
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: RNA-sequencing identified miR-3681 as a negative regulator in the proliferation and migration of cervical cancer cells via the posttranscriptional suppression of HGFR
Fan Shi,
Yingbing Zhang,
Juan Wang,
Jin Su,
Zi Liu and
Tao Wang *
*
Department of Radiation Oncology, The First Affiliated Hospital of Xi'an Jiaotong University, 277 Yanta West Road, Xi'an, Shaanxi 710061, P. R. China. E-mail: taowang_md@163.com; Tel: +86-029-85324019
First published on 18th July 2019
Abstract
In this study, RNA-sequencing was used to investigate the differentially expressed miRNAs between cervical cancer tissues and matched adjacent non-tumor tissues. Five miRNAs were sharply downregulated in the cancer tissue, including miR-199a, miR-22, miR-615, miR-3681-3p (miR-3681), and miR-1193. Among them, miR-3681 was uncharacterized. The results from qPCR analysis showed that miR-3681 expression was decreased in patients with cervical cancer compared with the control, and decreased in the human cervical cancer cell lines SiHa, HeLa, C4-1, C-33A and Caski, compared with the normal human cervical epithelial cell line HCerEpic. Then, different concentrations of miR-3681 mimic and miR-3681 inhibitor were respectively transfected into the human cervical cancer cell line C-33A, and the expression of miR-3681, cell proliferation, cell apoptosis and cell migration were measured after 48 h. The results showed that the miR-3681 mimic increased the miR-3681 level, suppressed cell proliferation and migration, and induced cell apoptosis in a dose-dependent manner. In contrast, the miR-3681 inhibitor decreased the miR-3681 level, promoted cell proliferation and migration, and inhibited cell apoptosis in a dose-dependent manner. Moreover, bioinformatics analysis showed that there was a miR-3681 binding site in the mRNA 3′UTR of HGFR, which was robustly upregulated in cervical cancer cell lines compared with HCerEpic cells. In addition, luciferase activity analysis demonstrated that miR-3681 could directly target HGFR, which promoted the proliferation and migration of C-33A cells via activation of the PI3K/Akt pathway in a dose-dependent manner. Furthermore, our results showed that knockdown of HGFR could antagonize the promotion of anti-miR-3681 on the activation of the PI3K/Akt pathway and cell proliferation and migration. In conclusion, MiR-3681 was identified as a negative regulator in the proliferation and migration of cervical cancer cells. This function is associated with the posttranscriptional suppression of HGFR and the deactivation of the PI3K/Akt pathway.
1. Introduction
Cervical cancer is one of the most common gynecological malignancies and the most lethal gynecological cancer. In recent years, an increase in the incidence of cervical cancer was found in younger people.1 Although its morbidity and mortality has been reduced during the past decade, total hysterectomy and hysterectomy for the treatment of cervical cancer still pose a great threat to the fertility of women.2 The early stages of cervical cancer are usually completely free of symptoms and in later stages pain and other symptoms are experienced, but are non-specific, including abnormal vaginal bleeding, pelvic pain, or pain during sexual intercourse.3 Most cervical cancers occur in developing countries where there is a lack of cervical screening programs.4 Therefore, it is still urgent to explore available biomarkers for diagnosis and therapy of cervical cancer.The development of high-throughput sequencing has greatly accelerated the discovery rate of disease biomarkers. Non-coding RNAs (ncRNAs) refer to RNA transcripts that do not have the capacity of protein coding. MicroRNAs (miRNAs) are small ncRNA molecules with a length of about 22 nucleotides and function in RNA silencing and post-transcriptional suppression of gene expression.5 Unlike most other ncRNAs, miRNAs have been widely found in plants, animals and some viruses.6 They have been involved in pathogenesis of multiple diseases including various malignancies.7,8 Many miRNAs have found functions in the diagnosis, prognosis, metastasis and treatment of cervical cancer. For example, miR-200a and miR-99 were identified as biomarkers for the prediction of cervical cancer patient survival, and they played important roles in the proliferation, invasion and epithelial–mesenchymal transition (EMT) of cervical cancer cells.9,10 Some other miRNAs, such as miR-139, miR-634, miR-375 and miR-125, were also recently found to be dysregulating in cervical cancer.11–14 They modulated behaviors of cervical cancer cells, including proliferation, apoptosis, invasion, radio sensitivity and drug sensitivity, via posttranscriptional suppression of oncogenes Nin one binding protein (NOB1), mammalian target of rapamycin complex 1 (mTOR), signal transducer and activator of transcription 3 (STAT3), and insulin-like growth factor 1 receptor (IGF-1R).
In this study, RNA-sequencing was used to investigate the differentially expressed miRNAs between cervical cancer tissues and matched adjacent non-tumor tissues. MiR-199a, miR-22, miR-615, miR-3681-3p (miR-3681), and miR-1193 were the top 5 downregulated miRNAs in the cancer tissues. Then, the role of miR-3681 in the proliferation and invasion of cervical cancer cells was investigated, the function of which has not yet been uncovered. Furthermore, we explored the mechanism through which miR-3681 regulated the proliferation and invasion of cervical cancer cells.
