55, 10948–10957 CrossRef CAS PubMed.
- G. Gosselin, M. C. Bergogne and J. L. Imbach, Nucleosides Nucleotides, 1990, 9, 81–87 CrossRef CAS.
- K. I. Imai, A. Nohara and M. Honjo, Chem. Pharm. Bull., 1966, 14, 1377–1381 CrossRef CAS PubMed.
- H. Iwamura, M. Miyakado and T. Hashizum, Carbohydr. Res., 1973, 27, 149–156 CrossRef CAS.
- M. J. Robins and R. K. Robins, J. Phys. Chem., 1969, 34, 2160–2163 CAS.
- F. Morvan, in Protocols for Oligonucleotides and Analogs, ed. S. Agrawal, Humana Press, Totowa, NJ, 1993, ch. 13, vol. 20, pp. 261–283 Search PubMed.
- E. M. Acton, R. N. Goerner, H. S. Uh, K. J. Ryan, D. W. Henry, C. E. Cass and G. A. Lepage, J. Med. Chem., 1979, 22, 518–525 CrossRef CAS PubMed.
- J. Liu, S. A. Ingale and F. Seela, Bioconjugate Chem., 2018, 29, 2265–2277 CrossRef CAS PubMed.
- R. A. Sanchez and L. E. Orgel, J. Mol. Biol., 1970, 47, 531–543 CrossRef CAS PubMed.
- H. Sawai, A. Nakamura, H. Hayashi and K. Shinozuka, Nucleosides Nucleotides, 1994, 13, 1647–1654 CrossRef CAS.
- J. J. Fox, M. Hoffer, I. Wempen and N. C. Yung, J. Am. Chem. Soc., 1961, 83, 4066–4070 CrossRef CAS.
- R. Duschinsky, T. Gabriel, M. Hoffer, J. Berger, E. Titsworth, E. Grunberg, J. H. Burchenal and J. J. Fox, J. Med. Chem., 1966, 9, 566–572 CrossRef CAS PubMed.
- E. Walton, S. R. Jenkins, R. F. Nutt and F. W. Holly, J. Med. Chem., 1969, 12, 306–309 CrossRef CAS PubMed.
- V. C. Solan, G. L. Szekeres, E. K. Ryu, H. Kung, Y. K. Ho and T. J. Bardos, Nucleosides Nucleotides, 1983, 2, 419–434 CrossRef CAS.
- D. Peters, A. B. Hornfeldt, S. Gronowitz and N. G. Johansson, Nucleosides Nucleotides, 1992, 11, 1151–1173 CrossRef CAS.
- M. Imazawa and F. Eckstein, J. Org. Chem., 1978, 43, 3044–3048 CrossRef CAS.
- T. Yamaguchi and M. Saneyoshi, Chem. Pharm. Bull., 1984, 32, 1441–1450 CrossRef CAS PubMed.
- Y. Sato, G. Tateno, K. Seio and M. Sekine, Tetrahedron Lett., 2002, 43, 3251–3254 CrossRef CAS.
- F. Seela, S. Menkhoff and S. Behrendt, J. Chem. Soc., Perkin Trans. 2, 1986, 525–530 RSC.
- D. I. Ward, S. M. Jeffs, P. L. Coe and R. T. Walker, Tetrahedron Lett., 1993, 34, 6779–6782 CrossRef CAS.
- S. Janardhanam and K. P. Nambiar, Tetrahedron Lett., 1994, 35, 3657–3660 CrossRef CAS.
- Z. W. Wang and C. J. Rizzo, Tetrahedron Lett., 1997, 38, 8177–8180 CrossRef CAS.
- J. Garcia, A. Diaz-Rodriguez, S. Fernandez, Y. S. Sanghvi, M. Ferrero and V. Gotor, J. Org. Chem., 2006, 71, 9765–9771 CrossRef CAS PubMed.
- A. Hatano, A. Harano, Y. Takigawa, Y. Naramoto, K. Toda, Y. Nakagomi and H. Yamada, Bioorg. Med. Chem., 2008, 16, 3866–3870 CrossRef CAS PubMed.
- U. Sequin, Helv. Chim. Acta, 1974, 57, 68–81 CrossRef CAS PubMed.
- F. Morvan, B. Rayner, J. L. Imbach, S. Thenet, J. R. Bertrand, J. Paoletti, C. Malvy and C. Paoletti, Nucleic Acids Res., 1987, 15, 3421–3437 CrossRef CAS PubMed.
- N. T. Thuong, U. Asseline, V. Roig, M. Takasugi and C. Helene, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 5129–5133 CrossRef CAS PubMed.
- T. A. Bacon, F. Morvan, B. Rayner, J. L. Imbach and E. Wickstrom, J. Biochem. Biophys. Methods, 1988, 16, 311–318 CrossRef CAS PubMed.
- F. Morvan, B. Rayner, J. L. Imbach, D. K. Chang and J. W. Lown, Nucleic Acids Res., 1987, 15, 4241–4255 CrossRef CAS PubMed.
- J. S. Sun, U. Asseline, D. Rouzaud, T. Montenay-Garestier, N. T. Thuong and C. Helene, Nucleic Acids Res., 1987, 15, 6149–6158 CrossRef CAS PubMed.
