
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the photo-efficacy of an organic visible-light-activated chromophore (alizarin red S) on zinc oxide with a Ag–Na electrolyte to photo-transform aromatic and aliphatic alcohols†
Timothy M. Underwood and
Ross S. Robinson*
and
Ross S. Robinson*
School of Chemistry and Physics, University of Kwazulu-Natal, Private Bag X01, Pietermaritzburg, 3209, South Africa. E-mail: robinsonr@ukzn.ac.za
First published on 5th August 2019
Abstract
The development of an aqueous silver-sodium/alizarin red sensitised zinc oxide system has been reported to oxidise a range of both aromatic and aliphatic alcohols to aldehydes. Furthermore, photoluminescence spectroscopy validated the electron quenching effect of zinc oxide's defect sites after surface sensitising the metal-oxide with alizarin red. Powder diffuse reflectance UV/Vis data further substantiated the visible-light attenuated properties of alizarin red sensitised zinc oxide, and hence justification for its visible light reactivity towards alcohol oxidations. Lastly, density functional theory calculations supported the intermolecular photo-electronic transfer between alizarin red organic and zinc oxide.
Introduction
Alcohol oxidations are ubiquitous in the literature with conventional techniques using 2-iodobenzoic acid (IBX),1 polymer bound dimethyl sulfoxide2 and various pyridinium chlorochromate3 reagents as primary oxidants. Literature reviews have also illustrated the requirement for nitric acid assisted oxidation of alcohols using Mn(VII) and Cr(VI) salts.4 However, with contemporary research directed towards ‘Eco-Friendly’ reactions, reducing waste has become of international importance.With a focus on working towards more ecologically friendly oxidants, photocatalysts have rapidly become the chosen standard to mediate alcohol oxidations ever since Fujishima and Honda's noteworthy photo-electrochemical article (1972).5
Besides the photo-oxidation of alcohols, researchers have adapted the science to numerous photo-applications. Zhang et al.,6 discovered an application of solvated electrons from chemical vapour deposition diamonds for the reduction of carbon dioxide to methane. This finding was noteworthy due to the long-term acceptance that synthetic diamonds were insulator materials, with little application in the photocatalytic division of materials science. Recently, Verlage et al.,7 designed, fabricated and tested a GaAs/lnGaP under a TiO2 stability film, capable of producing 48 μL O2/H2(g) min−1.
Apart from the advent of significant photo-electrocatalysis studies, photocatalysis (a contemporary alternative to photo-electrocatalysis that produces a photocurrent without an external bias) has exemplified the noteworthy potential towards alcohol oxidations. Multi-component semiconductors have proven to be suitable materials to oxidise a variety of alcohols. Kornarakis et al.,8 reported a three-component cadmium sulfide (CdS), silver(I) sulphide (Ag2S), Keggin structured polyoxometalate (POM) multi-component semiconductor that oxidised aryl alcohols with satisfactory results. The compound semiconductor mediated the alcohol oxidations from the cascading effect of exciton lifetime extension through photo-electron hopping from the CdS conduction band to the lower energy state POM.
A publication by Zavahir et al.,9 indicated that vanadium(IV and V) oxides activated by LED sources were capable photocatalysts for the oxidation of aromatic, saturated and unsaturated alcohols with conversions exceeding 70%. The data reported by the research groups of Fujishima and Honda, Zhang, Verlage, Kornarakis and Zavahir illustrate the need for photocatalysis as an applicant to meet the global requirements for environmentally friendly protocols, a sustainable future and clean chemical practises.5–9 As part of meeting these contemporary protocols, attenuating wide band gap semiconductors with visible light chromophores (alizarin red S, rhodamine B, indigo carmine and eosin Y) have demonstrated targeted visible-light-activated semiconductor examples that are capable of mediating the oxidation of organic sulfides and alcohols, and the synthesis of tetrahydroquinolines.10–13 As observed in the literature, low catalytic loadings of organic dyes offer the unique ability to facilitate viable visible-light-activating photocatalytic properties without the need for an ultraviolet component that typically induces the over-oxidation of feedstock materials with poor selectivity.14
One well researched organic chromophores is alizarin red S (AR).14–16 The UV/Vis absorption profile of AR has been reported in the literature to be highly pH dependent; 2–3 (yellow, absorption @ 261–424 nm), 6–7 (orange, absorption @ 424–510 nm), and lastly violet at pH 11–12 (absorption @ 556–596 nm).14 Stemming from the pH dependence of alizarin red, various coordination sites have been reported in the literature (Fig. 1).16,17
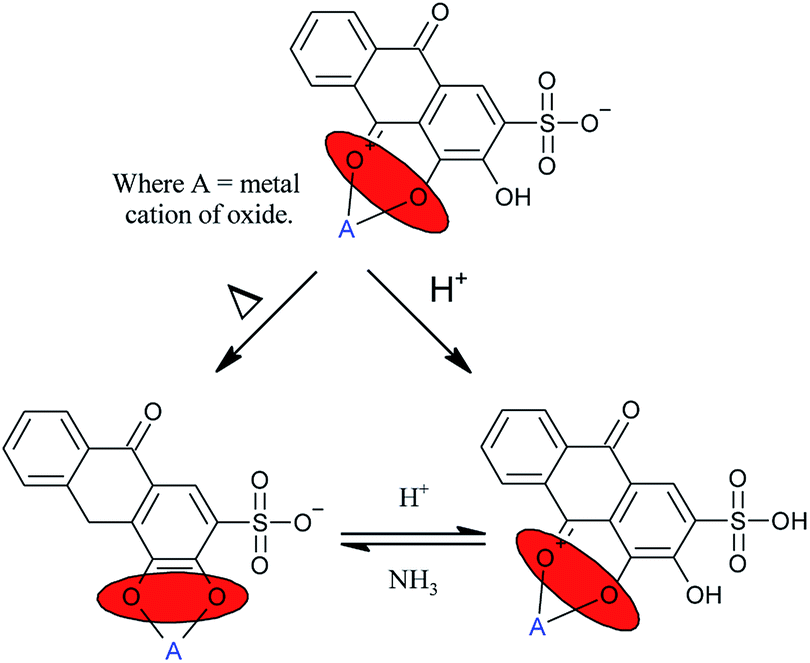 | ||
| Fig. 1 Reported orientations of alizarin red on metal oxides. | ||
Considering the protocol of organic dye mediated photocatalysis, the typical reaction scheme is initiated after the organic dye molecule is excited (Scheme 1, Step A) and electrons are ejected from the excited orbitals of the organic dye into the conduction band (CB) of the metal oxide (MO, Step B). The free electrons relax into the CB of the metal oxide, producing an active cationic dye radical and a free electron in the CB of the metal oxide (Step C). Terminal oxidants (TO) are subsequently reduced after accepting the conduction band electron (Step C), whereby the oxidative process proceeds, viz. oxidation of organic sulfides and alcohols, and the synthesis of tetrahydroquinolines (Step D).10–13
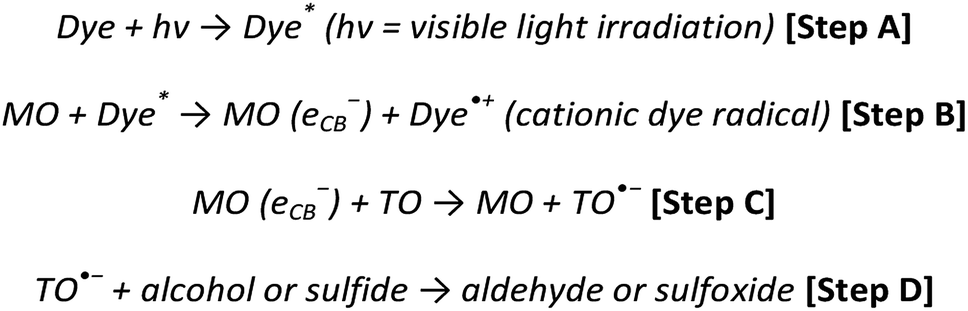 | ||
| Scheme 1 Recognised protocol of organic dye mediated photocatalysis.14 Four stages outline the process from photo-activating the organic dye molecule to facilitating the oxidation of the organic substrate. | ||
From previous work published within the group18,19 an alizarin red-silver zinc oxide system was developed to facilitate the oxidation of seven alcohol derivatives. During the aforementioned studies, experimental procedures (electron spin resonance spectroscopy) validated the necessity of silver nitrate and TEMPO, which were identified to partake in quenching photogenerated electrons (Fig. 2, [b]), and facilitating the oxidative cycle of alcohols to aldehydes, respectively (Fig. 2, [c–e]).
