
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA step-wise synthetic approach is necessary to access γ-conjugates of folate: folate-conjugated prodigiosenes†
Carlotta Figliola‡
,
Estelle Marchal‡,
Brandon R. Groves and
Alison Thompson *
*
Department of Chemistry, Dalhousie University, PO Box 15000, Halifax, NS, B3H 4R2, Canada. E-mail: Alison.Thompson@dal.ca; Tel: +1-902-494-3305
First published on 7th May 2019
Abstract
Despite the vast literature that describes reacting folic acid with a pharmacophore, this route is ineffective in providing the correct regioisomer of the resulting conjugate. We herein present a step-wise route to the preparation of nine folate conjugates of the tripyrrolic prodigiosene skeleton. The strict requirement for step-wise construction of the folate core is demonstrated, so as to achieve conjugation at only the desired γ-carboxylic acid and thus maintain the α-carboxylic site for folate receptor (FRα) recognition. Linkages via ethylenediamine, polyethylene glycol and glutathione are demonstrated.
Introduction
Prodigiosin is a tripyrrolic, red pigmented natural product, produced by Gram positive and Gram negative bacteria, including certain strains of Serratia marcescens (Fig. 1).1,2 Prodigiosin is well known to exhibit immunosuppressive, antimicrobial and anticancer properties, yet modern uses extend from sunscreen3 to antibacterial dyes for silk4 and cotton.5 Known mechanisms of bioactivity include H+/Cl− exchange, oxidative DNA cleavage through Cu(II) chelation and signal-transduction interference.6–15 Synthetic mimics of prodigiosin, named prodigiosenes,16 with modifications on the A-, B- or C-ring17–24 have been shown to maintain the biological activity of the parent compound yet with 100-fold improvements in selectivity towards malignant cells and with 100-fold reduction of systemic toxicity in mice compared to the natural product (R = CO2iPr, R1 = Me, Fig. 1).25 Furthermore, the study of these synthetic analogues has enriched the knowledge of structure–activity relationships (SAR) for a better understanding of the biological activity of prodigiosin.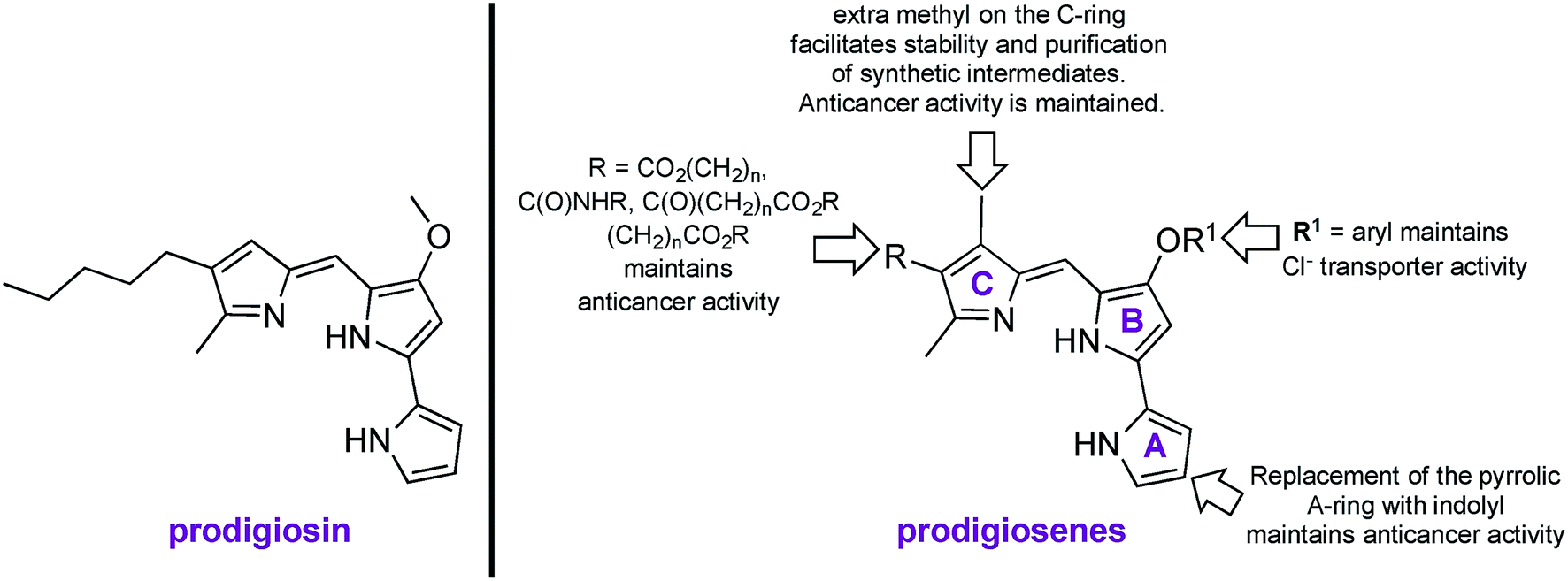 | ||
| Fig. 1 Naturally occurring prodigiosin, and SAR of prodigiosene analogues.17–25 | ||
Folic acid (vitamin B9, Fig. 2) is considered26 a promising biomarker for triple-negative breast cancer (TNBC).27 More than 50% of TNBCs overexpress folate receptor alpha (FRα), as do a limited number of other cancers28 (e.g. ovarian cancers)29,30 along with those tissues where TNBC is likely to result in metastasis, such as brain and lung.31 Importantly, FRα is present on only a few types of healthy cells (activated macrophages and proximal tubules of kidneys),32 and, crucially, the uptake of the vitamin folic acid (as a folate salt) is not mediated by FRα.33–35 Consequently, cognisant that folate-conjugated drugs do not generally enter normal cells,36 folic acid has been employed in the synthesis of multiple drug-delivery systems for chemotherapeutic treatment and imaging.
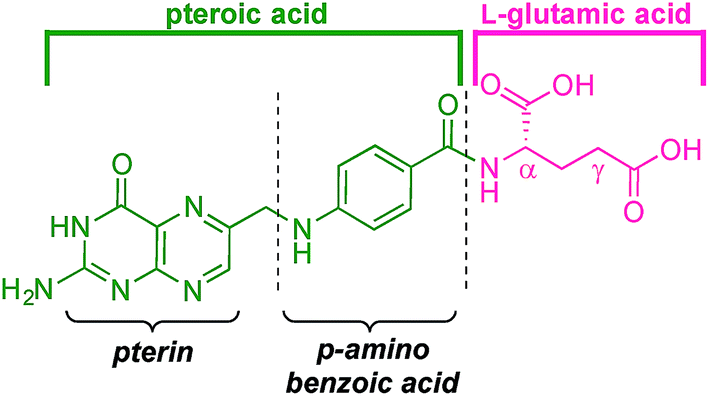 | ||
| Fig. 2 Folic acid, or vitamin B9. | ||
Given the potency of prodigiosenes and the targeting ability of folate, we herein report the synthesis of the first series of folate–prodigiosene conjugates. In doing so, we emphasize the ineffectiveness of the much-utilised direct synthetic approach to couple unmodified folic acid with the desired pharmacophore. Our observations and comments contrast with the vast literature that apparently describes the folate-targeted bioactivity of unpurified and uncharacterised material that results from such direct coupling. We categorically demonstrate that this direct approach, despite its wide use by others, gives rise to mixtures that include both regioisomers featuring the pharmacophore coupled to both carboxyl sites of folic acid. Instead, we demonstrate the requirement for step-wise construction of the folate core, so as to achieve conjugation at only the desired γ-carboxylic acid and thus maintenance of the α-carboxylic site for FRα recognition.37
Results and discussion
Our folate–prodigiosene conjugates (Fig. 3) feature prodigiosene 1 as the pharmacophore,38 with varied aliphatic spacer length (n = 2, 4, 8) leading to the folate moiety. The conjugated carbonyl group on the β-position of the C-ring has been shown to facilitate purification of prodigiosene intermediates21 and so it, and the adjacent β-methyl substituent, were incorporated into the synthetic strategy. Linkers were chosen to increase either the lipophilicity39 (ethylenediamine, DA) or the water-solubility and biocompatibility40,41 (polyethylene glycol, PEG) of the conjugates, or to enable intracellular redox-mediated cleavage by glutathione (disulfide, SS, Fig. 3).42–44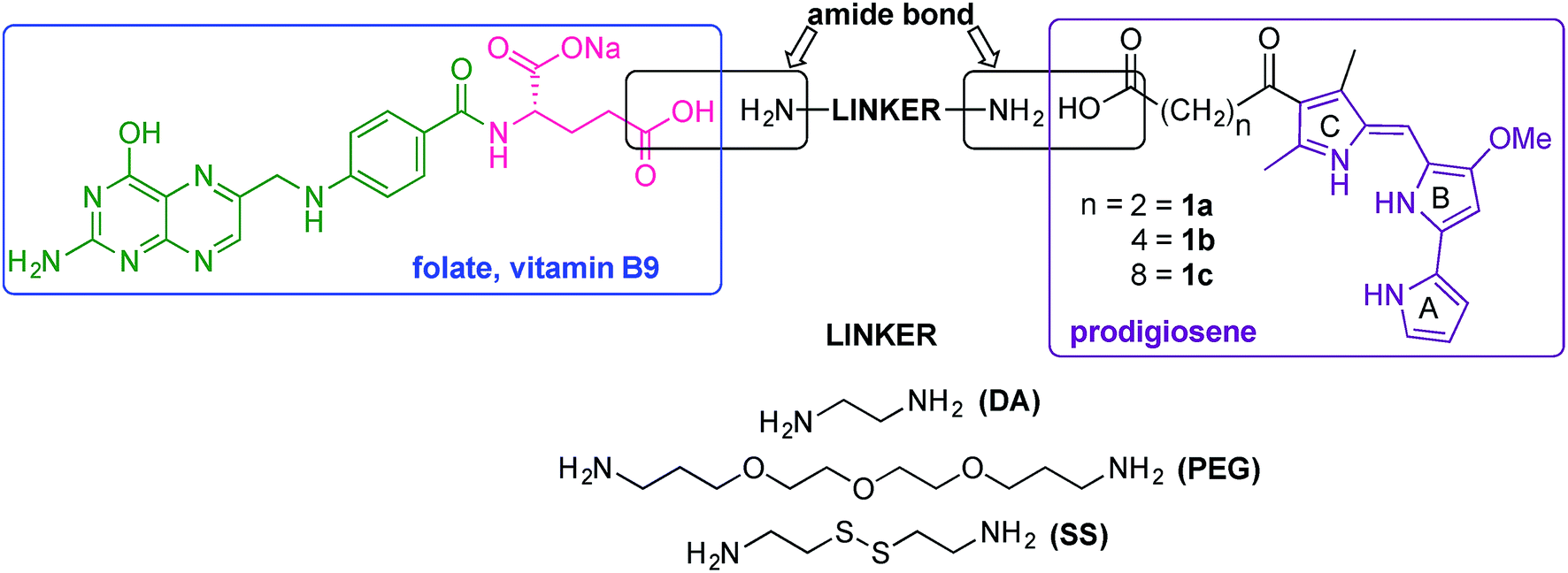 | ||
| Fig. 3 Approach to targeted folate–prodigiosene conjugates. | ||
Synthesis of linker-prodigiosene conjugates
In order to synthesise the linker-prodigiosene conjugates 2–4a–c, prodigiosenes 1a–c were coupled38 to the Boc-protected linkers 5–7 using HBTU in the presence of DMAP.39,40,45 Compounds 8–10 were thus obtained in moderate-to-excellent yields (Scheme 1, General procedure in experimental section). Deprotection, using HCl, afforded the targeted linker-prodigiosene adducts 2–4 after basic work-up to liberate the respective free-bases. However, prodigiosene-linker conjugates 4b·HCl and 4c·HCl were used as crude salts, as the S–S bond was found to be highly sensitive to basic work-up conditions and purification using flash chromatography, both of which caused cleavage of the disulfide.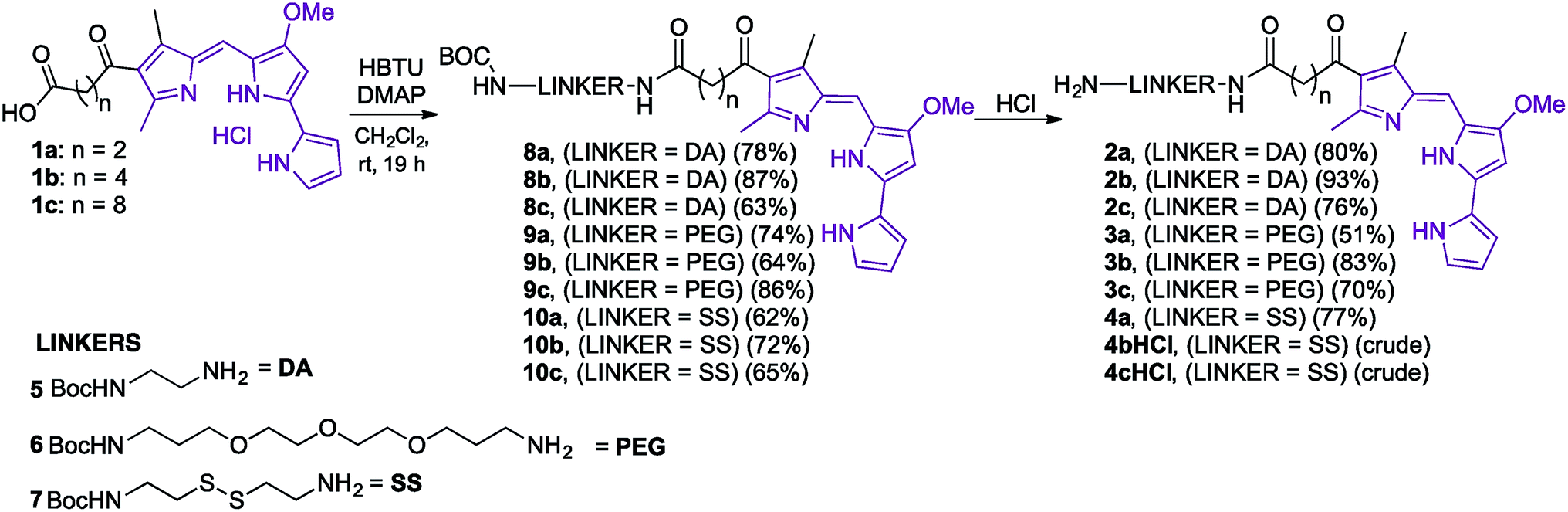 | ||
| Scheme 1 Synthesis of linker-prodigiosene adducts 2–4. | ||
γ-Coupling to folic acid and synthesis of folate–prodigiosene conjugates
