
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural analysis and antioxidant activity of an arabinoxylan from Malvastrum coromandelianum L. (Garcke)
Shanti Devi,
Ajeet K. Lakhera and
Vineet Kumar *
*
Chemistry and Bioprospecting Division, Forest Research Institute, Dehradun, India. E-mail: drvineet@gmail.com; Fax: +91-135-2756865; Tel: +91-9410555335
First published on 6th August 2019
Abstract
Malvastrum coromandelianum L. (Garcke) is extensively used in traditional medicinal systems to treat various ailments. In the present study, an alkali-soluble polysaccharide (MAP) was isolated from the leaves of M. coromandelianum in 1.15% (w/w) yield. MAP was composed of L-rhamnose, L-arabinose, D-xylose, D-glucose and D-galactose in a 1.00![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6.04:19.88:1.07:3.03 molar ratio along with D-glucuronic acid (1.95). Methylation/linkage analysis revealed a backbone of →4)-β-D-Xylp(1→ (30.09 mol%) with a side chain of →3)-α-L-Araf(1→ (15.21 mol%) residues. The structure of MAP was elucidated by a combination of degradative and derivatization techniques, including hydrolysis, alditol acetate derivatization, methylation, GC-MS, partial hydrolysis, ESI-MS and NMR (1D, 2D) spectral analysis. Based on correlation analysis, MAP was found to be an arabinoxylan comprising a backbone of →4)-β-D-linked Xylp(1→ with branching at O-2 by a →3)-α-L-Araf(1→ and →3)-β-D-Xylp(1→ chain. MAP also exhibited ferric ion reducing activity, with a reducing power of 0.914 ± 0.01 (R2 = 0.972) at 1 mg mL−1 concentration, which showed dose-dependent behavior. MAP can be utilized as a potential antioxidant.
:6.04:19.88:1.07:3.03 molar ratio along with D-glucuronic acid (1.95). Methylation/linkage analysis revealed a backbone of →4)-β-D-Xylp(1→ (30.09 mol%) with a side chain of →3)-α-L-Araf(1→ (15.21 mol%) residues. The structure of MAP was elucidated by a combination of degradative and derivatization techniques, including hydrolysis, alditol acetate derivatization, methylation, GC-MS, partial hydrolysis, ESI-MS and NMR (1D, 2D) spectral analysis. Based on correlation analysis, MAP was found to be an arabinoxylan comprising a backbone of →4)-β-D-linked Xylp(1→ with branching at O-2 by a →3)-α-L-Araf(1→ and →3)-β-D-Xylp(1→ chain. MAP also exhibited ferric ion reducing activity, with a reducing power of 0.914 ± 0.01 (R2 = 0.972) at 1 mg mL−1 concentration, which showed dose-dependent behavior. MAP can be utilized as a potential antioxidant.
1. Introduction
Reactive oxygen species (ROS), such as singlet oxygen (1O2), superoxide anions (O2−), hydroxyl, peroxyl and peroxynitrite, are generated during normal cellular respiration in the body.1,2 Under normal circumstances, these species regulate cell growth and control viruses and bacteria.3 However, excessive ROS in the body can attack tissues and disrupt redox homeostasis, which result in oxidative stress4 and eventually lead to T-cell damage, decreased immune function and aging, diabetes, immuno-suppression, neuro-degeneration, increasing cardiovascular and cerebrovascular disease risk, etc. ROS also act as strong carcinogens and damage DNA via altering the genes such as oncogenes and tumor suppressor, apoptosis-regulating, and DNA-repair genes; this causes numerous chronic ailments, e.g., cancer and arteriosclerosis.5,6 Antioxidants play noteworthy roles in the prevention of free radical damage to the body by quenching ROS. In this context, polysaccharides are gaining massive popularity as potent natural antioxidants from plants. Polysaccharides can inhibit lipid peroxidation, enhance the body's ability to scavenge free radicals, inhibit cardiovascular and cerebrovascular diseases, etc.7 Algal polysaccharides are found to prevent oxidative damage in living organisms.8 Astragalus9 and Rhizoma Dioscorea nipponica polysaccharide10 block the NF-κB pathway to inhibit ROS production. Lycium barbarum polysaccharides enhance the activities of superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px) in chickens.11 Ganoderma lucidum polysaccharides exhibit in vitro antioxidant activities.12 A polysaccharide from Cordyceps sinensis slows ROS generation by inhibiting PDGF-BB-induced inflammation.13 An extracellular polysaccharide from Morchella can significantly improve the activity of enzymes, viz., GSH-Px and SOD in the blood, spleen, heart, liver, and kidneys of mice and decrease the malondialdehyde content in these organs.14Therefore, polysaccharides from plants are a promising source of antioxidants, which can effectively prevent damage due to oxidative stress and hence prevent diseases. In our laboratory, the polysaccharides from plant species used in traditional medicine are being characterized, and their activity profiles are being studied to provide leads for future drug development based on their structure–activity relationships.15–19 In our continuous endeavor towards this goal, it was observed that a polysaccharide from Malvastrum coromandelianum (Garcke) Linn showed significant ferric ion reducing ability using the potassium ferricyanide assay. Interestingly, M. coromandelianum is an alien weed species found throughout India;20,21 it is known to possess diverse pharmacological and biological activities. The species is reported to have emollient, resolvent, bechic and antidysenteric properties, and it is widely used in traditional systems of medicine as an anti-inflammatory and analgesic agent and also to treat ulcers,22 stomachache,23 jaundice, etc. Its flowers are used to treat cough along with chest and lung diseases.24 Different parts of the species are employed in diverse herbal formulations to treat gout,25 rheumatic arthralgia,26 laying hen salpingitis,27 otitis,28 and tumors;29 it is also used in herbicidal compositions30 and medicinal wine.31
Pharmacological screening of the aqueous extract exhibited significant antidiabetic and antihyperlipidemic activities.32,33 The extract also inhibited inflammation induced by carrageenan and pain due to formalin stimulation.34 A crude water extract from the aerial parts exhibited antibacterial activity against methicillin-resistant strains of Staphylococcus aureus.35,36 Further, in view of the fact that natural plant polysaccharides are currently gaining significant popularity as potent natural antioxidants, concomitant with the vast pharmacological activity of the species, it was hypothesized that the polysaccharide constituting the major fraction of water extract of M. coromandelianum is responsible for its activity. However, to date, there have been no studies regarding the purification, characterization or antioxidant activities of polysaccharides from M. coromandelianum.
In the present study, a polysaccharide was isolated from leaves of M. coromandelianum using 1 M NaOH solution, and its ferric reducing power activity was examined using the potassium ferricyanide assay. Interestingly, the polysaccharide showed positive results and was found to significantly reduce ferric ions in a dose-dependent manner. Thus, it was thought to be worthwhile to explore the structure of the alkali soluble polysaccharide (MAP) from the species. The structural characterization was carried out via chemical degradation and derivatisation methods, viz. hydrolysis, alditol acetates, and methylation, concomitant with GC-MS and ESI-MS analysis. The results were further corroborated by 1D and 2D NMR spectroscopy (1H, 13C, HSQC and HMBC). Structural analysis of the polysaccharide may lead to enhanced utilization of this species in diverse biological and pharmaceutical applications in addition to other industrial applications based on its structure–activity relationship. The encouraging results of our investigation offer a strong basis for the development of novel polysaccharide-based antioxidants from this species.
2. Experimental
2.1. Materials
The leaves of M. coromandelianum were collected from the Forest Research Institute campus, Dehradun in July 2014. The species was authenticated by Dr H. B. Naithani, Systematic Botanist, Botany Division, and voucher specimen no. 170580 was deposited at the herbarium of Systematic Botany Discipline, Botany Division, Forest Research Institute, Dehradun.All the chemicals and reagents used were of analytical grade. The standard solutions were freshly prepared before experimentation. Sephadex G-200 was purchased from Pharmacia, Stockholm, Sweden. Dialysis membrane with a molecular weight cutoff (MWCO) of 12 to 14 kDa was procured from Himedia Laboratories Pvt. Ltd., Mumbai, India. UV-vis spectra were recorded on a Thermo Scientific Spectrascan UV 2700 spectrophotometer.
2.2. Isolation, purification and homogeneity analysis of the polysaccharide
The leaves of M. coromandelianum were washed thoroughly under running water and dried in shade at room temperature. The polysaccharide was extracted by following the method of Kumar and Kumar.16 Briefly, air dried leaves (100 g) were shredded and extracted with distilled water at 30 °C with continuous stirring for 3 h (3 times) followed by filtration. The obtained residue was stirred at 80 °C for 3 h (3 times) to isolate the hot water-soluble polysaccharide. Further, the residue left after hot water extraction was stirred with 1 M NaOH solution at ambient temperature for 1 h and filtered. The filtrate was neutralized using 0.5 M HCl and centrifuged at 2000 rpm for 15 minutes to remove all undissolved impurities. The supernatant was concentrated to a small volume and poured into 3 volumes of ethanol with constant stirring. The polysaccharide precipitated as a fluffy mass and was separated by centrifugation. The precipitates were again dissolved in a minimum quantity of water and re-precipitated with ethanol. This step was repeated two times. The obtained polysaccharide was washed sequentially with diethyl ether, acetone, and ethanol to eliminate impurities. The supernatant was decanted off every time. The precipitates were dried and dissolved in a minimum amount of distilled water (50 mL) and the undissolved mass was removed by ultracentrifugation at 12000 rpm.37 The supernatant was freeze dried to obtain the crude alkali-soluble polysaccharide.
A dilute solution (5 mg mL−1) of alkali-soluble polysaccharide was passed through a freshly regenerated cation exchange resin column [Dowex-50 W-X8] followed by an anion exchange resin column [Seralite SRA-400], and the eluents were analyzed by the Molisch carbohydrate test.38 The combined eluents were concentrated to a small volume and dialyzed against distilled water using a dialysis membrane (MWCO: 12 to 14 kDa) for 72 h. The dialyzed solution was concentrated and re-precipitated with four volumes of ethanol. The precipitates were separated, dried, dissolved in distilled water and lyophilized to obtain the crude polysaccharide as an off-white amorphous mass (1.15 g).
2.3. Gel permeation chromatography of MAP
The homogeneity analysis of the crude polysaccharide was performed using GPC. A Pharmacia column (1.6 × 40 cm) was packed with Sephadex G-200 as a gel permeation medium using double distilled water as the mobile phase, and 200 mg (MAP) polysaccharide was loaded. Fractions of 2.5 mL each at an elution rate of 0.30 mL min−1 were collected using a Gilson fraction collector (Model-202). The absorbance of the fractions was studied using the anthrone–sulphuric acid method39 at 620 nm, and a graph was plotted of absorbance vs. the fractions collected after elution. The fractions comprising the major peak were pooled, dialyzed and lyophilized to obtain the pure native alkali-soluble polysaccharide, which was designated as MAP.2.4. Complete hydrolysis and paper chromatography
MAP (5 mg) was completely hydrolyzed by refluxing with 2 N sulphuric acid (5 mL) for 25 h followed by neutralization using barium carbonate slurry; then, it was filtered and concentrated to a syrupy liquid. The resulting liquid was subjected to paper chromatography along with standard monosaccharides using 1 chr and 3 chr Whatman chromatography paper. The papers were run for 45 to 120 h using the following solvent systems; (a) n-butanol:formic acid:water; 12:1:7 (upper layer); (b) n-butanol:pyridine:water; 6:4:3; and (c) ethyl acetate:acetic acid:n-butanol:water; 4:3:2:2. The paper chromatograms were dried at room temperature, followed by detection of the monosaccharides (neutral and acidic) using aniline phthalate spray and ammoniacal silver nitrate complex independently by employing standard protocols.40
2.5. Carboxyl reduction of polysaccharide
MAP was reduced using the method by Taylor, Shively and Conard.41 The polysaccharide (50 mg) solution in distilled water (100 mL) was adjusted to pH 4.75 using 1 M HCl. To the resulting solution, solid 1-ethyl-3-(3-dimethylaminopropyl)carbodimide hydrochloride (167 mg, EDC·HCl) was added. The mixture was stirred for 45 min at pH 4.75, followed by dropwise addition of 3 M NaBH4 solution at 25 °C during a one hour period. During the addition of sodium borohydride, the pH was maintained at 7.0 using 4 M HCl solution. At this stage, the reaction mixture was further stirred for 1 h, and the pH of the solution was decreased to 4 by addition of dil. HCl (1 M) to destroy excess sodium borohydride. The resulting solution was dialyzed (12 to 14 kDa) exhaustively against distilled water for 72 h, concentrated to a small volume and precipitated by addition of ethanol. The precipitates were filtered, dissolved in a minimum quantity of water and lyophilized to obtain the reduced polysaccharide (MAP-R; 31 mg). The complete reduction of the polysaccharide was analyzed and confirmed by the negative carbazole–sulphuric acid color test for uronic acid.2.6. Monosaccharide analysis
MAP and the reduced polysaccharide (MAP-R, 10 mg each) were hydrolysed using 2 N H2SO4 for 6 h with pinitol (1 mg) as an internal standard. Alditol acetate as a standard monosaccharide and the sample were prepared by following the method by Jansson et al.42 and subjected to GLC analysis43 using an Agilent 7890B gas chromatograph equipped with a flame ionization detector (FID). The initial temperature of the column was 190 °C with a hold time of 1 min; the temperature was increased at a rate of 3 °C per minute until attainment of the final temperature of 260 °C, with holding for 10 min. Helium was used as a carrier gas with a flow rate of 1.2 mL min−1. The monosaccharide composition (for neutral as well as acidic sugar residues) was calculated by comparing the GLC chromatograms of native and reduced polysaccharides.The uronic acid was further quantified spectrophotometrically by the 3,5-DMP method.44 All the experiments were performed in triplicate.
2.7. Linkage analysis
Linkage analysis of MAP was performed by methylation of the reduced and native polysaccharides with the modified Hakomori method and derivatization to PMAA, followed by comparing the GC-MS results.45 Briefly, the polysaccharide (50 mg) was dissolved in DMSO (10 mL; dried over molecular sieves) in a stream of nitrogen. Alkoxide ions were formed by portionwise addition of sodium hydride at 60 °C, and the reaction mixture was left for 1 h with continuous stirring. At this stage, the nitrogen flow was stopped and the reaction mixture was cooled over an ice bath followed by addition of methyl iodide. The permethylated polysaccharide was extracted with chloroform (3 × 10 mL). The combined fractions were washed with distilled water, dried over anhydrous sodium sulphate and concentrated under vacuum.The permethylated derivatives were hydrolysed with 90% (v/v) formic acid (5 h) followed by hydrolysis with 2 M TFA for 6 h. The hydrolysates were reduced to alditols with sodium borohydride and converted to partially methylated alditol acetates (PMAA) using pyridine: acetic anhydride in a 1:1 ratio. The PMAAs were analyzed by GC-MS (Agilent 7890B gas chromatograph with a 5977A mass spectrometer with electron ionization (EI) and a quadrupole analyzer) using an RTX 2330 column and helium as a carrier gas. The program used to run the PMAA derivatives was as follows: the initial temperature of the column was 170 °C, and the temperature was increased at a rate of 6 °C per minute up to 240 °C with final holding for 10 min. 10 μL of sample was injected with a split ratio of 10:1. The linkages were identified by the relative retention times and fragmentation patterns of the mass spectra for individual PMAA peaks.
2.7. Partial hydrolysis and ESI-MS analysis
MAP (5 mg) was partially hydrolysed using 0.5 M and 1 M hydrochloric acid (5 mL) for 1 h separately. The partially hydrolysed solution was dried under vacuum on a rotary evaporator. Traces of acid were removed by passing a stream of nitrogen. ESI-MS analysis was performed on a Waters Micromass Q-TOF micro instrument equipped with the Waters Alliance 2795 separations module in unit resolution (MRM) acquisition mode. The injection volume of analyte was 20 μL and the flow rate was 0.4 mL min−1. MS was operated in positive and negative ionization mode, and the spectra were acquired with a m/z range of 0 to 2000. Other parameters used for the mass spectrometer were desolvation gas: 550 L h−1, cone gas: 30 L h−1, desolvation temperature: 300 °C, source temperature: 110 °C. The capillary and cone voltages were set at 3000 V and 30 V, respectively, and a collision energy of 4 eV was used. N2 (6 to 7 bar) and argon (5 to 6 bar) were used as drying gases.2.8. NMR spectral analysis
MAP (35 mg) was deuterium-exchanged with 99.9% D2O (3 × 650 μL) three times using freeze–thaw cycles prior to NMR analysis. The deuterium-exchanged polysaccharide was dissolved in D2O (650 μL) and the spectra were recorded on a JEOL JNM-ECS400 spectrometer. The NMR measurements included various experiments, viz. 1H, 13C, DEPT-135, COSY, HSQC and HMBC. The experiments were performed at 292 K. The raw data were processed with Mestrec Nova version 9.0.0.2.9. Reducing power
The reducing power of MAP was determined by the potassium ferricyanide assay according to the standard protocol.1 Briefly, 1 mL phosphate buffer (0.2 M, pH 6.6) was added to 0.5 mL potassium ferricyanide (1%, w/v). To the above solution, 1 mL of sample solution (ascorbic acid and MAP) with varying concentration (0.2 to 1.8 mg mL−1, w/v in distilled water) was added. The resulting mixtures were incubated at 50 °C in a water bath for 20 minutes and cooled to ambient temperature. This was followed by addition of 1 mL trichloroacetic acid (10%, w/v) and 0.2 mL freshly prepared ferric chloride solution (0.1%, w/v). The final solution was vortexed, and the absorbance was measured at 700 nm. The reducing power was calculated using the formula:| Reducing power = [As700 − Ac700] |
3. Results and discussion
3.1. Isolation of polysaccharide
The alkali-soluble polysaccharide (MAP) was isolated in 1.15% (w/w) yield. The ash content of MAP was 0.75% (w/w) on a dry weight basis. The aqueous solution (1%, w/v) of MAP has a pH value of 6.4.3.2. GPC of MAP
A graph of the fractions collected from GPC against absorbance was plotted using the anthrone-sulphuric acid method39 at 620 nm, as shown in Fig. 1.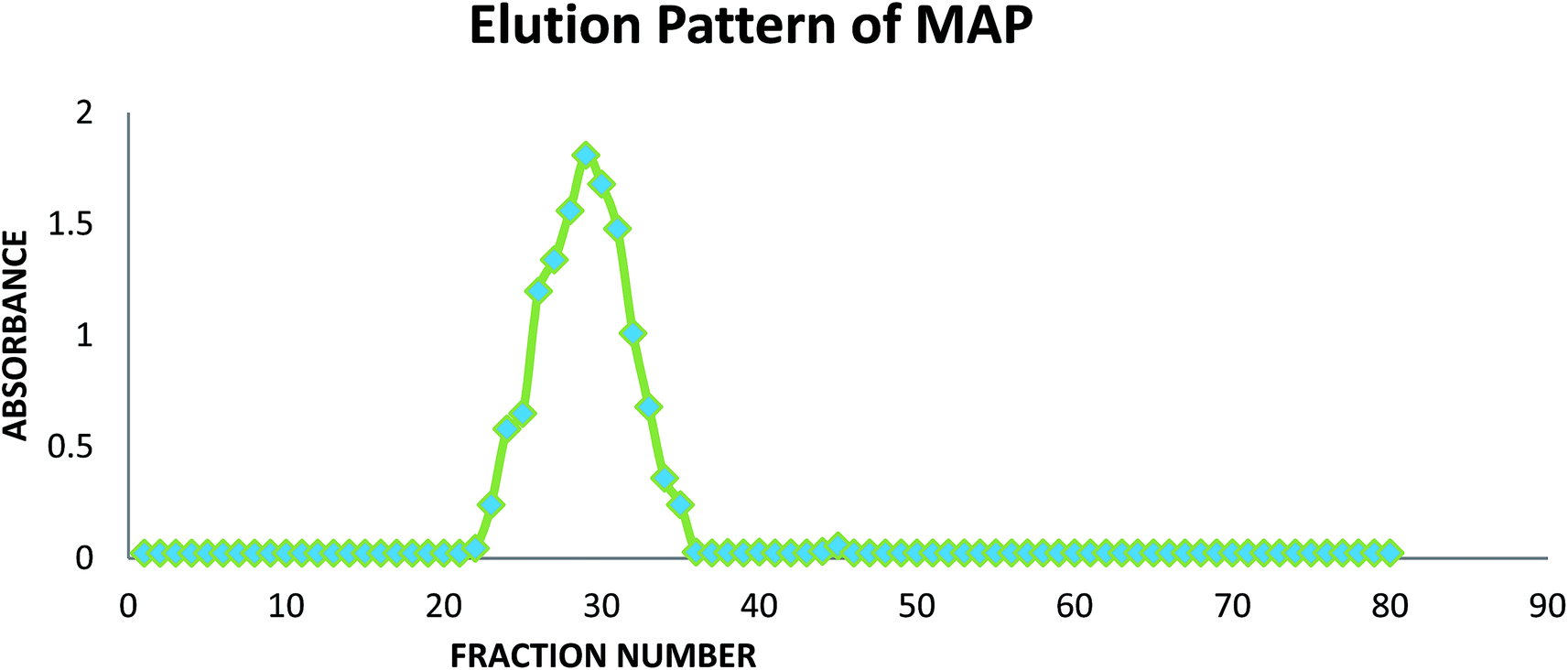 | ||
| Fig. 1 Elution pattern of MAP in GPC. | ||
The fractions of the major eluted peaks (23–34) were collected and pooled, dialyzed against distilled water (48 h) and lyophilized to obtain the homogeneous polysaccharide (146 mg).
3.3. Monosaccharide analysis
Qualitative evaluation of MAP with paper chromatography indicated the presence of five neutral monosaccharides and one acidic sugar residue. The sugar residues identified in MAP were rhamnose (Rha), arabinose (Ara), xylose (Xyl), glucose (Glc), galactose (Gal), and glucuronic acid (GlcA). The monosaccharide composition of MAP was quantitatively investigated by GLC. Based on comparison with monosaccharide standards, MAP was found to be composed of xylose (64.52 mol%) and arabinose (18.87 mol%). The other sugar residues were found to be rhamnose, glucose, and galactose in 3.27, 3.25 and 10.09 mole percentages, respectively. After complete reduction with EDC, HCl and NaBH4, the carboxyl-reduced polysaccharide MAP-R was obtained. MAP-R was found to be composed of Rha, Ara, Xyl, Glc and Gal in 3.04, 18.36, 60.42, 8.97, 9.21 mole percentages, respectively. The mol% of glucuronic acid was 5.76 based on the percentage increase of glucose in the reduced polysaccharide (MAP-R) vis-à-vis native MAP. The molar ratio of monosaccharides Rha, Ara, Xyl, Glc, Gal and GlcA in the alkali-soluble polysaccharide was 1.00:6.04:19.88:1.07:3.03:1.95. The arabinose/xylose ratio of MAP was found to be 0.3.
The glucuronic acid content was analyzed using a spectrophotometric method based on the 3,5-dimethyl phenol assay. It was found to be 6.78% (w/w) ± 0.51 (avg ± st dev) in MAP.44
3.4. Linkage analysis
The linkage pattern of MAP was revealed by complete methylation of native (MAP) and reduced (MAP-R) polysaccharides followed by hydrolysis, derivatisation to PMAA and GC-MS analysis, as explained in Section 2.6. The PMAAs were characterized by relative retention time and mass fragmentation pattern. The results of the methylation analysis of MAP are presented in Table 1. MAP was found to contain 32.26 mol% 2,3-O-Me2-1,4,5-O-Ac3-pentitol and 19.35 mol% 3-O-Me-1,2,4,5-O-Ac4-pentitol. According to the monosaccharide analysis, xylose is the most abundant sugar residue present in MAP, which indicates that 2,3-O-Me2-1,4,5-O-Ac3-pentitol and 3-O-Me-1,2,4,5-O-Ac4-pentitol are 1,4-linked Xylp and 1,2,4-linked Xylp, respectively; this was confirmed on the basis of the fragmentation pattern and relative retention times. According to the literature, both these linkages are assumed to be present in the backbone of xylan.46–48 Similarly, the other major linkages characterized in MAP were 16.19 mol% 1,3-linked Araf, 12.83 mol% 1,3-linked Xylp and 9.68 mol% 1,4-linked Galp with 3.11 mol% 1,3,4-linked Glcp; these probably form branches in MAP. The non-reducing terminal end linkages were found to be t-linked Rhap (3.23 mol%) and t-linked Araf (3.35 mol%) in MAP. Methylation analysis of MAP-R (reduced MAP) showed t-linked Glcp at 6.25 mol%, suggesting that glucuronic acid is present as t-linked GlcpA in the polysaccharide. The excess percentage of xylose followed by arabinose when combined with the linkage results reveals the arabinoxylan nature of the polysaccharide. The methylation results are in agreement with the monosaccharide composition analysis.| PMAA derivative | MAP (mol%) | MAPR (mol%) | Linkage identified | Observed mass fragments (m/z) |
|---|---|---|---|---|
| 1,5-Ac2-2,3,4-Me3-rhamnitol | 3.23 | 3.13 | t-Rhap | 43, 59, 87, 99, 89, 101, 117, 131, 161, 175 |
| 1,4-Ac2-2,3,5-Me3-arabinitol | 3.35 | 3.19 | t-Araf | 43, 45, 59, 87, 101, 117, 129, 145, 161 |
| 1,3,4-Ac3-2,5-Me2-arabinitol | 16.19 | 15.21 | 1,3-Araf | 43, 59, 71, 87, 99, 101, 117, 129, 173, 205, 233 |
| 1,3,5-Ac3-2,4-Me2-xylitol | 12.83 | 12.25 | 1,3-Xylp | 43, 59, 71, 87, 101, 117, 233 |
| 1,4,5-Ac3-2,3-Me2-xylitol | 32.26 | 30.09 | 1,4-Xylp | 43, 59, 71, 87, 99, 101, 117, 129, 161, 189 |
| 1,2,4,5-Ac4-3-Me-xylitol | 19.35 | 17.96 | 1,2,4-Xylp | 43, 74, 87, 101, 129, 145, 161, 189, |
| 1,5-Ac2-2,3,4,6-Me4-glucitol | — | 6.25 | t-GlcpA | 43, 45, 59, 71, 87, 101, 117, 129, 145, 161, 173, 205 |
| 1,4,5-Ac3-2,3,6-Me3-galactitol | 9.68 | 8.99 | 1,4-Galp | 43, 45, 58, 71, 87, 99, 101, 117, 129, 131, 157, 161, 173, 233 |
| 1,4,5,6-Ac4-2,3-Me2-glucitol | 3.11 | 2.93 | 1,4,6-Glcp | 43, 57, 71, 85, 99, 117, 127, 142, 161, 173, 201, 261 |
3.5. Partial hydrolysis and ESI-MS analysis
MAP was partially hydrolysed using 0.5 M and 1 M HCl to obtain a mixture of oligomers. Because the individual linkages between various monosaccharides were already identified by GC-MS investigation of the PMAA derivatives, ESI-MS (n = 1) of the partially degraded polysaccharide was performed to support the PMAA analysis and to establish the arrangement of monomer buildups in the oligomers to better understand the structure of the polysaccharide. The hydrolysed mixture was studied in positive as well as negative ion mode to obtain maximum information about the oligomeric pattern. Oligomers containing pentose (Pent), hexose (Hex), uronic acid (UA) and rhamnose (Rha) with DP7 to DP2 were identified in the mass spectrum. ESI-MS analysis revealed a pentose sugar with an anhydro mass unit of 132.0 to be the most abundant sugar residue in MAP.The hydrolysate prepared using 0.5 M HCl furnished oligomers up to DP4 in positive ion mode (Fig. 2a). The ions of partial hydrolysates were obtained as sodium adducts of oligomers. Two tetramers of [Pent3 + Hex + Na]+ and [Pent4 + Na]+ at m/z 599.07 and 569.11, respectively, were identified. Three trimers of m/z 497.19, 437.24 and 467.22 were attributed to sodium adducts of oligomers [Pent + Hex2 + Na]+, [Pent3 + Na]+ and [Pent2 + Hex + Na]+, respectively. The dimers with m/z 365.33, 335.33, and 305.35 were recognized as [Hex2 + Na]+, [Pent + Hex + Na]+ and [Pent2 + Na]+, respectively.
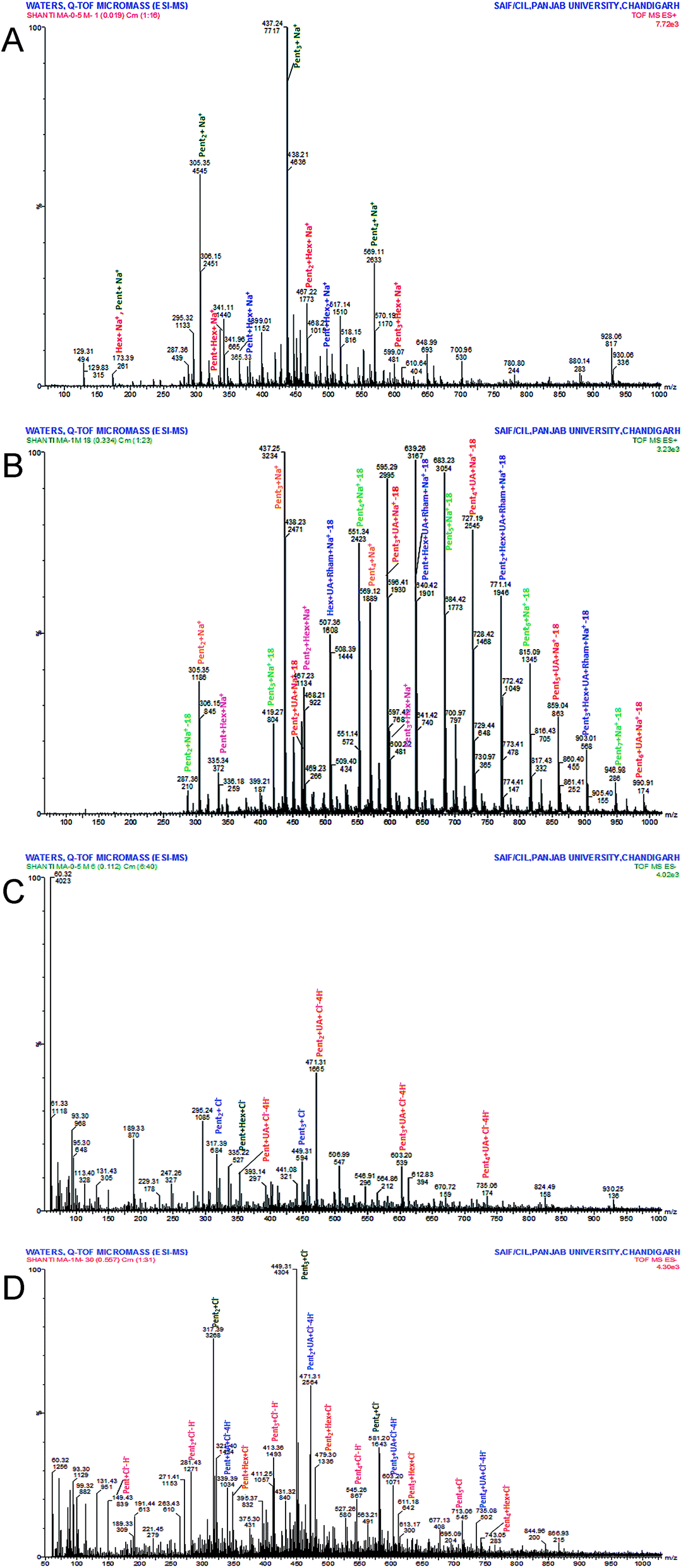 | ||
| Fig. 2 (A) ESI+ve mode spectrum of MAP hydrolysate (0.5 M HCl), (B) ESI+ve mode spectrum of MAP hydrolysate (1 M HCl), (C) ESI−ve mode spectrum of MAP hydrolysate (0.5 M HCl), (D) ESI−ve mode spectrum of MAP hydrolysate (1 M HCl). | ||
The hydrolysate formed using 1 M HCl exhibited spectra with good intensity and an increase in the number of peaks (Fig. 2b). The oligomers up to DP7 were identified in positive ion mode as sodium adducts. The oligomers with higher DP (7 to 5) include two heptamers, [Pent6 + UA + Na-18]+ (m/z 990.91) and [Pent7 + Na-18]+ (m/z 946.98); three hexamers, [Pent3 + Hex + UA + Rham + Na-18]+ (m/z 903.01), [Pent5 + UA + Na-18]+ (m/z 859.04) and [Pent6 + Na-18]+ (m/z 815.09); and three pentamers, [Pent2 + Hex + UA + Rham + Na-18]+ (m/z 771.14), [Pent4 + UA + Na-18]+ (m/z 727.19) and [Pent5 + Na-18]+ (m/z 683.23). The identified oligomers with lower DP include five tetramers, five trimers and three dimers. The observed tetramers were [Pent4 + Na]+ (m/z 569.12); [Pent3 + Hex + Na]+ (m/z 599.07); [Pent + Hex + UA + Rham + Na-18]+ (m/z 639.26); [Pent3 + UA + Na-18]+ (m/z 595.29); and [Pent4 + Na-18]+ (m/z 551.34). The ion fragments of trimers at m/z 507.36, 463.23, 419.27, 437.25, and 467.23 were attributed to [Hex + UA + Rham + Na-18]+, [Pent2 + UA + Na-18]+, [Pent3 + Na-18]+, [Pent3 + Na]+, and [Pent2 + Hex + Na]+, respectively. The recognized dimers were characterized as [Pent2 + Na-18]+, [Pent2 + Na]+, and [Pent + Hex + Na]+ with m/z 287.36, 305.35 and 335.34, respectively.