2. Materials and methods
2.1. Ethical statements
All experiments were performed in accordance with the Guidelines of The First Affiliated Hospital of Xi'an Jiaotong University (Xi'an, China), and experiments were approved by the ethics committee of The First Affiliated Hospital of Xi'an Jiaotong University. Informed consents were obtained from human participants of this study.2.2. RNA sequencing
Cervical carcinoma tissues and adjacent normal tissues were respectively obtained from six cervical cancer patients. Total RNA was isolated using the miRNeasy kit (Qiagen) and its integrity was checked by an Agilent 2100 Bioanalyzer. Strand-specific barcoded libraries were generated using the QIAseq miRNA Library Kits (QIAGEN) according to the manufacturer's protocol with the following modifications: PCR amplification was limited to 12 cycles to avoid bias from PCR “jackpot” mutations and RNA sequencing was performed on the HiSeq 2500 (Illumina). About 2 million filtered reads per sample uniquely mapped to the human genome. Transcripts were annotated using publicly available datasets from RefSeq for small noncoding genes. The enrichment of each RNA transcript was measured in fragments per kilobase of per million reads mapped (FPKM) using Cufflinks 2.1.1. Differential expression (DE) between the carcinoma and normal tissue was tested with Cuffdiff. miRNAs with a fold change of 2.0 and a false discovery rate-adjusted P value of <0.05 were considered as differentially expressed. Ingenuity Pathway Analysis software (Qiagen) was used to conduct Gene Ontology (GO) analysis.2.3. Tissue sampling
Cervical tissues were isolated from 48 volunteers (52 ± 6.9 years) and a total of 120 cervical cancer patients (51 ± 7.0 years) who underwent tumor resection during the last 2 years in our hospital. Patients who ever received chemotherapy or radiotherapy or suffered other serious reproductive disorders and chronic inflammation diseases, were excluded from this study.2.4. Cell culture and transfection
Human normal cervical epithelial cell line HCerEpic, cervical cancer cell lines SiHa, HeLa, C4-1, C-33A and Caski, and HEK293 cells were purchased from American Type Culture Collection Company (ATCC; Manassas, VA, USA). They were cultured in RPMI-1640 medium (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 U mL−1 penicillin (Gibco) and 100 U mL−1 streptomycin (Gibco). The cells were incubated at a humidified atmosphere at 37 °C in a 5% CO2 incubator.For transfection, C-33A cervical cancer cells were sub-grown in 6-well plates. On reaching to 70% of confluence, different doses of the miR-3681 mimic oligo, different doses of the miR-3681 antagomir oligo and relative control oligos, were respectively transfected into the cells by using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions.
2.5. Cell proliferation
The cells were seeded in 6-well culture plates at 105 per well. After transfection for 72 h, cell proliferation was evaluated using the Cell Counting Kit-8 (CCK-8; Sigma, St. Louis, MO, USA) assay according to the manufacturer's instructions.2.6. In vitro migration assay
Transwell migration assay was used to detect cell migration. 3.5 × 104 cells with serum-free medium were plated in the top chamber of the plates (24-well insert, 8 μm, Corning). Medium containing 10% serum was used as a chemoattractant in the lower chamber. After incubation for 24 h, a cotton swab was used to remove the non-migrated cells in the upper chamber, and the filters were individually stained with 1% crystal violet. The invasive cells were captured under a light cervical cancer microscope (×100; Olympus IX70; Olympus Corporation, Osaka, Japan) and counted.2.7. Real-time quantitative PCR
Real-time qPCR reactions were carried on in a 25 μL system, using SYBR Premix Ex Taq (Invitrogen), 0.4 mM of primers, and 200 ng of cDNA template. The primers of miR-3681 and U6 RNA (internal reference) were designed and produced by Ribobio Co., Ltd. PCR amplification cycles were performed using the SYBR Premix Ex Taq II Kit (Invitrogen). The reactions were initially denatured at 95 °C for 1 min, and followed by 35 cycles of 94 °C for 15 s and 55 °C for 1 min. The transcript abundance was calculated using the 2−ΔΔCt method.2.8. Western blotting
30 μg of total protein from each sample was separated by 12% SDS-PAGE gel. The protein was transferred onto a PVDF membrane (Millipore, Billerica, MA, USA). The primary antibodies HGFR (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :300, Abcam), PI3K (1:400, Abcam), Akt (1:400, Abcam), pPI3K (1:200, Abcam) and pAkt (1:300, Abcam) and β-actin (1:800, Abcam) were used to incubate with the membrane at 4 °C overnight. After incubation with the HRP-conjugate secondary antibody for 60 min at room temperature, proteins were detected with an ECL kit (Millipore) and analyzed with a UV Transilluminator (Bio-Rad).
:300, Abcam), PI3K (1:400, Abcam), Akt (1:400, Abcam), pPI3K (1:200, Abcam) and pAkt (1:300, Abcam) and β-actin (1:800, Abcam) were used to incubate with the membrane at 4 °C overnight. After incubation with the HRP-conjugate secondary antibody for 60 min at room temperature, proteins were detected with an ECL kit (Millipore) and analyzed with a UV Transilluminator (Bio-Rad).