Folic acid formally consists of pteroic acid linked to L-glutamic acid through a peptide bond (Fig. 2).46 The crystal structure of human FRα complexed with folic acid reveals a deep binding pocket with the pteroate element of folate buried inside the receptor.47 In contrast, the glutamate moiety of folate sits at the pocket entrance. The α-carboxylic acid of the amino-acid is involved in the interaction with the FRα and, thus, in the metabolism and function of folate.37 However, conjugates bound at the pendant γ-carboxylic acid, further away from the binding pocket, readily undergo binding to FRα. As such, in order to leave the α-position unmodified, conjugation of pharmacophores to folic acid must be specific to the γ-carboxylic acid in order to maintain ligand binding affinity.37As shown in Fig. 4, three retrosynthetic strategies can be envisaged to achieve the desired folate–prodigiosene conjugates (top). The simplest method, Strategy A, involves the direct coupling of folic acid with the pharmacophore. Following this approach, many literature reports appear to describe the direct activation of folic acid, followed by addition of the desired amine to afford conjugated folates.48–61 However, we met Strategy A with considerable scepticism as the presence of two carboxylic acid moieties within folic acid renders two regioisomeric coupling products clearly tangible. Given the nature of those two carboxylic acid groups, we were unable to find any rationale by which to convince ourselves that direct reaction would result in production of the required γ-conjugate. Furthermore, as conjugates to folic acid are often soluble only in DMSO the separation of the two regioisomers represents a considerable challenge. Adding to our skepticism, we found the literature regarding this direct synthetic approach to be incomplete and unreliable, due to either vagueness of purification techniques or lack of characterization such as to give assurance of purity. Indeed, upon close inspection it appears that there are many published reports that simply use the uncharacterised crude precipitate, obtained following the mixing of folic acid, coupling agent and pharmacophore, in studies relating to targeting using folate. Cognisant that coupling with DCC etc. involves ureas and other synthetic intermediates, and that these often persist in crude product mixtures, we remained unconvinced that Strategy A would be fruitful as we required a procedure that gave unequivocal access to the desired γ-conjugate of folic acid bearing prodigiosenes.
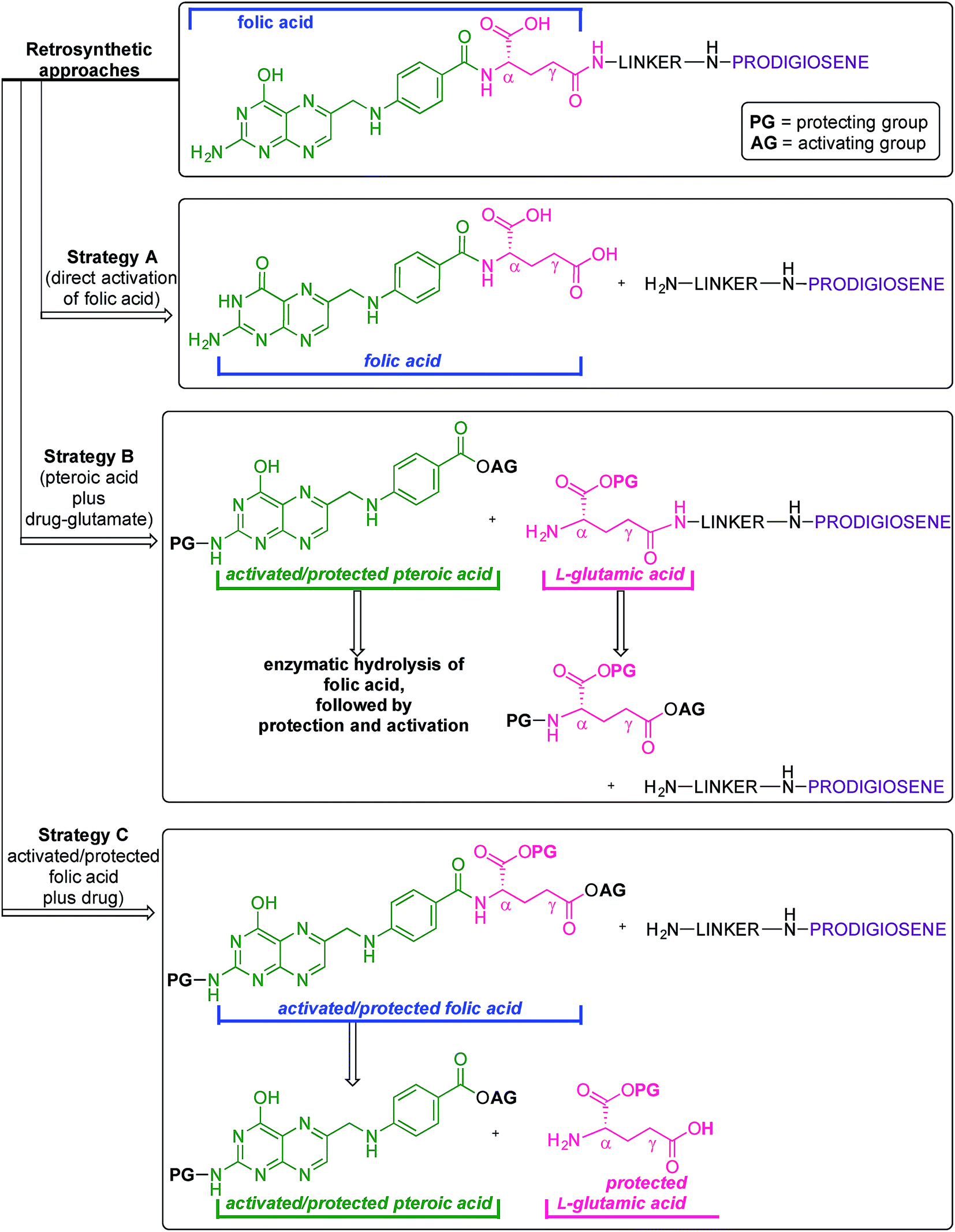 | ||
| Fig. 4 Retrosynthetic approaches for the synthesis of folate–prodigiosene conjugates. | ||
Retrosynthetic Strategies B and C (Fig. 4) involve the step-wise construction of suitably protected and activated folic acid from pteroic acid and L-glutamic acid, allowing for the selective conjugation of pharmacophore at the desired γ-carboxyl site. Strategy B requires that the γ-activated, α-protected glutamic acid moiety be coupled with the pharmacophore prior to reaction with the pteroate portion of what will become folate.62–64 Strategy C requires that the γ-activated, α-protected glutamic acid moiety first be coupled with activated pteroate portion to create γ-activated, α-protected folate prior to reaction with the pharmacophore.65 Solid-state resin approaches66,67 recognise the virtues of a step-wise approach yet, compared to the lure of Strategy A, both step-wise strategies obviously demand long synthetic sequences and tedious purification processes and have not enjoyed widespread adoption. Indeed, the vast majority of published routes involving folate conjugates steer away from these step-wise approaches.68
Given the evident popularity of the direct approach offered by Strategy A, we wanted to unequivocally assess the regioselective outcome of reacting folic acid with a coupling agent and an alcohol. We thus treated folic acid with DCC and NHS, and analysed the crude product mixture. Based on the phenyl group signals in the 1H NMR spectrum (see ESI†), the crude NHS-adduct was determined to contain α- and γ-adducts in an approx. 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :75 α:γ ratio. A solution of the adduct mixture in DMSO was treated with the linker-prodigiosene conjugate 2a in an attempt to form the desired amide linkage. Attempts to isolate pure material (12a) from the precipitate thus formed were completely unsuccessful (Scheme 2). Rather, the crude mixture was poorly soluble and decomposition occurred on chromatographic media (alumina and silica). Treatment of the NHS-adduct mixture with 2b and 2c also resulted in bulk precipitation, and again separation and purification of the highly insoluble material was not possible. Still intrigued by the apparent widespread success of Strategy A, we treated folic acid with DCC, NHS and a simple amine to thus employ another route by which to gain insight regarding the regioselectivity of activation.57 A solution of the 25:75 α:γ folate–NHS adduct in DMSO was treated with octyl amine, as a model amine, and the resulting precipitate analyzed using 1H NMR spectroscopy. Both α- and γ-regioisomers were present, along with the NHS-activated folic acid, and purification proved unsuccessful given the lack of solubility of the material. In short, direct activation following Strategy A was found to be wholly unsatisfactory in the quest towards isolated and pure γ-appended conjugates of folic acid.
:75 α:γ ratio. A solution of the adduct mixture in DMSO was treated with the linker-prodigiosene conjugate 2a in an attempt to form the desired amide linkage. Attempts to isolate pure material (12a) from the precipitate thus formed were completely unsuccessful (Scheme 2). Rather, the crude mixture was poorly soluble and decomposition occurred on chromatographic media (alumina and silica). Treatment of the NHS-adduct mixture with 2b and 2c also resulted in bulk precipitation, and again separation and purification of the highly insoluble material was not possible. Still intrigued by the apparent widespread success of Strategy A, we treated folic acid with DCC, NHS and a simple amine to thus employ another route by which to gain insight regarding the regioselectivity of activation.57 A solution of the 25:75 α:γ folate–NHS adduct in DMSO was treated with octyl amine, as a model amine, and the resulting precipitate analyzed using 1H NMR spectroscopy. Both α- and γ-regioisomers were present, along with the NHS-activated folic acid, and purification proved unsuccessful given the lack of solubility of the material. In short, direct activation following Strategy A was found to be wholly unsatisfactory in the quest towards isolated and pure γ-appended conjugates of folic acid.
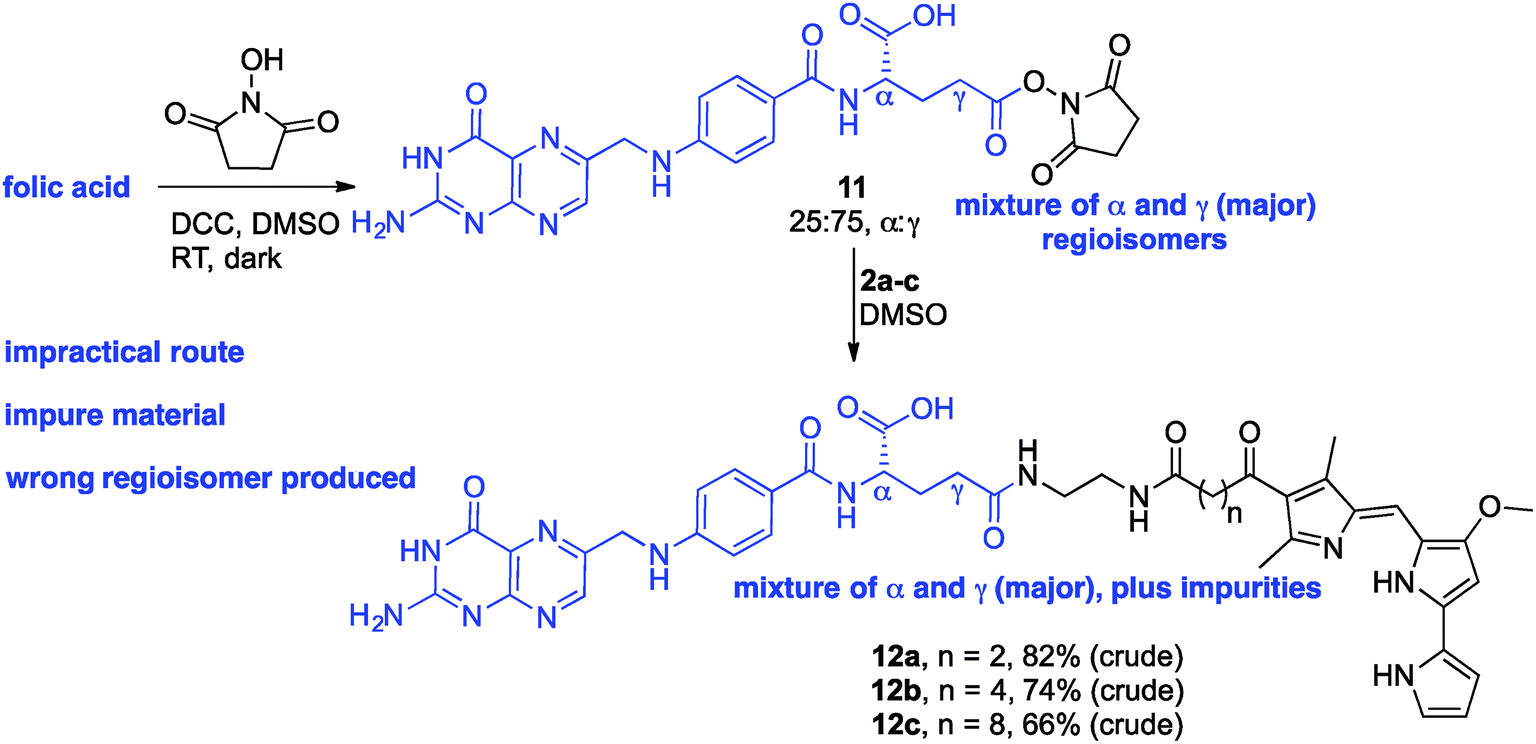 | ||
| Scheme 2 Activation of folic acid with NHS and conjugation to linker-prodigiosenes 2a–c. | ||
With firm evidence that the direct reaction of folic acid with coupling agent and alcohol/amine does not regioselectively provide the desired γ-regioisomer, we turned our attention to strategies that used sequential protection/activation. We first turned to the applicability of retrosynthetic Strategy B, involving step-wise construction of the folate moiety so as to ensure conjugation at the desired γ-carboxyl site. As shown in Fig. 4, the critical step entails coupling of suitably protected (the base portion) and activated (the acid portion) pteroic acid with a derivative of L-glutamic acid that has been pre-linked to the prodigiosene pharmacophore by way of the γ-position. This key coupling would result in the folate–prodigiosene conjugate as a single isomer, courtesy of the previous regiospecific activation of L-glutamic acid.62–65,68,69 To embark upon Strategy B, pteroic acid was prepared following literature procedure through hydrolysis of folic acid using carboxypeptidase G in the presence of ZnCl2 at 30 °C.65 Carboxypeptidase G was purchased from Sigma and used as is (7 mg; 20 units, since 1 unit = 1 μmol min−1 at optimal conditions of 30 °C and pH 7.3). The enzyme operates optimally at pH = 7.3. However, in our hands the reported 0.1 M Tris–HCl buffer was insufficient to maintain stable pH after the addition of folic acid. Therefore, the initial buffer concentration was increased to 1 M and the pH subsequently stabilised in the range of 7.2–7.3 via the addition of solid buffer. After 5 days, 1H NMR spectroscopic analysis of the crude product mixture revealed 75% conversion of folic acid to pteroic acid. The carboxylic acid of the crude pteroic acid was activated using CDI and the primary amine then protected with a trimethylsilylethoxycarbonyl (Teoc) group, to give 13 after purification using column chromatography on silica (Scheme 3). This purification also allowed the removal of folic acid remaining from the initial step.
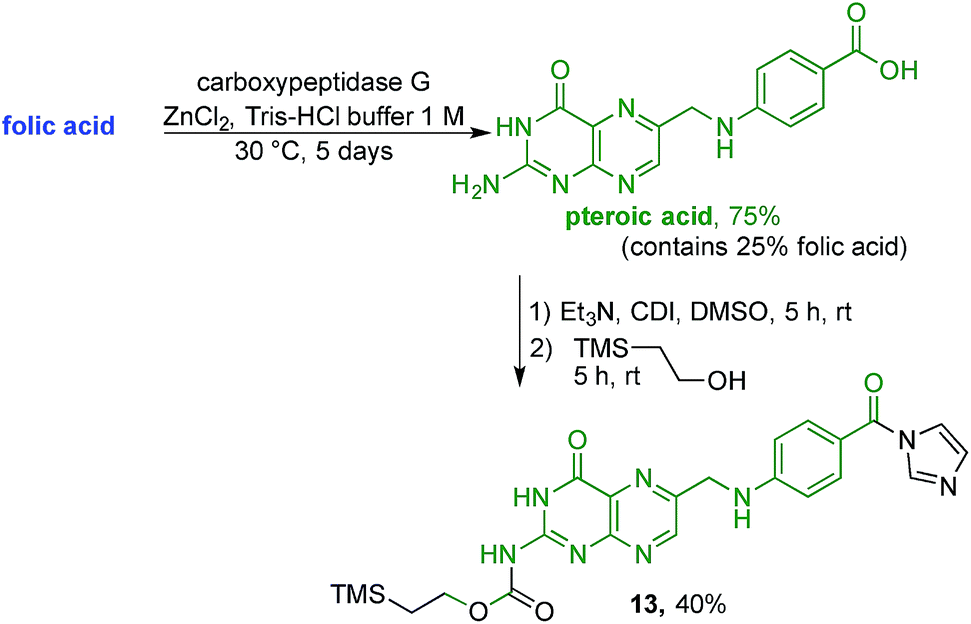 | ||
| Scheme 3 Preparation of the activated Teoc-protected pteroic acid 13. | ||
The L-glutamic acid moiety of folic acid was prepared from its commercially available Boc-L-glutamic acid 5-benzyl ester derivative (14), which was first esterified at the α-position using CDI and 2-(trimethylsilyl)ethanol, in order to obtain 15. Subsequent hydrogenolysis of the benzyl ester at the γ-position gave the protected amino acid 16, ready for coupling with prodigiosene (Scheme 4).65
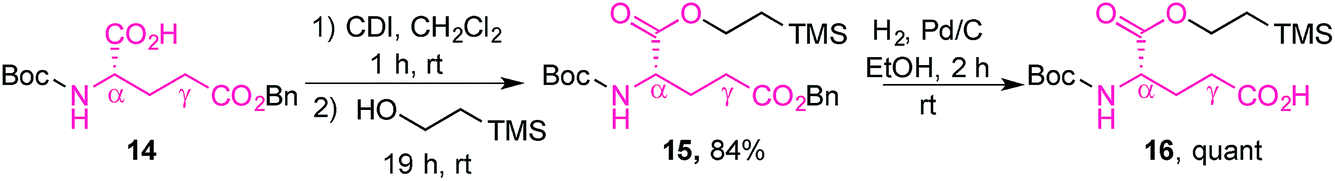 | ||
| Scheme 4 Preparation of the protected glutamic acid derivative 16. | ||
Attempts to couple 16 with the dithio linker-prodigiosene conjugate 4c·HCl in the presence of HBTU and DMAP were unsuccessful and only starting material was recovered (Scheme 5). However, coupling between 3b–c and the protected amino-acid 16 successfully afforded substrates 18b–c, thus crucially connecting the prodigiosene pharmacophore with the γ-position of L-glutamic acid, by way of a PEG linker. Boc-deprotection in the presence of TsOH in a mixture of dioxane/water gave the amines 19b–c. Strategy B (Fig. 4) then requires coupling of the drug-appended glutamic acid moiety with the activated pteroic acid, but attempts to couple 19b–c to 13 resulted in only partial recovery of the substrate prodigiosenes 19b–c (Scheme 5).