ESI-MS in negative mode produced chlorine and hydrogen ion adducts of oligomers. Oligomers up to DP5 were found in the partial hydrolysate mixture obtained using 0.5 M HCl (Fig. 2c). The oligosaccharides at m/z 735.08, 603.20, 471.31, 339.39, 449.3, 317.39 and 335.22 were identified as [Pent4 + UA + Cl–4H]− (pentamer); [Pent3 + UA + Cl–4H]− (tetramer); [Pent2 + UA + Cl–4H]− (trimer); [Pent + UA + Cl–4H]− (dimer); [Pent3 + Cl]− (trimer); [Pent3 + Cl]− (dimer); and [Pent + Hex + Cl]− (dimer).
The hydrolysate obtained with 1 M HCl (Fig. 2d) produced intense peaks up to DP6 which include one hexamer [Pent5 + UA + Cl–4H]− (m/z 866.93); four pentamers, [Pent4 + Hex + Cl]− (m/z 743.05), [Pent4 + UA + Cl–4H]− (m/z 735.08), [Pent5 + Cl]− (m/z 713.06), and [Pent5–H]− (m/z 677.13); four tetramers, [Pent3 + Hex + Cl]− (m/z 611.18), [Pent3 + UA + Cl–4H]− (m/z 603.20), [Pent4 + Cl]− (m/z 581.20), and [Pent4–H]− (m/z 545.26); four trimers, [Pent2 + Hex + Cl]− (m/z 479.30), [Pent2 + UA + Cl–4H]− (m/z 471.31), [Pent3 + Cl]− (m/z 449.31), and [Pent3–H]− (m/z 413.36); and four dimers, [Pent + Hex + Cl]− (m/z 347.37), [Pent + UA + Cl–4H]− (m/z 339.39), [Pent2 + Cl]− (m/z317.39), and [Pent2–H]− (m/z 281.43). One monomer, [Pent–H]−, was observed at m/z 149.43.
The abundance of pentose-containing oligomers in the ESI-MS spectra supported the monosaccharide and methylation results, which showed xylose (a pentose sugar) to be the most abundant residue in MAP. The complete oligomeric patterns identified are presented in Table 2.
| DP | |||||||
|---|---|---|---|---|---|---|---|
| Oligomers | 7 | 6 | 5 | 4 | 3 | 2 | 1 |
| ESI+ve mode, 1 M HCl (1 h) | |||||||
| [Pentn + Hex + UA + Rham + Na-18]+ (n = 3 to 0) | — | 903.01 | 771.14 | 639.26 | 507.36 | — | — |
| [Pentn + UA + Na-18]+ (n = 6 to 2) | 990.91 | 859.04 | 727.19 | 595.29 | 463.23 | — | — |
| [Pentn + Na-18]+ (n = 7 to 2) | 946.98 | 815.09 | 683.23 | 551.34 | 419.27 | 287.36 | — |
| [Pentn + Na]+ (n = 4 to 2) | — | — | — | 569.12 | 437.25 | 305.35 | — |
| [Pentn + Hex + Na]+ (n = 3 to 1) | — | — | — | 599.07 | 467.23 | 335.34 | — |
|
|||||||
| ESI+ve mode, 0.5 M HCl (1 h) | |||||||
| [Pentn + Hex + Na]+ (n = 3 to 1) | — | — | — | 599.07 | 467.22 | 335.33 | — |
| [Pentn + Na]+ (n = 4 to 1) | — | — | — | 569.11 | 437.24 | 305.35 | 173.39 |
| [Pentn + Hex2 + Na]+ (n = 1 to 0) | — | — | — | — | 497.19 | 365.33 | — |
|
|||||||
| ESI−ve mode, 1 M HCl (1 h) | |||||||
| [Pentn + Hex + Cl]− (n = 4 to 1) | — | — | 743.05 | 611.18 | 479.30 | 347.37 | — |
| [Pentn + UA + Cl–4H]− (n = 5 to 1) | — | 866.93 | 735.08 | 603.20 | 471.31 | 339.39 | — |
| [Pentn + Cl]− (n = 5 to 2) | — | — | 713.06 | 581.20 | 449.31 | 317.39 | — |
| [Pentn–H]− (n = 5 to 1) | — | — | 677.13 | 545.26 | 413.36 | 281.43 | 149.43 |
|
|||||||
| ESI−ve mode, 0.5 M HCl (1 h) | |||||||
| [Pentn + UA + Cl–4H]− (n = 4 to 1) | — | — | 735.08 | 603.20 | 471.31 | 339.39 | — |
| [Pentn + Cl]− (n = 3 to 2) | — | — | — | — | 449.31 | 317.39 | — |
| [Pent + Hex + Cl]− | — | — | — | — | — | 335.22 | — |
3.6. NMR analysis and structure of MAP
NMR spectra of MAP were recorded on a JEOL JNM-ECS400 spectrometer. The completely deuterium-exchanged MAP (35 mg dissolved in 650 μL of D2O) was subjected to 1D (1H, 13C, DEPT-135) and 2D (HSQC, HMBC, TOCSY, COSY) experiments to acquire significant information for the elucidation of the structure of MAP. The anomeric protons appeared between 4.20 and 5.20 ppm in the 1H NMR spectrum at 5.01, 4.90, 4.59, 4.83, 4.44, 4.29, 4.23, 5.11, 4.39 and 5.05 ppm (Fig. 3a). In the 1H NMR spectrum, one signal was found at 1.01 ppm which was highly shielded; this was assigned to the methyl protons of rhamnose residues. The 13C NMR spectrum (Fig. 3b) showed ten anomeric carbon signals at 107.48, 107.24, 104.03, 101.62, 101.47, 101.68, 101.67, 97.44, 96.39, and 91.91 ppm; these are labeled as A, B, C, D, E, F, G, H, I and J, respectively. The corresponding anomeric proton signals in the 1H NMR spectrum were marked with the contribution of HSQC. The signal at 16.49 ppm was assigned to the methyl carbon of rhamnose, and the signal at 174.75 ppm was assigned to C-6 of GlcpA. All 1H and 13C NMR assignments were marked with the assistance of COSY, HSQC, HMBC and TOCSY [Fig. 3c–e] spectra. The chemical shifts of all the residues of MAP were assigned completely and are shown in Table 3.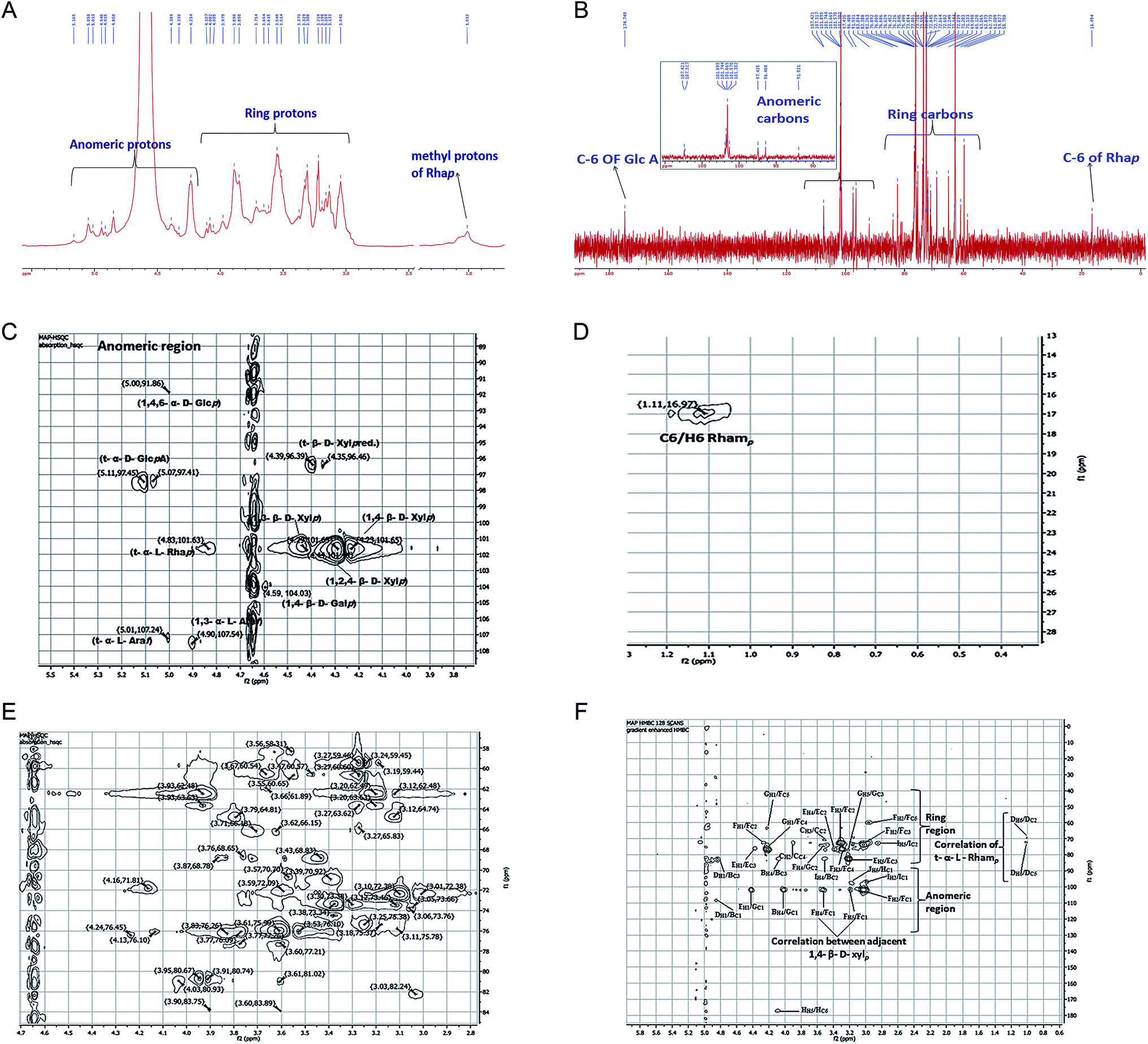 | ||
| Fig. 3 (A) 1H NMR spectrum of MAP, (B) 13C NMR spectrum of MAP, (C) HSQC spectrum of the anomeric region, (D) HSQC spectrum of the rhamnose region, (E) HSQC spectrum of the ring region, (F) HMBC spectrum with inter and intra-residue connectivities. | ||
| S. no. | Monosaccharide moiety | C1/H1 | C2/H2 | C3/H3 | C4/H4 | C5/H5,5′ | C6/H6,6′ | References |
|---|---|---|---|---|---|---|---|---|
| A | α-Araf(1→ | 107.24/5.01 | 76.10/4.13 | 80.74/3.90 | 80.67/3.95 | 60.65/3.55 | — | Fischer et al., 2004; Kang et al., 2011; Yin et al., 2012 |
| B | →3)-α-Araf(1→ | 107.48/4.90 | 76.45/4.24 | 83.75/3.89 | 80.93/4.03 | 58.31/3.56 | — | Yin et al., 2012; Kang et al., 2011 |
| C | →4)-β-Galp(1→ | 104.03/4.59 | 70.92/3.39 | 72.09/3.59 | 68.78/3.87 | 76.26/3.83 | 66.18/3.71,3.62 | Habibi, Y., Mahrouz, M., and Vignon, M. R. 2005 |
| D | α-Rhap (1→ | 101.62/4.83 | 68.78/3.87 | 70.70/3.57 | 73.38/3.30 | 71.81/4.16 | 16.49/1.01 | Kang et al., 2011 |
| E | →3)-β-Xylp(1→ | 101.47/4.44 | 75.38/3.25 | 81.02/3.62 | 73.34/3.38 | 63.63/3.93, 3.20 | — | Hoije et al., 2006; Yin et al., 2011; Yin et al., 2012; Sun et al., 2011 |
| F | →4)-β-Xylp(1→ | 101.68/4.29 | 72.38/3.06 | 74.49/3.36 | 75.99/3.61 | 62.48/3.93, 3.12 | — | Hoije et al., 2006; Yin et al., 2011; Yin et al., 2012; Sun et al., 2011 |
| G | →2,4)-β-Xylp(1→ | 101.67/4.23 | 74.49/3.36 | 77.21/3.60 | 81.02/3.61 | 62.49/3.93, 3.12 | — | Hoije et al., 2006; Yin et al., 2011; Yin et al., 2012; Sun et al., 2011 |
| H | α-GlcpA(1→ | 97.44/5.11 97.41/5.07 | 70.92/3.39 | 73.34/3.38 | 80.74/3.91 | 71.81/4.14 | 174.75 | Peng et al., 2012; Dobruchowska, Gerwig, Babuchowski, and Kamerling, 2008 |