2.9. Luciferase gene reporter assay
The 3′UTR sequences of human KLF6 and KLF15 mRNAs were amplified by PCR. The sequences were respectively purified and inserted into a psiCHECK™-2 Vector (Promega, Madison, WI, USA). 20 nM control mimic or miR-3681 mimic were respectively co-transfected with 2 μg of Luc-3′UTR-WT or Luc-3′UTR-MUT into the HEK293 cells by using Lipofectamine 3000 (Invitrogen). The cells were then incubated for 72 h and luciferase activity was measured using a Microplate Reader (PT-3502, Potenov, Beijing, China).2.10. Statistical analysis
All measurements were obtained from >three independent experiments. The data were expressed as means ± SEM. Statistics were calculated with SPSS v23.0. Multiple comparisons were assessed by one-way ANOVA followed by Dunnett's tests. P < 0.05 was considered statistically significant.3. Results
3.1. MiR-3681 was significantly downregulated in human cervical cancer tissue and cell lines
We used the RNA-seq data from six cervical cancer patients to identify the miRNAs differentially expressed between the cancer tissues and adjacent normal tissues. A total of 9132 unique miRNAs were identified, among which 6705 miRNA candidates were supported by at least two reads spanning a head-to-tail splice junction. We noticed that 1242 miRNAs were upregulated, and 965 miRNAs were downregulated in the cancer tissue. Among the downregulated miRNAs, there were 395 miRNAs with a log2 fold change ≤ −2. To further explore the potential function of these 395 miRNAs during tumorigenesis, we analyzed the GO enrichment of them and found that they were enriched in the regulation of cell death, cell migration and angiogenesis (Fig. 1A). Then, qPCR analysis was used to confirm the expression of the top 5 downregulated miRNAs, including miR-199a, miR-22, miR-615, miR-3681 and miR-1193. The results showed that their log2 fold changes were all <−3 (Fig. 1B). Since the functions of miR-199a, miR-22, miR-615 and miR-1193 in tumorigenesis have been reported, we selected miR-3681 as a new candidate in this study. The qPCR analysis for the expression of miR-3681 among the control group and cervical cancer patients at different stages showed that miR-3681 was significantly downregulated in primary cervical cancer patients, sharply decreased in patients with early cervical cancer and further decreased in patients with late cervical cancer (Fig. 1C). Moreover, we detected expression of miR-3681 in normal cervical epithelial cell line HCerEpic and cervical cancer cell lines SiHa, HeLa, C4-1, C-33A and Caski. The results showed that miR-3681 expression was significantly decreased in all of these cell lines (Fig. 1D). These data above suggested that miR-3681 might play a role in the progression of cervical cancer.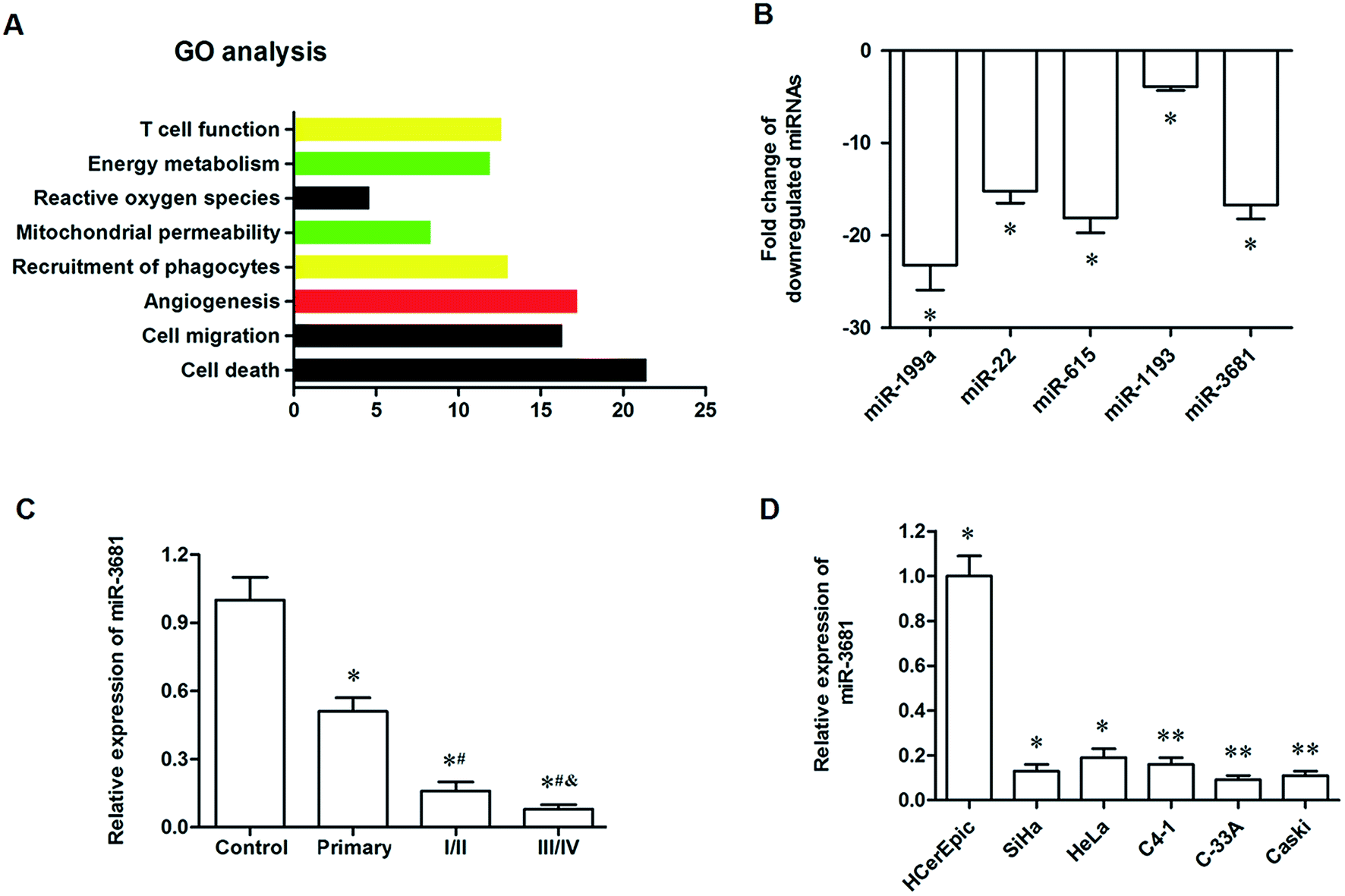 | ||
| Fig. 1 MiR-3681 was identified as a potential regulator in human cervical cancer. (A) Gene ontology (GO) analysis for the downregulated miRNAs in cervical cancer. (B) Confirmation of the expression of the top 5 downregulated miRNAs by qPCR. (C) MiR-3681 was significantly upregulated in cervical cancer tissues. (D) MiR-3681 was significantly elevated in cervical cancer cell lines. RNA-seq was used to identify the miRNAs differentially expressed between the cancer tissues and adjacent normal tissues in six cervical cancer patients. Among the downregulated miRNAs, there were 395 miRNAs with a log2 fold change ≤ −2, and GO analysis was used to evaluate the potential biological processes in which they were involved. The level of miR-3681 was detected in 48 controls and 120 patients with different stages of cervical cancer (including 30 primary cervical cancer patients, 50 patients at I and II stages, and 40 patients at III and IV stages). Then, the expression level of miR-3681 was detected in the human normal cervical epithelial cell line HCerEpic and the cervical cell lines SiHa, HeLa, C4-1, C-33A and Caski. *P < 0.05 vs. control, #P < 0.05 vs. I/II, &P < 0.05 vs. III/IV. | ||
3.2. MiR-3681 regulated proliferation and migration of cervical cancer cells
To explore the role of miR-3681 in the progression of cervical cancer, different doses of miR-3681 mimic were transfected into C-33A cervical cancer cells. After incubation for 72 h, their overexpression efficacies were detected. As shown in Fig. 2A, miR-3681 mimic at the concentration of 10 nM significantly increased the miR-3681 level (by about 4.5 fold), and miR-3681 mimic at the concentrations of 20 nM or 40 nM further increased the miR-3681 level (both by more than 10 fold). CCK-8 assay indicated that 10 nM miR-3681 mimic significantly suppressed cell proliferation, and 20 nM or 40 nM further suppressed cell proliferation (Fig. 2B). AnnexinV-PI staining combined with flow cytometry assays showed that 10 nM miR-3681 mimic significantly increased cell apoptosis, and 20 nM or 40 nM further increased cell apoptosis (Fig. 2C). Transwell Cell Migration assay indicated that 10 nM miR-3681 mimic significantly suppressed cell migration, and 20 nM or 40 nM further suppressed cell migration (Fig. 2D). In addition, different doses of miR-3681 antagomir (anti-3681) were transfected into C-33A cervical cancer cells. After incubation for 72 h, their interference efficiencies were measured. As shown in Fig. 2E, miR-3681 antagomir at a concentration of 20 nM significantly decreased the miR-3681 level (by about 60%), and miR-3681 antagomir at the concentrations of 40 nM or 