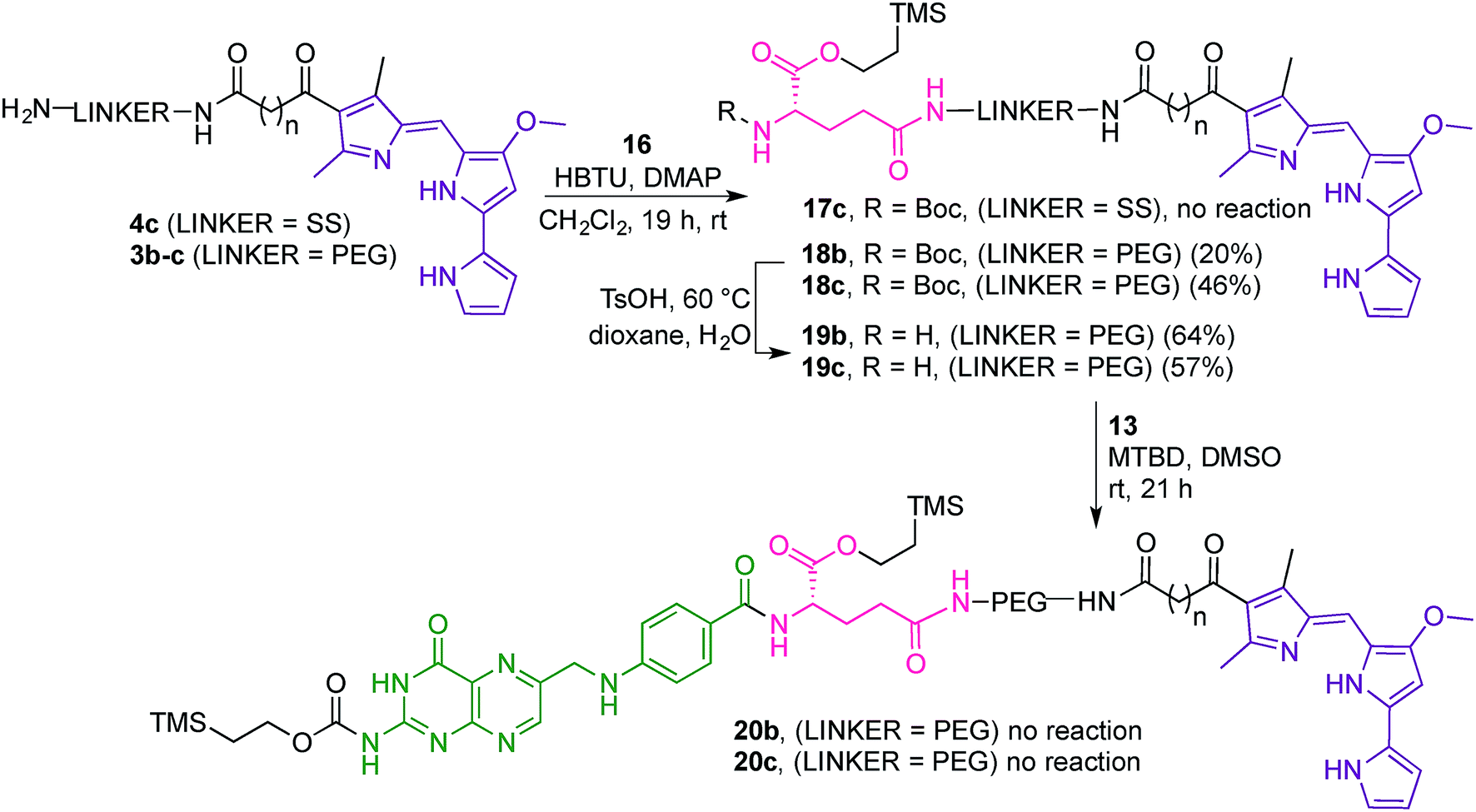 | ||
| Scheme 5 Attempts to synthesise Teoc-protected folic acid-prodigiosene conjugates 20. | ||
In turning our attention to Strategy C (Fig. 4), the L-glutamic acid derivative 16 was first deprotected at the amino position, and then coupled to the activated pteroic acid to successfully afford 22. This γ-carboxylic acid was then reacted with NHS to give 23 which is activated in the desired position of what will become folate (Scheme 6).65 The amines 2–4a–c were reacted with a solution of 23, to provide conjugates 24–26a–c. Subsequently, the Teoc protecting group was removed with TBAF, and the resulting tetrabutylammonium salt precipitated via the addition of water. Dissolution of the salt in DMSO, followed by reaction with NaOAc, afforded eight sodium folate–prodigiosene conjugates 27–28a–c and 29a–b. However, addition of TBAF to 26c caused the cleavage of the S–S bond and the corresponding folate–prodigiosene conjugate 29c could not be isolated (Scheme 6). Clearly the step-wise approach to building folate acid, such that only the γ-carboxyl site is available for coupling, requires more synthetic effort that the direct approach. However, in contrast to Strategy A, the step-wise approaches of Strategies B and C tolerate substantiation of purity and characterization at each synthetic step and should thus be the approaches adopted for the preparation of regioisomerically pure desired conjugates of folate.
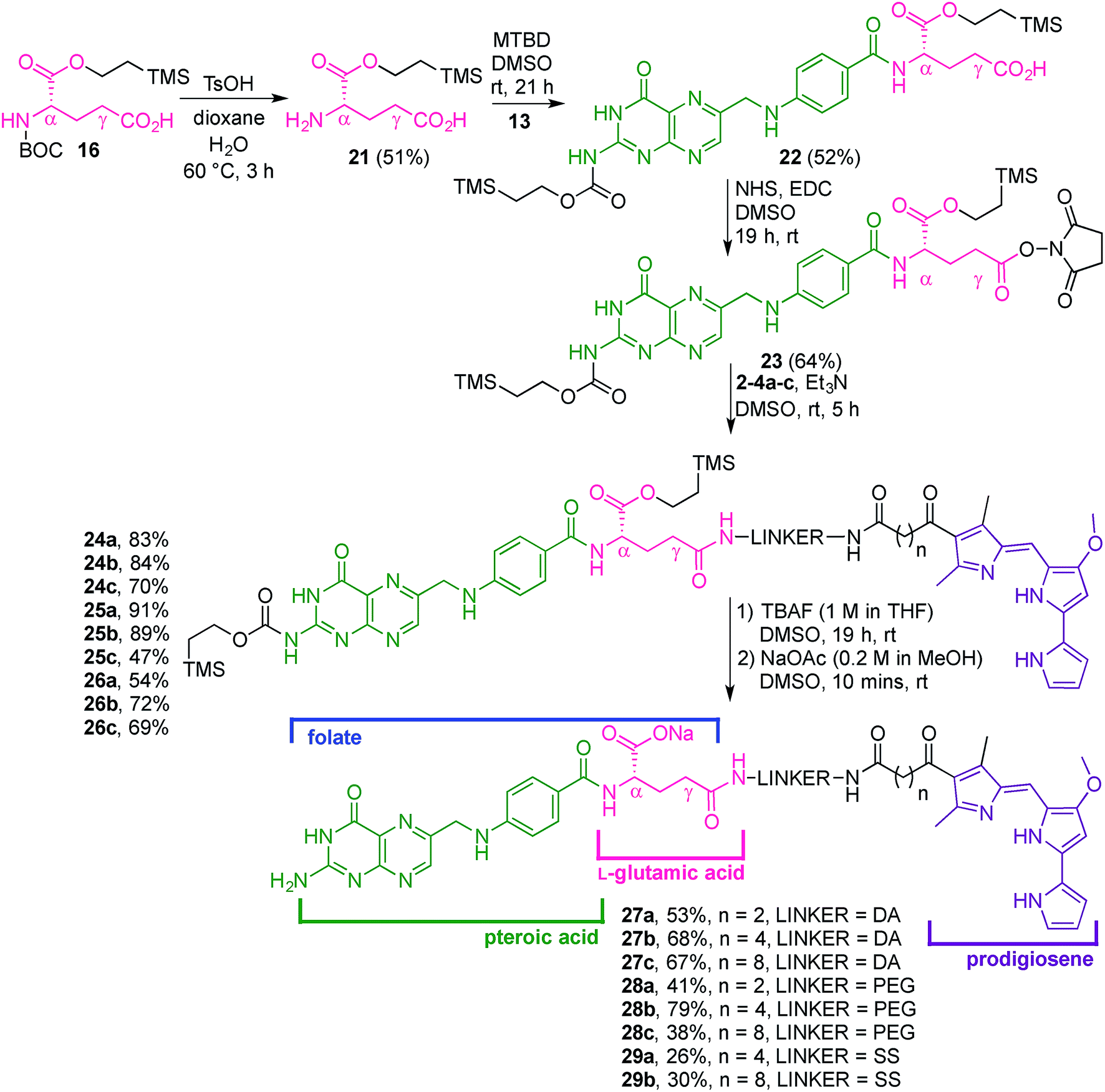 | ||
| Scheme 6 Synthesis of folate–prodigiosene conjugates 27–28a–c and 29a–b. | ||
Conclusions
In summary, the first series of folate–prodigiosene conjugates have been synthesised, and full characterisation acquired. In order to increase the lipophilicity, the water-solubility and the ease of cleavage, three linkers were chosen to link the folate and the prodigiosene moieties (DA, PEG and SS respectively). Cognisant that the α-carboxylate of folate is involved in the interaction with FRα, three synthetic approaches were investigated for the preparation of the targeted γ-adduct. The direct and most commonly used literature method involves coupling the unprotected folic acid, activated in situ, to the amino-counterpart. Despite the widespread use of this direct approach, often presented along with unsubstantiated claims as to purity and constitution of the product(s) thus obtained, we determined it to be wholly unsatisfactory as it gives mixtures of α- and γ-regioisomers, along with various ureas and other synthetic intermediates presenting as impurities, which are challenging to separate. We evaluated this shortcoming at the activation stage, using simply NHS, as well as through reaction with octyl amine and the prodigiosene pharmacophore. In order to access the required γ-adduct, the hydrolysis of folic acid into its two core components (pteroic acid and L-glutamic acid) was necessary. The subsequent step-wise synthesis of folic acid, with the α-carboxylic acid protected, allowed synthesis of only the desired regioisomer thus, for the first time, connecting folic acid with the prodigiosene pharmacophore. Next steps will involve evaluation of the cytotoxicity of the conjugates, alongside optimization of drug release and internalisation.Experimental section
All chemicals were purchased and used as received unless otherwise indicated. Moisture sensitive reactions were performed in oven-dried glassware and under a positive pressure of nitrogen. Air- and moisture-sensitive compounds were introduced via syringe or cannula through a rubber septum. Flash chromatography was performed using either Silicycle ultra-pure silica (230–400 mm) or 150 mesh Brockmann III activated neutral/basic alumina oxide, as indicated. The NMR spectra were recorded using a 500 MHz spectrometer instrument using CDCl3, MeOD or DMSO as solvents and are reported in part per million (ppm). Internal solvents were referenced at 7.26 ppm for 1H and at 77.16 ppm for 13C when using CDCl3, at 3.31 ppm for 1H and at 49.00 ppm for 13C when using MeOD and at 2.50 ppm for 1H and at 39.5 ppm for 13C when using DMSO-d6. Coupling constants (J) are given in Hertz (Hz). Mass spectra were obtained using TOF and LCQ Duo ion trap instruments operating in ESI+/− mode, as indicated. Compounds 1a–c,21 5–7,39,40,45 11,57 pteroic acid,65 13,65 15–16,65 and 21–23 (ref. 65) were prepared following literature procedures.General procedure 1 (GP1)
Amines 5–7 (2 eq.), HBTU (1.2 eq.) and DMAP (1.2 eq.) were added to a solution of prodigiosene 1 (1 eq.) in anhydrous CH2Cl2 (0.01 M) and the reaction mixture was stirred at room temperature for 16 h under N2 atmosphere. The solvent was removed under reduced pressure and the crude mixture was partitioned between EtOAc and NaHCO3 (aq. sat solution). The aqueous layer was extracted with EtOAc (×3). The combined organic layers were washed with water (×5), brine, dried over Na2SO4 and concentrated under reduced pressure.:MeOH 100:0, 99:1), followed by A2O3 type III neutral (CH2Cl2:MeOH, 100:0, 99:1) to afford 8a (0.098 g, 78%) as dark red glass. 1H NMR (CDCl3, 500 MHz) 1.41 (s, 9H), 2.21 (s, 3H), 2.38 (s, 3H), 2.47 (t, J = 6.1 Hz, 2H), 3.02 (t, J = 6.3 Hz, 2H), 3.24 (br s, 2H), 3.31 (br s, 2H), 3.96 (s, 3H), 5.14 (br s, 1H), 6.04 (s, 1H), 6.20–6.21 (m, 1H), 6.35 (br s, 1H), 6.72 (d, J = 3.4 Hz, 1H), 6.76 (s, 1H), 6.89 (s, 1H). 13C NMR (CDCl3, 125 MHz) 12.6, 14.6, 28.5, 29.8, 30.7, 38.3, 40.4, 58.7, 79.5, 96.1, 110.8, 111.9, 113.9, 122.7, 123.3, 126.4, 128.2, 129.8, 141.0, 142.8, 156.7, 160.9, 169.0, 173.7, 196.2. HRMS (TOF) (m/z): [M + H]+ calcd for C27H36N5O5, 510.2711; found, 510.2719.:MeOH 100:0, 99:1), precipitated with CH2Cl2:hexanes (1:9), filtered and washed with hexanes to afford 8b (0.100 g, 87%) as orange glass. 1H NMR (CD3OD, 500 MHz) 1.42 (s, 9H), 1.68–1.70 (m, 4H), 2.22–2.26 (m, 2H), 2.39 (s, 3H), 2.66 (s, 3H), 2.81 (t, J = 6.6 Hz, 2H), 3.15 (t, J = 6.0 Hz, 2H), 3.25 (t, J = 6.1 Hz, 2H), 3.92 (s, 3H), 6.08 (s, 1H), 6.26–6.27 (m, 1H), 6.76 (s, 2H), 7.02 (s, 1H). 13C NMR (CD3OD, 125 MHz) 12.3, 15.4, 25.1, 26.7, 28.8, 30.2, 37.0, 40.5, 41.0, 43.1, 59.0, 96.7, 111.2, 111.3, 114.1, 123.6, 123.7, 128.3, 128.8, 129.8, 142.6, 144.3, 162.1, 169.6, 176.3, 200.0 (1 carbon atom unaccounted for). HRMS (TOF) (m/z): [M + H]+ calcd for C29H40N5O5, 538.3024; found, 538.3012.:MeOH 99:1) and Al2O3 type III basic (CH2Cl2:MeOH 99.5:0.5, 99:1, 98.5:0.5) to afford 8c as a red glass (0.116 g, 63%). 1H NMR (CDCl3, 300 MHz) 1.26 (s, 8H), 1.42 (s, 9H), 1.58–1.60 (m, 4H), 2.10–2.13 (m, 5H), 2.38 (s, 3H), 2.62 (t, J = 7.2 Hz, 2H), 3.24 (br s, 2H), 3.31 (br s, 2H), 3.97 (s, 3H), 5.05 (br s, 1H), 6.06 (s, 1H), 6.18 (s, 1H), 6.25 (br s, 1H), 6.71–6.72 (s, 2H), 6.93 (s, 1H). 13C NMR (CDCl3, 100 MHz) 12.4, 14.0, 24.3, 25.8, 28.5, 29.2, 29.3, 29.4, 36.8, 40.5, 40.7, 42.8, 58.7, 79.7, 96.1, 110.7, 112.1, 113.8, 123.4, 123.6, 126.2, 128.3, 129.7, 132.3, 140.5, 142.2, 157.1, 161.1, 169.1, 174.0, 198.3. HRMS (TOF) (m/z): [M + Na]+ calcd for C33H47N5NaO5, 616.3436; found, 616.3490.:hexanes 50:50, 70:30 EtOAc 100:0, CH2Cl2:MeOH 100:0, 99:1) to afford 9a as an orange glass (0.124 g, 74%). 