| I | β-Xylp(1→ | 96.39/4.39 96.46/4.35 | 72.76/3.06 | 73.38/3.30 | 76.10/3.53 | 59.45/3.27,2.95 | — | Schendel, Becker, Tyl and Bunzel, 2015; Zhang, 2016 |
| J | →4,6)-α-Glcp(1→ | 91.91/5.05 | 70.92/3.39 | 72.09/3.59 | 75.99/3.61 | 65.62/3.27 | 58.31/3.56 | Jiang, Kong, Li, Zhang, Yan and Lv, 2016 |
Residue A showed an anomeric carbon chemical shift at 107.24 ppm, and its anomeric proton shift was observed at 5.01 ppm. Other proton chemical shifts were assigned from the COSY and TOCSY spectra. The other carbon signals of residue A were observed at 76.10, 80.74, 80.67 and 60.65, corresponding to C-2, C-3, C-4 and C-5, respectively. The intensity of the anomeric carbon chemical shift is weaker and was assigned to α-linked Araf(1→; this is supported by the methylation results, which show a lower percentage of α-linked Araf(1→ compared to →3)-α-linked Araf(1→. The proton and carbon chemical shifts were in agreement with literature values.46,48–50
Residue B showed a carbon chemical shift at 107.48 ppm with a proton shift at 4.90 ppm. The C-3 chemical shift is also deshielded, with a chemical shift value of 83.75 ppm. Methylation analysis showed that the percentage of →3)-α-linked Araf(1→ is much higher than that of α-linked Araf(1→. The anomeric signal for residue B is more intense than that for residue A in the HSQC spectrum. All these factors suggest that this residue is →3)-α-linked Araf(1→. The carbon signals of residue B were observed at 76.45, 80.93, and 58.31, corresponding to C-2, C-4 and C-5 from the HSQC spectrum. These assignments were consistent with previous data.48,49
Residue C showed anomeric carbon and proton chemical shifts at 104.03 and 4.59 ppm, respectively. Its anomeric proton chemical shift indicates that it is β-linked. According to methylation fragmentation analysis and comparison of the NMR data, residue C was assigned as →4)-β-linked Galp(1→, which was found to be consistent with the literature.51 The other carbon chemical shifts were 70.92, 72.09, 68.78, 72.26 and 66.18, which correspond to C-2, C-3, C-4, C-5 and C-6, respectively.
The anomeric carbon chemical shift of residue D was found at 101.62 ppm, with the anomeric proton chemical shift at 4.83 ppm. The chemical shifts of other protons were assigned through COSY, and the corresponding carbon shift values were recognized with HSQC. The chemical shift values of the other carbons were found at 68.78, 70.70, 73.38, 71.81 and 16.49 ppm and were assigned to C-2, C-3, C-4, C-5 and C-6, respectively. The NMR data was in agreement with literature data.49 The spectral assignments and methylation results indicate that residue D is α-linked Rhap(1→.
Residues E, F and G showed anomeric carbon/proton shift values at 101.47/4.44, 101.68/4.29, and 101.67/4.23 ppm, respectively. →4)-β-linked Xylp(1→, →2,4)-β-linked Xylp(1→ and →3)-β-linked Xylp(1→ are the most abundant linkages found in the methylation study, and they were found to be 32.26, 19.35 and 12.83 mol%, respectively. The anomeric carbon and proton intensities of residues E, F and G were found to be the most abundant peaks in the spectra. The carbon chemical shift C-3 (81.02 ppm) of residue E was found to be deshielded, and C-4 (75.99 ppm) of residue F was also deshielded. Similarly, C-2 (74.49 ppm) and C-4 (81.02 ppm) of residue G showed deshielding. The chemical shifts of C-2, C-4, and C-5 of residue E were observed at 75.38, 81.02 and 63.63 ppm, respectively. The chemical shifts of C-2, C-3 and C-5 of residue F were observed at 72.38, 74.49, and 62.48 ppm, respectively. The chemical shifts of C-3 and C-5 of residue G were observed at 77.21 and 62.49 ppm, respectively. The spectral assignments and methylation results revealed residues E, F, and G to be →3)-β-linked Xylp(1→, →4)-β-linked Xylp(1→ and →2,4)-β-linked Xylp(1→. The other proton and carbon chemical shifts were assigned completely from the HSQC, COSY and TOCSY spectra, and the values were found to be in agreement with literature data.48,50,52,53
The anomeric carbon signal of residue H was found at 97.44 ppm, and the anomeric proton shift was 5.11 ppm. The C-6 of residue H was found at 174.75 ppm, which is highly deshielded due to the carboxylic group of uronic acid. Other carbon signals were found at 70.92, 73.34, 80.74 and 71.81 ppm and were assigned to C-2, C-3, C-4 and C-5, respectively. These values were assigned to β-linked GlcpA(1→ and compared with the literature.54,55 Sometimes, GlcpA is also found in C-6 methyl ester form; however, in the present case, uronic acid was not methyl-esterified because no signal was found near 53 ppm for the –OMe carbon.48,56 Also, no ester carbonyl peak was found in the FT-IR spectra, confirming that the GlcpA present in MAP is not methyl esterified.
Residue I showed anomeric C1/H1 chemical shifts at 96.39/4.39 ppm with significant intensity. The other proton and carbon shift values were assigned with the aid of the COSY and HSQC spectra. The carbon chemical shift values at 72.76, 73.38, 76.10, and 59.45 ppm were assigned to C-2, C-3, C-4 and C-5, respectively. In conformity with the literature,57,58 the anomeric chemical shift value was found to be consistent with β-linked Xylp(1→ with a free reducing end, which is well reported in xylans from different plant sources. No evidence was found regarding β-linked Xylp(1→ residue in the methylation analysis; however, based on the NMR analysis, residue I was assigned to β-linked Xylp(1→ with a free reducing end.
Residue J showed anomeric C1/H1 chemical shifts at 91.91/5.05 ppm with weak intensity. The other carbon signals of residue J were observed at 70.92, 72.09, 75.99, 65.62 and 58.31 ppm for C-2, C-3, C-4, C-5 and C-6, respectively. All the 1H and 13C chemical shift assignments of residue J were achieved using the COSY and HSQC spectra. Compared with previous reports59 on methylation data and NMR peak intensity, residue J was assigned to →4,6)-β-linked Glcp(1→.