80 nM further decreased the miR-3681 level (both by more than 80%). CCK-8 assay indicated that 20 nM miR-3681 antagomir significantly increased cell proliferation, and 40 nM or 80 nM antagomir further increased cell proliferation (Fig. 2F). AnnexinV-PI staining combined with flow cytometry assays showed that 20 nM miR-3681 mimic significantly suppressed cell apoptosis, and 40 nM or 80 nM further suppressed cell apoptosis (Fig. 2G). The Transwell Cell Migration assay indicated that 20 nM miR-3681 mimic significantly increased cell migration, and 40 nM or 80 nM further increased cell migration (Fig. 2H). These data, combined with the results from miR-3681 mimic transfection experiments, demonstrated that miR-3681 could suppressed proliferation and migration, and induced apoptosis in cervical cancer cells.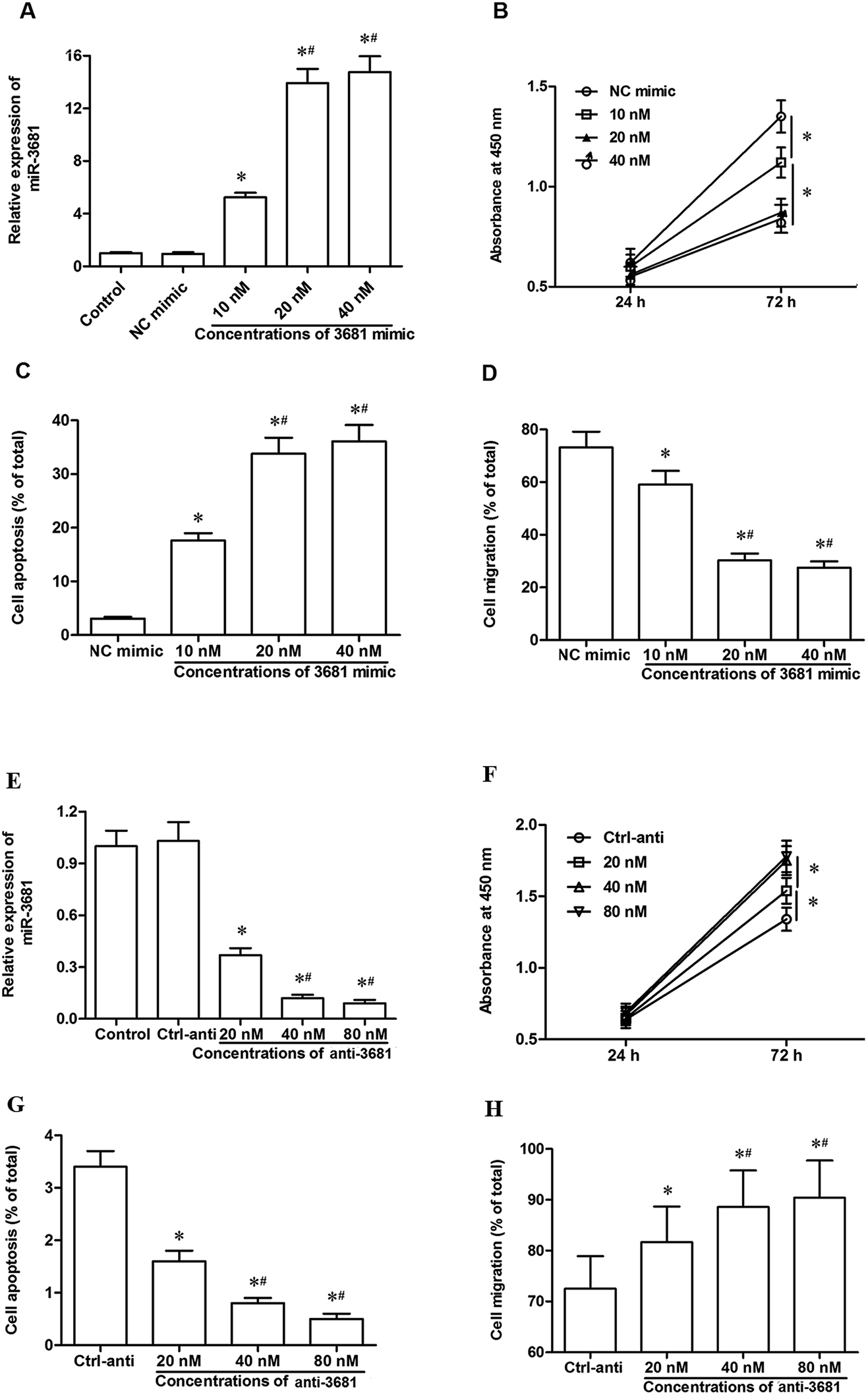 | ||
| Fig. 2 MiR-3681 regulated proliferation and migration of C-33A cervical cancer cells. (A) MiR-3681 expression was increased greatly by miR-3681 mimic transfection in a dose-dependent manner. (B) The miR-3681 mimic significantly suppressed the proliferation of C-33A cells in a dose-dependent manner. (C) The miR-3681 mimic significantly increased apoptosis of C-33A cells in a dose-dependent manner. (D) The miR-3681 mimic significantly reduced the migration of C-33A cells in a dose-dependent manner. The negative control mimic (NC mimic) and different doses of miR-3681 mimic (10, 20 and 40 nM) were respectively transfected into C-33A human cervical cancer cells. After incubation for 72 h, miR-3681 expression was measured with real-time qPCR, cell proliferation was measured with the CCK-8 method, cell apoptosis was measured with AnnexinV-PI staining combined with flow cytometry assays, and cell migration was measured with the Transwell migration assay. *P < 0.05 vs. NC mimic, #P < 0.05 vs. 10 nM. (E) MiR-3681 expression was significantly decreased by the miR-3681 antagomir transfection in a dose-dependent manner. (F) The miR-3681 antagomir significantly promoted the proliferation of C-33A cells in a dose-dependent manner. (G) The miR-3681 antagomir significantly repressed apoptosis of C-33A cells in a dose-dependent manner. (H) The miR-3681 antagomir significantly increased the migration of C-33A cells in a dose-dependent manner. The negative control antagomir (Ctrl-anti) and different doses of miR-3681 antagomir (20, 40 and 80 nM) were respectively transfected into C-33A human cervical cancer cells. After incubation for 72 h, miR-3681 expression was measured with real-time qPCR, cell proliferation was measured with the CCK-8 method, cell apoptosis was measured with AnnexinV-PI staining combined with flow cytometry assays, and cell migration was measured with the Transwell migration assay. *P < 0.05 vs. Ctrl-anti, #P < 0.05 vs. 20 nM. | ||
3.3. MiR-3681 directly targeted the oncogene HGFR and suppressed its protein expression
To explore the potential mechanism of miR-3681 suppressing proliferation and invasion of breast cancer cells, bioinformatics analysis was applied to seek its potential targets. The results showed that seven consecutive bases in the 3′ untranslated region (3′UTR) of HGFR mRNA sequence completely matched with the seed of miR-3681 (Fig. 3A). Western blotting analysis showed HGFR protein was robustly upregulated in cervical cancer cell lines, which displayed an opposite expression pattern to that of miR-3681 (Fig. 3B). Then, 3′UTR luciferase reporter gene reporters containing the wild type (WT) HGFR 3′UTR sequence and the mutated (MUT) HGFR 3′UTR sequence at the miR-3681 binding site were constructed (Fig. 3C). They were respectively transfected into the cells together with NC mimic or miR-3681 mimic. The 3′UTR luciferase reporter gene assay showed that miR-3681 markedly suppressed the luciferase activity of the WT HGFR 3′UTR luciferase reporter gene, but not the MUT (Fig. 3D). To further verify that HGFR were target genes of miR-3681, the miR-3681 mimic, miR-3681 antagomir and their negative control oligos were respectively transfected into C-33A cells. Western blotting indicated that HGFR protein expression was sharply reduced by the miR-3681 mimic transfection, and significantly elevated by the miR-3681 antagomir (Fig. 3E). These results indicated that miR-3681 directly targeted the oncogene HGFR in cervical cancer cells.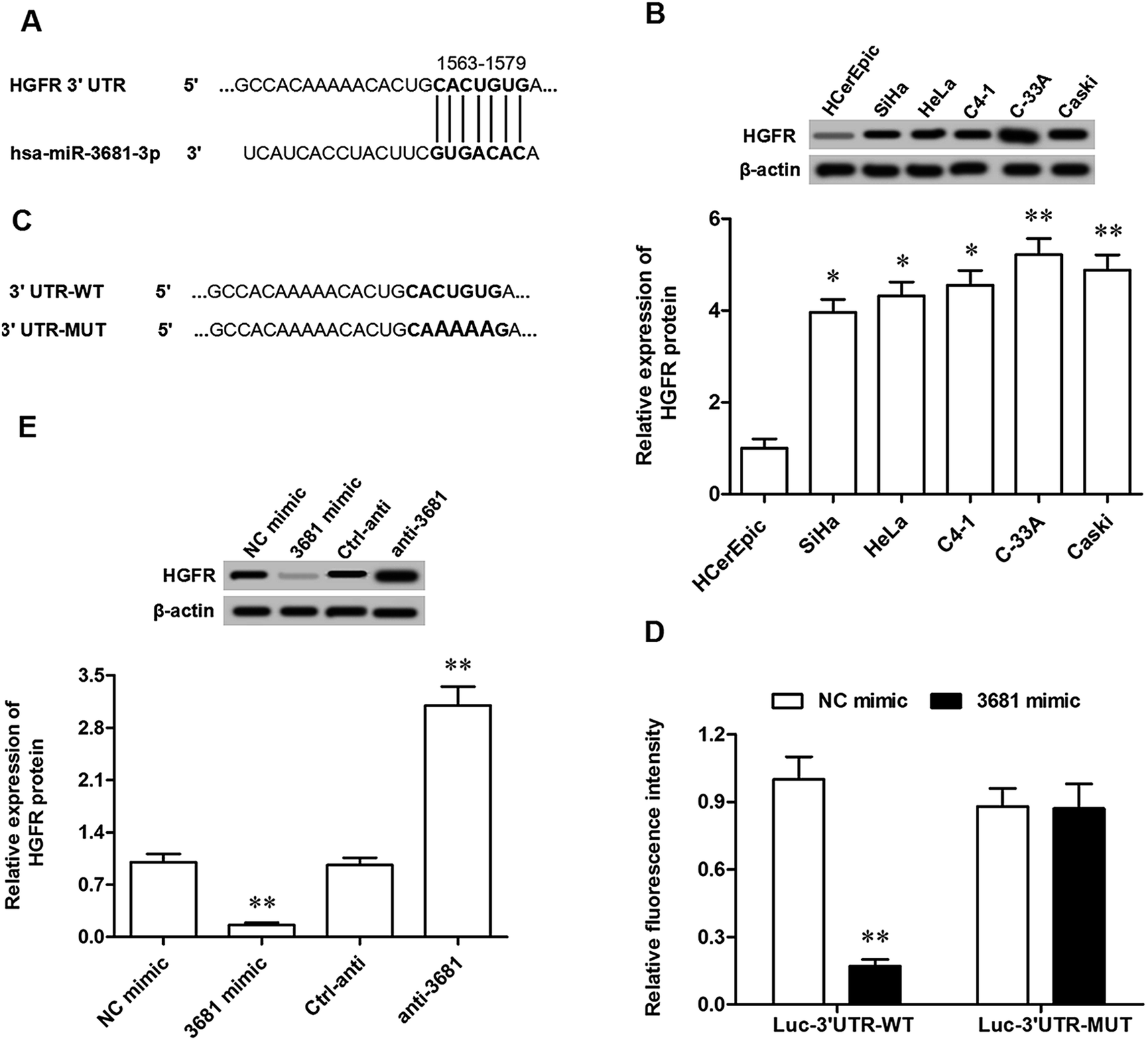 | ||
| Fig. 3 MiR-3681 targeted HGFR mRNA at the 3′UTR region and suppressed its expression. (A) The output of the bioinformatics analysis on the binding of miR-3681 with the 3′UTR region of HGFR mRNA. (B) HGFR protein was greatly upregulated in cervical cancer cell lines determined by Western blotting. (C) Details of the WT and MUT 3′UTR luciferase reporter gene reporters at the miR-3681 binding cite. (D) Validation of the binding of miR-3681 with HGFR mRNA by the 3′UTR luciferase reporter gene assay. (E) MiR-3681 negatively regulated the expression of HGFR protein. 20 nM miR-3681 mimic, 40 nM antagomir **P < 0.01. | ||
3.4. HGFR mediated miR-3681 function in the regulation of proliferation and migration in cervical cancer cells