1H NMR (CDCl3, 500 MHz) 1.42 (s, 9H), 1.73–1.79 (m, 4H), 2.25 (br s, 3H), 2.38 (s, 3H), 2.49 (t, J = 6.5 Hz, 2H), 3.02 (t, J = 6.5 Hz, 2H), 3.19–3.20 (m, 2H), 3.34 (q, J = 6.0 Hz, 2H), 3.51–3.59 (m, 8H), 3.62–3.64 (m, 4H), 3.96 (s, 3H), 5.02 (br s, 1H), 6.02 (s, 1H), 6.22 (br s, 1H), 6.42 (br s, 1H), 6.72 (br s, 1H), 6.78 (br s, 1H), 6.88 (s, 1H). 13C NMR (CDCl3, 125 MHz) 12.5, 14.7, 28.6, 29.1, 29.8, 30.5, 37.8, 38.2, 38.6, 58.6, 69.6, 69.9, 70.3 (×2), 70.6, 79.0, 95.9, 110.8, 111.9, 113.8, 122.8, 123.4, 126.4, 128.2, 129.9, 140.7, 142.6, 156.2, 160.6, 168.8, 172.8, 196.0 (1 carbon atom unaccounted for). HRMS (TOF) (m/z): [M + Na]+ calcd for C35H51N5Na1O8, 692.3630; 692.3627.:EtOAc 50:50, 40:60, 30:70, 20:80, 10:90, 0:100, then CH2Cl2:MeOH 99:1), followed by twice A2O3 type III basic (CH2Cl2:MeOH, 100:0, 99:1) to afford 9b (0.104 g, 64%) as dark red solid. 1H NMR (CDCl3, 500 MHz) 1.42 (s, 9H), 1.63–1.66 (m, 4H), 1.70–1.80 (m, 4H), 2.14–2.19 (m, 5H), 2.38 (s, 3H), 2.65–2.69 (m, 2H), 3.19 (d, J = 5.5 Hz, 2H), 3.34 (dd, J = 12.0 and 6.0 Hz, 2H), 3.48–3.62 (m, 12H), 3.97 (s, 3H), 5.02 (m, 1H), 6.04 (s, 1H), 6.19–6.21 (m, 1H), 6.39 (br s, 1H), 6.72 (d, J = 3.6 Hz, 1H), 6.76 (s, 1H), 6.91 (s, 1H). 13C NMR (CDCl3, 125 MHz) 12.3, 13.9, 23.7, 25.4, 28.5, 29.1, 29.7, 36.6, 37.8, 38.5, 42.3, 58.6, 70.0, 70.2 (×2), 70.6, 78.9, 96.1, 110.6, 111.9, 113.9, 123.3, 123.5, 126.1, 128.1, 129.7, 140.3, 142.3, 156.2, 161.0, 169.1, 172.9, 197.5 (2 carbon atoms unaccounted for). HRMS (TOF) (m/z): [M + H]+ calcd for C37H55N5NaO5, 698.4123; found, 698.4102.:hexanes 60:40, 80:20, EtOAc:MeOH 100:0, 99:1) followed by Al2O3 type III neutral (EtOAc:hexanes 60:40, 80:20, EtOAc:MeOH 100:0, 99:1, 98:2) and Al2O3 type III neutral (EtOAc 100%) to afford 9c as a red glass (0.200 g, 86%). 1H NMR (CDCl3, 500 MHz) 1.28 (br s, 8H), 1.43 (s, 9H), 1.56–1.63 (m, 4H), 1.75 (quint., J = 6.0 Hz, 4H), 2.12 (t, J = 7.5 Hz, 2H), 2.20 (s, 3H), 2.39 (s, 3H), 2.63 (t, J = 7.5 Hz, 2H), 3.19–3.23 (m, 2H), 3.34 (q, J = 6.0 Hz, 2H), 3.49–3.64 (m, 12H), 3.97 (s, 3H), 4.97 (br s, 1H), 6.04 (s, 1H), 6.19–6.21 (m, 2H), 6.72 (d, J = 3.6 Hz, 1H), 6.75 (br s, 1H), 6.91 (s, 1H). 13C NMR (CDCl3, 125 MHz) 12.4, 14.1, 24.3, 25.9, 28.6, 29.1, 29.3, 29.4, 29.5, 29.8, 36.9, 38.0, 38.6, 42.8, 58.6, 69.7, 70.3 (×2), 70.6, 70.7, 77.4, 77.6, 79.0, 96.1, 110.7, 112.1, 113.8, 123.3, 126.2, 128.3, 129.6, 140.5, 142.1, 156.2, 160.9, 169.0, 173.2, 198.2 (1 carbon atom unaccounted for). HRMS (TOF) (m/z): [M + Na]+ calcd for C41H63N5Na1O8, 776.4569; found, 776.4558.:hexanes 20:80, 30:70, 40:60, 50:50, 60:40, 70:50, 100:0, EtOAc:MeOH 99:1) to afford 10a as a red glass (0.052 g, 72%). 1H NMR (CDCl3, 300 MHz) 1.43 (s, 9H), 2.28 (s, 3H), 2.38 (s, 3H), 2.55 (t, J = 6.4 Hz, 2H), 2.75–2.82 (m, 4H), 3.04 (t, J = 6.4 Hz, 2H), 3.41–3.45 (m, 2H), 3.55 (q, J = 6.4 Hz, 2H), 3.97 (s, 3H), 5.05 (br s, 1H), 6.03 (s, 1H), 6.23 (dd, J = 3.6, 2.4 Hz, 1H), 6.54 (t, J = 5.1 Hz, 1H), 6.23 (dd, J = 3.6, 2.4 Hz, 1H), 6.52–6.56 (m, 1H), 6.73 (dd, J = 3.6, 1.2 Hz, 1H), 6.81 (br s, 1H), 6.88 (s, 1H). 13C NMR (CDCl3, 125 MHz) 12.6, 14.5, 28.5, 30.5, 38.2, 38.4, 39.6, 58.6, 77.4, 79.7, 96.0, 110.8, 111.9, 113.9, 122.7, 123.5, 126.3, 128.1, 130.0, 140.5, 142.8, 156.0, 160.7, 168.9, 173.2, 195.9 (1 carbon atom unaccounted for). HRMS (TOF) (m/z): [M + H]+ calcd for C29H40N5O5S2, 602.2465; found, 602.2456.:hexanes 20:80, 30:70, 40:60, 50:50, 60:40, 70:30, 100:0 then EtOAc:MeOH 99/1) to afford 10b as a red-brown glass (0.090 g, 75%). 1H NMR (CDCl3, 300 MHz) 1.43 (s, 9H), 1.66–1.68 (m, 4H), 2.21–2.26 (m, 5H), 2.39 (s, 3H), 2.69 (t, J = 6.3 Hz, 2H), 2.75 (t, J = 6.6 Hz, 2H), 2.82 (t, J = 6.3 Hz, 2H), 3.37–3.43 (m, 2H), 3.55 (q, J = 6.1 Hz, 2H), 3.98 (s, 3H), 5.02 (br s, 1H), 6.05 (s, 1H), 6.21 (dd, J = 3.6, 2.4 Hz, 1H), 6.59 (br s, 1H), 6.73 (dd, J = 3.6, 1.2 Hz, 1H), 6.77 (br s, 1H), 6.92 (s, 1H). 13C NMR (CDCl3, 125 MHz) 12.4, 14.0, 23.7, 25.3, 28.5, 36.4, 38.0, 38.3, 38.4, 39.6, 42.3, 58.7, 79.7, 96.2, 110.7, 112.0, 114.0, 123.5, 126.2, 128.2, 129.7, 140.5, 142.3, 156.0, 161.1, 169.1, 173.3, 197.6 (1 carbon atom unaccounted for). HRMS (TOF) (m/z): [M + H]+ calcd for C31H44N5O5S2, 630.2778; found, 630.2770.:hexanes 50:50, 70:30 EtOAc 100%) followed by Al2O3 type III neutral (EtOAc:hexanes 50:50, 60/40) to afford 10c as a red glass (0.138 g, 65%). 1H NMR (CDCl3, 300 MHz) 1.28 (s, 8H), 1.43 (s, 9H), 1.58–1.62 (m, 4H), 2.14 (s, 3H), 2.18 (t, J = 7.4 Hz, 2H), 2.39 (s, 3H), 2.62 (t, J = 7.4 Hz, 2H), 2.75 (t, J = 6.7 Hz, 2H), 2.81 (t, J = 5.8 Hz, 2H), 3.39–3.43 (m, 2H), 3.54 (q, J = 6.1 Hz, 2H), 3.98 (s, 3H), 5.06 (br s, 1H), 6.06 (s, 1H), 6.18 (t, J = 3.0 Hz, 1H), 6.41 (br s, 1H), 6.72 (d, J = 3.0 Hz, 2H), 6.93 (s, 1H). 13C NMR (CDCl3, 100 MHz) 12.4, 14.0, 24.3, 25.8, 28.5, 29.3, 29.5, 36.7, 37.9, 38.2, 38.6, 39.7, 42.8, 58.7, 79.7, 96.1, 110.7, 112.1, 113.9, 123.4, 123.6, 126.2, 128.2, 129.8, 140.3, 142.2, 156.1, 161.0, 169.1, 173.7, 198.2 (2 carbon atoms unaccounted for). HRMS (TOF) (m/z): [M + H]+ calcd for C35H52N5O5S2, 686.3404; found, 686.3386.:CHCl3 (1:1, 4 mL) was treated with HCl (3.3 mL, 2 M solution in Et2O, 6.60 mmol) and the reaction mixture was stirred at room temperature for 18 h. The solvent was removed under reduced pressure and the crude mixture was dissolved in MeOH (3.8 mL). NaOH (10% aq. solution) was added to the solution until a colour change occurred (dark red to orange). The solvent was removed under reduced pressure and the precipitate was filtered and washed with H2O to afford 2b (0.076 g, 93%) as orange solid. 1H NMR (CD3OD, 500 MHz) 1.68–1.72 (m, 4H), 2.24–2.27 (m, 2H), 2.40 (s, 3H), 2.66 (s, 3H), 2.79–2.83 (m, 2H), 3.28–3.32 (m, 2H), 3.92 (s, 3H), 6.08 (s, 1H), 6.25–6.27 (m, 1H), 6.76–6.78 (m, 2H), 7.02–7.03 (m, 1H). 13C NMR (CD3OD, 125 MHz) 12.3, 15.4, 25.1, 26.6, 37.0, 41.5, 41.7, 43.1, 59.00, 96.7, 111.2, 111.3, 114.1, 123.5, 123.7, 128.2, 128.9, 129.8, 142.7, 144.2, 162.0, 169.6, 176.6, 199.9. HRMS (TOF) (m/z): [M + H]+ calcd for C24H32N5O3, 438.2500; found, 438.2488.:MeOH (1:1, 6 mL) was treated with HCl 12 N (0.024 mL, 0.29 mmol) and the reaction mixture was stirred at room temperature for 7 h. The solvent was removed under vacuum and NaOH (10% aq. solution) was added drop-wise under stirring until the red solution turned orange-brown. The solvent was removed under vacuum and the precipitate was washed with water to give a dark red solid (0.066 g, 76%). Mp 88 °C. 1H NMR (DMSO-d6, 500 MHz) 1.22–1.32 (m, 8H), 1.47–1.49 (m, 2H), 1.56–1.59 (m, 2H), 2.03–2.06 (m, 2H), 2.34 (s, 3H), 2.67 (s, 3H), 2.72–2.74 (m, 2H), 2.99–3.03 (m, 2H), 3.25–3.35 (m, 2H), 3.88 (s, 3H), 6.19 (s, 1H), 6.25 (s, 1H), 6.70 (s, 1H), 6.79 (s, 1H), 7.14 (s, 1H), 7.68 (br s, 1H), 11.81 (br s, 1H). 