The sequence of glycosyl residues in MAP was determined with the assistance of long-range HMBC coupling, as shown in Fig. 3f. The cross peaks of both anomeric carbons and the protons of the glycosyl residues were examined for both intra- and inter-residual connectivity. Cross-peaks were found between H-1 (5.01 ppm) of residue A and C-1 (107.48 ppm) of residue B, representing →3)-α-linked Araf(1→ residues linked to O-1 of α-linked Araf(1→. Cross-peaks were found between H-1 (4.23 ppm) of residue E and C-1 (101.67 ppm) of residue G, which indicates that the →3)-β-linked Xylp(1→ residues are linked to O-2 of →2,4)-β-linked Xylp(1→. There were cross connections of H-1 (4.23 ppm) of residue G with C-4 and C-5 of residue F, showing that →2,4)-β-linked Xylp(1→ is connected with →4)-β-linked Xylp(1→. The methylation results show that →4)-β-linked Xylp(1→ is the most abundant linkage in MAP; this was further supported by the HMBC spectra because the cross peak intensity for →4)-β-linked Xylp(1→ residues in HMBC is the maximum. Also, HMBC showed the greatest number of cross connecting linkages for →4)-β-linked Xylp(1→. The inter-connections between →4)-β-linked Xylp(1→ residues were found to be H-4 (3.06 ppm) with C-1 (101.68 ppm) and H-5 (3.93 ppm) with C-1 (101.68 ppm), indicating connections between →4)-β-linked Xylp(1→. The cross connection of H-4 (3.06 ppm) of residue I with C-3 (73.38 ppm) of residue B indicates that β-linked Xylp(1→ is connected to O-3 of →3)-α-linked Araf(1→. Intra-residual connectivities were found for residue F →4)-β-linked Xylp(1→at H-1 (3.06 ppm)/C-2 (72.38 ppm); H-3 (3.36 ppm)/C-4 (75.99 ppm); C-2 (72.38 ppm); H-2 (3.61 ppm)/C-5 (62.48 ppm); C-1 (101.68 ppm); and C-3 (74.49 ppm). Other intra-residual connectivities observed for residue H were α-linked GlcpA (1→ at H-5/C-6; for residue I, β-linked Xylp(1→ at H-2/C-1 and H-5/C-2; and for residue J, →4,6) α-linked Glcp (1→ at H-5/C-1. All the HMBC connectivities are summarized in Table 4.
| Residue | Linkage | Observed Connectivities | ||
|---|---|---|---|---|
| Proton (ppm) | Residue | Carbon | ||
| A | α-Araf(1→ | H-1 (5.01) | B | C-1 |
| B | →3)-α-Araf(1→ | H-4 (4.03) | B | C-3 |
| G | C-1 | |||
| C | →4)-β-Galp(1→ | H-2 (3.39) | C | C-4 |
| H-3 (3.59) | C | C-2 | ||
| D | α-Rhap (1→ | H-1 (4.83) | D | C-5 |
| H-5 (4.16) | D | C-2 | ||
| E | →3)-β-Xylp(1→ | H-1 (4.44) | E | C-3 |
| G | C-1 | |||
| H-4 (3.38) | E | C-2 | ||
| H-5 (4.14) | E | C-3 | ||
| F | →4)-β-Xylp(1→ | H-1 (4.29) | F | C-2 |
| H-2 (3.06) | F | C-1, C-3, C-5 | ||
| H-3 (3.36) | F | C-2, C-4 | ||
| H-4 (3.61) | F | C-1 | ||
| G | C-2 | |||
| H-5 (3.93) | F | C-1 | ||
| G | →2,4)-β-Xylp(1→ | H-1 (4.23) | F | C-4, C-5 |
| H-5 (3.93) | G | C-3 | ||
| H | α-GlcpA(1→ | H-5 (4.14) | H | C-6 |
| I | β-Xylp(1→ | H-2 (3.06) | I | C-1 |
| H-4 (3.53) | B | C-2 | ||
| J | →4,6)-α-Glcp(1→ | H-5 (3.27) | H | C-1 |
Summarizing the linkage results and the NMR and partial acid hydrolytic analysis, a tentative structure (Fig. 5) is proposed for alkali-soluble polysaccharide MAP isolated from the leaves of M. coromandelianum. MAP was found to comprise a backbone of →4)-β-linked Xylp(1→ with branching at O-2 mostly by →3)-α-linked Araf(1→ and →3)-β-linked Xylp(1→ with a few residues of →4)-β-linked Galp (1→. The structure was found to be similar to that of arabinoxylans. It is presumed that →3)-α-linked Araf(1→ and α-linked Araf(1→ are present in the L-configuration of MAP and →3)-β-linked Xylp(1→, →4)-β-linked Xylp(1→ and →2,4)-β-linked Xylp(1→ are present in its D-configuration because similar linkages have been reported in the literature for plant-based arabinoxylan-type polysaccharides.60,61
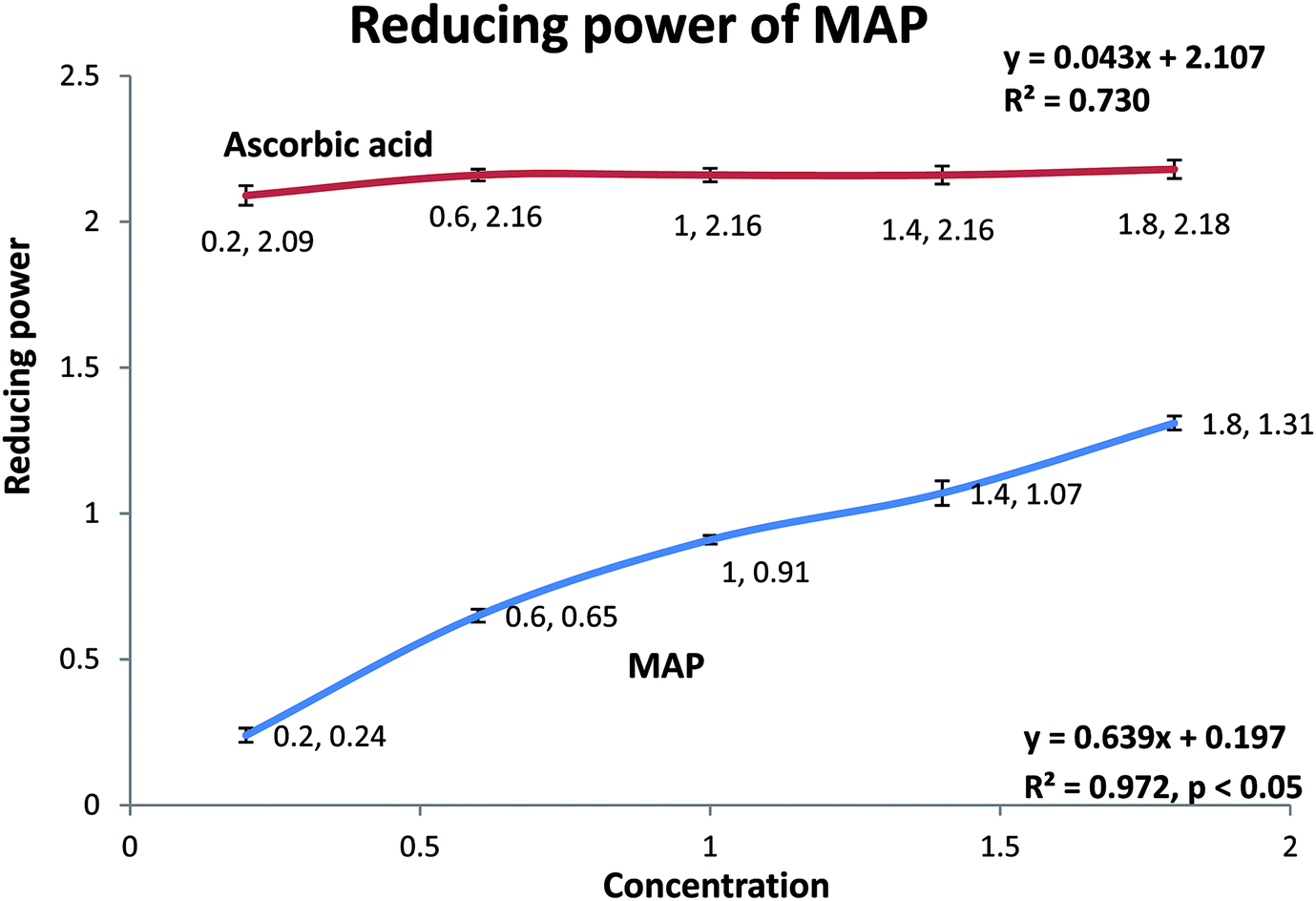 | ||
| Fig. 4 Reducing power of MAP (R2 = 0.972, p < 0.05). | ||
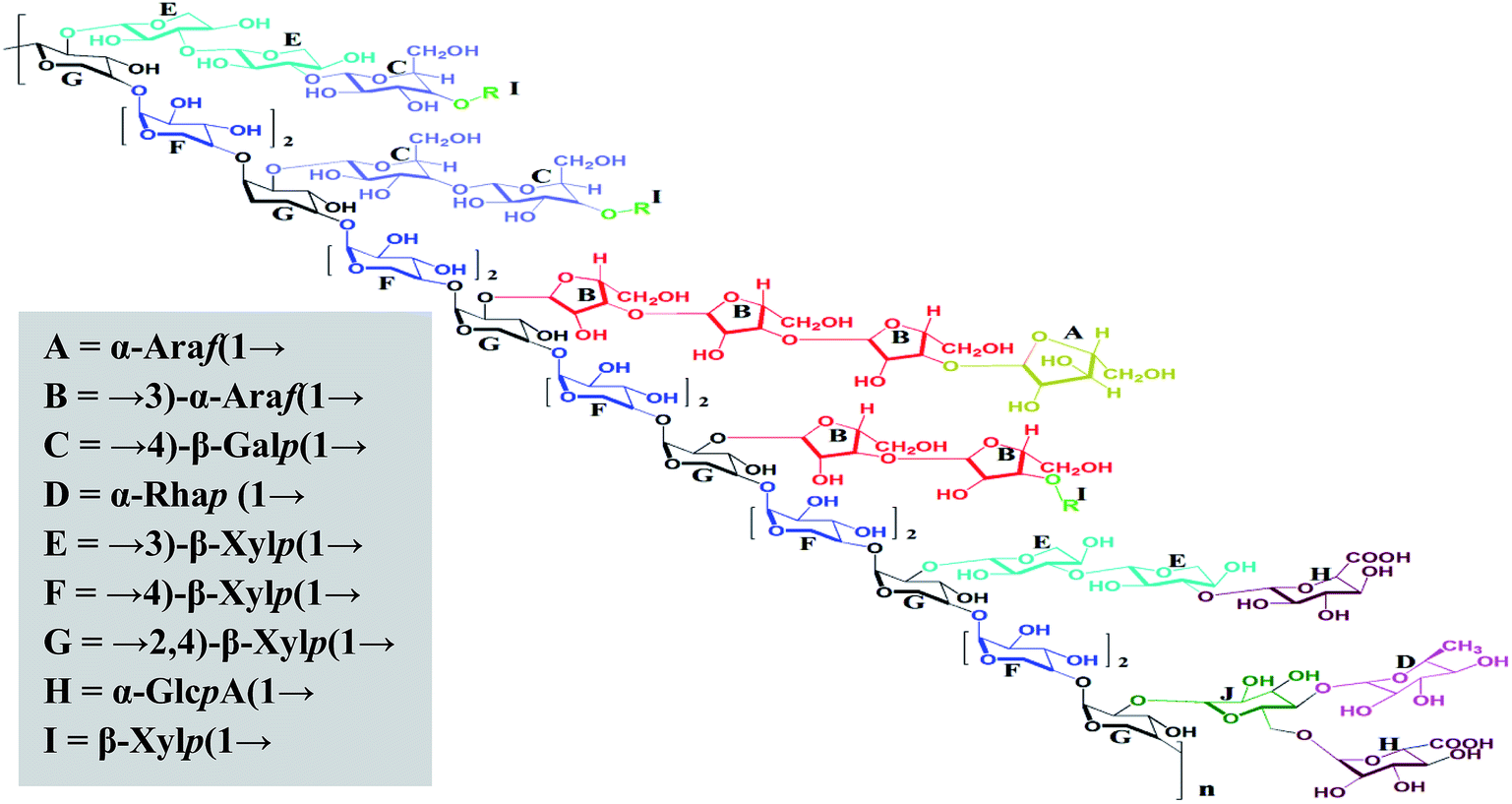 | ||
| Fig. 5 The proposed structure of MAP, R = t-Xylp with a free reducing end. Note: stereochemistry was not considered when drawing the structure. | ||
3.7. Reducing power of MAP
MAP was analyzed for its ferric ion reducing ability using the potassium ferricyanide assay.1 Solutions of MAP and standard ascorbic acid were prepared in five concentrations, viz. 0.2, 0.6, 1.0, 1.4, and 1.8 mg mL−1. Both MAP and the standard showed dose-dependent behavior and increased reducing ability with increasing concentration of the sample. MAP and the standard showed ferric ion reducing powers of 0.914 ± 0.01 (R2 = 0.972, p < 0.05) and 2.178 ± 0.004 (R2 = 0.730), respectively, at 1 mg mL−1 concentration, as shown in Fig. 4.The antioxidant activities of carbohydrates, including monosaccharides, oligosaccharides and polysaccharides, have been discussed and validated by different researchers. Nishizawa et al.62 determined the abilities of galactinol and raffinose to scavenge hydroxyl radicals in vitro. It was observed that the second-order rate constants for the reactions between galactinol or raffinose and hydroxyl radicals were higher than those reported for typical antioxidants, such as ascorbate and reduced glutathione or citrulline. The studies concluded that galactinol and raffinose scavenge hydroxyl radicals as a novel function to protect plant cells from oxidative damage caused by methyl viologen treatment, salinity, chilling or drought. In another study, while comparing the antioxidant activities of monosaccharides and disaccharides, Morell et al.63 observed that the free radical scavenging activity of disaccharides, viz. maltose and sucrose, was greater than that of deoxyribose. It was proposed that the difference between the free radical scavenging activities and stabilities of disaccharides and monosaccharides is due to the variation in the number of hydroxyl residues, which are essential for HO˙ (hydroxyl radical) quenching. Wehmeier and Mooradian64 reported that the antioxidant potency of deoxyribose was comparable to that of ascorbate using the 2,2-azobis(2-amidinopropane) dihydrochloride (AAPH) assay. Further, it was suggested that because glucose and ascorbate are highly similar in their molecular structures and share identical transporters, glucose and other related molecules can act as antioxidants under certain conditions. Van den et al.65 reported that sucrose and sucrosyl oligosaccharides (SOS), including fructans and raffinose family oligosaccahrides (RFOs), fulfill various functional roles in plant metabolism. SOS may either directly detoxify ROS in chloroplasts and vacuoles or indirectly stimulate the