Finally, the involvement of HGFR in the regulation of miR-3681-mediated cell proliferation and migration was investigated. Different doses of pcDNA-HGFR overexpression vector and HGFR siRNA were individually transfected into C-33A cervical cancer cells. Following incubation for 72 h, their overexpression and interference efficacies were confirmed by Western blotting analysis. The results showed that 0.5 μg mL−1 pcDNA-HGFR significantly increased and 1 μg mL−1 pcDNA-HGFR, the level of HGFR protein (Fig. 4A), and 40 nM HGFR siRNA, significantly reduced and 80 nM HGFR siRNA further reduced the level of HGFR protein (Fig. 4B). 1 μg mL−1 pcDNA-HGFR and 80 nM HGFR siRNA were applied in the following experiments: C-33A cells were treated respectively with miR-3681 mimic, miR-3681 mimic plus pcDNA-HGFR, miR-3681 antagomir, and miR-3681 antagomir plus HGFR siRNA. Western blotting analysis revealed that miR-3681 mimic suppressed expression of HGFR and activation of the downstream pathway PI3K/Akt, which could be antagonized by pcDNA-HGFR (Fig. 4C). Moreover, inhibition of miR-3681 increased expression of HGFR and activation of the PI3K/Akt pathway, which could be eliminated by HGFR siRNA (Fig. 4C). Additionally, pcDNA-HGFR antagonized the suppression effects of miR-3681 mimic on cell progression/migration (Fig. 4D/E) and the promotion on cell apoptosis (Fig. 4F), and HGFR siRNA eliminated the promotion effects of miR-3681 antagomir on cell progression/migration (Fig. 4D/E) and the promotion on cell apoptosis (Fig. 4F). These data demonstrated that HGFR mediated miR-3681 function in the regulation of proliferation and migration in cervical cancer cells.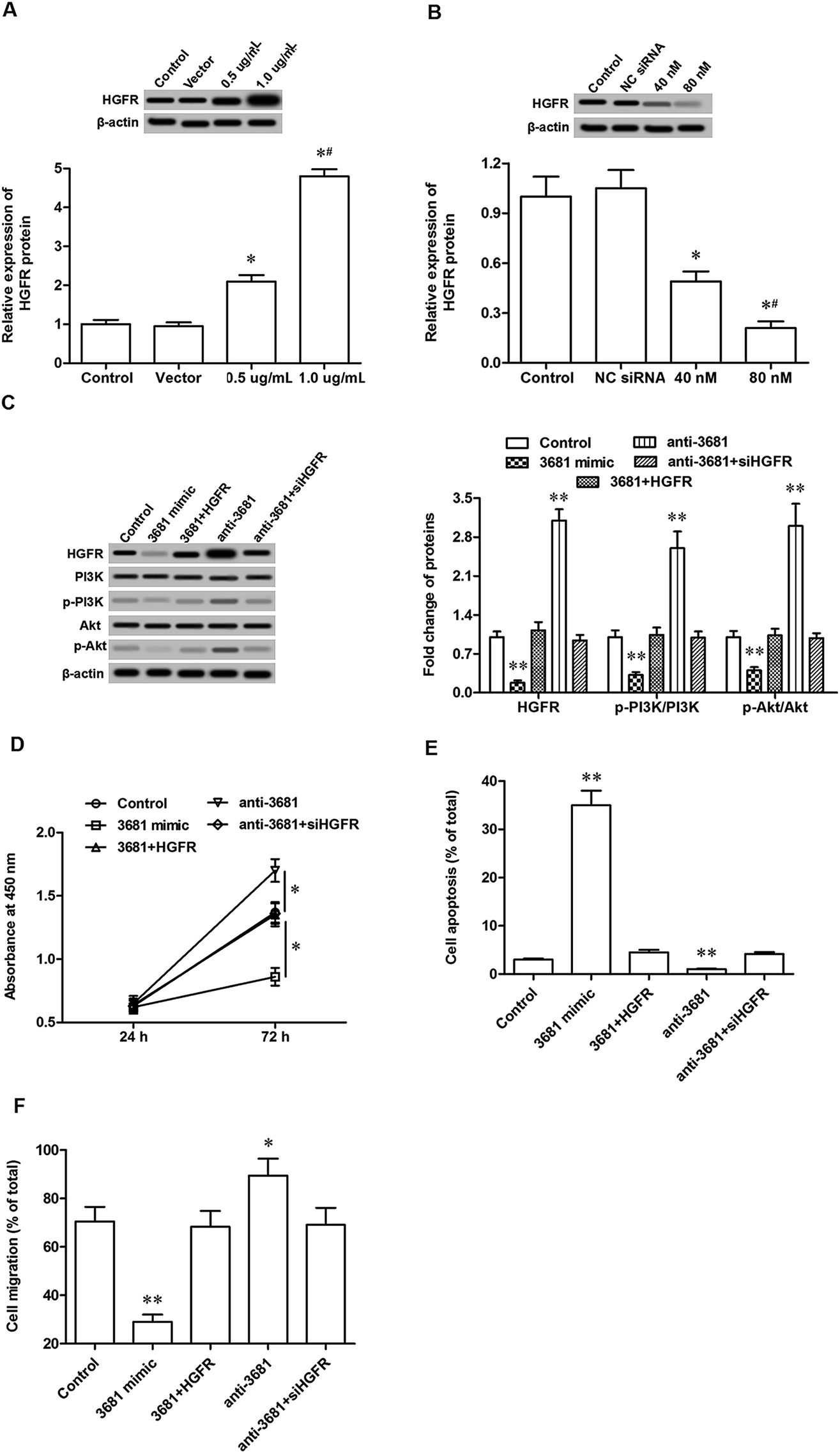 | ||
| Fig. 4 MiR-3681 regulated proliferation and migration in cervical cancer cells in a HGFR-dependent manner. (A) The overexpression efficacies of pcDNA-HGFR determined by Western blotting. (B) The interference efficacies of HGFR siRNA determined by Western blotting. (C) MiR-3681 negatively regulated HGFR protein expression and its downstream pathway PI3K/Akt. (D) HGFR mediated miR-3681 function in the regulation of proliferation in cervical cancer cells. (E) HGFR mediated miR-3681 function in the regulation of apoptosis in cervical cancer cells. (F) HGFR mediated miR-3681 function in the regulation of migration in cervical cancer cells. The C-33A cells were transfected with 20 nM NC mimic, 20 nM miR-3681 mimic, 20 nM miR-3681 mimic plus 1 μg of pcDNA-HGFR, 40 nM antagomir, or 40 nM of miR-3681 antagomir plus 80 nM HGFR siRNA. After incubation for 72 h, Western blotting was used to detect the protein levels of HGFR, PI3K, pPI3K, Akt and pAkt. Cell proliferation was detected with the CCK-8 method, cell apoptosis was detected with AnnexinV-PI staining combined with flow cytometry assays, and cell migration was detected with the Transwell migration assay. *P < 0.05, **P < 0.01 vs. control; #P < 0.05 vs. 20 nM mimic or 40 nM antagomir. | ||
4. Discussion