13C NMR (DMSO-d6, 125 MHz) 11.7, 15.3, 23.7, 25.3, 28.6, 28.7, 28.8, 28.9, 35.4, 41.4, 41.8, 42.2, 58.4, 96.2, 110.1 (2C), 113.0, 122.4, 122.9, 126.3, 127.4, 128.2, 140.6, 142.1, 159.3, 167.3, 172.1, 196.6. HRMS (TOF) (m/z): [M + H]+ calcd for C28H40N5O3, 494.3126; found, 494.3129.:1, 4 mL) was treated with HCl 12 N (0.031 mL, 0.37 mmol) at room temperature. After 30 min additional HCl 12 N (0.030 mL, 0.36 mmol) was added and the reaction mixture stirred for an additional 3 hours. The solvent was removed under reduced pressure and the crude mixture was dissolved in MeOH (2 mL). NaOH (10% aq. solution) was added drop-wise under stirring until the red solution turned orange-brown. The solvent was removed under vacuum and the crude solid was purified using flash chromatography on Al2O3 basic type III (EtOAc 100%, CH2Cl2:MeOH 98:2, 95:5, 90:10 then MeOH 100%) to afford 3a as a dark red glass (0.105 g, 51%). 1H NMR (CDCl3, 500 MHz) 1.68–1.73 (m, 4H), 2.20 (s, 3H), 2.34 (s, 3H), 2.48 (t, J = 6.5 Hz, 2H), 2.84 (t, J = 6.5 Hz, 2H), 2.97 (t, J = 6.5 Hz, 2H), 3.28 (q, J = 6.5 Hz, 2H), 3.47–3.60 (m, 12H), 3.95 (s, 3H), 6.03 (s, 1H), 6.18–6.19 (s, 1H), 6.68–6.71 (m, 2H), 6.76 (s, 1H), 6.86 (s, 1H). 13C NMR (CDCl3, 125 MHz) 12.5, 14.5, 29.3, 30.4, 30.7, 37.4, 38.0, 40.1, 58.6, 69.3, 69.9, 70.0 (×2), 70.2, 70.5, 96.0, 110.7, 111.7, 113.9, 122.7, 123.5, 126.4, 128.1, 129.8, 140.6, 142.7, 160.7, 168.9, 173.2, 196.1. HRMS (TOF) (m/z): [M + H]+ calcd for C30H44N5O6, 570.3286; found, 570.3295.:CHCl3 (1:1, 3.4 mL) was treated with HCl 12 N (0.55 mL, 6.60 mmol) and the reaction mixture was stirred at room temperature for 25 h. The solvent was removed under reduced pressure and the crude mixture was dissolved in MeOH (2 mL). NaOH (6 M aq. solution) was added to the solution until a colour change occurred (dark red to orange). The solvent was removed under reduced pressure and the crude mixture was purified using flash chromatography on Al2O3 basic type III (CH2Cl2:MeOH, 9:1) to afford 3b (0.074 g, 83%) as a dark red glass. 1H NMR (CDCl3, 500 MHz) 1.63 (br s, 4H), 1.66–1.71 (m, 2H), 1.72–1.77 (m, 2H), 2.15 (s, 3H), 2.17 (br s, 2H), 2.37 (s, 3H), 2.65 (br s, 2H), 2.76 (t, 2H, J = 6.6 Hz), 3.30–3.34 (m, 2H), 3.52 (t, 4H, J = 5.8 Hz), 3.56 (ddd, 12H, J = 24.1, 10.1, 8.0 Hz), 3.96 (s, 3H), 6.04 (s, 1H), 6.18–6.19 (m, 1H), 6.63 (br s, 1H), 6.71 (d, 1H, J = 3.5 Hz), 6.74 (br s, 1H), 6.91 (s, 1H). 13C NMR (CDCl3, 125 MHz) 12.4, 14.0, 23.8, 25.5, 29.1, 29.8, 31.0, 33.3, 36.7, 37.7, 39.7, 42.4, 58.6, 69.6, 69.9, 70.2, 70.6, 96.2, 110.7, 112.0, 113.9, 123.4 (×2), 126.2, 128.2, 129.7, 140.5, 142.3, 161.1, 169.1, 173.0, 197.6. HRMS (TOF) (m/z): [M + H]+ calcd for C32H48N5O6, 598.3599; found, 598.3577.:CHCl3 (1:1, 3.2 mL) was treated with HCl 12 N (0.65 mL of 12 M aqueous solution, 7.80 mmol) and the reaction mixture was stirred at room temperature for 5 h. The solvent was removed under reduced pressure and the crude mixture was dissolved in MeOH (2 mL). NaOH (2 M aq. solution) was added to the solution until a colour change occurred (dark red to orange). The solvent was removed under reduced pressure and the crude mixture was purified by flash chromatography using Al2O3 basic type III (CH2Cl2:MeOH, 9:1) to afford 3c (0.068 g, 70%) as a dark red glass. 1H NMR (CDCl3, 500 MHz) 1.24–1.27 (m, 8H), 1.56–1.60 (m, 4H), 1.63 (br s, 4H), 1.65–1.79 (m, 4H), 2.11 (d, 2H, J = 7.8 Hz), 2.15 (s, 3H), 2.38 (s, 3H), 2.62 (t, 2H, J = 7.4 Hz), 2.77 (t, 2H, J = 6.7 Hz), 3.33 (dd, 2H, J = 12.2, 6.0 Hz), 3.51–3.63 (m, 12H), 3.97 (s, 3H), 6.05 (s, 1H), 6.17–6.19 (m, 1H), 6.39–6.43 (br m, 1H), 6.70–6.73 (m, 2H), 6.92 (s, 1H). 13C NMR (CDCl3, 125 MHz) 12.4, 14.1, 24.3, 25.9, 29.1, 29.3, 29.4, 29.5, 33.4, 36.9, 37.8, 39.8, 58.6, 69.6 (2C), 70.1, 70.3 (2C), 70.7, 96.1, 110.7, 112.1, 113.8, 123.3, 123.6, 126.2, 128.2, 129.7, 140.4, 142.2, 160.9, 169.1, 173.2, 198.1 (2 carbons uncounted for). HRMS (TOF) (m/z): [M + H]+ calcd for C36H56N5O6, 654.4225; found, 654.4203.:CHCl3 (1:1, 4 mL) was treated with HCl 12 N (0.024, 0.077 mmol). The reaction was stirred overnight at room temperature and additional HCl 12 N (0.024, 0.077 mmol) was added. The solution was concentrated under reduced pressure and the crude mixture dissolved in MeOH (3 mL). NaOH (10% aq. solution) was added dropwise under stirring until the red solution turned orange-brown. The solvent was removed under vacuum and the crude solid was purified using flash chromatography on Al2O3 basic type III (EtOAc 100%, CH2Cl2:MeOH, 95:5) to afford 4a as a dark red glass (0.033 g, 77%) 1H NMR (CDCl3, 300 MHz) 2.25 (s, 3H), 2.38 (s, 3H), 2.53 (t, J = 6.3 Hz), 2.73–2.80 (m, 4H), 2.97–3.05 (m, 4H), 3.56 (q, J = 6.8 Hz, 2H), 3.97 (s, 3H), 6.03 (s, 1H), 6.22 (dd, J = 3.8, 2.7 Hz, 1H), 6.38 (t, J = 5.7 Hz, 1H), 6.73 (dd, J = 3.8, 1.2 Hz, 1H), 6.79 (s, 1H), 6.89 (s, 1H). 13C NMR (CDCl3, 125 MHz) 12.6, 14.8, 30.6, 38.0, 38.2, 38.5, 40.8, 42.7, 58.7, 96.0, 110.9, 111.9, 113.8, 122.7, 123.2, 126.5, 128.2, 129.8, 142.6, 160.7, 168.9, 173.1, 196.0 (1 carbon atom unaccounted for). HRMS (TOF) (m/z): [M + H]+ calcd for C24H32N5O3S2, 502.1941; found, 502.1933. HRMS (TOF) (m/z): [M + H]+ calcd for C24H32N5O3S2, 502.1933; found, 502.1941.:water (1:1, 1 mL) and PTSA (0.016 g, 0.052 mmol) was added. The reaction was stirred at 60 °C for 3 hours and additional PTSA (0.016 g, 0.052 mmol) was added. After 16 h the solvent was removed under reduced pressure the crude solid was purified using Al2O3 type III basic (CH2Cl2/MeOH 99/1) to afford 19b as an orange glass (0.011 g, 64%). 1H NMR (CDCl3, 300 MHz) 0.04 (s, 9H), 0.96–1.02 (m, 2H), 1.65–1.68 (m, 4H), 1.73–1.82 (m, 5H), 2.04–2.13 (m, 1H), 2.15–2.29 (m, 2H), 2.32–2.40 (m, 2H), 2.68 (s, 3H), 2.72 (s, 3H), 3.30–3.43 (m, 2H), 3.50–3.64 (m, 5H), 3.96 (s, 3H), 4.15–4.21 (m, 2H), 6.01 (s, 1H), 6.23–6.25 (m, 1H), 6.45 (t, 1H, J = 5.1 Hz), 6.51 (t, 1H, J = 4.8 Hz), 6.72 (d, 1H, J = 3.6 Hz), 6.84 (br s, 1H), 6.89 (s, 1H). 13C NMR (CDCl3, 100 MHz), −1.4 (3C), 12.5, 14.9, 17.6, 23.9, 25.5, 29.2, 29.3, 30.5, 33.0, 36.7, 37.8, 42.4, 54.2, 58.6, 63.5, 70.0, 70.1, 70.2, 70.6 (4C), 95.9, 110.8, 111.9, 113.5, 123.0, 123.2, 126.7, 128.4, 129.2, 141.4, 141.7, 160.6, 168.8, 172.5, 173.0, 175.9, 197.6.0. HRMS (TOF) (m/z): [M + Na]+ calcd for C42H66N6Na1O9Si1, 849.4553; found, 849.4557.:water (1:1, 6 mL) and PTSA (0.063 g, 0.32 mmol) was added. The reaction was stirred at 60 °C for 3 hours and additional PTSA (0.063 mg, 0.32 mmol) was added. After 16 h the reaction mixture was quenched by addition of NaHCO3 solid until the dark red solution turned dark brown. The solvent were removed under vacuum and the crude solid was purified using Al2O3 type III basic (CH2Cl2/MeOH 98/2, 95/5) to afford 19c as an orange glass (0.066 g, 57%). 1H NMR (CDCl3, 300 MHz) 0.03 (s, 9H), 0.96–1.01 (m, 2H), 1.24–1.27 (m, 11H), 1.58–1.60 (m, 4H), 1.74 (quint., 4H, J = 6.1 Hz), 2.09–2.14 (m, 3H), 2.19 (s, 3H), 2.29 (t, 2H, J = 7.3 Hz), 2.38 (s, 3H), 2.63 (t, 2H, J = 7.3 Hz), 3.32 (q, 4H, J = 6.1 Hz), 3.38–3.43 (m, 1H), 3.50–3.64 (m, 13H), 3.96 (s, 3H), 4.15–4.21 (m, 2H), 6.04 (s, 1H), 6.19 (dd, 1H, J = 3.6, 2.7 Hz), 6.24 (br s, 1H), 6.49 (br s, 1H), 6.71 (dd, 1H, J = 3.6, 1.1 Hz), 6.74–6.75 (m, 1H), 6.91 (s, 1H). 13C NMR (CDCl3, 125 MHz), −1.4 (3C), 12.4, 14.3, 17.6, 24.3, 25.9, 29.2, 29.3, 29.4, 29.5, 29.8, 30.5, 32.9, 36.9, 37.8 (×2), 42.8, 54.1, 58.6, 63.4, 70.0 (×2), 70.2, 70.6 (4C), 96.0, 110.7, 112.1, 113.7, 123.2, 123.5, 126.3, 128.3, 129.5, 140.8, 142.0, 160.8, 179.0, 172.4, 173.3, 175.9, 198.2. HRMS (TOF) (m/z): [M + Na]+ calcd for C46H74N6Na1O9Si1, 905.5179; found, 905.5145.General procedure 2 (GP2)
A solution of linker-prodigiosene conjugate (1 eq.) and 23 (1 eq.) in anhydrous DMSO (0.05 M) was treated with NEt3 (2 eq.). The resulting reaction mixture was stirred at room temperature for 5 h under N2 atmosphere. Water was added to the reaction mixture until a precipitate formed, which was isolated via microfiltration, then washed 3 times with water and dried under vacuum.:MeOH, 9:1) and 25c was obtained as dark red glass (0.065 g, 47%). 1H NMR (CDCl3, 500 MHz) 0.01 (s, 9H), 0.05 (s, 9H), 0.82–0.89 (m, 1H), 0.96–1.07 (m, 4H), 1.25 (br s, 8H), 1.57–1.58 (br m, 4H), 1.68–1.77 (m, 4H), 2.09–2.14 (m, 2H), 2.19–2.25 (m, 2H), 2.29–2.33 (m, 2H), 2.36 (s, 6H), 2.61 (t, 2H, J = 7.1 Hz), 3.27–3.34 (m, 4H), 3.47–3.58 (m, 12H), 3.94 (s, 3H), 4.17–4.22 (m, 2H), 4.26–4.31 (m, 2H), 4.62 (br s, 3H), 5.43 (br s, 1H), 5.98 (s, 1H), 6.24 (br s, 1H), 6.44–6.48 (m, 1H), 6.60 (d, 2H, J = 8.3 Hz), 6.72 (br s, 1H), 6.81–6.85 (m, 2H), 6.90 (br s, 1H), 7.40 (d, 1H, J = 7.2 Hz), 7.67 (d, 2H, J = 8.3 Hz), 8.77 (s, 1H). 13C NMR (CDCl3, 125 MHz), −1.4 (6C), 12.4, 15.2, 17.5, 17.7, 24.3, 25.9, 27.9, 29.0, 29.3 (×2), 29.4, 29.5 (×2), 29.8 (2C), 32.9, 36.9, 37.7, 38.0 (2C), 42.8, 46.9, 53.1, 58.7, 64.0, 66.1 (2C), 69.9, 70.0, 70.1, 70.2, 70.6, 95.7, 110.9, 111.9, 112.3 (2C), 114.0, 122.8, 123.5, 126.6, 127.9, 129.2, 130.1 (2C), 140.2, 142.2, 149.4, 150.3, 151.8, 154.0, 154.8, 159.6, 161.0, 167.3, 168.5, 172.6, 172.7, 173.5, 198.1. HRMS (TOF) (m/z): [M + H]+ calcd for C66H97N12O13Si2, 1321.6831; found, 1321.6807.:CHCl3 (1:1, 4 mL) was treated with HCl 12 N (1.2 mL, 14.4 mmol) and the reaction mixture was stirred at room temperature for 4 h. The solvent was removed under reduced pressure to obtain 4b as dark purple solid (0.124 g), which was used in the next step without any further purification. HRMS (TOF) (m/z): [M + H]+ calcd for C26H36N5O3S2, 530.2254; found, 530.2264. Compound 26b was obtained according to GP2. The crude mixture was purified by column chromatography using Al2O3 type III neutral (CH2Cl2:MeOH, 9:1) and 26b was obtained as an orange solid (0.176 g, 72%). Mp 135–137 °C. 1H NMR (CDCl3, 500 MHz) 0.02 (s, 9H), 0.06 (s, 9H), 0.81–0.90 (m, 1H), 0.96–1.06 (m, 4H), 1.58 (br s, 4H), 2.09–2.19 (m, 4H), 2.26 (br m, 3H), 2.31 (s, 3H), 2.34–2.40 (m, 2H), 2.57 (br s, 2H), 2.69–2.78 (m, 4H), 3.40–3.53 (m, 4H), 3.93 (s, 3H), 4.16–4.32 (m, 4H), 4.58 (d, 2H, J = 4.0 Hz), 4.65–4.72 (m, 1H), 5.45–5.46 (m, 1H), 5.95 (s, 1H), 6.25–6.27 (m, 1H), 6.56 (d, 2H, J = 8.6 Hz), 6.72 (br s, 1H), 6.80 (s, 1H), 6.89–6.95 (m, 2H), 7.14 (t, 1H, J = 5.6 Hz), 7.32 (d, 1H, J = 6.7 Hz), 7.65 (d, 2H, J = 8.6 Hz), 8.75 (s, 1H). 13C NMR (CDCl3, 125 MHz), −1.4 (6C), 12.4, 15.4, 17.5, 17.7, 23.6, 25.3, 27.3, 29.8, 32.9, 36.4, 37.9, 38.1, 38.6, 38.8, 42.2, 46.8, 53.0, 58.7, 64.0, 66.0, 95.6, 111.0, 111.6, 112.2 (2C), 114.2, 122.5, 123.1, 124.0, 126.5, 127.5, 129.2 (2C), 129.9, 139.8, 142.3, 149.2, 149.9, 150.4, 151.8, 154.0, 154.7, 159.3, 161.5, 167.7, 168.3, 172.6, 173.2, 173.8, 197.5. HRMS (TOF) (m/z): [(M + H + Na)/2]+ calcd for C56H77N12NaO10S2Si2, 610.2376; found, 610.2356.:CHCl3 (1:1, 4 mL) was treated with HCl 12 N (0.036 mL, 0.44 mmol). After an additional 30 min HCl 12 N (0.030 mL, 0.36 mmol) was added and the reaction mixture stirred for a further 12 hours. The solvent was removed under reduced pressure to afford 4c as a red glass (0.130 g), which was used in the next step without any further purification. HRMS (TOF) (m/z): [M + H]+ calcd for C30H44N5O3S2, 586.2880; found, 586.2856. Compound 26c was obtained according to GP2 as a dark red glass (0.086 g, 69%). 1H NMR (CDCl3, 500 MHz) 0.00 (s, 9H), 0.04 (s, 9H), 0.96–1.00 (m, 4H), 1.02–1.04 (m, 8H), 1.21–1.24 (m, 4H), 1.53–1.55 (m, 4H), 2.10–2.13 (m, 3H), 2.21 (app br s, 4H), 2.34–2.36 (m, 5H), 2.57–2.58 (m, 2H), 2.68–2.69 (m, 2H), 2.72–2.75 (m, 2H), 3.40–3.52 (m, 4H), 3.92 (m, 3H), 4.17–4.20 (m, 2H), 4.26 (t, J = 8.2 Hz, 2H), 4.58 (s, 2H), 4.64–4.68 (m, 1H), 5.59 (br s, 1H), 5.97 (s, 1H), 6.21 (s, 1H), 6.56 (d, J = 7.5 Hz, 2H), 6.70 (s, 2H), 6.83 (s, 2H), 7.25–7.26 (m, 2H), 7.35 (d, J = 6.0 Hz, 1H), 7.62 (d, J = 7.5 Hz, 2H), 8.74 (s, 1H). 13C NMR (CDCl3, 125 MHz), −1.4 (6C), 12.4, 14.9, 17.5, 17.7, 24.3, 25.8, 28.1, 29.1, 29.2, 29.4 (×2), 32.8, 36.6, 37.9, 38.0, 38.6, 42.8, 46.9, 52.9, 58.7, 64.1, 66.0, 95.7, 110.9, 111.8, 112.2 (2C), 114.0, 122.5, 123.4, 123.8, 126.4, 127.7, 129.2 (2C), 130.0, 139.9, 142.1, 149.3, 149.8, 150.4, 151.8, 154.1, 154.8, 159.8, 161.3, 167.6, 168.5, 168.6, 172.6, 173.2, 174.0, 198.1 (1 carbon atom unaccounted for). HRMS (TOF) (m/z): [(M + H + Na)/2]2+ calcd for C60H85N12Na1O10S2Si2, 638.2689; found, 638.2663.General procedure 3 (GP3)
A solution of (1 eq.) Teoc-FA-LINKERS-prod. (1 eq.) in anhydrous DMSO (0.1 M) was treated with TBAF (1 M solution in THF, 2 eq.) and AcOH (1.25/0.1 mmol). The resulting reaction mixture was stirred at room temperature for 18 h under N2 atmosphere. EtOAc was added to the reaction mixture until a precipitate formed, which was filtered using Milipore® and washed 3 times with water. The obtained dark red solid was dissolved in DMSO (0.1 M), treated with NaOH (0.2 M solution in MeOH) and stirred at room temperature for 10 min. Water was added and a precipitate was formed. The resulting sodium salt was isolated via microfiltration, then washed 3 times with water and dried under vacuum.Conflicts of interest
There is no conflict to declare.Acknowledgements
During this work, E. M. was supported by a trainee award from The Beatrice Hunter Cancer Research Institute (BHCRI) with funds provided by Cancer Care Nova Scotia as part of The Terry Fox Foundation Strategic Health Research Training Program in Cancer Research at Canadian Institutes of Health Research (CIHR). This work was funded by research grants to A. T. from: CIHR (Grant # 133110), Canadian Breast Cancer Foundation-Atlantic, Breast Cancer Society of Canada/QEII Foundation Awards for Breast Cancer Research, BHCRI and Nova Scotia Health Research Foundation. A. T. is a Senior Scientist of the BHCRI.References
- A. Fürstner, Angew. Chem., Int. Ed., 2003, 42, 3582–3603 CrossRef PubMed.
- N. R. Williamson, P. C. Fineran, F. J. Leeper and G. P. Salmond, Nat. Rev. Microbiol., 2006, 4, 887–899 CrossRef CAS PubMed.
- R. K. Suryawanshi, C. D. Patil, H. P. Borase, C. P. Narkhede, A. Stevenson, J. E. Hallsworth and S. V. Patil, Int. J. Cosmet. Sci., 2015, 37, 98–107 CrossRef CAS PubMed.
- Y. Kim and J. Choi, Fibers Polym., 2015, 16, 1981–1987 CrossRef CAS.
- Y. Ren, J. Gong, R. Fu, Z. Li, Q. Li, J. Zhang, Z. Yu and X. Cheng, Dyes Pigm., 2017, 138, 147–153 CrossRef CAS.
- A. Fürstner and E. J. Grabowski, ChemBioChem, 2001, 2, 706–709 CrossRef.
- A. Fürstner, K. Reinecke, H. Prinz and H. Waldmann, ChemBioChem, 2004, 5, 1575–1579 CrossRef PubMed.
- M. S. Melvin, J. T. Tomlinson, G. Park, C. S. Day, G. R. Saluta, G. L. Kucera and R. A. Manderville, Chem. Res. Toxicol., 2002, 15, 734–741 Search PubMed.
- M. S. Melvin, J. T. Tomlinson, G. R. Saluta, G. L. Kucera, N. Lindquist and R. A. Manderville, J. Am. Chem. Soc., 2000, 122, 6333–6334 CrossRef CAS.