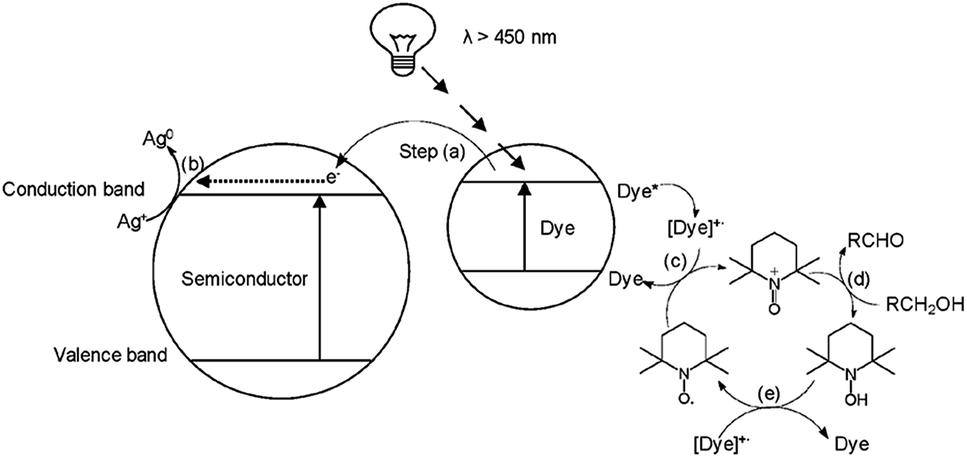 | ||
| Fig. 2 Alizarin red-sensitised ZnO mechanism proposed by Jeena and Robinson.18 Reproduced from V. Jeena and R. S. Robinson, Convenient photooxidation of alcohols using dye sensitised zinc oxide in combination with silver nitrate and TEMPO, Chemical Communications, 2012, 48(2), 299–301 with permission from The Royal Society of Chemistry. | ||
Despite the photocatalytic system yielding good alcohol conversions, the high ionic strength of the aqueous silver nitrate solution (18 molar equivalents) was undesirable and required further investigation (Scheme 2).
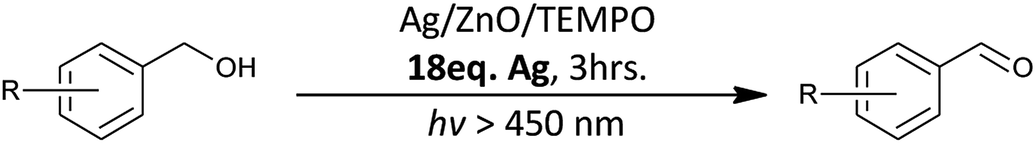 | ||
| Scheme 2 Previous research conducted within the group.18,19 | ||
In this research, two refinement strategies are explored. Firstly, the high ionic concentration of silver nitrate necessitated a review to lower the eighteen equivalents required to stabilise the physi-adsorbed alizarin red, while still achieving high alcohol conversions. Previous ESR research18,19 monitored the free radical processes that existed between alizarin red and TEMPO (Fig. 2, [c–e]), although no mechanistic study of the photo-electronic interaction between alizarin red and zinc oxide in Fig. 2, Step (a) was investigated. To probe the photo-physical nature between alizarin red and zinc oxide under irradiative conditions (as highlighted in Fig. 3), photoluminescence and powder diffuse reflectance spectroscopic techniques were utilised in conjunction with density functional theory modelling to further insight into the organic dye-metal oxide interface.
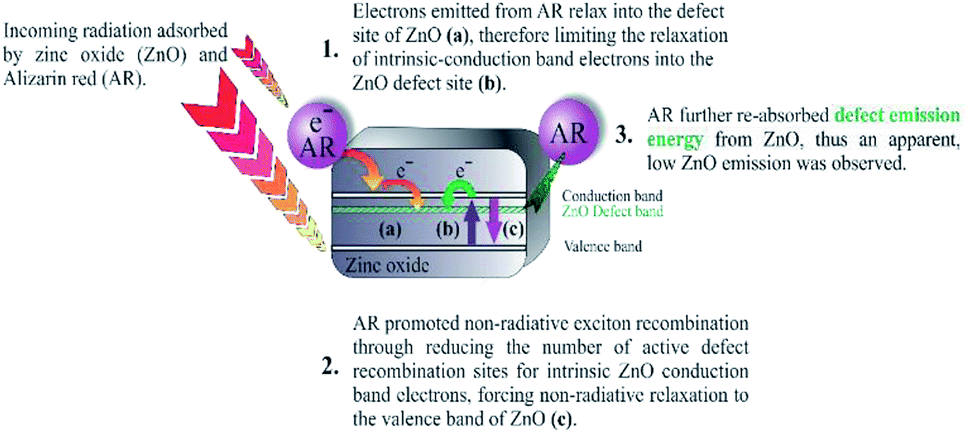 | ||
| Fig. 3 Proposed research of alizarin red sensitised zinc oxide. Adapted from V. Jeena and R. S. Robinson, Convenient photooxidation of alcohols using dye sensitised zinc oxide in combination with silver nitrate and TEMPO, Chemical Communications, 2012, 48(2), 299–301 with permission from The Royal Society of Chemistry. | ||
Within the alizarin red-zinc oxide interface, electrons were hypothesised of being transferred from the highest occupied molecular orbital (HOMO) of alizarin red, and into the defect sites of zinc oxide. After which, the electrons would relax into zinc oxide's defect site (Fig. 2). Spectroscopic studies to monitor the proposed photo-electronic process was monitored using photoluminescence spectroscopy, powder diffuse reflectance spectroscopy and density functional theory modelling.
Results and discussion
After recognising that silver ions not only functioned as electron acceptors in Fig. 2 but also as ionic mediators (to inhibit the hydrolysis of the physi-absorbed alizarin red on zinc oxide),18 sodium nitrate was introduced into the catalytic cycle. Due to sodium's negative standard reduction potential (−2.17 V), its only function was to stabilise the alizarin red organic dye. Therefore, an electrolytic solution of silver nitrate (one equivalent) and sodium nitrate (three equivalents, 1.2 M) afforded a supporting electrolyte of higher ionic strength (1.24 M) that was adequate to stabilise the alizarin red dye on the surface of zinc oxide (Fig. 4).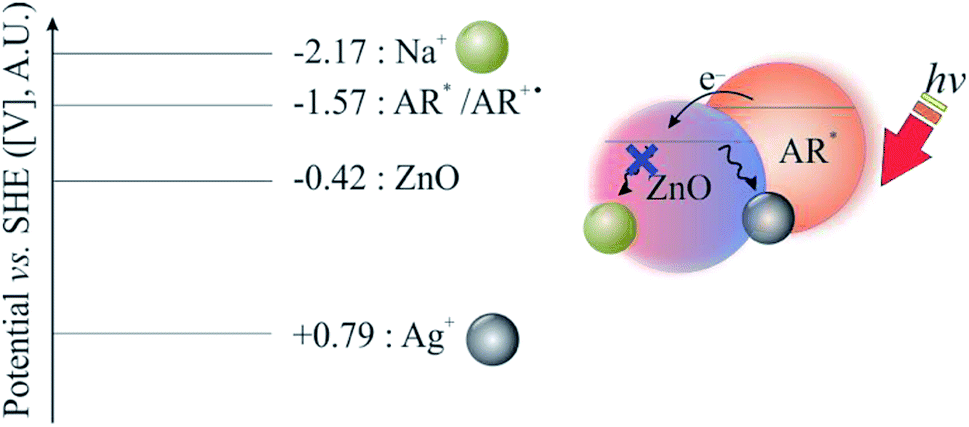 | ||
| Fig. 4 Relative band position of ZnO vs. metal ions. | ||
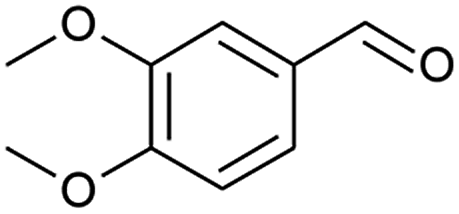
The literature has noted that silver solutions are known to stabilise dye sensitised photocatalytic systems, while in other reports, anthocyanin extracts have been noticeably stabilised in the presence of Mg, Fe, K, Cu, Al and Sn cations due to the presence of hydroxy groups that co-ordinate and stabilise the organic dye.18,20 Therefore, an optimisation study was conducted to investigate the ionic strength affect by varying the sodium electrolyte concentration in the reaction solution, while attempting to oxidise 3,4-dimethoxybenzyl alcohol under photo-irradiation conditions using alizarin red sensitised zinc oxide and 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO, Table 1). Note, a detailed experimental procedure outlining the synthesis of alizarin red sensitised zinc oxide has been made available in the ESI (Chapter S3).†
| Entry | Ag equivalents | Na equivalents | Conversion (%) |
|---|---|---|---|
a Reaction conditions: 0.2 mmol 3,4-dimethoxybenzyl alcohol, 20 mg dye-ZnO, 1.24 M Ag–Na solution electrolyte, 0.096 mmol TEMPO, 0.5 mL de-ionised H2O, 7 hours OSRAM® VIALOX 70W incandescent lamp irradiation (65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 lumens). 600 lumens). |
|||
| 1 | 0 | 18 | 18 |
| 2 | 1 | 15 | >99 |
| 3 | 1 | 10 | >99 |
| 4 | 1 | 5 | >99 |
| 5 | 1 | 3 | 99 |
| 6 | 1 | 1 | 97 |
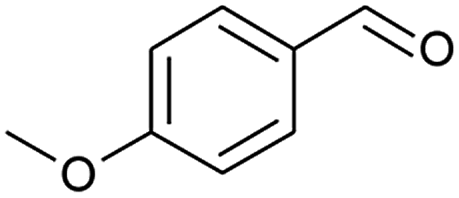
After obtaining the optimised reaction conditions (Table 1, entry 5), the oxidative potential of alizarin red sensitised zinc oxide was evaluated by oxidising a table of fourteen alcohol derivatives (Table 2). The reactions' conversions were quantitatively calculated using 4-octylphenol as an internal standard on a Shimadzu QP-2010SE in conjunction with the established equation noted below.21–23
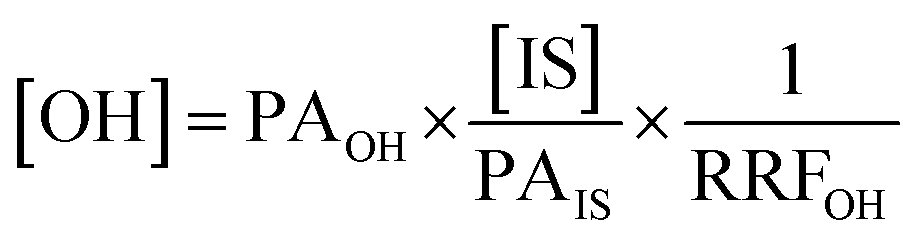
| Entry | Alcohol | Aldehyde | Conv. (%) |
|---|---|---|---|
| a Reaction conditions: 0.2 mmol alcohol, 20 mg dye-ZnO, 1.24 M Na/Ag solution electrolyte, 9.6 × 10−5 mol TEMPO, 0.5 mL H2O, 7 hours. OSRAM® VIALOX 70W incandescent lamp irradiation (65600 lumens). Aldehyde product conversion was determined by GC-MS.b No dimethylmalonaldehyde was detected. |
|||
| 1 |  |
 |
≥99 |
| 2 | 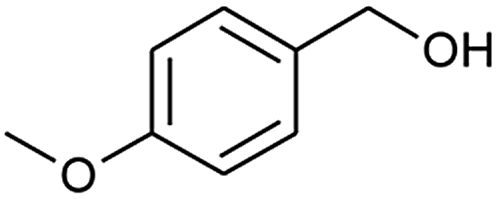 |
 |
59 |
| 3 | 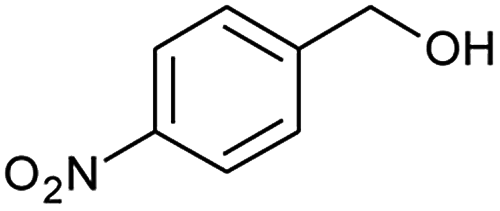 |
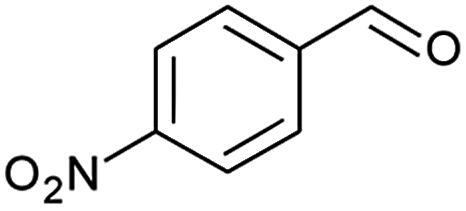 |
97 |
| 4 | 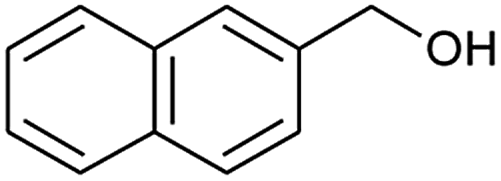 |
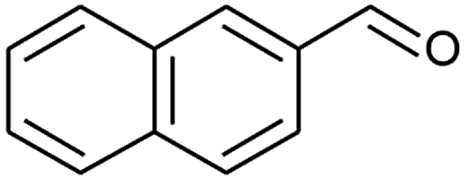 |
70 |
| 5 | 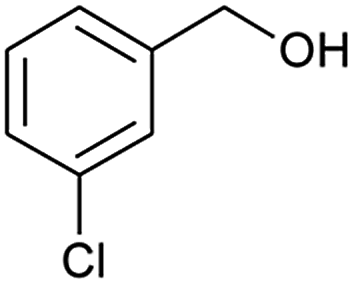 |
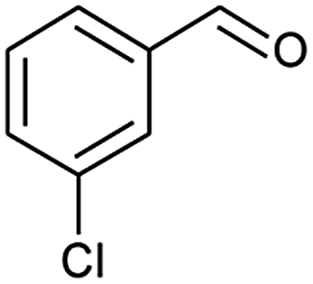 |
66 |
| 6 | 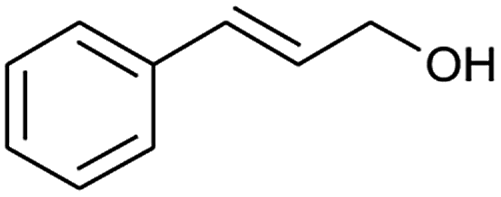 |
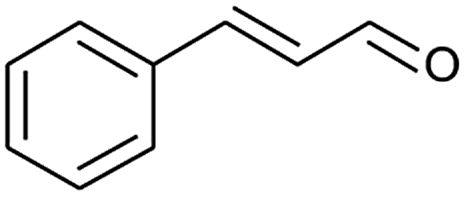 |
69 |
| 7 |  |
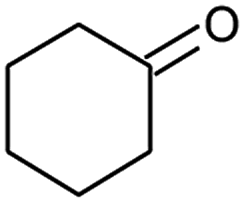 |
72 |
| 8 | 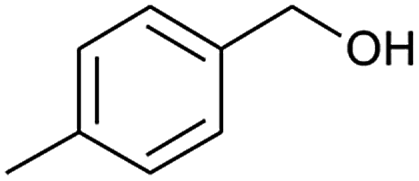 |
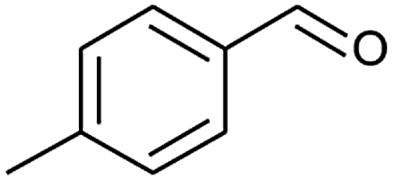 |
70 |
| 9 | 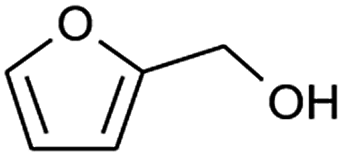 |
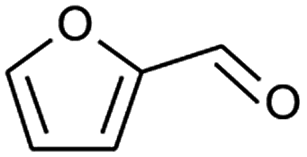 |
73 |
| 10 | 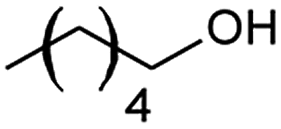 |
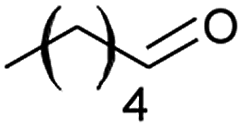 |
66 |
| 11 | 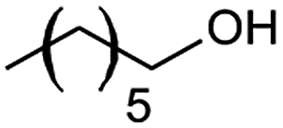 |
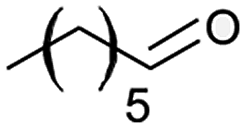 |
75 |
| 12 | 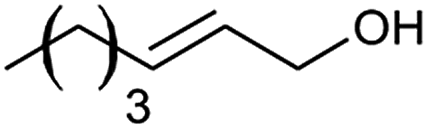 |
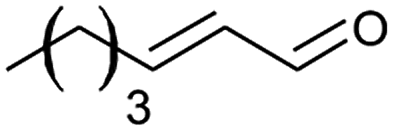 |
60 |
| 13 | 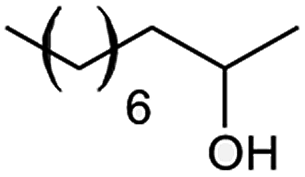 |
 |
10 |
| 14b | 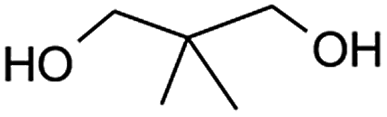 |
 |
88 |
The alizarin red sensitised zinc oxide was noted to favour the oxidation of both electron withdrawing and electron donating benzylic alcohols (Table 2, entries 1–5, 7, and 8). In addition, alizarin red sensitised zinc oxide mediated the oxidation of phenyl-substituted and non-aromatic-substituted alkenes with fair to excellent conversions (Table 2, entries 6 and 12).
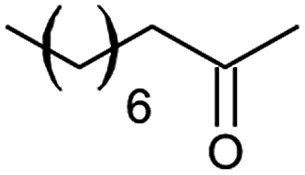
However, we were delighted to note that alizarin red sensitised zinc oxide successfully achieved the partial oxidation of aliphatic alcohols with high selectivity to their respective aldehydes (Table 2, entries 10–14). Table 2, entry 13 demonstrated that alizarin red sensitised zinc oxide was less suited towards secondary aliphatic alcohols. Although similar reports of secondary alcohol oxidations have also proven difficult to convert under comparable conditions using TEMPO as the oxidant.24
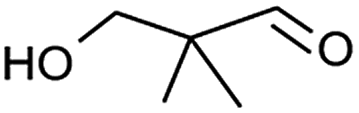
Table 2, entry 14 identified the unexpected potential (!) of alizarin red sensitised zinc oxide to selectively control the single site oxidation of 2,2-dimethyl-1,3-propandiol to 2,2-dimethyl-3-hydroxypropionaldehyde (96%) without the production of dimethylmalonaldehyde. The observed conversions in Table 2 were slightly lower than the previous alizarin red sensitised zinc oxide that utilised eighteen equivalents of silver nitrate,18 although the significant reduction of the metallic salt to one equivalent was highly favourable. Future studies are underway to evaluate the potential of the photocatalytic system in chiral alcohol photo-oxidation studies and assess the stereoselective oxidation of either R- or S-enantiomers.
The aliphatic oxidation results were indeed of significant interest as their controlled oxidations are notoriously difficult to undertake; especially whilst using mild oxidative conditions to retain the selectivity of the aldehyde intermediate.9
Wiles et al.25 reported a Jones' reagent bound to silica gel and loaded into a pressurised flow reactor to selectively convert aliphatic and aromatic alcohols to either aldehydes (at 650 μL min−1) or carboxylic acids (50 μL min−1) with near quantitative yields. However, chromium derivatives (Jones' reagent, PCC, PDC) that typically require organic solvents (Jones reagent [explosive], chromium in concentrated sulphuric acid [corrosive], hypervalent iodine derivatives [DMP, IBX, explosive])26–28 and expensive transition metals (ruthenium and palladium),29,30 are indeed capable of selectively controlling aldehyde synthesis, yet their potentially explosive and/or toxic nature retard their contemporary applications.
Similar literature has reported alcohol oxidations in the absence of TEMPO. Raji et al.31 used (001) faceted titanium dioxide in trifluorotoluene, an oxygen atmosphere, and irradiation from a CFL lamp, which afforded 86% of heptanal. However, a prolonged 24 hour reaction period limited its practicality. A nitroxyl radical derivative (a porous co-ordination polymer [PCP] with a long-lived isoindoline nitroxide radical) was later developed by Li et al.32 to transform benzylic and aliphatic alcohols. However, 24–96 hour reaction periods and 0.016 mmol reaction scales have established the superiority of alizarin red sensitised zinc oxide towards mediating alcohol oxidations in seven hours with high conversions. Photocatalytically active metal organic frameworks (MOFs) have also demonstrated as plausible oxidants of organic materials. MIL-125/Ag/g-C3N4 was previously prepared by Yang et al.33 and shown to facilitate 4-methyl benzyl alcohol (72% conversion) and 4-nitrobenzyl alcohol (12% conversion) in an oxygen environment. Although, the low conversion of an electron withdrawing substituted benzylic alcohol and a 0.2 mmol reaction scale with visible light irradiation (>400 nm) after six hours have further established the efficiency of alizarin red sensitised zinc oxide to mediate a diversified array of alcoholic reactants (Table 2).
Various photocatalytic procedures have attempted to oxidise aliphatic alcohols using dye sensitised titanium dioxide,34 β-cyclodextrin loaded on iron oxide (Fe3O4),35 gaseous alcohols using titanium dioxide,36 and nickel modified gold nanoparticles loaded onto hydrotalcite37 yet, with varied success. The presented research establishes a new photocatalytic opportunity to selectively convert aliphatic alcohols with good to high conversions, well above those reported in the literature vide supra. The use of earth abundant reagents and water as a reaction solvent further elevates its application in contemporary photocatalytic studies.
In the previous studies,19 electron spin resonance (ESR) spectroscopy was utilised to probe plausible mechanistic electron pathways between the organic dye (alizarin red) and TEMPO (primary oxidant). The findings illustrated that the duality of alizarin red and TEMPO were vital for generating and recycling the active N-oxoammonium salt of TEMPO which was responsible for mediating the conversion of alcohols to aldehydes. The ESR characterisation amply validated the proposed mechanism (Fig. 2, [c–e]) but instrumental studies were required to obtain an in-depth understanding of the photo-physical processes that were occurring at the alizarin red-zinc oxide interface (Fig. 2, [a]). Photoluminescence spectroscopy, powder diffuse reflectance spectroscopy and density functional theory modelling were therefore used to further investigate the Step (a) of the mechanism (Fig. 2).
In efforts to further examine the complex photo-electronic pathways that were occurring in the dye-sensitised zinc oxide system, photoluminescence spectroscopic studies (Fig. 5) were investigated to further refine the proposed mechanism (Fig. 3).
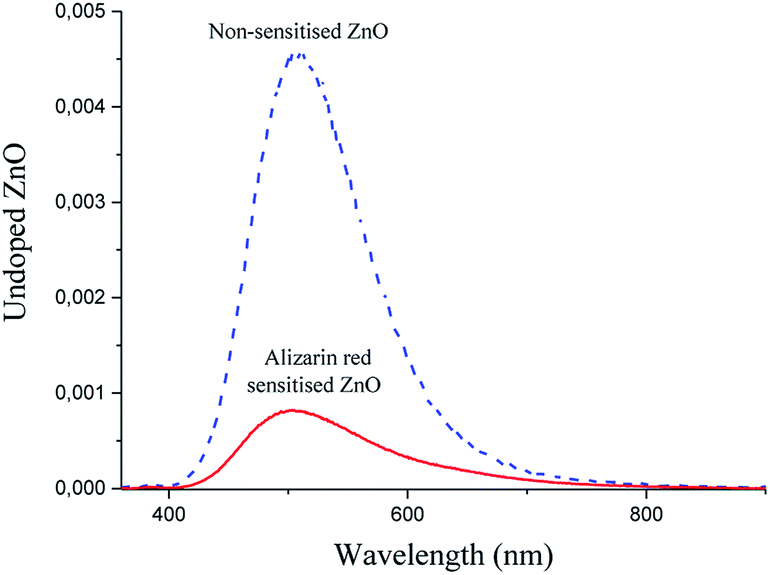 | ||
| Fig. 5 Photoluminescence spectra of alizarin red sensitised zinc oxide (solid red line) and non-sensitised ZnO (dashed blue). The lowered peak intensity of alizarin red sensitised zinc oxide (solid red line) has indicated fewer electron–hole recombinations between the conduction band and intrinsic defect site of zinc oxide. | ||
The reduced peak intensity signal in the photoluminescence spectrum of alizarin red-sensitised ZnO (Fig. 5, solid red line) to non-sensitised ZnO (Fig. 5, dashed blue line) illustrated a decrease of approximately 500% for the zinc oxide's surface-exciton defect recombination process. After sensitising the surface of zinc oxide with alizarin red, the 500% decrease in peak intensity at 507 nm was suggested to arise from a photoelectronic transfer of electrons from the HOMO orbital of alizarin red and into the defect site on zinc oxide, which is in agreement with the literature on similar studies where alizarin red has been utilised as a visible light chromophore.12,38 Despite zinc oxide having a band gap of 3.2 eV, Han et al.,39 also reported strong 500 nm emission signals for zinc oxide, which arose from intrinsic defect sites within the zinc oxide's crystal lattice.
Exciton emission spectra (Fig. 6a–c) of zinc oxide were further studied to assess the influence of electrons trapped by the defect site on exciton state emissions.
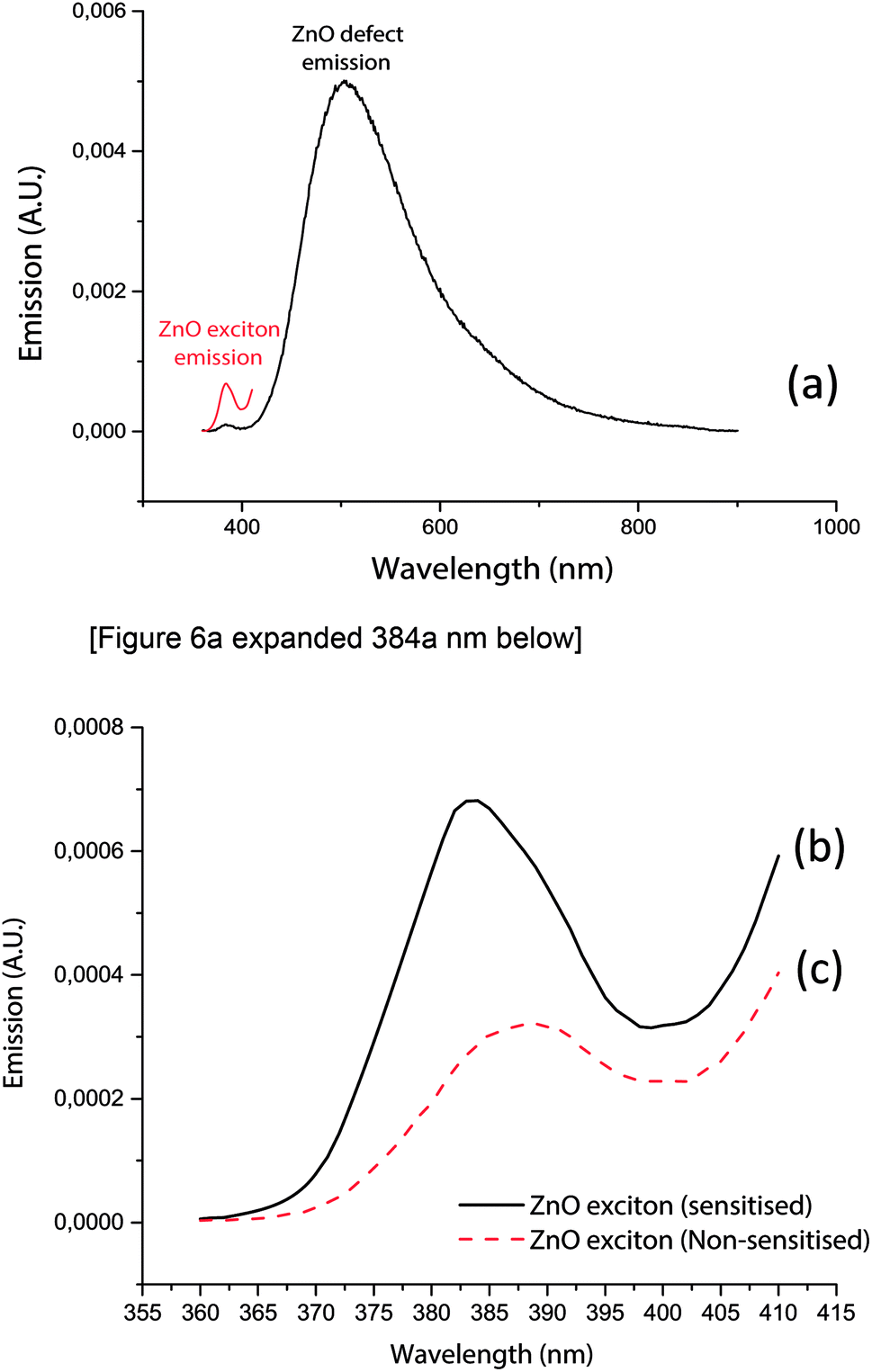 | ||
| Fig. 6 (a) and (b): Photoluminescence signals from zinc oxide's intrinsic band gap (387 nm) and significantly larger defect emission (507 nm). The intense peak emission at the defect site wavelength (507 nm) indicated that electron–hole recombinations were dominant at the defect site and not the intrinsic band gap. (c) A comparison of the exciton emission intensities between alizarin red sensitised zinc oxide (solid black line) and non-sensitised zinc oxide (dotted red line). The larger exciton intensity of alizarin red sensitised zinc oxide indicated more exciton recombinations occurred when alizarin red S was adsorbed onto the surface of zinc oxide. | ||
Fig. 6a demonstrate emission signatures that were detectable from both the band gap and intrinsic defect sites of zinc oxide, yet zinc oxide's intrinsic defects were the dominant source of recombination centres. The exciton emission energies (Fig. 6c) of zinc oxide (solid black line) were 240% higher than alizarin red sensitised zinc oxide (dashed red line), which theorised that alizarin red promoted more exciton recombinations on zinc oxide.
From the photoluminescence results presented in Fig. 5, an intense 507 nm emission peak for non-sensitised zinc oxide suggested an intermolecular electron transfer process between the conduction band of zinc oxide and an intrinsic defect site; which subsided after alizarin red was used as a sensitising agent. The fivefold reduction in the 507 nm peak intensity (after zinc oxide was sensitised with alizarin red) was intriguing and potentially understood to originate from a re-absorption of emitted energy from the intrinsic defect of zinc oxide by alizarin red, thus producing the observed reduction in emission intensity. Further investigations to monitor the fivefold reduction and subsequent absorption of emitted energy was studied with powder diffuse reflectance UV/Vis spectroscopy (Fig. 7).
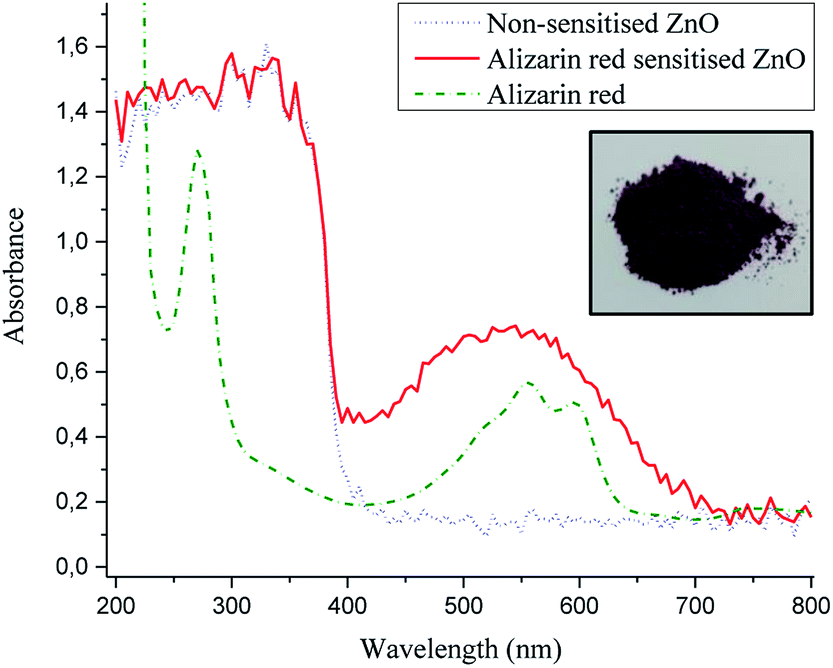 | ||
| Fig. 7 UV/Vis absorbance spectra of zinc oxide (dotted blue line), alizarin red sensitised zinc oxide (solid red line) and alizarin red S (dashed green line). Inset of alizarin red sensitised zinc oxide. The spectra depict the visible light absorbance from alizarin red S at 500 nm. | ||
Alizarin red sensitised zinc oxide (Fig. 7, solid red line) depicted an absorption profile that matched well with non-sensitised zinc oxide (Fig. 7, dotted blue line) up till the band gap of zinc oxide (384 nm), after which alizarin red sensitised zinc oxide had a strong 500 nm absorption shoulder which could be assigned to alizarin red (at red shifted wavelengths >420 nm) on sensitised zinc oxide.
At the absorbance maximum wavelength of 507 nm (Fig. 7), alizarin red-sensitised zinc oxide demonstrated a 500% absorbance increase (0.71) compared to non-sensitised zinc oxide (0.15). This absorbance excess was in favourable agreement with the 500% emission decrease at 507 nm (Fig. 5), which lent support to identify that electrons were re-absorbed by alizarin red.
After the assessment of alizarin red sensitised zinc oxide using photoluminescence spectroscopy and powder diffuse UV/Vis data, density functional theory (DFT) simulations were applied towards further understanding the electronic migration between alizarin red and alizarin red sensitised zinc oxide. The simulations were performed using Gaussian (09W40 at the CAM-B3LYP/6-31G[d,p]41) level of theory. Single first polarization functions were added to the basis set to increase the accuracy of the simulations. Both the prepared input files and analysed output files were prepared using GaussView 5.0.42 The molecular orbitals were assigned by studying the spatial distribution of their isosurfaces. Only singlet excited states were considered. Geometry optimisations and frequency calculations were carried out on both species using the above-mentioned level of theory. The absence of negative frequency eigen values suggests that the geometry-optimised structures are true minima on the global potential energy surface.
Modelling the coordination of alizarin red to zinc oxide through the two hydroxy groups confirmed the preliminary findings as depicted in Fig. 8 below. The basis for the modelling study was developed on similar research in the literature of chatecolate scaffolds that were bound to the surface of TiO2.43
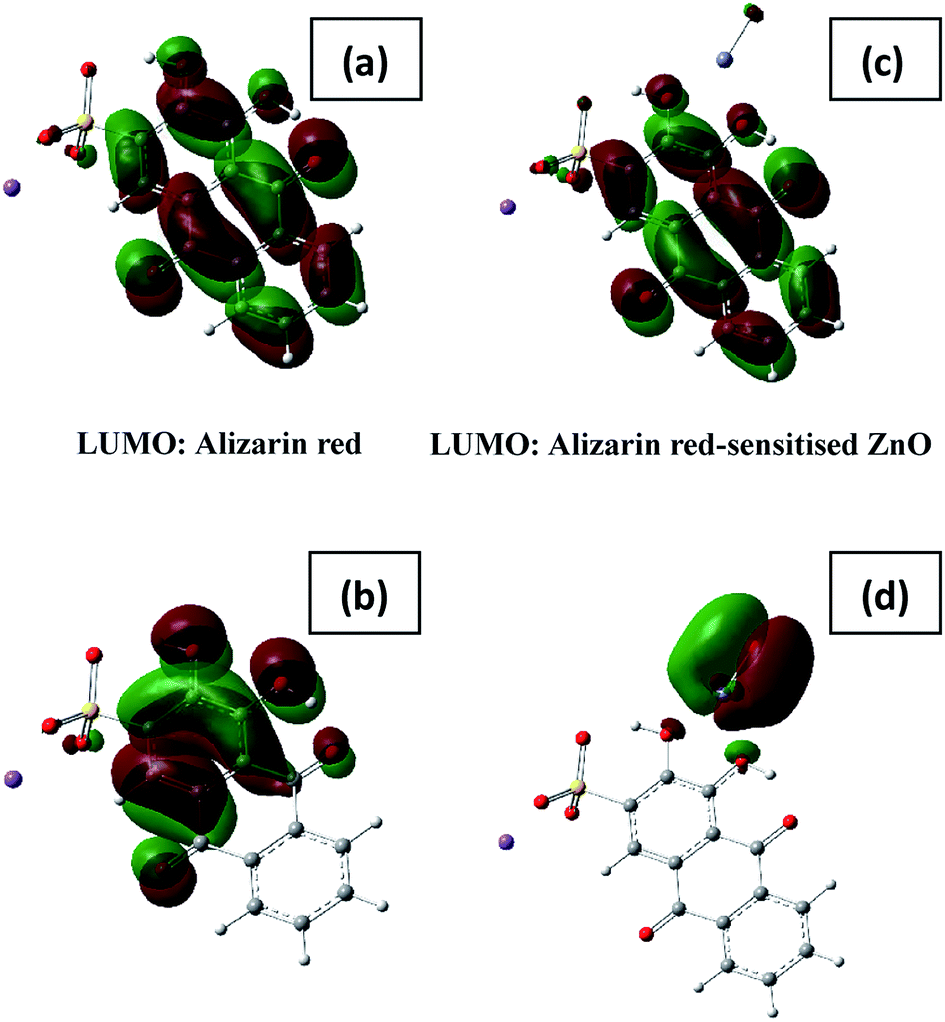 | ||
| Fig. 8 HOMO and LUMO orbitals of alizarin red and the likely structure of the alizarin red-sensitised ZnO. A key feature of the molecular orbitals is the migration of the HOMO from the dye to the ZnO, this neatly illustrates the mixing of the molecular orbitals. The HOMO–LUMO energy gaps measure 5.65 and 2.77 eV, respectively. HOMO and LUMO states of alizarin red and alizarin red-sensitised ZnO species. | ||
From the initial assumptions, alizarin red was photo-excited with hν > 450 nm (note the shift in electronic density between the LUMO and HOMO states of alizarin red above, Fig. 8a and b). After physi-adsorption of alizarin red onto zinc oxide, alizarin red's electronic density resided over the entire molecular (see ‘LUMO: Alizarin red-sensitised ZnO in Fig. 8c’).
However, after photoexcitation (hν > 450 nm), intramolecular electric charge transfer from alizarin red's HOMO state to zinc oxide rapidly ensued (Fig. 8d). Subsequently, electrons transferred to the conduction band of zinc oxide were either terminally quenched with ionic silver or relaxed into the zinc oxide's intrinsic defect site. The DFT study exemplified the HOMO state of the uncoordinated alizarin red species to exist predominately over the aromatic component of the molecule. However, upon coordination of alizarin red to zinc oxide, a rapid shift of the HOMO state resided over the zinc oxide-alizarin red coordination site as seen elsewhere in similar TiO2-pendant catechol studies.17,43 This confirmed favourable photoelectron ejection into the conduction band of zinc oxide while the photocatalytic system was irradiated.
Emanating from the photoluminescence, powder diffuse reflectance UV/Vis spectroscopy, and DFT modelling, a mechanism of the complex photo-system between alizarin red and zinc oxide has been proposed below for the oxidative cycle of alcohols to aldehydes, which is in agreement with similar literature of alizarin red and TEMPO mediated systems.16
Excitation of photo-generated electrons into the HOMO state of alizarin red proceeded after illumination (Fig. 9, Step a). Electrons entered into the conduction band of zinc oxide are either quenched by silver ions (Ag+) or subsequently fall into the sub band-gap defect site of zinc oxide (Fig. 9, Step b). If a back-electron transfer from zinc oxide to alizarin red did occur, the sub band-gap defect site on zinc oxide would have an equally high recombination rate prior to sensitising zinc oxide with alizarin red (see Fig. 5). However, this was not experimentally observed. The alizarin red dye cationic radical proceeds to oxidise TEMPO to the N-oxoammonium salt (the active oxidant of the photocatalytic system, Fig. 9, Step c), which in-turn facilitates the alcohol oxidation (Fig. 9, Step d). Consequently, the N-oxoammonium salt of TEMPO is reduced to the hydroxylamine (Fig. 9, Step e) and concurrently oxidised back to the TEMPO radical by the alizarin red dye cationic radical (Fig. 9, Step f).
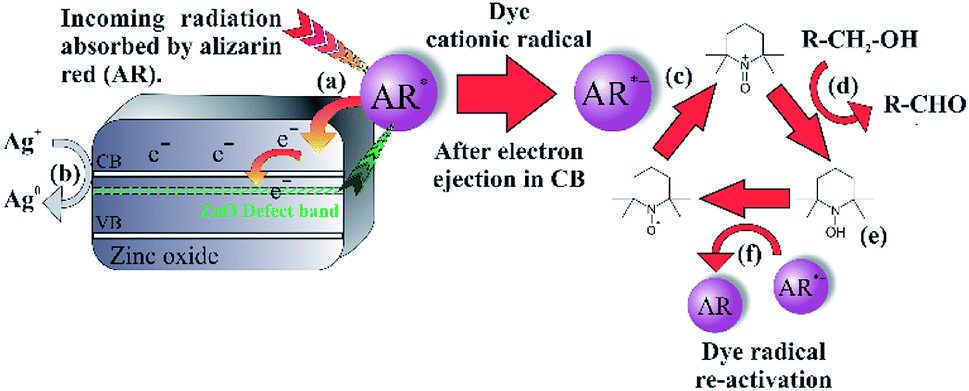 | ||
| Fig. 9 Proposed photoelectron transfer within alizarin red-sensitised zinc oxide. | ||
Conclusions
The presented dye-sensitised zinc oxide photo-oxidation system has demonstrated a refined procedure to drastically lower the silver nitrate content (from 18 to 1 equivalent), while retaining the high photo-capacity to mediate the oxidation of alcohols to aldehydes under visible light irradiation (conversions up to and exceeding 99%).Furthermore, the controlled photo-oxidation of aliphatic alcohols yielding aldehyde intermediates and regio-selectivity for 2,2-dimethyl-3-hydroxypropionaldehyde synthesis illustrates the potential for alizarin red sensitised zinc oxide to function as a prominent photo-oxidising agent in contemporary photo-redox chemistry practises.
In conjunction to the previously obtained electron spin resonance studies, the photoluminescence and powder diffuse reflectance UV/Vis spectroscopy studies, and DFT modelling have arguably substantiated a refined mechanism to provide further insight into understanding the photo-mechanistic pathways in the alizarin red/Ag–Na/ZnO/TEMPO system.
Future investigations to recycle the aqueous sodium, extend the aliphatic scope to more complex natural product systems in various electrolytic solvents and understand the regio-selective oxidation of 2,2-dimethyl-1,3-propandiol are currently underway within our laboratories.
Declaration
The financial assistance of the National Research Foundation (NRF) towards this research is hereby acknowledged. Opinions expressed and conclusions arrived at, are those of the author and are not necessarily to be attributed to the NRF.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge ESKOM TESP and the NRF for their generous financial assistance and kindly thank Mr Craig Grimmer, Professor Matthew Akerman and Mrs Caryl Janse van Rensburg for their support and density functional theory knowledge throughout the study.References
- V. Satam, A. Harad, R. Rajule and H. Pati, Tetrahedron, 2010, 66, 7659–7706 CrossRef CAS.
- T. S. R. Prasanna and K. Mohanaraju, Tetrahedron Lett., 2011, 52, 6971–6973 CrossRef CAS.
- M. Hunsen, Tetrahedron Lett., 2005, 46, 1651–1653 CrossRef CAS.
- C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547–564 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- L. Zhang, D. Zhu, G. M. Nathanson and R. J. Hamers, Angew. Chem., Int. Ed., 2014, 53, 9746–9750 CrossRef CAS PubMed.
- E. Verlage, S. Hu, R. Liu, R. J. R. Jones, K. Sun, C. Xiang, N. S. Lewis and H. A. Atwater, Energy Environ. Sci., 2015, 8, 3166–3172 RSC.
- I. Kornarakis, I. N. Lykakis, N. Vordos and G. S. Armatas, Nanoscale, 2014, 6, 8694–8703 RSC.
- S. Zavahir, Q. Xiao, S. Sarina, J. Zhao, S. Bottle, M. Wellard, J. Jia, L. Jing, Y. Huang, J. P. Blinco, H. Wu and H.-Y. Zhu, ACS Catal., 2016, 6, 3580–3588 CrossRef CAS.
- X. Lang, J. Zhao and X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4697–4700 CrossRef CAS PubMed.
- M. Hosseini-Sarvari, M. Koohgard, S. Firoozi, A. Mohajeri and H. Tavakolian, New J. Chem., 2018, 42, 6880–6888 RSC.
- Y. Zhang, Z. Wang and X. Lang, Catal. Sci. Technol., 2017, 7, 4955–4963 RSC.
- E. L. Tae, S. H. Lee, J. K. Lee, S. S. Yoo, E. J. Kang and K. B. Yoon, J. Phys. Chem. B, 2005, 109, 22513–22522 CrossRef CAS PubMed.
- R. Giovannetti, C. A. D. Amato, M. Zannotti, E. Rommozzi, R. Gunnella, M. Minicucci and A. Di Cicco, Sci. Rep., 2015, 5, 17801 CrossRef CAS PubMed.
- J. Kurepa, T. Paunesku, S. Vogt, H. Arora, B. M. Rabatic, J. Lu, M. B. Wanzer, G. E. Woloschak and J. A. Smalle, Nano Lett., 2010, 10, 2296–2302 CrossRef CAS PubMed.
- X. Lang, J. Zhao and X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4697–4700 CrossRef CAS PubMed.
- P. M. Jayaweera and T. A. U. Jayarathne, Surf. Sci., 2006, 600, L297–L300 CrossRef CAS.
- V. Jeena and R. S. Robinson, Chem. Commun., 2012, 48, 299–301 RSC.
- V. Jeena and R. S. Robinson, Dalton Trans., 2012, 41, 3134–3137 RSC.
- R. Cortez, D. A. Luna-Vital, D. Margulis and E. Gonzalez de Mejia, Compr. Rev. Food Sci. Food Saf., 2017, 16, 180–198 CrossRef CAS.
- C. Zheng, G. He, X. Xiao, M. Lu, H. Zhong, X. Zuo and J. Nan, Appl. Catal., B, 2017, 205, 201–210 CrossRef CAS.
- A. Braithwaite and F. J. Smith, Chromatographic Methods, Chapman and Hall Ltd, United States of America, 4th edn, 1985 Search PubMed.
- G. Guiochon and C. L. Guillemin, in Journal of Chromatography Library ed. G. Guiochon and C. L. Guillemin, Elsevier, 1988, vol. 42, pp. 629–659 Search PubMed.
- J. Einhorn, C. Einhorn, F. Ratajczak and J.-L. Pierre, J. Org. Chem., 1996, 61, 7452–7454 CrossRef CAS PubMed.
- C. Wiles, P. Watts and S. J. Haswell, Tetrahedron Lett., 2006, 47, 5261–5264 CrossRef CAS.
- G. Tojo and M. I. Fernandez, Oxidation of Alcohols to Aldehydes and Ketones: A Guide to Current Common Practice, Springer, US, 2006 Search PubMed.
- D. G. Lee and R. Stewart, J. Org. Chem., 1967, 32, 2868–2871 CrossRef CAS.
- T. Hirofumi and K. Yasuyuki, Adv. Synth. Catal., 2004, 346, 111–124 CrossRef.
- C. Bilgrien, S. Davis and R. S. Drago, J. Am. Chem. Soc., 1987, 109, 3786–3787 CrossRef CAS.
- B. Delina and N. Ronny, Adv. Synth. Catal., 2010, 352, 293–298 CrossRef.
- V. Raji, B. A. Kalam, S. Tapan, P. Provas, G. Bishwajit, G. S. Chandra and P. A. Baran, Chem.–Asian J., 2016, 11, 3084–3089 CrossRef PubMed.
- L. Li, R. Matsuda, I. Tanaka, H. Sato, P. Kanoo, H. J. Jeon, M. L. Foo, A. Wakamiya, Y. Murata and S. Kitagawa, J. Am. Chem. Soc., 2014, 136, 7543–7546 CrossRef CAS PubMed.
- Z. Yang, X. Xu, X. Liang, C. Lei, Y. Cui, W. Wu, Y. Yang, Z. Zhang and Z. Lei, Appl. Catal., B, 2017, 205, 42–54 CrossRef CAS.
- M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2008, 47, 9730–9733 CrossRef CAS PubMed.
- J. Zhu, P.-c. Wang and M. Lu, J. Braz. Chem. Soc., 2013, 24, 171–176 CrossRef CAS.
- U. R. Pillai and E. Sahle-Demessie, J. Catal., 2002, 211, 434–444 CrossRef CAS.
- D. Guo, Y. Wang, P. Zhao, M. Bai, H. Xin, Z. Guo and J. Li, Catalysts, 2016, 6, 64 CrossRef.
- G. Ramakrishna, A. K. Singh, D. K. Palit and H. N. Ghosh, J. Phys. Chem. B, 2004, 108, 1701–1707 CrossRef CAS.
- N. S. Han, H. S. Shim, J. H. Seo, S. Y. Kim, S. M. Park and J. K. Song, J. Appl. Phys., 2010, 107, 084306 CrossRef.
- M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (c) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS; (d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS; (e) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS; (f) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- R. Dennington, T. Keith and J. Millam, Gauss View, Version 5, Semichem Inc., Shawnee Mission KS, 5th edn, 2009, vol. 29.
- P. Kar, S. Verma, A. Sen, A. Das, B. Ganguly and H. N. Ghosh, Inorg. Chem., 2010, 49, 4167–4174 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03474a |
| This journal is © The Royal Society of Chemistry 2019 |