classic antioxidative defense system. The in vitro antioxidant studies of polysaccharides also indicate that they are effective antioxidants.66 However, the underlying mechanism is still being investigated because the relationships between antioxidant activity and physicochemical properties or structural features are very complex and therefore have not been comprehensively elucidated and confirmed.67–69 The main physicochemical properties which determine the antioxidant activity of polysaccharides are dependent upon the monosaccharides constituting the main backbone and side chains as well as their linkages, functional groups, molecular weights, water solubility, degrees of branching, triple helical structures, etc.68,70–75 It was also observed that the free radical scavenging activity increased when the monosaccharides were arrayed in polymeric form.76 A number of researchers reported that the structural properties of polysaccharides may affect the complex interactions of monosaccharides in polysaccharides to improve their hydrogen atom-donating capability, which in turn plays an important role in their antioxidant activity.71,77,78 In continuum, other researchers reported that the antioxidant activity of polysaccharides increases with increasing uronic acid content, which is due to the capability of uronic acids to chelate metal ions and in turn scavenge DPPH radicals.75,79,80 In addition, recent research reports have indicated that pure polysaccharides have remarkable antioxidant activity.81,82
In brief, the antioxidant activity of polysaccharides is dependent on their hydrogen atom donating ability and the capability of uronic acids to chelate metal ions. Thus, in the present study, the antioxidant activity of MAP is due to the uronic acids in the arabinoxylan backbone.
4. Conclusion
In the present study, an alkali-soluble polysaccharide (MAP) was isolated from the leaves of M. coromandelianum using 1 M NaOH in 1.15% (w/w) yield and was found to reduce Fe+3 ions in a dose-dependent manner. Further, structural analysis showed that the polysaccharide is mainly composed of xylose (60.42 mol%) and arabinose (18.36 mol%), indicating that MAP is an arabinoxylan with an A/X ratio of 0.3. Based on GLC analysis, the molar ratios of monosaccharides L-rhamnose, L-arabinose, D-xylose, D-glucose and D-galactose are 1.00, 6.04, 19.88, 1.07 and 3.03, respectively, and that of D-glucuronic acid is 1.95. The glycosidic linkage analysis showed that MAP consists of →4)-β-Xylp(1→ (30.09 mol%) as a backbone with monosubstitution at O-2 of →2,4)-β-Xylp(1→ (17.96 mole%) with other sugar residues. →3)-β-Xylp(1→ (12.25 mol%) and →3)-α-Araf(1→ (15.21 mol%) are the main side chain residues. The terminal residues in MAP are α-Araf(1→ (3.19 mol%), α-GlcpA (1→ (6.25 mol%) and α-Rhmp(1→ (3.13 mol%), respectively. The oligomeric pattern consisting of xylose residues was abundantly clarified from ESI-MS analysis of partial hydrolysates (0.5 M and 1 M HCl) of MAP clearly indicated that xylose units are linked as oligomers (heptamer, hexamer, etc.). Based on the monosaccharide composition, methylation and partial hydrolysis studies concomitant with NMR analysis, MAP was found to be an arabinoxylan. Further, the reducing power of MAP was determined using the potassium ferricyanide assay, which showed dose-dependent behavior. These results may be useful to understand the biological and medicinal activities of the polysaccharide based on further structure–activity relationship studies, thereby offering an opportunity for better utilisation of a species widely used in traditional systems of medicine.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The authors are grateful to the Director, Forest Research Institute (FRI), Dehradun for providing laboratory facilities. Thanks are also due to Dr H. B. Naithani, Systematic Botanist, Forest Research Institute for authentication of the plant material.References
- V. Rana, M. K. Das, S. Gogoi and V. Kumar, Carbohydr. Polym., 2014, 102, 341–350 CrossRef CAS PubMed.
- L. Cai, S. Zou, D. Liang and L. Luan, Carbohydr. Polym., 2018, 184, 354–365 CrossRef CAS PubMed.
- Y. Chen, F. K. Yao, K. Ming, D. Y. Wang, Y. L. Hu and J. G. Liu, Molecules, 2016, 21, 1705–1728 CrossRef PubMed.
- J. Ghosh and C. E. Myers, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13182–13187 CrossRef CAS PubMed.
- S. Devi and V. Kumar, Arabian J. Chem., 2018 Feb 22 DOI:10.1016/j.arabjc.2018.01.009.
- M. A. Pahlavani and M. D. Harris, Free Radic. Biol. Med., 1998, 25, 903–913 CrossRef CAS PubMed.
- Z. J. Wang, J.-H. Xie, S.-P. Nie and M.-Y. Xie, Food Funct., 2017, 8, 915–926 RSC.
- A. C. Díaz, M. L. Espino, N. S. Arzoz, S. M. Velurtas, N. M. A. Ponce, C. A. Stortz and J. L. Fenucci, Latin American Journal of Aquatic Research, 2017, 45(1), 104–112 CrossRef.
- H. Xue, F. Gan, Z. Zhang, J. Hu, X. Chen and K. Huang, Int. J. Biol. Macromol., 2015, 81, 22–30 CrossRef CAS PubMed.
- Y. Jin, K. X. Liu, J. Y. Peng, C. Y. Wang, L. Kang, N. Chang and H. Sun, Toxicol. Lett., 2015, 232, 149–158 CrossRef CAS PubMed.
- S. L. Qiu, J. Chen, X. Chen, Q. Fan, C. S. Zhang, D. Wang, X. Li, X. Chen, X. Chen, C. Liu and Z. Gao, Carbohydr. Polym., 2014, 103, 148–153 CrossRef CAS PubMed.
- L. Fan, J. Li, K. Deng and L. Ai, Carbohydr. Polym., 2012, 87, 1849–1854 CrossRef CAS.
- Y. Wang, Y. Wang, D. Liu, W. Wang, H. Zhao, M. Wang and H. Yin, Carbohydr. Polym., 2015, 125, 135–145 CrossRef CAS PubMed.
- F. Meng, B. Zhou, R. S. Lin, L. Jia, X. N. Liu, P. Deng, K. Fan, G. L. Wang and J. Zhang, Bioresour. Technol., 2010, 101, 4564–4569 CrossRef CAS PubMed.
- S. Bisht, R. Kant and V. Kumar, Int. J. Biol. Macromol., 2013, 59, 214–220 CrossRef CAS PubMed.
- P. Kumar and V. Kumar, Carbohydr. Polym., 2017, 165, 205–212 CrossRef CAS PubMed.
- A. K. Lakhera and V. Kumar, Int. J. Biol. Macromol., 2017, 94, 45–50 CrossRef CAS PubMed.
- S. Nagar, A. Hensel, P. Mischnick and V. Kumar, Carbohydr. Polym., 2018, 193, 326–335 CrossRef CAS PubMed.
- V. Kumar, S. Nagar and Y. C. Tripathi, J. Biol. Macromol., 2014, 70, 360–363 CrossRef CAS PubMed.
- Anonymous, The Wealth of India: a dictionary of Indian raw materials and industrial products–raw material series, Publications and Information Directorate, Council of Scientific and Industrial Research, New Delhi, 1962, vol. VI, p. 251 Search PubMed.
- K. R. Kirtikar and B. D. Basu, Indian Medicinal Plants, 2nd edn, International Book Distributors, New Delhi, 1935, p. 306 Search PubMed.