- F. Morvan, B. Rayner, J. L. Imbach, M. Lee, J. A. Hartley, D. K. Chang and J. W. Lown, Nucleic Acids Res., 1987, 15, 7027–7044 CrossRef CAS PubMed.
- D. Praseuth, L. Perrouault, T. L. Doan, M. Chassignol, N. Thuong and C. Helene, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 1349–1353 CrossRef CAS PubMed.
- J. S. Sun, C. Giovannangeli, J. C. Francois, R. Kurfurst, T. Montenay-Garestier, U. Asseline, T. Saisonbehmoaras, N. T. Thuong and C. Helene, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 6023–6027 CrossRef CAS PubMed.
- F. Debart, B. Rayner, G. Degols and J. L. Imbach, Nucleic Acids Res., 1992, 20, 1193–1200 CrossRef CAS PubMed.
- M. Koga, A. Wilk, M. F. Moore, C. L. Scremin, L. Zhou and S. L. Beaucage, J. Org. Chem., 1995, 60, 1520–1530 CrossRef CAS.
- M. W. Germann, J. M. Aramini, B. W. Kalisch, R. T. Pon and J. H. Van de Sande, Nucleosides Nucleotides, 1997, 16, 1481–1485 CrossRef CAS.
- C. Gagnor, J. R. Bertrand, S. Thenet, M. Lemaitre, F. Morvan, B. Rayner, C. Malvy, B. Lebleu, J. L. Imbach and C. Paoletti, Nucleic Acids Res., 1987, 15, 10419–10436 CrossRef CAS PubMed.
- J. Paoletti, D. Bazile, F. Morvan, J. L. Imbach and C. Paoletti, Nucleic Acids Res., 1989, 17, 2693–2704 CrossRef CAS PubMed.
- M. Bobek, A. Bloch, R. Parthasarathy and R. L. Whistler, J. Med. Chem., 1975, 18, 784–787 CrossRef CAS PubMed.
- M. T. Migawa, J. L. Girardet, J. A. Walker 2nd, G. W. Koszalka, S. D. Chamberlain, J. C. Drach and L. B. Townsend, J. Med. Chem., 1998, 41, 1242–1251 CrossRef CAS PubMed.
- E. Bloch, M. Lavignon, J. R. Bertrand, F. Pognan, F. Morvan, C. Malvy, B. Rayner, J. L. Imbach and C. Paoletti, Gene, 1988, 72, 349–360 CrossRef CAS PubMed.
- K. Shinozuka, N. Yamada, A. Nakamura, H. Ozaki and H. Sawai, Bioorg. Med. Chem. Lett., 1996, 6, 1843–1848 CrossRef CAS.
- K. B. Lesiak and K. T. Wheeler, Radiat. Res., 1990, 121, 328–337 CrossRef CAS PubMed.
- C. N. Johnson, A. M. Spring, S. Desai, R. P. Cunningham and M. W. Germann, J. Mol. Biol., 2012, 416, 425–437 CrossRef CAS PubMed.
- N. J. Amato, Q. Q. Zhai, D. C. Navarro, L. J. Niedernhofer and Y. S. Wang, Nucleic Acids Res., 2015, 43, 8314–8324 CrossRef CAS PubMed.
- A. A. Ishchenko, H. Ide, D. Ramotar, G. Nevinsky and M. Saparbaev, Biochemistry, 2004, 43, 15210–15216 CrossRef CAS PubMed.
- N. A. Timofeyeva and O. S. Fedorova, Mol. BioSyst., 2016, 12, 3435–3446 RSC.
- N. L. Williams, N. J. Amato and Y. S. Wang, Chem. Res. Toxicol., 2017, 30, 1127–1133 Search PubMed.
- C. Boiziau, F. Debart, B. Rayner, J. L. Imbach and J. J. Toulme, FEBS Lett., 1995, 361, 41–45 CrossRef CAS PubMed.
- J. M. Aramini, B. W. Kalisch, R. T. Pon, J. H. vandeSande and M. W. Germann, Biochemistry, 1996, 35, 9355–9365 CrossRef CAS PubMed.
- J. M. Aramini, J. H. Van de Sande and M. W. Germann, Biochemistry, 1997, 36, 9715–9725 CrossRef CAS PubMed.
- F. Morvan, F. Debart and J. J. Vasseur, Chem. Biodiversity, 2010, 7, 494–535 CrossRef CAS PubMed.
- J. Müller, D. Bohme, P. Lax, M. Morell Cerda and M. Roitzsch, Chemistry, 2005, 11, 6246–6253 CrossRef PubMed.
- J. Muller, D. Bohme, N. Dupre, M. Mehring and F. A. Polonius, J. Inorg. Biochem., 2007, 101, 470–476 CrossRef PubMed.
- X. R. Guo and F. Seela, Chem.–Eur. J., 2017, 23, 11776–11779 CrossRef CAS PubMed.
- H. Sawai, T. Itoh, K. Kokaji and K. Shinozuka, J. Mol. Evol., 1997, 45, 209–215 CrossRef CAS PubMed.
- T. Bhattacharyya, P. Saha and J. Dash, ACS Omega, 2018, 3, 2230–2241 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |