
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntiprotozoal dimeric naphthylisoquinolines, mbandakamines B3 and B4, and related 5,8′-coupled monomeric alkaloids, ikelacongolines A–D, from a Congolese Ancistrocladus liana†
Jean-Pierre Mufusamaab,
Doris Feineisa,
Virima Mudogoc,
Marcel Kaiserde,
Reto Brunde and
Gerhard Bringmann *a
*a
aInstitute of Organic Chemistry, University of Würzburg, Am Hubland, D-97074 Würzburg, Germany. E-mail: bringman@chemie.uni-wuerzburg.de
bFaculté des Sciences Pharmaceutiques, Université de Kinshasa, B.P. 212, Kinshasa XI, Democratic Republic of the Congo
cFaculté des Sciences, Université de Kinshasa, B.P. 202, Kinshasa XI, Democratic Republic of the Congo
dSwiss Tropical and Public Health Institute, Socinstrasse 57, CH-4002 Basel, Switzerland
eUniversity of Basel, Petersplatz 1, CH-4003 Basel, Switzerland
First published on 16th April 2019
Abstract
From the leaves of a botanically and phytochemically as yet unexplored Ancistrocladus liana discovered in the rainforests of the Central region of the Democratic Republic of the Congo in the vicinity of the town of Ikela, six new naphthylisoquinoline alkaloids were isolated, viz., two constitutionally unsymmetric dimers, the mbandakamines B3 (3) and B4 (4), and four related 5,8′-linked monomeric alkaloids, named ikelacongolines A–D (5a, 5b, 6, and 7). The dimers 3 and 4 are structurally unusual quateraryls comprising two 5,8′-coupled monomers linked via a sterically strongly constrained 6′,1′′-connection between their naphthalene units. These compounds contain seven elements of chirality, four stereogenic centers and three consecutive chiral axes. They were identified along with two known related compounds, the mbandakamines A (1) and B2 (2), which had so far only been detected in two Ancistrocladus species indigenous to the Northwestern Congo Basin. In addition, five known monomeric alkaloids, previously found in related Central African Ancistrocladus species, were isolated from the here investigated Congolese liana, three of them belonging to the subclass of 5,8′-coupled naphthylisoquinoline alkaloids, whereas two compounds exhibited a less frequently occurring 7,8′-biaryl linkage. The stereostructures of the new alkaloids were established by spectroscopic (in particular HRESIMS, 1D and 2D NMR), chemical (oxidative degradation), and chiroptical (electronic circular dichroism) methods. The mbandakamines B3 (3) and B4 (4) displayed pronounced activities in vitro against the malaria parasite Plasmodium falciparum and the pathogen of African sleeping sickness, Trypanosoma brucei rhodesiense.
Introduction
The evergreen rain forests and swamp regions in Central Africa, in particular in the Democratic Republic of the Congo, are one of the main centers of distribution of lianas belonging to the Ancistrocladaceae plant family.1–3 Comprehensive data about the natural diversity of the genus Ancistrocladus within the Congo region, however, are still lacking.1,3 Recent field trips in combination with phylogenetic and phytochemical investigations clearly suggested the presence of further, hitherto unrecognized taxa,3–11 along with the four botanically acknowledged Ancistrocladus species,1,2,12,13 A. congolensis J. Léonard, A. likoko J. Léonard, A. ealaensis J. Léonard, and A. ileboensis Heubl, Mudogo & G. Bringmann. The latter species was discovered in the Southern Congo Basin, and described for the first time in 2010,12 whereas the three other Congolese taxa were botanically acknowledged for the first time in 1949.13 These three species are widely distributed in the North-Western and North-Central region of the Democratic Republic of the Congo.It is all the more remarkable that phytochemical investigations on Central African Ancistrocladus plants have picked up speed only quite recently, during the past decade,4–11,14–21 considering that intense isolation work on Asian as well as West and East African taxa had started much earlier, leading to the discovery of structurally unique mono- and dimeric naphthylisoquinoline alkaloids with promising antiparasitic and antiviral activities.22–27 More recent phytochemical studies on Ancistrocladus lianas from the Congo region, including all of the four valid taxa14–21 mentioned above, but also botanically as yet unidentified plants,4–11 furnished a whole series of further novel-type naphthylisoquinoline alkaloids, among them N,C-coupled compounds9,10 and, in particular, dimeric representatives with complex molecular architectures.4–8,14–16,18,21 Some of these secondary metabolites attracted attention due to their extraordinary antiplasmodial6,8,15,16 or antileishmanial10 activities or, according to their individual structures, to their strong antiproliferative effects against human leukemia,16,18,20 cervical HeLa,7,21 or pancreatic cancer cells.7,9,17,19,21
Structurally most fascinating are the highly unsymmetric mbandakamine-type dimers like e.g., mbandakamines A (1)4,16 and B2 (2)6 (Fig. 1), which were discovered in a botanically as yet undescribed liana growing in the rainforests near the town of Mbandaka in the Northwestern part of the Democratic Republic of the Congo. The dimers 1 and 2 consist of two constitutionally and stereochemically nearly identical 5,8′-coupled monomeric halves, which are linked via the sterically highly crowded 6′,1′′-positions of their naphthalene units. The central axis of 1 and 2 is thus located in one of the peri-positions of the ‘northwestern’ naphthalene portion, neighboring one of the outer axes in the adjacent peri-position, which gives rise to a rotationally strongly hindered central binaphthalene linkage.4,6,16 With the two outer axes being chiral, too, 1 and 2 possess three consecutive stereogenic axes, and, thus, together with four stereogenic centers in their two tetrahydroisoquinoline subunits, a total of seven elements of chirality.4,6,16 The two mbandakamines exerted promising antiplasmodial and antitrypanosomal activities in vitro,4,6 and mbandakamine A (1) was also found to display strong cytotoxic activities against drug-sensitive (CCRF-CEM) and multidrug-resistant (CEM/ADR5000) human leukemia cells.16
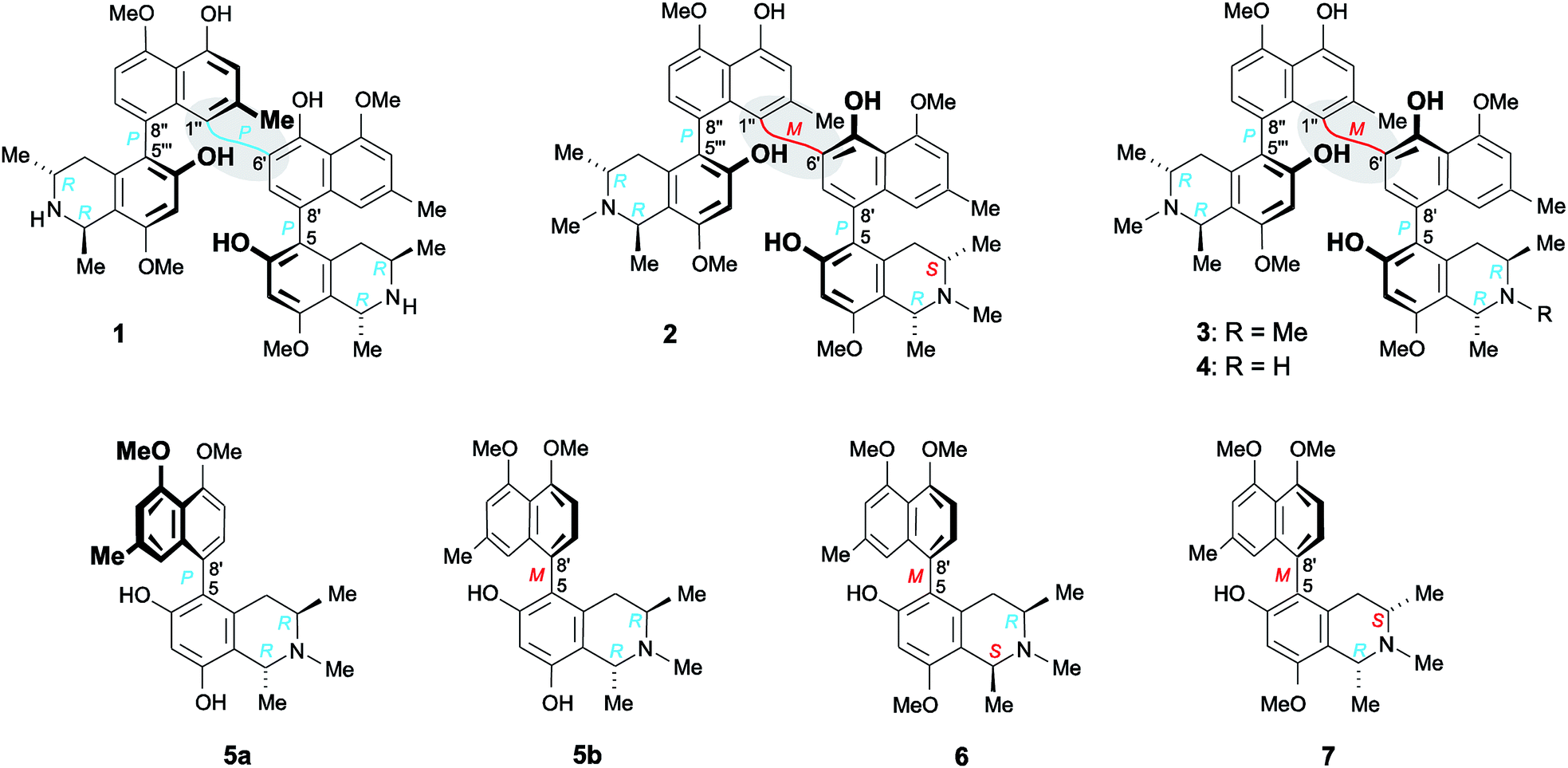 | ||
| Fig. 1 Mbandakamine-type dimers and related 5,8′-coupled monomeric naphthylisoquinolines from the leaves of an unexplored Congolese Ancistrocladus liana: the two previously known4,6 mbandakamines A (1) and B2 (2) and the two new mbandakamines B3 (3) and B4 (4), which were isolated along with four new alkaloids, ikelacongolines A–D (5a, 5b, 6, and 7). | ||
Here, we describe the isolation and structural elucidation of two new mbandakamine-type dimers, mbandakamines B3 (3) and B4 (4) (Fig. 1), from the leaves of an as yet unidentified Congolese Ancistrocladus liana. These two metabolites were found to co-occur in the plant with the mbandakamines A (1) and B2 (2). Such mbandakamine-type dimers have so far been known to be present only in two Ancistrocladus species, viz., in a botanically yet undescribed liana (three representatives)4,6 and in A. ealaensis (four examples).16 Both plants grow in the Mbandaka region.
Furthermore, we report on the identification of four new monomeric 5,8′-coupled naphthyltetrahydroisoquinolines, named ikelacongolines A–D (5a, 5b, 6, and 7) (Fig. 1), in the leaves of this undescribed Central Congolese Ancistrocladus liana, along with three known and likewise 5,8′-linked alkaloids previously detected in the two Central African species A. congolensis and A. likoko, ancistrocongoline C (8)28 and the ancistrolikokines C (9)29 and F2 (10)17 (Fig. 2). In addition, two naphthylisoquinoline alkaloids with a rare 7,8′-linkage, the ealamines A (11) and G (12)30 (Fig. 2), which had been isolated for the first time from the twigs and leaves of A. ealaensis only recently, have now been found to be part of the metabolite pattern of the here investigated liana.
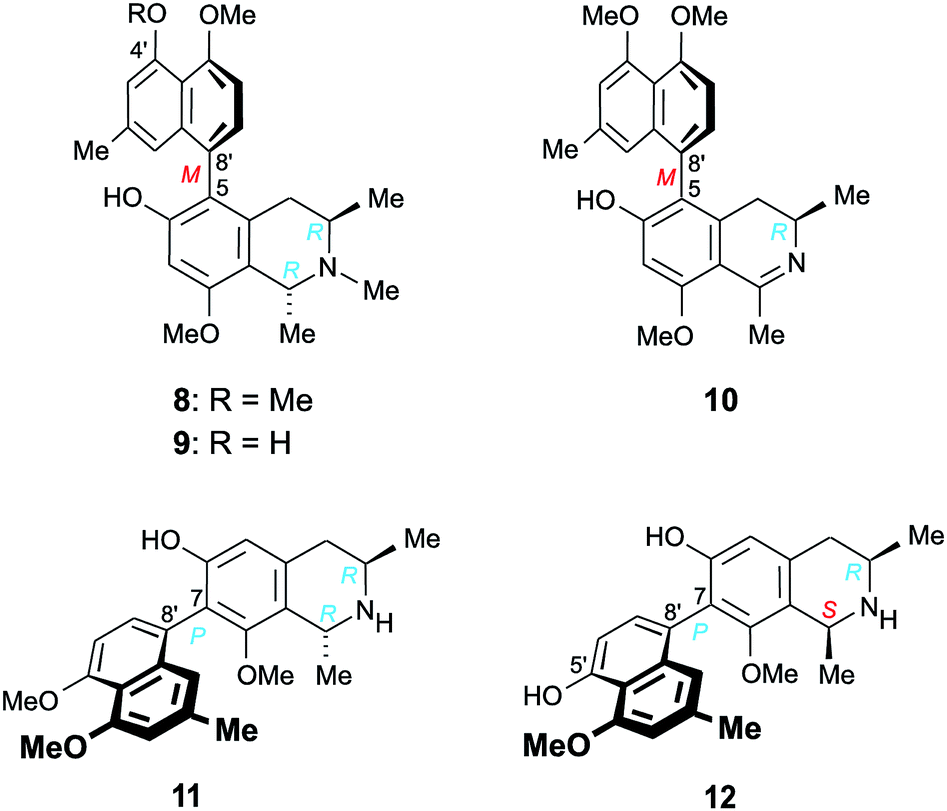 | ||
| Fig. 2 Five known17,28–30 5,8′- and 7,8′-coupled naphthylisoquinoline alkaloids isolated from the as yet unidentified Congolese Ancistrocladus liana: ancistrocongoline C (8), ancistrolikokines C (9) and F2 (10), and ealamines A (11) and G (12). | ||
As part of our ongoing studies on the antiparasitic potential of naphthylisoquinoline alkaloids, the mbandakamines B3 (3) and B4 (4) and the ikelacongolines A–C (5a, 5b, and 6) were tested against the pathogens causing malaria tropica, leishmaniasis, Chagas' disease, and African sleeping sickness.
Results and discussion
Isolation and structural elucidation of mono- and dimeric naphthylisoquinoline alkaloids
HPLC-UV-MS-guided analysis of leaf samples of a botanically yet undescribed Ancistrocladus liana from the Central Congo Basin, revealed the presence of secondary metabolites with UV and MS profiles typical of mono- and dimeric naphthylisoquinoline alkaloids. For the isolation of these compounds, ground material of air-dried leaves was exhaustively extracted with CH2Cl2–MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). The crude extract was suspended in H2O, and, after decantation and filtration, the solid was dissolved in MeOH. Fractionation and purification of the organic layer by reversed-phase HPLC provided six new naphthylisoquinoline alkaloids, along with seven known ones,4,6,16,17,28–30 which had already been isolated from other Congolese Ancistrocladus species before.
:1, v/v). The crude extract was suspended in H2O, and, after decantation and filtration, the solid was dissolved in MeOH. Fractionation and purification of the organic layer by reversed-phase HPLC provided six new naphthylisoquinoline alkaloids, along with seven known ones,4,6,16,17,28–30 which had already been isolated from other Congolese Ancistrocladus species before.
One of the monomeric compounds was readily identified as the 5,8′-linked ancistrocongoline C (8) (Fig. 2). This alkaloid had so far been known only from the Central African species A. congolensis.28 It is the 4′-O-methyl analog of the co-occurring alkaloid ancistrolikokine C (9), previously detected as a main constituent in the roots and twigs of A. likoko.17,29 These two N-methylated naphthyltetrahydroisoquinoline alkaloids have now been isolated along with ancistrolikokine F2 (10), which had been found for the first time in the twigs of A. likoko as one further example of the very small group of 5,8′-coupled naphthyldihydroisoquinolines with R-configuration at C-3.18
In addition, two known30 7,8′-linked alkaloids were detected in that unexplored Central African Ancistrocladus liana, viz., ealamines A (11) and G (12) (Fig. 2). They had previously been discovered in the leaves and twigs of the Congolese liana A. ealaensis.30 7,8′-Coupled compounds with R-configuration at C-3 have been detected in Ancistrocladus plants only rarely.15,30–32 Up to now, such an entity has been found in a total of just 13 (out of over 250) naphthylisoquinoline alkaloids, among them four dimers. Likewise noteworthy is the fact that these metabolites have so far been identified only in two species, namely in A. korupensis from Cameroon (three compounds)31,32 and in A. ealaensis (ten representatives).15,30
With their R-configuration at C-3 and an oxygen function at C-6, these five monomeric alkaloids 8–12 are typical ‘hybrid-type’, i.e., mixed Ancistrocladaceae/Dioncophyllaceae-type naphthylisoquinoline alkaloids.24 This finding is of special interest from a geo- and chemotaxonomic point of view. The metabolite profiles of most of the Ancistrocladus plants from the Northwestern Congo Basin are likewise dominated by the presence of such hybrid-type alkaloids.4–7,14–18,28–30 Asian and East African Ancistrocladaceae plants, by contrast, exclusively produce 3S-configured and 6-oxygenated compounds (known as ‘Ancistrocladaceae-type’ alkaloids),22–25 whereas the only other plant family that likewise contains naphthylisoquinolines, the West African Dioncophyllaceae, solely forms alkaloids with R-configuration at C-3, always lacking an oxygen function at C-6 (hence categorized as ‘Dioncophyllaceae-type’ alkaloids).24,25 Remarkably, from some of the species endemic to the Southern, Central, and Eastern Congo, metabolites exclusively belonging to the subclass of Ancistrocladaceae-type naphthylisoquinoline alkaloids were isolated,9–11 whereas other species from these regions mainly produce Dioncophyllaceae-type compounds, along with a certain number of Ancistrocladaceae- and hybrid-type alkaloids.8,20,21
Isolation work on the leaf extract of this phytochemically unexplored Central Congolese Ancistrocladus liana also furnished two known,4,6,16 constitutionally highly unsymmetric dimers with a 6′,1′′-coupled central biaryl axis, the mbandakamines A (1) and B2 (2) (Fig. 1). These structurally unusual quateraryls had already been identified as constituents of the above-mentioned as yet undescribed Congolese Ancistrocladus liana from the rainforests in the Mbandaka region in the Northwestern Congo Basin.4,6 Furthermore, only one further species, A. ealaensis, was known to likewise produce mbandakamine-type dimers, among them being mbandakamine A (1).16 This finding further supports the close chemotaxonomic relationship of that as yet unidentified Ancistrocladus plant from the Ikela region in Central Africa to other Congolese Ancistrocladus species, in particular to those indigenous to the Northwestern part of the Democratic Republic of the Congo.
Furthermore, six new mono- and dimeric naphthylisoquinoline alkaloids were discovered in the leaves of that Central Congolese Ancistrocladus liana.
The molecular formula of the first new alkaloid, obtained as a pale-yellow amorphous solid, was C50H56N2O8, as deduced from HRESIMS. It was, thus, identical to that of the co-occurring dimer mbandakamine B2 (2). The 1H and 13C NMR data (Table 1) of the isolated compound were almost identical to those of 2,6 except for some slight shift differences, again showing a full set of signals, which indicated that this new dimer was – similar to 2 – unsymmetric. The 1H NMR spectrum revealed the presence of eight aromatic protons, two methylene groups with diastereotopic protons, six methyl groups, two N-methyl functions, and four aromatic O-methyl groups. The ‘southeastern’ half of the new dimer displayed chemical shifts typical of an N-methylated naphthyltetrahydroisoquinoline with a 5,8′-biaryl linkage. It was established to be coupled via C-6′ (δC 136.2) in the naphthalene subunit with a second, ‘northwestern’ part, which likewise consisted of a naphthyl-1,3-dimethyl-N-methyltetrahydroisoquinoline of the 5,8′-coupling type.
| No. | 3 | 4 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1/1′′′ | 4.68, q (6.6)/4.55, q (6.8) | 58.9/58.1 | 4.70, q (6.7)/4.51, q (6.7) | 49.0/59.0 |
| 3/3′′′ | 3.95, m/3.67, m | 50.1/50.0 | 3.65, m/3.61, m | 44.7/50.1 |
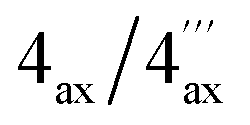 |
1.74, dd (18.2, 11.2)/1.84, dd | 32.2/29.3 | 1.75, dd (17.9, 11.4)/1.86, dd | 33.4/29.3 |
| (18.2, 11.8) | (17.9, 5.1) | |||
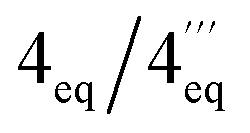 |
2.77, dd (18.3, 4.5)/1.84, dd | 2.84, dd (17.8, 4.7)/1.86, dd | ||
| (18.2, 5.1) | (17.9, 11.5) | |||
| 5/5′′′ | 119.5/123.0 | 119.7/122.9 | ||
| 6/6′′′ | 156.3/155.7 | 156.7/155.7 | ||
| 7/7′′′ | 6.65, s/5.64, s | 97.9/98.3 | 6.66, s/5.63, s | 98.0/98.3 |
| 8/8′′′ | 158.0/157.0 | 157.8/157.1 | ||
| 9/9′′′ | 115.6/112.4 | 114.8/112.3 | ||
| 10/10′′′ | 134.0/130.8 | 134.0/130.6 | ||
| 1′/1′′ | 6.56, s/— | 118.7/126.5 | 6.54, s/— | 118.8/126.5 |
| 2′/2′′ | 137.0/139.5 | 136.9/139.4 | ||
| 3′/3′′ | 6.81, s/6.87, s | 108.1/114.8 | 6.81, s/6.88, s | 108.1/114.8 |
| 4′/4′′ | 158.0/155.6 | 158.2/155.6 | ||
| 5′/5′′ | 152.6/158.2 | 152.6/158.2 | ||
| 6′/6′′ | —/7.01, d (8.1) | 136.2/104.8 | —/7.02, d (8.0) | 136.2/104.9 |
| 7′/7′′ | 6.83, s/6.97, d (8.0) | 132.8/132.8 | 6.83, s/6.97, d (8.0) | 132.7/132.8 |
| 8′/8′′ | 126.5/126.5 | 126.5/127.3 | ||
| 9′/9′′ | 137.0/137.9 | 136.9/137.9 | ||
| 10′/10′′ | 115.6/115.9 | 115.6/115.9 | ||
| 1-CH3/1′′′-CH3 | 1.49, d (6.5)/1.42, d (6.7) | 17.3/18.9 | 1.56, d (6.7)/1.42 (6.6) | 18.7/18.9 |
| 3-CH3/3′′′-CH3 | 1.08, d (6.4)/1.08, d (6.4) | 17.5/16.9 | 1.14, d (6.4)/1.08 (6.5) | 19.2/16.9 |
| N-CH3/N′′′-CH3 | 2.54, s/2.41, s | 35.7/34.7 | —/2.42, s | —/34.6 |
| 8-OCH3/8′′′-OCH3 | 3.93, s/3.42, s | 56.1/55.7 | 3.94, s/3.41, s | 56.2/55.7 |
| 2′-CH3/2′′-CH3 | 2.30, s/2.06, s | 22.1/22.7 | 2.29, s/2.07, s | 22.1/22.7 |
| 4′-OCH3 | 4.06, s | 57.0 | 4.06, s | 57.0 |
| 5′′-OCH3 | 4.16, s | 56.9 | 4.16, s | 56.9 |
The first, ‘southeastern’ molecular portion (Fig. 3B) was found to possess four aromatic protons, with two singlets (δH 6.65 and 6.83) and a two-proton spin system in the naphthalene part exhibiting a meta-coupling pattern (δH 6.56 and 6.81). In the aliphatic region, it exhibited a quartet [H-1, (δH 4.68)], a multiplet [H-3, (δH 3.95)], two diastereotopic protons, H-4ax (δH 1.74) and H-4eq (δH 2.77), and two O-methyl groups (δH 3.93 and 4.06). In that part of the 1H NMR spectrum, also two three-proton doublets were monitored, typical of Me-1 (δH 1.49) and Me-3 (δH 1.08), and two three-proton singlets, one corresponding to an aryl-methyl group {Me-2′, (δH 2.30)} in the naphthalene part, and the other one evidencing the presence of an N-methyl group (δH 2.54) in the isoquinoline subunit (Table 1). This finding was confirmed by HMBC interactions of the methyl protons with both, C-1 and C-3 (Fig. 3B). The location of the biaryl linkage at C-8′ was established from an HMBC cross peak between H-1′ (δH 6.56) and the quaternary carbon atom C-8′ (δC 126.5). The attribution of H-1′ was, in turn, corroborated by a NOESY interaction with H-4ax and by the NOESY correlation sequence {H-1′ ↔ Me-2′ ↔ H-3′ ↔ OMe-4′}. In the tetrahydroisoquinoline portion, the coupling position was deduced to be C-5 (δC 119.5), as obvious from HMBC interactions from H-7 (δH 6.65), H-7′ (δH 6.83), and H-4eq to C-5. The two methoxy groups at δH 3.93 and 4.06 showed NOESY interactions with H-7 and H-3′ (δH 6.81), respectively, thus being located at C-8 in the isoquinoline subunit and at C-4′ in the naphthalene part. Consequently, the remaining two oxygen functions had to be free hydroxy groups at C-6 and C-5′ (Fig. 3B).
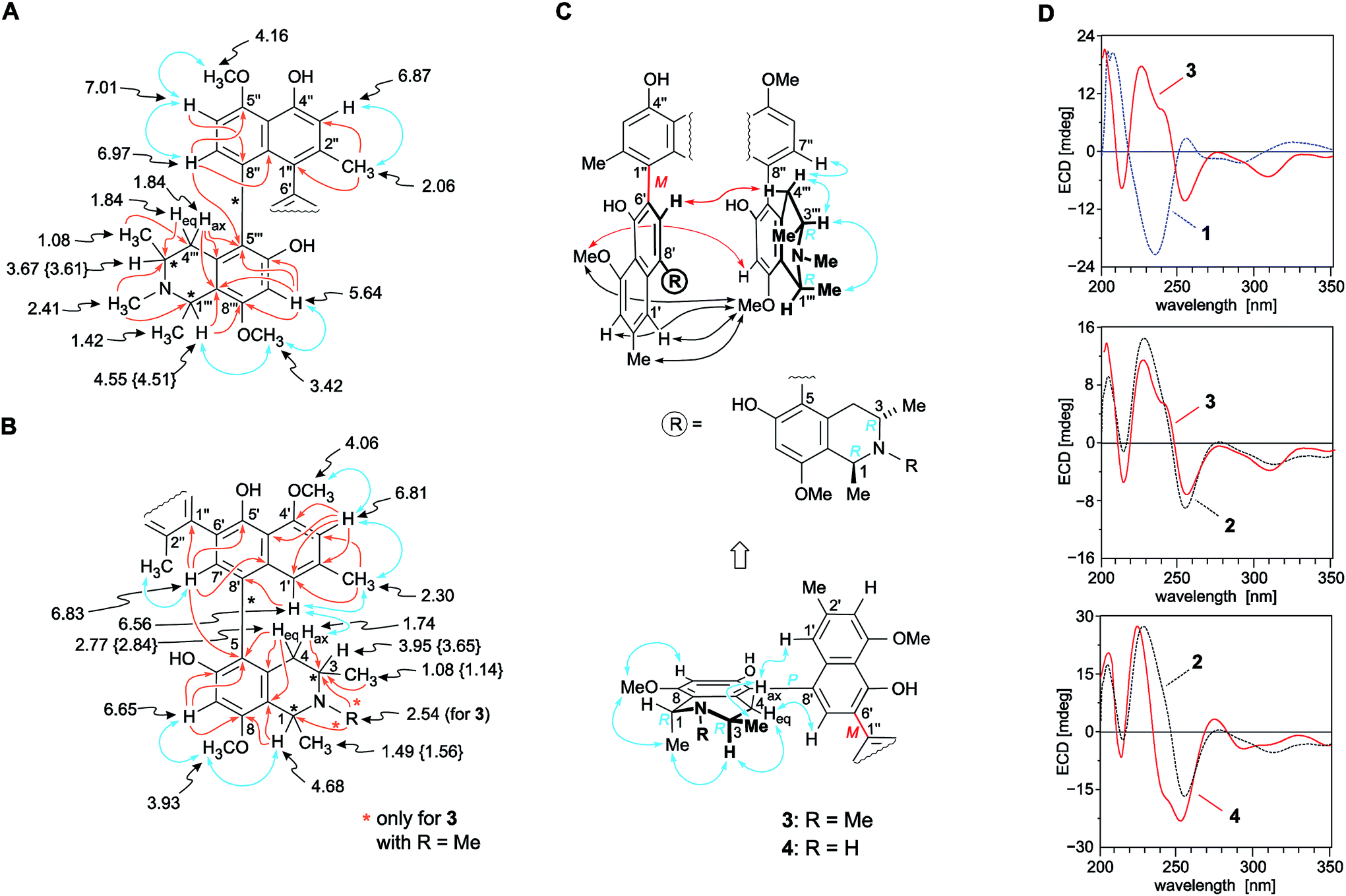 | ||
| Fig. 3 Selected 1H NMR data (in methanol-d4, δ in ppm), HMBC (single orange arrows) and NOE (double blue arrows) interactions indicative of the constitutions of the (A) ‘northwestern’ and (B) ‘southeastern’ naphthylisoquinoline portions of the mbandakamines B3 (3) and B4 (4): the values of 4 that are different from those of 3 are given in {}. All other NMR data of 3 and 4 are nearly identical (±0.02 ppm); (C) selected ROESY correlations between the naphthalene subunit of the ‘northwestern’ half and the isoquinoline part of the ‘southeastern’ moiety establishing the 6′,1′′-coupling sites in the central binaphthalene core (black arrows) and the relative configuration at the centers and the two outer axes (blue arrows) and at the central biaryl axis (red arrows) of 3 and 4; (D) confirmation of the absolute configuration of 3 and 4 at the central biaryl axis by comparison of their ECD spectra with those of the known4,6,16 related mbandakamines A (1, central axis: P) and B2 (2, central axis: M): in the case of 3 with the ECD curves of 1 (top) and 2 (center), and, in the case of 4, with the ECD spectrum of 2 (bottom); for the structures of 1 and 2, see Fig. 1. | ||
The 1H NMR spectrum (Table 1) of the northwestern molecular portion again displayed the typical chemical shifts of a 5,8′-coupled N-methylated naphthyltetrahydroisoquinoline with a methoxy group (δH 3.42) at C-8′′′ (Fig. 3A). This was confirmed by NOESY correlations in the series {H-7′′′ ↔ OMe-8′′′ ↔ Me-1′′′ ↔ H-3′′′}, and by HMBC interactions from H-7′′ (δH 6.97), Heq-4′′′ (δH 1.84), and H-7′′′ (δH 5.64) to C-5′′′ (δC 123.0), evidencing the coupling position of the biaryl axis in the isoquinoline subunit to be located at C-5′′′. In contrast to the naphthalene part of the southeastern half, the signal pattern in the 1H NMR spectrum showed an ortho coupling of the two aromatic protons H-7′′ and H-6′′ (δH 7.01), no meta-coupled doublets next to the high-field shielded methyl group (δH 2.06) at C-2′′, but only a singlet at H-3′′ (δH 6.87), thus suggesting C-1′′ and C-8′′ to be axis-bearing carbon atoms. ROESY correlations between H-6′′ and MeO-5′′ (δH 4.16) revealed the naphthalene unit of the northwestern naphthylisoquinoline to possess a methoxy function at C-5′. HMBC interactions from H-7′′ to C-5′′ (δC 158.2) and C-9′′ (δC 137.9) and from H-6′′ to C-8′′ established C-8′′ to be quaternary and, thus, proving that the isoquinoline and the naphthalene portions of the northwestern molecular half were coupled through C-5′′′ and C-8′′ (Fig. 3A). The proximity of a second aryl substituent to the methyl group (δH 2.06) of the naphthalene subunit in the northwestern part of the dimer became evident from the strongly upfield-shifted signal of Me-2′′, which revealed the central biaryl axis to connect the two molecular halves of the isolated alkaloid by a 6′,1′′-coupling of the two naphthalene subunits. This assignment was further proven by 3J HMBC interactions from H-3′′, Me-2′′, and H-7′ to C-1′′ (Fig. 3A and B), and from ROESY interactions between H-1′, Me-2′, H-3′, and MeO-4′ in the naphthalene unit of the northwestern naphthylisoquinoline and OMe-8′′′ in the isoquinoline portion of the southeastern half of the dimer (Fig. 3C). Thus, the central axis was located at C-1′′, i.e., in the peri-position neighboring one of the outer axes (i.e., C-8′′), which led to an extremely high steric hindrance around that 6′,1′′-linked central biaryl axis.
ROESY cross peaks of the protons of Me-1 (δH 1.49) to H-3 (δH 3.95), and of Me-1′′′ (δH 1.42) to H-3′′′ (δH 3.67) assigned the relative configuration of the methyl group at C-1 versus the one at C-3, and that at C-1′′′ versus the one at C-3′′′, to be trans to each other in both cases (Fig. 3C). The absolute configurations at C-3 and C-3′′′ in the two tetrahydroisoquinoline portions were established by ruthenium-mediated oxidative degradation with subsequent GC-MSD analysis of the resulting chiral products according to a procedure developed by our group earlier.33 In the case of the isolated alkaloid dimer, this analytical method delivered only the R-enantiomers of 3-aminobutyric acid and of N-methyl-3-aminobutyric acid (both derived from C-3), thus, together with the relative trans-configuration assigned above, the absolute configurations at C-3, C-3′′′, C-1, and C-1′′′ were attributed to be all R. The configurations at the two outer axes in the monomeric halves of the new dimer relative to the stereocenters became evident from specific long-range ROESY interactions between H-1′ and H-4ax and between H-7′ and H-4eq in the southeastern portion and between H-7′′ and 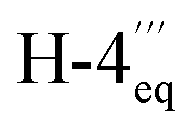 in the northwestern moiety (Fig. 3C). This, in combination with the absolute configurations at C-1 and C-3 and at C-1′′′ and C-3′′′, as again determined by the oxidative degradation, clearly showed that the naphthalene–isoquinoline linkages both had to be P-configured.
in the northwestern moiety (Fig. 3C). This, in combination with the absolute configurations at C-1 and C-3 and at C-1′′′ and C-3′′′, as again determined by the oxidative degradation, clearly showed that the naphthalene–isoquinoline linkages both had to be P-configured.
ROESY interactions were found across the central biaryl axis between H-7′ and Hax-4′′′ and between OMe-4′ and H-7′′′ (Fig. 3C), which were identical to those earlier observed for mbandakamine B2 (2),6 but complementary to those in mbandakamine A (1).4 These interactions established the central biaryl axis of the new dimer to be M-configured.
This assignment was confirmed by the fact that the ECD spectrum of the new metabolite was virtually identical to that of the structurally closely related mbandakamine B2 (2) (Fig. 3D, center), but nearly mirror-imaged to that of mbandakamine A (1) (Fig. 3D, top), which is P-configured at the central biaryl axis. Previous investigations4,8,16,21,34 had already revealed that the chiroptical properties of such naphthylisoquinoline dimers are dominated by the orientation of the two strong naphthalene chromophores to each other, and, thus, by the configuration at the central biaryl linkage, although – as in the case of 1 and the new dimer – the stereoarrays at all the other six stereogenic elements (two outer axes and four centers) were identical. Consequently, the new mbandakamine-type dimer isolated from the ‘new’ Central Congolese Ancistrocladus species had to possess the full stereostructure 3, and – as displayed in Fig. 1 – to be 1R,3R,5P,6′M,5′′′P,3′′′R,1′′′R-configured. It was, hence, the 3-epi-analog of the co-occurring mbandakamine B2 (2). According to its close structural relationship to 2, the new dimer 3 was given the name mbandakamine B3.
Resolution of a further alkaloid-enriched subfraction of the leaf extract provided a second new dimeric naphthylisoquinoline alkaloid. In 1H and 13C NMR analysis the compound showed full sets of signals (Table 1) similar to those of 3, again hinting at the presence of a mbandakamine-type dimer consisting of two 5,8′-coupled naphthyltetrahydroisoquinoline halves connected to each other via C-6′ and C-1′′. According to HRESIMS, the new compound possessed a molecular formula of C49H54N2O8, suggesting the presence of an O- or N-demethylated derivative of 3. The 1D and 2D NMR data clearly revealed the missing methyl group to be the one attached to the nitrogen atom in the southeastern portion of mbandakamine B3 (3) (Table 1 and Fig. 3B), which was confirmed by the upfield shifts of C-1 (δC 49.0) and C-3 (δH 44.7) compared to those in 3 (δC 58.9 and 50.1, respectively).
By the above-employed methods – the oxidative degradation, NMR investigations showing specific NOESY and ROESY interactions across the two outer biaryl axes and between the isoquinoline subunit of the southeastern molecular half and the naphthalene portion of the northwestern molecular moiety (Fig. 3B), and ECD spectroscopy (Fig. 3C, bottom) – the new dimer was assigned to be R-configured at all four stereogenic centers, P-configured at the two outer axes, and M at the central biaryl linkage, similar to mbandakamine B3 (3). Thus, this second new dimer had the absolute stereostructure 4 as shown in Fig. 1. It was henceforth named mbandakamine B4.
In addition to these two mbandakamine-type dimers 3 and 4, four new monomeric 5,8′-coupled naphthylisoquinolines were isolated from the leaf extract. According to the 1H and 13C NMR data (Tables 2 and 3), these alkaloids showed a great constitutional similarity, having in common a 4′,5′-dimethoxynaphthalene portion and an N-methyl function in their isoquinoline halves, but clearly differing by the OH/OMe substitution patterns of their tetrahydroisoquinoline subunits, and by their elements of central and axial chirality.
| No. | 5a | 5b | 6 | 7 |
|---|---|---|---|---|
| 1 | 4.72, q (6.7) | 4.72, q (6.7) | 4.65, q (6.6) | 4.64, q (6.7) |
| 3 | 3.96, m | 3.92, m | 3.15, m | 3.23, m |
| 4ax | 2.03, dd (18.6, 11.9) | 2.49, dd (18.6, 12.2) | 2.58, dd (17.3, 11.5) | 2.23, dd (17.3, 11.0) |
| 4eq | 2.61, dd (18.5, 4.9) | 2.20, dd (18.8, 5.0) | 2.21, dd (17.3, 2.9) | 2.45, dd (17.3, 3.4) |
| 7 | 6.50, s | 6.48, s | 6.60, s | 6.62, s |
| 1′ | 6.65, s | 6.77, s | 6.69, s | 6.73, s |
| 3′ | 6.78, s | 6.79, s | 6.80, s | 6.79, s |
| 6′ | 6.95, d (8.0) | 6.93, d (8.0) | 6.94, d (8.0) | 6.94, d (8.0) |
| 7′ | 7.18, d (7.9) | 7.15, d (7.9) | 7.16, d (7.9) | 7.17, d (8.0) |
| 1-CH3 | 1.72, d (6.7) | 1.74, d (7.9) | 1.73, d (6.6) | 1.75, d (6.6) |
| 3-CH3 | 1.24, d (6.6) | 1.24, d (6.6) | 1.27, d (6.5) | 1.28, d (6.5) |
| N-CH3 | 2.72, s | 2.77, s | 3.02, s | 3.01, s |
| 8-OCH3 | 3.92, s | 3.92, s | ||
| 2′-CH3 | 2.30, s | 2.32, s | 2.31, s | 2.32, s |
| 4′-OCH3 | 3.93, s | 3.93, s | 3.93, s | 3.93, s |
| 5′-OCH3 | 3.95, s | 3.95, s | 3.96, s | 3.96, s |
| No. | 5a | 5b | 6 | 7 |
|---|---|---|---|---|
| 1 | 60.0 | 60.1 | 62.1 | 61.7 |
| 3 | 50.8 | 50.8 | 60.3 | 59.9 |
| 4 | 29.4 | 29.9 | 34.0 | 33.1 |
| 5 | 119.7 | 119.6 | 119.9 | 120.0 |
| 6 | 157.2 | 157.2 | 157.5 | 157.2 |
| 7 | 102.6 | 102.6 | 99.0 | 99.3 |
| 8 | 156.4 | 156.4 | 157.2 | 157.5 |
| 9 | 110.9 | 111.0 | 114.3 | 114.0 |
| 10 | 132.2 | 132.1 | 134.7 | 134.7 |
| 1′ | 118.0 | 118.4 | 118.3 | 118.3 |
| 2′ | 138.2 | 138.0 | 137.9 | 137.9 |
| 3′ | 110.0 | 110.1 | 109.9 | 109.8 |
| 4′ | 159.0 | 158.8 | 158.7 | 158.8 |
| 5′ | 158.4 | 158.4 | 158.4 | 158.4 |
| 6′ | 107.0 | 106.9 | 106.6 | 106.6 |
| 7′ | 130.4 | 130.8 | 130.9 | 130.4 |
| 8′ | 126.6 | 126.7 | 126.1 | 126.0 |
| 9′ | 138.0 | 137.8 | 137.5 | 138.1 |
| 10′ | 117.8 | 117.8 | 117.4 | 117.6 |
| 1-CH3 | 19.1 | 19.3 | 19.8 | 20.0 |
| 3-CH3 | 16.7 | 16.9 | 17.9 | 17.9 |
| N-CH3 | 34.2 | 34.2 | 41.4 | 41.4 |
| 2′-CH3 | 22.2 | 22.2 | 22.0 | 22.1 |
| 8-OCH3 | 56.1 | 56.1 | ||
| 4′-OCH3 | 57.1 | 57.1 | 56.9 | 56.9 |
| 5′-OCH3 | 56.9 | 56.9 | 56.7 | 56.7 |
The first new compound within this series of monomeric alkaloids was obtained as a yellow amorphous solid. It corresponded to a molecular formula of C25H29NO4, as deduced from HRESIMS. Given a spin system of five aromatic protons, with three singlets (δH 6.50, 6.65, and 6.78) and two doublets due to the presence of a pair of adjacent protons (δH 6.95 and 7.18) in the naphthalene part (Fig. 4A), this substitution pattern hinted either at a 6′- or an 8′-coupling site of the biaryl axis. HMBC interactions from H-1′ (δH 6.65) and H-6′ (δH 6.95) to the quaternary carbon atom C-8′ (δC 126.6) in combination with NOESY correlations in the series {H-1′ ↔ Me-2′ ↔ H-3′ ↔ OMe-4′} and a cross peak between H-6′ and H-7′ (δH 7.18) evidenced the tetrahydroisoquinoline moiety to be located at C-8′ (Fig. 4A). The high-field shifted signals of the diastereotopic protons at C-4 (δH 2.03 and 2.61) suggested the naphthalene half to be located at C-5 of the isoquinoline part. This assignment was confirmed by HMBC interactions of H-7′, H-7 (δH 6.50), and Heq-4 (δH 2.61) to C-5 (δC 119.7) (Fig. 4A). The 1H NMR spectrum showed the chemical shifts of two methoxy functions (δ 3.93 and 3.95), which were attributed to be located at C-4′ and C-5′ in the naphthalene subunit due to NOESY interactions to H-3′ and H-6′, respectively. The two other oxygen functions thus had to be free phenolic hydroxy groups at C-6 and C-8 in the isoquinoline part. From the 1H NMR signal at δH 2.72, corresponding to three protons, and from the HMBC interactions of this methyl group with both C-1 and C-3, it was evident that the new compound was N-methylated.
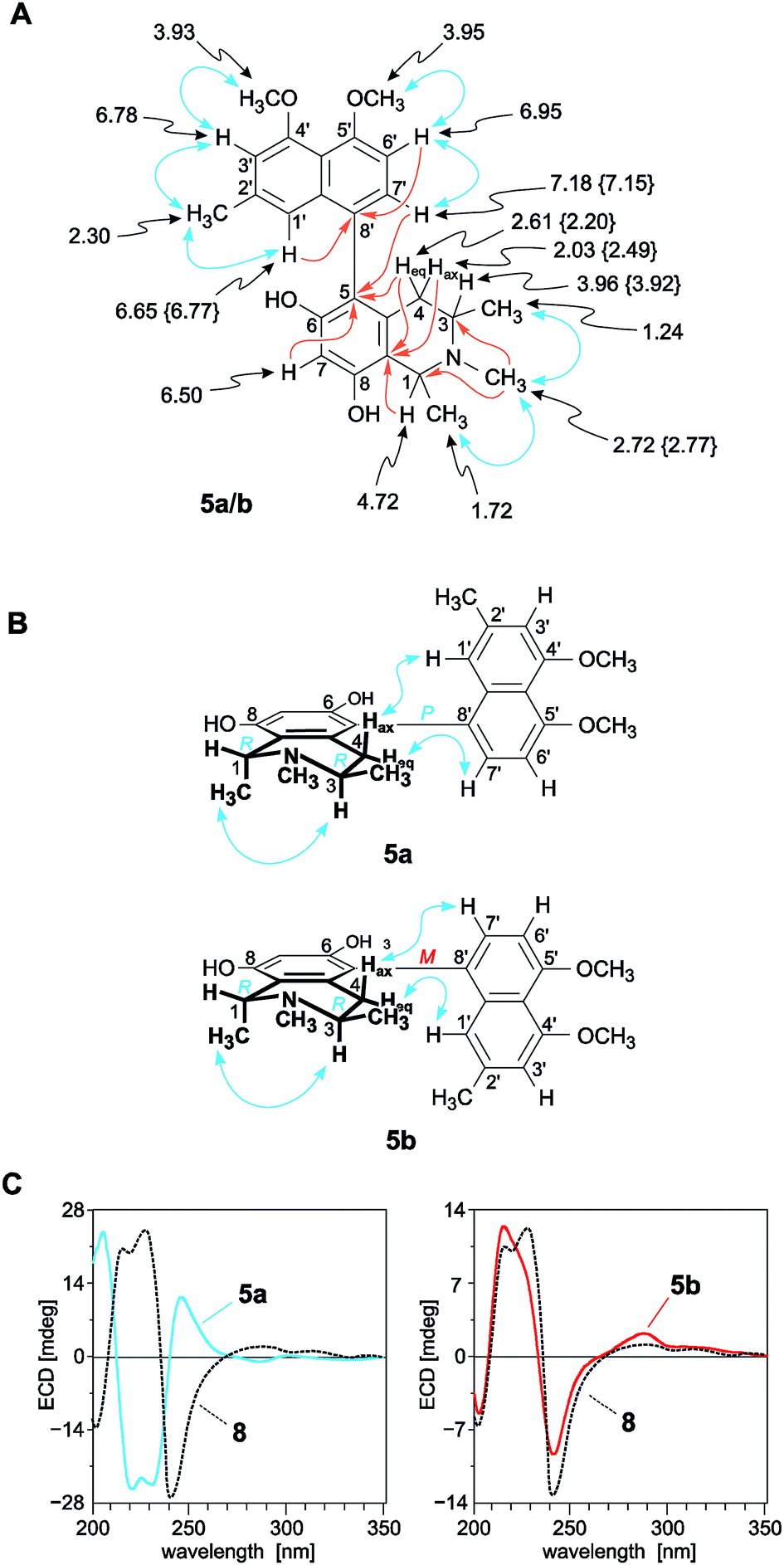 | ||
| Fig. 4 (A) 1H NMR data (in methanol-d4, δ in ppm), selected HMBC (single orange arrows) and NOE interactions (double blue arrows) of ikelacongolines A (5a) and B (5b): the values of 5b that are different from those of 5a are given in {}. All other values of 5a and 5b are nearly identical (±0.02 ppm); (B) NOESY correlations evidencing the relative configuration of 5a and 5b at the biaryl axis and at the stereogenic centers C-1 and C-3 in the tetrahydroisoquinoline subunit; (C) assignment of the absolute axial configuration of 5a and 5b, by comparison of their ECD spectra with that of the known,28 co-occurring and structurally related alkaloid ancistrocongoline C (8); for the structure of 8, see Fig. 1. | ||
From a NOESY correlation between H-3 (δH 3.96) and Me-1 (δH 1.72) (Fig. 4B, top), a relative trans-configuration of the two stereocenters at C-1 and C-3 was established. The absolute configurations, determined by ruthenium-mediated oxidative degradation,33 were found to be R at both stereocenters. The relative and, thus, absolute axial configuration was deduced to be P, according to NOESY interactions between H-4ax (δH 2.03) and H-1′ on the one hand, and between H-4eq (δH 2.61) and H-7′ on the other (Fig. 4B). The results were in agreement with the ECD spectrum of the new metabolite, which was opposite to that of the known28 (and co-occurring) 5,8′-coupled, but M-configured alkaloid ancistrocongoline C (8) (Fig. 4C, left). Thus, the new compound possessed the stereostructure 5a as presented in Fig. 1. It was named ikelacongoline A, after the Congolese town of Ikela, where the plant material had been collected. This alkaloid may also be referred to as the N-methylated analog of korupensamine C from the Cameroonian liana A. korupensis,26 or as the 5′-O-methyl derivative of ancistrocongoline A,17,28 a main metabolite of both of the two Congolese species A. congolensis and A. likoko.
The second new monomeric alkaloid had the same molecular formula of C25H29NO4 as ikelacongoline A (5a), which was evidenced by HRESIMS measurements. It was again a 5,8′-linked N-methylated naphthyltetrahydroisoquinoline alkaloid, with 1H and 13C NMR data almost identical to those of 5a, suggesting that the isolated compound possessed the same constitution as 5a. Significant differences in the NMR spectra of these two alkaloids were monitored only for the chemical shifts of the diastereotopic protons Hax-4 (δH 2.49) and Heq-4 (δH 2.20) (see Fig. 4A and Table 2) located next to the biaryl axis, thus hinting at the presence of two probably atropo-diastereomeric alkaloids in the leaves of this ‘new’ Ancistrocladus plant.
NOESY cross peaks of the protons of Me-1 (δH 1.74) to H-3 (δH 3.92) established the methyl groups at C-1 and C-3 to be trans to each other (Fig. 4B, bottom). The oxidative degradation procedure again furnished only the R-enantiomers of 3-aminobutyric acid and N-methyl-3-aminobutyric acid. In conjunction with the relative trans-configuration, the absolute configuration of the tetrahydroisoquinoline subunit had to be 1R,3R, similar as in the case of 5a. Given the identical constitution and the same configurations at C-1 and C-3, 5a and the new metabolite had to differ only by an opposite axial configuration. Long-range NOESY correlations between Hax-4 and H-7′ and between H-4eq and H-1′ (Fig. 4B, bottom) clearly revealed the new compound to be M-configured at the biaryl linkage. This finding was confirmed by the fact that the ECD spectrum was virtually opposite to that of 5a, but nearly identical to that of the likewise M-configured and 5,8′-coupled ancistrocongoline C (8) (Fig. 4C, left). Consequently, the new alkaloid possessed the stereostructure 5b and was hence the atropo-diastereomer of 5a. It was named ikelacongoline B.
According to HRESIMS analysis, the third new alkaloid had a molecular formula of C26H31NO4, thus possessing 14 mass units more than 5a and 5b. Similar to the latter, the compound again displayed NMR signals (see Tables 2 and 3) typical of an N-methylated 5,8′-coupled naphthyltetrahydroisoquinoline with two methoxy groups in the naphthalene part (δH 3.93 and 3.96). Unlike 5a and 5b, this alkaloid possessed a third methoxy function (δH 3.92) at C-8 in the isoquinoline portion as deduced from NOESY interactions between H-7 (δH 6.60) and OMe-8.
From an NOE correlation between H-1 (δH 4.65) and H-3 (δH 3.15) (Fig. 5A), the relative configuration at C-1 versus C-3 was established to be cis. Ruthenium-mediated oxidative degradation yielded 3-aminobutyric acid and its N-methyl derivative as their R-enantiomers, thus clearly establishing C-3 to be R-configured. In view of the above-assigned cis-array of the two methyl groups at C-1 and C-3, the absolute configuration of the stereogenic center at C-1 was deduced to be S.
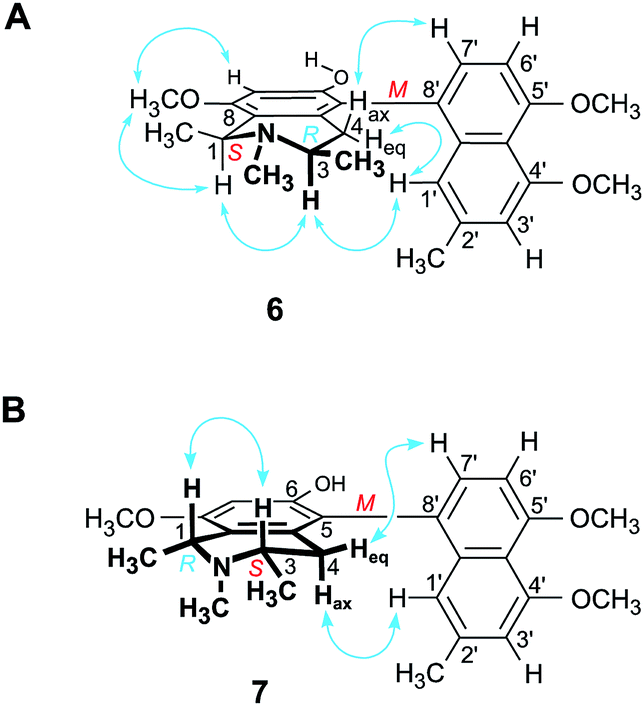 | ||
| Fig. 5 NOESY interactions indicative of the configuration at the biaryl axis of (A) ikelacongoline C (6) and (B) ikelacongoline D (7) relative to the stereogenic centers in the tetrahydroisoquinoline subunit. | ||
Based on the long-range NOESY interactions from H-4ax (δH 2.58) to H-7′ (δH 7.16), and from H-1′ (δH 6.69) to H-4eq (δH 2.21) and H-3, the axial configuration was determined to be M (Fig. 5A). The assignment was further corroborated by the fact that the ECD spectrum of the isolated alkaloid matched the ECD curves of the two related, likewise M-configured metabolites ikelacongoline B (5b) and ancistrocongoline C (8) nearly perfectly (see ESI†). This monomeric naphthylisoquinoline thus possessed the absolute stereostructure 6, as presented in Fig. 1, and, hence, was the new 1-epi analog of 8. It was named ikelacongoline C.
According to HRESIMS, the fourth isolated new metabolite had the same molecular formula of C26H31NO4 as the above-investigated ikelacongoline C (6). Moreover, 1D and 2D NMR measurements hinted at a great structural similarity with 6, indicating that the two alkaloids differed only in the stereochemical orientation of the methyl groups at C-1 and C-3. The formation of S-N-methyl-3-aminobutyric acid by oxidative degradation proved the new alkaloid to be S-configured at C-3 (instead of R as assigned for 6). The relative configuration of C-1 versus C-3 was again deduced to be cis, due to a NOESY interaction of H-1 (δH 4.64) with H-3 (δH 3.23), which was axial, as obvious from the coupling constants of Hax-4 [δH 2.23, (1H, J = 17.3, 11.0 Hz)] and Heq-4 [δH 2.45, (1H, J = 17.3, 3.4 Hz)]. Thus, given the relative cis-configuration at C-1 and C-3, the absolute configuration at C-1 was determined to be R. On the basis of the resulting 1R,3S-stereoarray in the tetrahydroisoquinoline part, the absolute axial configuration was assigned to be M due to long-range NOE cross peaks between H-7′ (δH 7.17) in the naphthalene subunit and Heq-4 in the isoquinoline portion, and also between H-1′ (δH 6.73) and Hax-4 (Fig. 5B). Comparison of the ECD spectrum of the isolated alkaloid with that of 6, which was similar (see ESI†), further confirmed this new metabolite to possess the stereostructure 7, as shown in Fig. 1. The compound was named ikelacongoline D. With its constitution being identical to that of ancistroyafungine A,9 an alkaloid isolated earlier from a botanically likewise as yet unidentified Ancistrocladus liana from the North-Central part of the Congo Basin, and with the same configuration at C-1 and C-3, but with M-configuration at the biaryl axis, ikelacongoline D might also be addressed as 5-epi-ancistroyafungine A.
The alkaloid pair 6/7 is most remarkable because their tetrahydroisoquinoline parts are enantiomeric to each other (1S,3R versus 1R,3S), while the axes are identically configured. The occurrence of such a pair of naphthylisoquinolines within one plant is as yet unprecedented.
Antiprotozoal activities of the mbandakamines B3 and B4 and the ikelacongolines A–C
Parasitic diseases such as malaria, African sleeping sickness, Chagas' disease, and leishmaniasis are still a major threat for millions of people in tropical and developing countries and, as a consequence, represent a serious obstacle to the social-economic progress in affected regions of Asia and Africa. The dramatic increase of parasite resistance to routinely used chemotherapeutics and the absence of vaccines aggravate the situation dramatically.35–37 For this reason, several research groups from medicinal chemistry, pharmacy, and infection biology focus on the search of new effective agents for the treatment of tropical infectious diseases. In the recent years, intense phytochemical studies on African plants have led to the discovery of terpenoids, alkaloids, flavonoids, quinones, lignans, coumarins, and many other natural products displaying excellent antiprotozoal activities,38–45 thus providing templates and new scaffolds for the development of a new generation of more potent drugs. Most promising sources for such lead compounds are Ancistrocladus species from Central Africa. Some of the known4–6,8,10,11,15,16,20,21,25,28,29,46,47 mono- and dimeric naphthylisoquinolines exhibited pronounced inhibitory activities against Plasmodium, Trypanosoma, and Leishmania parasites, thus suggesting to likewise investigate the alkaloids of this Central Congolese liana presented here for their biological potential.The two newly discovered mbandakamine-type naphthylisoquinoline dimers 3 and 4 exhibited excellent activities in vitro against both the chloroquine-sensitive (NF54) and the chloroquine-resistant (K1) strain of the malaria parasite Plasmodium falciparum, with half-maximum inhibition concentration values (IC50) of 39 nM (NF54) and even 6 nM (K1) for 3, and 66 nM (NF54) and 26 nM (K1) for 4 (Table 4). Thus, their antiplasmodial activities were comparable to those of the structurally closely related – and likewise strongly active – mbandakamine B2 (2),6 and were in the same order of magnitude as those of the standard agents chloroquine and artemisinin (Table 4). Noteworthy was in particular the pronounced antiplasmodial activities of 2–4 especially against the chloroquine-resistant K1 strain, without showing any cross-resistance. The new mbandakamine B3 (3) was found to be one of the most active naphthylisoquinolines against the chloroquine-resistant strain K1 ever investigated. In addition, given the comparatively low cytotoxicity of 3 (6.64 μM), this dimer possessed a high selectivity index of ca. 1110. These findings made the mbandakamines B3 (3) and B4 (4) promising candidates for further biological evaluations, also with respect to the fact that they displayed good to strong activities against the pathogen of African sleeping sickness, Trypanosoma brucei rhodesiense, with IC50 values of 0.476 μM for 3 and 0.211 μM for 4 (Table 4). Their inhibitory potentials, however, were slightly weaker than that of mbandakamine B2 (2), which showed a pronounced effect in vitro in the lower nanomolar range (IC50 = 5.0 nM),6 stronger than that of any other of the numerous alkaloids tested so far.4–8,10,15,16,20,21,25,46,47
| Compound | IC50a [μM] | ||||||
|---|---|---|---|---|---|---|---|
| P. falciparum (K1 strain) | P. falciparum (NF54 strain) | T. cruzi | T. brucei rhodesiense | L. donovani | L6 cell (cytotoxicity) | Selectivity indexb (K1/NF54) | |
| a The IC50 values are the means of two independent assays; the individual values vary by a factor of less than 2.b The selectivity index is calculated as the ratio of the IC50 values concerning L6 cells to the IC50 data relative to P. falciparum.c Chloroquine.d Artemisinin.e Benznidazole.f Melarsoprol.g Miltefosine.h Podophyllotoxin.i Values reported earlier, see ref. 6. | |||||||
| Standard | 0.293c | 0.009c | 6.109e | 0.030f | 1.077g | 0.0096h | —/—b |
| 0.010d | 0.021d | ||||||
| 2i | 0.004i | 0.017i | 2.98i | 0.005i | 94.2i | 1.37i | 342/80.6 |
| 3 | 0.006 | 0.039 | 15.1 | 0.476 | 65.5 | 6.64 | 1107/170 |
| 4 | 0.026 | 0.066 | 19.7 | 0.211 | 59.5 | 6.68 | 257/101 |
| 5a | 2.87 | 6.38 | 130 | 47.2 | >100 | 182 | 63.4/28.5 |
| 5b | 2.75 | 8.10 | 72.2 | 15.3 | >100 | 60.5 | 22.0/7.47 |
| 6 | 1.41 | 4.51 | 92.9 | 15.1 | >100 | 49.9 | 35.4/11.1 |
The inhibitory effects of the new monomeric alkaloids ikelacongolines A (5a), B (5b), and C (6) against the strains NF54 and K1 of P. falciparum were drastically lower compared to those of the dimers 2–4. For lack of material, ikelacongoline D (7) was not tested. The compounds 5a, 5b, and 6 displayed only moderate antiplasmodial activities, with micromolar IC50 values ranging from 1.41 to 8.10 (Table 4), and thus being 115 to 480 times less effective than the mbandakamines 2–4.
These test results clearly demonstrated that the dimeric architecture of the mbandakamines had a significant impact on the bioactivity of 5,8′-linked naphthylisoquinoline alkaloids. A similar effect had previously also been observed for jozimine-type naphthylisoquinoline dimers of natural8,21 and synthetic48–50 origin consisting of two 7,1′-coupled – far less active – monomeric halves.
Against Leishmania donovani, the causative agent of visceral leishmaniasis, only very weak or virtually no activities were recorded for any of the investigated naphthylisoquinolines, while for the pathogen of Chagas' disease, Trypanosoma cruzi, a low, but unspecific inhibitory effect (SI < 1) was observed for the mbandakamines 2–4 (Table 4).
Experimental
General experimental procedures
Optical rotations were determined on a Jasco P-1020 polarimeter operating with a sodium light source (λ = 589 nm). IR spectra were recorded on a Jasco FT/IR-410 spectrometer, UV spectra on a Shimadzu UV-1800 spectrophotometer, and ECD measurements were performed on a Jasco J-715 spectropolarimeter. The ECD data are reported in Δε values (cm2 mol−1) at the given wavelength λ (nm), they were processed using SpecDis.51,52 NMR spectra (600 MHz for 1H and 150 MHz for 13C) were taken on a Bruker DMX 600 instrument, using methanol-d4, with the 1H or 13C signals of methanol (1H, δ 3.31 ppm, 13C, δ 49.0 ppm) in the deuterated solvent as the internal reference. Chemical shifts (δ) are reported in parts per million (ppm), and coupling constants (J) are given in hertz (Hz). NMR signal multiplicities are denoted as singlet (s), doublet (d), doublet of doublets (dd), quartet (q), or multiplet (m). Spectra were acquired and processed using the Topspin 3.5 software (Bruker Daltonics). HRESIMS measurements were performed in positive mode on a Bruker Daltonics micrOTOF-focus mass instrument. GC-MSD was done on a GCMS-QP 2010SE System (Shimadzu). Preparative HPLC was carried out on a Jasco System (PU-1580 Plus) in combination with UV/Vis detection at 200–680 nm (Jasco MD-2010 Plus diode array detector) at room temperature. For the isolation and purification of the constituents of the plant extracts, unless otherwise stated, SymmetryPrep™ C18 column (19 × 300 mm, 7 μm, Waters) was used, flow rate 10 mL min−1; mobile phases: (A) 90% H2O with 10% MeCN (0.05% trifluoroacetic acid) and (B) 90% MeCN with 10% H2O (0.05% trifluoroacetic acid), using linear gradients (details given below). All organic solvents were of analytical-grade quality. Ultra-pure water was obtained from an Elga Purelab Classic system. (S)-MTPA was purchased from Sigma-Aldrich (Steinheim, Germany).Plant material
Leaves of the botanically as yet unidentified Ancistrocladus plant were collected by one of us (V. M.) in July 2006, in the vicinity of the village Leeke near the town of Ikela, Province Tshuapa, Democratic Republic of the Congo. A voucher specimen (No. 114) has been deposited at the Herbarium Bringmann, Institute of Organic Chemistry, University of Würzburg.Extraction and isolation
Air-dried leaves (ca. 300 g) were ground and exhaustively extracted with CH2Cl2/MeOH (1:1, v/v) at room temperature. The combined extracts were concentrated under reduced pressure to give 22.1 g of a crude residue. The solid extract was macerated in H2O, with mechanical shaking (160 rpm), then filtered and dissolved in MeOH. After filtration, the raw methanolic extract was directly subjected to preparative HPLC on a SymmetryPrep™ C18 column, applying a linear gradient (0 min 20% B, 28 min 45% B), finally providing 11 fractions (F1–F11).
Fractions F1 (3.1 mg) and F2 (4.2 mg) were resolved by preparative HPLC, using an isocratic solvent system consisting of the mobile phases A and B (16:84, v/v) to give pure ikelacongoline B (5b) (2.7 mg) (retention time 25.4 min) and ikelacongoline A (5a) (3.5 mg) (retention time 28.9 min). From fraction F3 (2.6 mg), ealamine A (11) (1.7 mg) (retention time 18.9 min) was obtained, by isocratic HPLC using the solvents A and B (80:20, v/v).
Fraction F5 (8.5 mg) was subjected to preparative HPLC on a Chromolith SemiPrep RP-18e column (Merck, 10 × 100 mm) applying a linear gradient (0 min 5% B, 6 min 15% B, 9 min 25% B, 12 min 35% B, 15 min 45% B) at a flow rate of 5 mL min−1, affording pure mbandakamine A (1) (2.1 mg) (retention time 10.4 min) and an alkaloid-containing subfraction, which was further purified by preparative HPLC on a SymmetryPrep™ C18 column. Using an isocratic solvent system similar to the one described above, but with MeOH instead of MeCN, consisting of the solvents A′ and B′ (4.5:5:5, v/v), resolution of this subfraction at a flow rate of 10 mL min−1 furnished pure ikelacongoline C (6) (4.9 mg) (retention time 11.9 min) and ikelacongoline D (7) (1.3 mg) (retention time 14.0 min).
Purification of fraction F6 (9.1 mg) by preparative HPLC on a Chromolith SemiPrep RP-18e column under the conditions described above again yielded mbandakamine A (1) (3.6 mg) (retention time 10.4 min) and, additionally, ancistrocongoline C (8) (5.1 mg) (retention time 7.9 min).
From fractions F7 (8.7 mg) and F8 (8.3 mg), ealamine G (12) and ancistrolikokine F2 (10) were isolated by preparative HPLC on a Chromolith SemiPrep RP-18e column under the conditions given above; the two compounds were further purified by semi-preparative HPLC on a Waters XSelect HSS PFP column (10 × 250 mm, 5 μm) using a mobile phase system consisting of the solvents A′ and B′ under linear-gradient conditions (0 min 50% B, 20 min 100% B) at a flow rate of 5 mL min−1, thus finally giving rise to pure 12 (1.5 mg) (retention time 7.9 min) and pure 10 (1.3 mg) (retention time 10.2 min).
Fraction F9 (8.1 mg) was resolved on a Chromolith SemiPrep RP-18e column under the conditions described above, furnishing pure ancistrolikokine C (9) (5.1 mg) (retention time 9.5 min) and mbandakamine B4 (4) (3.0 mg) (retention time 11.2 min).
Resolution of fraction F10 (9.3 mg) by preparative HPLC on a Chromolith SemiPrep RP-18e column, followed by chromatography on a SymmetryPrep C18 column, using the solvent systems A and B, with a linear gradient (0 min 20% B, 20 min 45% B), at a flow rate of 10 mL min−1 gave pure mbandakamine B3 (3) (3.8 mg) (retention time 18.9 min) and mbandakamine B2 (2) (5.1 mg) (retention time 19.4 min).
ε): λmax 226 (3.9) nm; ECD (c 0.1, MeOH) λmax (Δε): 205 (−3.0), 218 (+6.1), 233 (+2.9), 249 (−3.9), 270 (−0.3), 289 (−1.0), 305 (−2.1), 329 (−0.4), 345 (−0.7), and 376 (+0.1) nm; IR (ATR) νmax: 3357, 2923, 2851, 1976, 1672, 1598, 1418, 1313, 1256, 1200, 1132, 1076, 1039, 833, 800, and 720 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS: m/z 813.4108 [M + H]+ (calcd for C50H57N2O8, 813.4114).ε): λmax 229 (3.9) nm; ECD (c 0.1, MeOH) λmax (Δε): 207 (−6.7), 218 (+9.9), 235 (−9.1), 240 (−9.6), 246 (−11.3), 271 (+1.6), 288 (−2.1), 302 (−1.8), 329 (−0.7), 350 (−1.8), 368 (+0.2), and 400 (+0.5) nm; IR (ATR) νmax: 33534, 2940, 2848, 2013, 1983, 1674, 1446, 1334, 1201, 1134, 838, 800, and 723 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS: m/z 799.3957 [M + H]+ (calcd for C49H55N2O8, 799.3958).ε): λmax 229 (3.5) nm; ECD (c 0.1, MeOH) λmax (Δε): 196 (+15.1), 211 (−15.9), 217 (−14.6), 223 (−15.5), 238 (+7.2), 266 (+0.1), 280 (−0.6), 300 (+0.1), 334 (−0.4), and 350 (−0.1) nm; IR (ATR) νmax: 3373, 2943, 2015, 1673, 1604, 1585, 1524, 1490, 1456, 1434, 1392, 1298, 1264, 1201, 1137, 1091, 1041, and 722 cm−1; 1H and 13C NMR data, see Tables 2 and 3; m/z 408.21699 [M + H]+ (calcd for C25H30NO4, 408.21748).ε): λmax 230 (3.6) nm; ECD (c 0.1, MeOH) λmax (Δε): 198 (−6.7), 211 (+11.1), 215 (+9.8), 224 (+6.5), 237 (−8.8), 261 (+0.1), 284 (+1.7), 300 (+0.3), and 350 (−0.2) nm; IR (ATR) νmax: 3220, 2941, 2839, 1672, 1584, 1456, 1200, 1137, 837, 800, and 722 cm−1; 1H and 13C NMR data, see Tables 2 and 3; HRESIMS: m/z 408.21747 [M + H]+ (calcd for C25H30NO4, 408.21748).ε): λmax 231 (3.6) nm; ECD (c 0.1, MeOH) λmax (Δε): 196 (−4.1), 210 (+12.4), 218 (+8.2), 222 (+8.6), 239 (−12.4), 277 (−0.3), 300 (+0.3), 318 (+0.5), and 351 (−0.1) nm; IR (ATR) νmax: 3379, 2941, 2839, 1671, 1583, 1542, 1449, 1363, 1198, 1021, 832, 800, and 720 cm−1; 1H and 13C NMR data, see Tables 2 and 3; HRESIMS: m/z 422.23133 [M + H]+ (calcd for C26H32NO4, 422.23313).ε): λmax 231 (3.6) nm; ECD (c 0.1, MeOH) λmax (Δε): 200 (−15.5), 214 (+14.7), 227 (+31.8), 243 (−11.2), 268 (+0.3), 285 (+0.9), 301 (−0.7), and 310 (−0.6) nm; IR (ATR) νmax: 3415, 2927, 2851, 2273, 1674, 1585, 1437, 1204, 1137, 841, 802, and 724 cm−1; 1H and 13C NMR data, see Tables 2 and 3; HRESIMS: m/z 422.23227 [M + H]+ (calcd for C26H32NO4, 422.23313).Known alkaloids isolated
The likewise isolated known monomeric and dimeric naphthylisoquinoline alkaloids mbandakamines A (1) and B2 (2) (Fig. 1), ancistrocongoline C (8), ancistrolikokine C (9), ancistrolikokine F2 (10), and ealamines A (11) and G (12) (Fig. 2), now isolated for the first time from the leaves of this as yet unidentified Central Congolese Ancistrocladus liana, were found to be identical in their spectroscopic, physical, and chromatographic behavior with the data reported previously.4,6,16,17,28–30Oxidative degradation
Following a procedure described earlier,33 ca. 0.8 mg of mbandakamines B3 (3) or B4 (4), or ikelacongolines A (5a), B (5b), C (6), or D (7) were subjected to ruthenium(VIII)-catalyzed periodate oxidation, followed by conversion of the resulting chiral amino with MeOH/HCl to the respective methyl esters and Mosher-type derivatization with (R)-α-methoxy-α-trifluoromethylphenylacetyl chloride [(R)-MTPA-Cl, prepared from (S)-MTPA]. The absolute configurations of 3, 4, 5a, 5b, 6, and 7 at C-3 were assigned by gas chromatography coupled to mass-selective detection (GC-MSD) and comparison with the corresponding derivatives of the authentic amino acids of known absolute configuration.Antiprotozoal assay
The in vitro antiparasitic activities of the mbandakamines B3 (3) and B4 (4), and the ikelacongolines A (5a), B (5b), and C (6) (Table 4) against the pathogens Plasmodium falciparum (NF54 and K1 strains), Trypanosoma cruzi (Tulahuen C4 strain, amastigotes), Trypanosoma brucei rhodesiense (STIB 900 strain, trypomastigotes), and Leishmania donovani (MHOM-ET-67/L82, amastigotes), and the cytotoxicity against mammalian host cells (rat skeletal myoblast L6 cells) were determined as described previously.53Conclusions
Prior to this work, mbandakamine-type naphthylisoquinoline dimers4,16 had been known only from two Ancistrocladus species indigenous to the Northwestern region of the Democratic Republic of the Congo. The as yet unidentified liana on which we report in this paper is the first Ancistrocladus plant discovered in the rainforests of the Central Congo Basin that was found to produce such mbandakamine-type compounds, too. From its leaves, four representatives were isolated, among them two new dimers, the mbandakamines B3 (3) and B4 (4). They were identified together with a series of four new 5,8′-coupled monomeric naphthylisoquinoline alkaloids, named ikelacongolines A–D (5a, 5b, 6, and 7), and along with five alkaloids (8–12) known from previous phytochemical studies on related Congolese Ancistrocladus species;17,28–30 three of them likewise belonged to the subclass of 5,8′-linked alkaloids, whereas two compounds were representatives of the quite rare 7,8′-coupling type. Particularly striking was the considerable number of N-methylated naphthylisoquinolines in this botanically and phytochemically so far unexplored plant, among them the two new mbandakamines B3 (3) and B4 (4) and the four new monomeric ikelacongolines A–D. More than half of the metabolites isolated from the leaf extract showed the presence of such an N-methyl group. The structures of these mono- and dimeric naphthylisoquinolines are chemotaxonomically quite significant, indicating a close phylogenetic relationship of this Ancistrocladus plant to other Ancistrocladus species from the Congo Basin, in particular to A. ealaensis15,16,30 and another yet unidentified Ancistrocladus taxon4–6 from the Northwestern part of the Democratic Republic of the Congo. These two species are likewise known to produce a high number of 5,8′-linked monomeric alkaloids along with mbandakamine-type dimers. Similar to the Ancistrocladus species presented here, their metabolite profiles are also dominated by the presence of mixed Ancistrocladaceae/Dioncophyllaceae, i.e., hybrid-type alkaloids, i.e., with R-configuration at C-3 and an oxygen function at C-6.Furthermore, the new mbandakamines 3 and 4 have proven to be potent bioactive compounds displaying good to strong inhibitory effects against the pathogen of African sleeping sickness, Trypanosoma brucei rhodesiense, and, in particular, exerting excellent activities against chloroquine-sensitive (NF54) and chloroquine-resistant (K1) strains of the malaria parasite Plasmodium falciparum. With respect to the fact that the efficiency of many recommended therapeutic agents routinely used for the treatment of infectious diseases is threatened by the formation of an increasing resistance, the discovery of new agents is urgently needed. The promising activities of mbandakamine-type dimers warrant further investigations on their antiparasitic potential in particular, and on that of mono- and dimeric naphthylisoquinoline alkaloids in general. This work is in progress.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, Individual Research Grant Br 699/14-2 “Molecular Phylogeny and Chemotaxonomy of the Ancistrocladaceae Plant Family”; SFB 630 “Agents against Infectious Diseases”, project A2), and by grants from the German Academic Exchange Service (Deutscher Akademischer Austauschdienst, DAAD) and the Excellence Scholarship Program BEBUC (http://www.foerderverein-uni-kinshasa.de) to one of us (J.-P. M.). This publication was funded by the DFG and the University of Würzburg within the funding programme Open Access Publishing. We acknowledge experimental assistance from Dr M. Büchneri and Mrs J. Adelmann (MS), from Dr Grüne and Mrs P. Altenberger (NMR), and from Mrs M. Michel and Mrs S. Fayez (oxidative degradation), all from the University of Würzburg. We also thank Mrs M. Cal, Mrs S. Keller-Märki, and Mrs R. Rocchetti for the help with parasitic assays.Notes and references
- C. M. Taylor, R. E. Gereau and G. M. Walters, Ann. Mo. Bot. Gard., 2005, 92, 360–399 Search PubMed.
- M. Cheek, Kew Bull., 2000, 55, 871–882 CrossRef.
- F. G. Turini, C. Steinert, G. Heubl, G. Bringmann, B. K. Lombe, V. Mudogo and H. Meimberg, Taxon, 2014, 63, 329–341 CrossRef.
- G. Bringmann, B. K. Lombe, C. Steinert, K. Ndjoko Ioset, R. Brun, F. Turini, G. Heubl and V. Mudogo, Org. Lett., 2013, 15, 2590–2593 CrossRef CAS PubMed.
- B. K. Lombe, T. Bruhn, D. Feineis, V. Mudogo, R. Brun and G. Bringmann, Org. Lett., 2017, 19, 1342–1345 CrossRef CAS PubMed.
- B. K. Lombe, T. Bruhn, D. Feineis, V. Mudogo, R. Brun and G. Bringmann, Org. Lett., 2017, 19, 6740–6743 CrossRef CAS PubMed.
- B. K. Lombe, D. Feineis, V. Mudogo, R. Brun, S. Awale and G. Bringmann, RSC Adv., 2018, 8, 5243–5254 RSC.
- G. Bringmann, G. Zhang, T. Büttner, G. Bauckmann, T. Kupfer, H. Braunschweig, R. Brun and V. Mudogo, Chem.–Eur. J., 2013, 19, 916–923 CrossRef CAS PubMed.
- S. M. Kavatsurwa, B. K. Lombe, D. Feineis, D. F. Dibwe, V. Maharaj, S. Awale and G. Bringmann, Fitoterapia, 2018, 130, 6–16 CrossRef CAS PubMed.
- G. Bringmann, I. Kajahn, M. Reichert, S. E. H. Pedersen, J. H. Faber, T. Gulder, R. Brun, S. B. Christensen, A. Ponte-Sucre, H. Moll, G. Heubl and V. Mudogo, J. Org. Chem., 2006, 71, 9348–9356 CrossRef CAS PubMed.
- G. Bringmann, J. Spuziak, J. H. Faber, T. Gulder, I. Kajahn, M. Dreyer, G. Heubl, R. Brun and V. Mudogo, Phytochemistry, 2008, 69, 1065–1075 CrossRef CAS PubMed.
- G. Heubl, F. Turini, V. Mudogo, I. Kajahn and G. Bringmann, Plant Ecol. Evol., 2010, 143, 63–69 CrossRef.
- J. Léonard, Bull. Soc. R. Bot. Belg., 1949, 82, 27–40 Search PubMed.
- G. Bringmann, C. Steinert, D. Feineis, V. Mudogo, J. Betzin and C. Scheller, Phytochemistry, 2016, 128, 71–81 CrossRef CAS PubMed.
- D. T. Tshitenge, D. Feineis, V. Mudogo, M. Kaiser, R. Brun and G. Bringmann, Sci. Rep., 2017, 7, 5767 CrossRef PubMed.
- D. T. Tshitenge, D. Feineis, V. Mudogo, M. Kaiser, R. Brun, E.-J. Seo, T. Efferth and G. Bringmann, J. Nat. Prod., 2018, 81, 918–933 CrossRef CAS PubMed.
- S. Fayez, D. Feineis, V. Mudogo, S. Awale and G. Bringmann, RSC Adv., 2017, 7, 53740–53751 RSC.
- S. Fayez, D. Feineis, V. Mudogo, E.-J. Seo, T. Efferth and G. Bringmann, Fitoterapia, 2018, 129, 114–125 CrossRef CAS PubMed.
- S. Awale, D. F. Dibwe, C. Chandrasekar Balachadran, S. Fayez, D. Feineis, B. K. Lombe and G. Bringmann, J. Nat. Prod., 2018, 81, 2282–2291 CrossRef CAS PubMed.
- J. Li, R. Seupel, D. Feineis, V. Mudogo, M. Kaiser, R. Brun, D. Brünnert, M. Chatterjee, E.-J. Seo, T. Efferth and G. Bringmann, J. Nat. Prod., 2017, 80, 443–458 CrossRef CAS PubMed.
- J. Li, R. Seupel, T. Bruhn, D. Feineis, M. Kaiser, R. Brun, V. Mudogo, S. Awale and G. Bringmann, J. Nat. Prod., 2017, 80, 2807–2817 CrossRef CAS PubMed.
- T. R. Govindachari and P. C. Parthasarathy, Tetrahedron, 1971, 27, 1013–1026 CrossRef CAS.
- T. R. Govindachari and P. C. Parthasarathy, Heterocycles, 1977, 7, 661–684 CrossRef CAS.
- G. Bringmann and F. Pokorny, The naphthylisoquinoline alkaloids, in The Alkaloids, ed. G. A. Cordell, Academic Press Inc, New York, 1995, vol. 46, pp. 127–271 Search PubMed.
- S. R. M. Ibrahim and G. A. Mohamed, Fitoterapia, 2015, 106, 194–225 CrossRef CAS PubMed.
- Y. F. Hallock, K. P. Manfredi, J. W. Blunt, J. H. Cardellina II, M. Schäffer, K. P. Gulden, G. Bringmann, A. Y. Lee, J. Clardy, G. François and M. R. Boyd, J. Org. Chem., 1994, 59, 6349–6355 CrossRef CAS.
- M. R. Boyd, Y. F. Hallock, J. H. Cardellina ΙΙ, K. P. Manfredi, J. W. Blunt, J. B. McMahon, R. W. Buckheit Jr, G. Bringmann, M. Schäffer, G. M. Cragg, D. W. Thomas and J. G. Jato, J. Med. Chem., 1994, 37, 1740–1745 CrossRef CAS PubMed.
- G. Bringmann, K. Messer, R. Brun and V. Mudogo, J. Nat. Prod., 2002, 65, 1096–1101 CrossRef CAS.
- G. Bringmann, C. Günther, W. Saeb, J. Mies, A. Wickramasinghe, V. Mudogo and R. Brun, J. Nat. Prod., 2000, 63, 1333–1337 CrossRef CAS.
- D. T. Tshitenge, PhD thesis, Julius-Maximilians-Universität Würzburg, 2017.
- Y. F. Hallock, J. H. Cardellina II, M. Schäffer, M. Stahl, G. Bringmann, G. François and M. R. Boyd, Tetrahedron, 1997, 53, 8121–8128 CrossRef CAS.
- Y. F. Hallock, J. H. Cardellina II, M. Schäffer, G. Bringmann, G. François and M. R. Boyd, Bioorg. Med. Chem. Lett., 1998, 8, 1729–1734 CrossRef CAS PubMed.
- G. Bringmann, R. God and M. Schäffer, Phytochemistry, 1996, 43, 1393–1403 CrossRef CAS.
- M. Xu, T. Bruhn, B. Hertlein, R. Brun, A. Stich, J. Wu and G. Bringmann, Chem.–Eur. J., 2010, 16, 4206–4216 CrossRef CAS PubMed.
- World malaria report 2015, http://apps.who.int/iris/bitstream/10665/200018/1/9789241565158_eng.pdf, accessed February 6, 2017.
- A. Degarege, K. Fennie, D. Degarege, S. Chennupati and P. Madhivanan, PLoS One, 2019, 14, e0211205 CrossRef CAS PubMed.
- T. K. Mackey, B. A. Liang, R. Cuomo, R. Hafen, K. C. Brouwer and D. E. Lee, Clin. Microbiol. Rev., 2014, 27, 949–979 CrossRef PubMed.
- K. C. Chinsembu, Acta Trop., 2015, 152, 32–48 CrossRef PubMed.
- P. A. Onguéné, F. Ntie-Kang, L. Likowo Lifongo, J. C. Ndom, W. Sippl and L. Meva'a Mbaze, Malar. J., 2013, 12, 449 CrossRef PubMed.
- C. Ramahete, F. P. da Cruz, S. Mulhovo, I. J. Sousa, M. X. Fernandes, M. Prudêncio and M. J. U. Ferreira, Bioorg. Med. Chem., 2014, 22, 3887–3890 CrossRef PubMed.
- C. N. Muthaura, J. M. Keriko, S. Derese and G. M. Rukunga, Exp. Parasitol., 2011, 127, 609–626 CrossRef CAS PubMed.
- M. A. Ibrahim, A. Mohammed, M. B. Isah and A. B. Aliyu, J. Ethnopharmacol., 2014, 154, 26–54 CrossRef PubMed.
- C. J. D. Obbo, S. T. Kariuki, J. W. Gathirwa, W. Olaho-Mukani, P. K. Cheplogoi and E. M. Mwangi, J. Ethnopharmacol., 2019, 229, 127–136 CrossRef CAS PubMed.
- D. Zofou, F. Ntie-Kang, W. Sippl and S. M. N. Efange, Nat. Prod. Rep., 2013, 30, 1098–1120 RSC.
- C. V. Simoben, F. Ntie-Kang, S. H. Akone and W. Sippl, Nat. Prod. Bioprospect., 2015, 8, 151–169 CrossRef PubMed.
- E. Izumi, T. Ueda-Nakamura, B. Prado Dias Filho, V. F. Veiga Júnior and C. V. Nakamura, Nat. Prod. Rep., 2011, 28, 809–823 RSC.
- M. M. Salem and K. A. Werbovetz, Curr. Med. Chem., 2006, 13, 2571–2598 CrossRef CAS PubMed.
- G. Bringmann, W. Saeb, D. Koppler and G. François, Tetrahedron, 1996, 52, 13409–13418 CrossRef CAS.
- G. Bringmann, W. Saeb, M. Wohlfarth, R. Messer and R. Brun, Tetrahedron, 2000, 56, 5871–5875 CrossRef CAS.
- G. Bringmann and S. Tasler, Tetrahedron, 2001, 57, 331–343 CrossRef CAS.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Pescitelli, SpecDis, Version 1.71, www.specdis-software.jimdo.com, Berlin, Germany, 2017 Search PubMed.
- I. Orhan, B. Şener, M. Kaiser, R. Brun and D. Tasdemir, Mar. Drugs, 2010, 8, 47–58 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data include NMR (1H, 13C, 1H, 1H-COSY, HSQC, HMBC, NOESY, and ROESY), HRESIMS, IR, and ECD spectra of compounds 3, 4, 5a, 5b, 6, and 7. See DOI: 10.1039/c9ra01784d |
| This journal is © The Royal Society of Chemistry 2019 |