- N. N. Sirkar, Pharmacological basis of Ayurvedic therapeutics, in Cultivation and utilization of Medicinal plants, Published by PID CSIR, 1989 Search PubMed.
- K. K. Ajibesin, B. A. Ekpo, D. N. Bala, E. E. Essien and S. A. Adesanya, Ethnobotanical survey of Akwa Ibom State of Nigeria Kola, J. Ethnopharmacol., 2008, 115, 387–408 CrossRef PubMed.
- S. N. Srivastava, L. D. Kapoor, A. Singh and S. L. Kapoor, Survey of Indian plants for saponins, alkaloids and flavonoid, Lloydia, 1969, 32, 297–304 Search PubMed.
- M. Jiaying, Method for manufacturing traditional Chinese medicine preparation for treating gout, CN 102302706 A 20120104, 2012.
- F. Z. Shenqing, Traditional Chinese medicine preparation for treating rheumatic arthralgia, CN 102366576 A 20120307, 2012.
- L. Xin and H. He, Veterinary Chinese medicinal composition for treating laying hen salpingitis and its preparation method, CN 103735832 A 20140423, 2014.
- B. Jialin, Chinese medicine for treatment of otitis media and its preparation method, CN 103656297 A 20140326, 2014.
- N. Jiqiang, Traditional Chinese medicine composition for treating tumor, CN 103393743 A 20131120, 2013.
- C. Hanting, F. Zhiwei, S. Yide, H. Qiaoqiao and L. Xiaoxia, Herbicidal composition for Manihot esculenta field, CN 103392703 A 20131120, 2013.
- P. Changding, Medicinal wine and its preparation method, CN 103301401 A 20130918, 2013.
- A. B. Deore, V. D. Sapakal and N. S. Naikwade, PharmacologyOnLine, 2011, 3, 147–154 Search PubMed.
- J. Sukanya, J. Apijade, J. Mongkol and W. Kamol, Plant compositions having oral insulin-like hypoglycemic activity and hypolipidemic and antimicrobial activities, Eur. Pat. Appl. EP, 1723960 A1 20061122, 2006.
- P. Khonsung, S. Nantsupawat, S. N. Jesadanont, V. Chantharateptawan and A. Panthong, Thai Journal of Pharmacology, 2006, 28, 3 Search PubMed.
- S. Chaiyasit, J. Sukanya, P. Pintip, N. Penphun and P. Sunanta, Curr. Res. Bacteriol., 2008, 1, 42–45 CrossRef.
- P. Jain, D. Bansal, P. Bhasin and A. Anjali, J. Res. Pharm., 2010, 3, 1260–1262 CAS.
- C. L. Leivas, M. Iacomini and L. M. C. Cordeiro, Carbohydr. Polym., 2015, 121, 224–230 CrossRef CAS PubMed.
- J. O. Schreck and W. M. Loffredo, Qualitative Testing for Carbohydrates, ed. H. Anthony Neidig, Che. Edu. Reso., 1994, vol. 3 Search PubMed.
- R. Katoch, Analytical Techniques in Biochemistry and Molecular Biology, Springer, 2011, p. 69 Search PubMed.
- W. E. Trevelyan, D. P. Proctor and J. S. Harrison, Nature, 1950, 166, 444–445 CrossRef CAS PubMed.
- R. L. Taylor, J. E. Shively and H. E. Conard, Stoichiometric reduction of uronic acid carboxyl groups in polysaccharide, in Meth. in Carbo. Chem., ed. R. L. Whistler and J. N. Be Miller, 1976, vol. 7, pp. 149–151 Search PubMed.
- P. E. Jansson, L. Kenne, H. Liedgren, B. Lindberg and J. Lonngren, Chem. Commun., 1976, 8, 1–76 Search PubMed.
- C. T. Bishop, Gas liquid chromatography of Carbohydrates derivatives, in Adv. in Carbohydr. Chem. and Biochem., ed. M. L. Wolfrom, 1964, 19, p. 95–157 Search PubMed.
- R. L. Scott, Anal. Chem., 1979, 51, 936–941 CrossRef CAS.
- T. Narui, K. Takahashi, M. Kobayashi and S. Shibata, Carbohydr. Res., 1982, 103, 293–295 CrossRef CAS.
- M. H. Fischer, N. Yu, G. R. Gray, J. Ralph, L. Anderson and J. A. Marlett, Carbohydr. Res., 2004, 339, 2009–2017 CrossRef CAS PubMed.
- Q. Guo, S. W. Cui, Q. Wang and J. C. Young, Carbohydr. Polym., 2008, 73, 35–43 CrossRef CAS.
- J. Y. Yin, H. X. Lin, S. P. Nie, S. W. Cui and M. Y. Xie, Carbohydr. Polym., 2012, 88, 1395–1401 CrossRef CAS.
- J. Kang, S.W. Cui, G.O. Phillips, J. Chen, Q. Guo and Q. Wang, Food Hydrocolloids, 2011, 25(8), 1991–1998 CrossRef CAS.
- J. Yin, H. Lin, J. Li, Y. Wang, S. W. Cui, S. Nie and M. Xie, Carbohydr. Polym., 2012, 87, 2416–2424 CrossRef CAS.
- Y. Habibi, M. Mahrouz and M. R. Vignon, Carbohydr. Polym., 2005, 60, 319–329 CrossRef CAS.
- A. Hoije, C. Sandstrom, J. P. Roubroeks, R. Andersson, S. Gohil and P. Gatenholm, Carbohydr. Res., 2006, 341, 2959–2966 CrossRef PubMed.
- Y. Sun, S. W. Cui, X. Gu and J. Zhang, Carbohydr. Polym., 2011, 85, 615–621 CrossRef CAS.
- F. Peng, J. Bian, P. Peng, Y. Guan, F. Xu and R. C. Sun, BioResources, 2012, 7, 4744–4759 CrossRef.
- J. M. Dobruchowska, G. J. Gerwig, A. Babuchowski and J. P. Kamerling, Carbohydr. Res., 2008, 343, 726–745 CrossRef CAS PubMed.
- A. B. Samuelsen, I. Lund, J. M. Djahromi, B. S. Paulsen, J. K. Wold and S. H. Knutsen, Carbohydr. Polymer, 1999, 38, 133–143 CrossRef CAS.
- R. Schendel, A. Becker, C. E. Tyl and M. Bunzel, Carbohydr. Res., 2015, 407, 16–25 CrossRef CAS PubMed.
- Y. Zhang, G. Yu, B. Li, X. Mu and H. Wang, Carbohydr. Polym., 2016, 141, 238–243 CrossRef CAS PubMed.
- J. Jiang, F. Kong, N. Li, D. Zhang, C. Yan and H. Lv, Carbohydr. Polym., 2016, 147, 365–371 CrossRef CAS PubMed.
- R. A. Hoffmann, M. Roza, J. Maat, J. P. Kamerling and J. F. G. Vliegenthart, Carbohydr. Polym., 1991, 15, 415–430 CrossRef CAS.
- R. A. Hoffmann, J. P. Kamerling and J. F. G. Vliegenthart, Carbohydr. Res., 1992, 226, 303–311 CrossRef CAS.
- A. Nishizawa, Y. Yabuta and S. Shigeoka, J. Plant Physiol., 2008, 147, 1251–1263 CrossRef CAS PubMed.
- R. Morelli, S. Russo-Volpe, N. Bruno and R. Lo Scalzo, J. Agric. Food Chem., 2003, 51, 7418–7425 CrossRef CAS PubMed.
- K. R. Wehmeier and A. D. Mooradian, Free Radic. Biol. Med., 1994, 17, 83–86 CrossRef CAS PubMed.
- W. Van den Ende and R. Valluru, J. Exp. Bot., 2008, 60, 9–18 CrossRef PubMed.
- Y. X. Sun and J. F. Kennedy, Carbohydr. Polym., 2010, 82, 510–514 CrossRef CAS.
- H. Wang, Y. M. Liu, Z. M. Qi, S. Y. Wang, S. X. Liu, X. Li and X. C. Xia, Curr. Med. Chem., 2013, 20, 2899–2913 CrossRef CAS PubMed.
- L. N. Malunga and T. Beta, Cereal Chem., 2015, 92, 29–36 CrossRef CAS.
- L. N. Malunga, M. Izydorczyk and T. Beta, Bioact. Carbohydr. Diet. Fibre, 2017, 10, 20–26 CrossRef CAS.
- T. C. T. Lo, C. A. Chang, K. H. Chiu, P. K. Tsay and J. F. Jen, Carbohydr. Polym., 2011, 86, 320–327 CrossRef CAS.
- W. C. Zeng, Z. Zhang and L. R. Jia, Carbohydr. Polym., 2014, 108, 58–64 CrossRef CAS PubMed.
- R. S. P. Rao and G. Muralikrishna, Phytochemistry, 2006, 67, 91–99 CrossRef CAS PubMed.
- J. Wang, S. Hu, S. Nie, Q. Yu and M. Xie, Oxid. Med. Cell. Longevity, 2016, 2016 CAS.
- H. Wang, Y. M. Liu, Z. M. Qi, S. Y. Wang, S. X. Liu, X. Li and X. C. Xia, Curr. Med. Chem., 2013, 20, 2899–2913 CrossRef CAS PubMed.
- J. Li, Y. Liu, L. Fan, L. Ai and L. Shan, Carbohydr. Polym., 2011, 84, 390–394 CrossRef CAS.
- E. Tsiapali, S. Whaley, J. Kalbfleisch, H. E. Ensley, I. W. Browder and D. L. Williams, Free Radical Biol. Med., 2001, 30, 393–402 CrossRef CAS PubMed.
- L. N. Malunga and T. Beta, Food Chem., 2015, 167, 311–319 CrossRef CAS PubMed.
- C. Hu, Y. Zhang and D. D. Kitts, J. Agric. Food Chem., 2000, 48, 3170–3176 CrossRef CAS PubMed.
- H. Wu, T. Min, X. Li, L. Li, F. Lai, Y. Tang and X. Yang, Int. J. Biol. Macromol., 2013, 59, 90–95 CrossRef CAS PubMed.
- S. H. Al-Sheraji, A. Ismail, M. Y. Manap, S. Mustafa, R. M. Yusof and F. A. Hassan, LWT–Food Sci. Technol., 2012, 48, 291–296 CrossRef CAS.
- J. Chen, L. Li, X. Zhou, P. Sun, B. Li and X. Zhang, Food and functions, 2018, 9, 6337–6348 RSC.
- Y. Tang, Z. Y. Zhu, Y. Liu, H. Sun, Q. Y. Song and Y. Zhang, Food and functions, 2018, 9, 2300–2312 RSC.
| This journal is © The Royal Society of Chemistry 2019 |