- B. Montaner, W. Castillo-Avila, M. Martinell, R. Oellinger, J. Aymami, E. Giralt and R. Perez-Tomas, Toxicol. Sci., 2005, 85, 870–879 CrossRef CAS PubMed.
- S. Ohkuma, T. Sato, M. Okamoto, H. Matsuya, K. Arai, T. Kataoka, K. Nagai and H. H. Wasserman, Biochem. J., 1998, 334, 731–741 CrossRef CAS PubMed.
- G. Park, J. T. Tomlinson, M. S. Melvin, M. W. Wright, C. S. Day and R. A. Manderville, Org. Lett., 2003, 5, 113–116 CrossRef CAS PubMed.
- R. I. Sáez Díaz, S. M. Bennett and A. Thompson, ChemMedChem, 2009, 4, 742–745 CrossRef PubMed.
- T. Sato, H. Konno, Y. Tanaka, T. Kataoka, K. Nagai, H. H. Wasserman and S. Ohkuma, J. Biol. Chem., 1998, 273, 21455–21462 CrossRef CAS PubMed.
- J. L. Seganish and J. T. Davis, Chem. Commun., 2005, 5781–5783 RSC.
- W. R. Hearn, M. K. Elson, R. H. Williams and J. Medina-Castro, J. Org. Chem., 1970, 35, 142–146 CrossRef CAS PubMed.
- E. Marchal, S. Rastogi, A. Thompson and J. T. Davis, Org. Biomol. Chem., 2014, 12, 7515–7522 RSC.
- E. Marchal, D. A. Smithen, I. M. Uddin, A. W. Robertson, D. L. Jakeman, V. Mollard, C. D. Goodman, K. S. MacDougall, S. A. McFarland, G. I. McFadden and A. Thompson, Org. Biomol. Chem., 2014, 12, 4132–4142 RSC.
- E. Marchal, M. I. Uddin, D. A. Smithen, L. A. Hawco, M. Lanteigne, D. P. Overy, R. G. Kerr and A. Thompson, RSC Adv., 2013, 3, 22967–22971 RSC.
- S. Rastogi, E. Marchal, I. Uddin, B. Groves, J. Colpitts, S. A. McFarland, J. T. Davis and A. Thompson, Org. Biomol. Chem., 2013, 11, 3834–3845 RSC.
- J. Regourd, A. Al-Sheikh Ali and A. Thompson, J. Med. Chem., 2007, 50, 1528–1536 CrossRef CAS PubMed.
- R. I. Sáez Díaz, J. Regourd, P. V. Santacroce, J. T. Davis, D. L. Jakeman and A. Thompson, Chem. Commun., 2007, 2701–2703 RSC.
- D. A. Smithen, A. M. Forrester, D. P. Corkery, G. Dellaire, J. Colpitts, S. A. McFarland, J. N. Berman and A. Thompson, Org. Biomol. Chem., 2013, 11, 62–68 RSC.
- M. I. Uddin, S. Thirumalairajan, S. M. Crawford, T. S. Cameron and A. Thompson, Synlett, 2010, 2561–2564 CAS.
- C. Ferrario and G. Batist, Expert Opin. Drug Discovery, 2014, 9, 647–668 CrossRef CAS PubMed.
- A. Cheung, H. J. Bax, D. H. Josephs, K. M. Ilieva, G. Pellizzari, J. Opzoomer, J. Bloomfield, M. Fittall, A. Grigoriadis, M. Figini, S. Canevari, J. F. Spicer, A. N. Tutt and S. N. Karagiannis, Oncotarget, 2016, 7, 52553–52574 Search PubMed.
- R. Paulmurugan, R. Bhethanabotla, K. Mishra, R. Devulapally, K. Foygel, T. Sekar, J. Ananta, T. Massoud and A. Joy, Mol. Cancer Ther., 2016, 15, 221–231 CrossRef CAS PubMed.
- Y. Lu and P. S. Low, Adv. Drug Delivery Rev., 2002, 54, 675–693 CrossRef CAS PubMed.
- J. Sudimack and R. J. Lee, Adv. Drug Delivery Rev., 2000, 41, 147–162 CrossRef CAS PubMed.
- F. Zagouri, M. A. Dimopoulos, E. Bournakis and C. A. Papadimitriou, Eur. J. Gynaecol. Oncol., 2010, 31, 268–277 CAS.
- Y.-S. Yi, Immune Netw., 2016, 16, 337–343 CrossRef PubMed.
- W. Xia, A. R. Hilgenbrink, E. L. Matteson, M. B. Lockwood, J. X. Cheng and P. S. Low, Blood, 2009, 113, 438–446 CrossRef CAS PubMed.
- L. H. Matherly, Z. Hou and Y. Deng, Cancer Metastasis Rev., 2007, 26, 111–128 CrossRef CAS PubMed.
- J. M. Scott and D. G. Weir, J. Cardiovasc. Risk, 1998, 5, 223–227 CrossRef CAS PubMed.
- R. Zhao, S. H. Min, Y. Wang, E. Campanella, P. S. Low and I. D. Goldman, J. Biol. Chem., 2009, 284, 4267–4274 CrossRef CAS PubMed.
- P. S. Low and S. A. SuKularatne, Curr. Opin. Chem. Biol., 2009, 13, 256–262 CrossRef CAS PubMed.
- S. Wang and P. S. Low, J. Controlled Release, 1998, 53, 39–48 CrossRef CAS PubMed.
- C. L. A. Hawco, E. Marchal, M. I. Uddin, A. E. G. Baker, D. P. Corkery, G. Dellaire and A. Thompson, Bioorg. Med. Chem., 2013, 21, 5995–6002 CrossRef CAS PubMed.
- G. Huang, D. Pemp, P. Stadtmüller, M. Nimczick, J. Heilmann and M. Decker, Bioorg. Med. Chem. Lett., 2014, 24, 4209–4214 CrossRef CAS PubMed.
- S. Dhar, Z. Liu, J. Thomale, H. Dai and S. J. Lippard, J. Am. Chem. Soc., 2008, 130, 11467–11476 CrossRef CAS PubMed.
- S. Zalipsky, Adv. Drug Delivery Rev., 1995, 16, 157–182 CrossRef CAS.
- M. P. Grillo, Expert Opin. Drug Metab. Toxicol., 2015, 11, 1281–1302 CrossRef CAS PubMed.
- H. Joyce, A. McCann, M. Clynes and A. Larkin, Expert Opin. Drug Metab. Toxicol., 2015, 11, 795–809 CrossRef CAS PubMed.
- A. Sullivan, A. Gibson, B. K. Park and D. J. Naisbitt, Expert Opin. Drug Metab. Toxicol., 2015, 11, 357–368 CrossRef CAS PubMed.
- R. S. Shirazi, K. K. Ewert, C. Leal, R. N. Majzoub, N. F. Bouxsein and C. R. Safinya, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2156–2166 CrossRef CAS PubMed.
- I. R. Vlahov and C. P. Leamon, Bioconjugate Chem., 2012, 23, 1357–1369 CrossRef CAS PubMed.
- C. Chen, J. Ke, X. E. Zhou, W. Yi, J. S. Brunzelle, J. Li, E.-L. Yong, H. E. Xu and K. Melcher, Nature, 2013, 500, 486–489 CrossRef CAS PubMed.
- M. P. Carrasco, E. A. Enyedy, N. I. Krupenko, S. A. Krupenko, E. Nuti, T. Tuccinardi, S. Santamaria, A. Rossello, A. Martinelli and M. A. Santos, Med. Chem., 2011, 7, 265–274 CrossRef CAS.
- S. K. Choi, T. Thomas, M.-H. Li, A. Kotlyar, A. Desai and J. R. Baker Jr, Chem. Commun., 2010, 46, 2632–2634 RSC.
- A. Galbiati, C. Tabolacci, B. M. D. Rocca, P. Mattioli, S. Beninati, G. Paradossi and A. Desideri, Bioconjugate Chem., 2011, 22, 1066–1072 CrossRef CAS PubMed.
- L. Jiang, Z.-M. Gao, L. Ye, A.-Y. Zhang and Z.-G. Feng, Polymer, 2013, 54, 5188–5198 CrossRef CAS.
- H. Jing, Z. Guo, W. Guo, W. Yang, P. Xu and X. Zhang, Bioorg. Med. Chem. Lett., 2012, 22, 3418–3424 CrossRef CAS PubMed.
- C. Lainé, E. Mornet, L. Lemiègre, T. Montier, S. Cammas-Marion, C. Neveu, N. Carmoy, P. Lehn and T. Benvegnu, Chemistry, 2008, 14, 8330–8340 CrossRef PubMed.
- P. Li, Y. Wang, F. Zeng, L. Chen, Z. Peng and L. X. Kong, Carbohydr. Res., 2011, 346, 801–806 CrossRef CAS PubMed.
- M. A. Santos, E. A. Enyedy, E. Nuti, A. Rossello, N. I. Krupenko and S. A. Krupenko, Bioorg. Med. Chem., 2007, 15, 1266–1274 CrossRef CAS PubMed.
- R. K. Singh, D. Rai, D. Yadav, A. Bhargava, J. Balzarini and E. De Clercq, Eur. J. Med. Chem., 2010, 45, 1078–1086 CrossRef CAS PubMed.
- A. F. Trindade, R. F. M. Frade, E. M. S. Macoas, C. Graca, C. A. B. Rodrigues, J. M. G. Martinho and C. A. M. Afonso, Org. Biomol. Chem., 2014, 12, 3181–3190 RSC.
- P. M. Valencia, M. H. Hanewich-Hollatz, W. Gao, F. Karim, R. Langer, R. Karnik and O. C. Farokhzad, Biomaterials, 2011, 32, 6226–6233 CrossRef CAS PubMed.
- Y. Yang, Y. M. Zhang, Y. Chen, D. Zhao, J. T. Chen and Y. Liu, Chemistry, 2012, 18, 4208–4215 CrossRef CAS PubMed.
- H. Zhang, Z. Cai, Y. Sun, F. Yu, Y. Chen and B. Sun, J. Biomed. Mater. Res., Part A, 2012, 100, 2441–2449 Search PubMed.
- A. Bettio, M. Honer, C. Müller, M. Brühlmeier, U. Müller, R. Schibli, V. Groehn, A. P. Schubiger and S. M. Ametamey, J. Nucl. Med., 2006, 47, 1153–1160 CAS.
- I. R. Vlahov, H. K. Santhapuram, P. J. Kleindl, S. J. Howard, K. Stanford and C. P. Leamon, Bioorg. Med. Chem. Lett., 2006, 16, 5093–5096 CrossRef CAS PubMed.
- W. A. Henne, S. A. Kularatne, J. Hakenjos, J. D. Carron and K. L. Henne, Bioorg. Med. Chem. Lett., 2013, 23, 5810–5813 CrossRef CAS PubMed.
- T. Suzuki, S. Hisakawa, Y. Itoh, N. Suzuki, K. Takahashi, M. Kawahata, K. Yamaguchi, H. Nakagawa and N. Miyata, Bioorg. Med. Chem. Lett., 2007, 17, 4208–4212 CrossRef CAS PubMed.
- M. Nomura, S. Shuto and A. Matsuda, J. Org. Chem., 2000, 65, 5016–5021 CrossRef CAS PubMed.
- C. P. Leamon, M. A. Parker, I. R. Vlahov, L. C. Xu, J. A. Reddy, M. Vetzel and N. Douglas, Bioconjugate Chem., 2002, 13, 1200–1210 CrossRef CAS PubMed.
- J. D. Seitz, J. G. Vineberg, E. Herlihy, B. Park, E. Melief and I. Ojima, Bioorg. Med. Chem., 2015, 23, 2187–2194 CrossRef CAS PubMed.
- E. Vlashi, L. E. Kelderhouse, J. E. Sturgis and P. S. Low, ACS Nano, 2013, 7, 8573–8582 CrossRef CAS PubMed.
- J. Luo, M. D. Smith, D. A. Lantrip, S. Wang and P. L. Fuchs, J. Am. Chem. Soc., 1997, 119, 10004–10013 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01435g |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2019 |