MiR-3681 was found by a deep sequencing of small RNA libraries in human leukocytes.15 Up to now, there have not been reports revealing the exact role of miR-3681 in any biological processes. A couple of studies showed that miR-3681 was dysregulated in progression of some diseases including cancer and coeliac disease.15,16 In this study, we performed a miRNA sequencing analysis in human cervical cancer tissues and matched adjacent non-tumor tissues. MiR-3681 and other 4 characterized miRNAs were found sharply downregulated in the cancer tissue. We confirmed their expression by qPCR analysis and determined miR-3681 as a candidate regulator in cervical cancer. Our gain-and-loss-function experiments demonstrated that miR-3681 negatively regulated proliferation/migration and promoted apoptosis of cervical cancer cells.Hepatocyte growth factor receptor (HGFR) gene is also known as the c-met protooncogene (MET), a relatively conserved gene among mammalians.17 HGFR protein is a single-pass tyrosine kinase receptor, which is required in embryonic development and organogenesis.18 Recent studies revealed that HGFR played important roles in progression of wound healing. HGFR has been demonstrated to be greatly upregulated or activated in many kinds of cancers, such as leukemia, lymphoma, sarcoma, ovarian cancer, renal carcinoma, etc.19–22 In this study, we found that HGFR was robustly increased in human cervical cancer cell lines compared with normal cervical epithelial cells. Moreover, HGFR mediated the function of miR-3681 in proliferation, apoptosis and migration of cervical cancer cells. Similar to the results of our study, Comunoğlu and his group reported that HGFR was overexpressed in cervical intraepithelial neoplasia and could be regarded as an important parameter in cervical early oncogenesis.23 Another study by Tsai et al. showed that HGFR expression was positively correlated with the International Federation of Gynecology and Obstetrics stage classification and was an independent predictor for the overall survival of cervical patients.24
HGFR is expressed on the surfaces of various cells, the activation of which by HGF or other stimuli initiates a series of intracellular signals.25 In cancer cells, aberrant activation of HGFR signaling promotes tumor development and progression by stimulating multiple carcinogenic pathways, including the PI3K/AKT, Ras/MAPK, JAK/STAT, Wnt/β-catenin and other signaling pathways. The PI3K/AKT pathway has been applied as a therapeutic target for cervical cancer, which plays a crucial role in the regulation of proliferation, apoptosis, invasion and drug resistance in cervical cancer.26–28 In this study, we showed that HGFR downregulation by miR-3681 reduced activation of the PI3K/AKT pathway and cell proliferation/migration in cervical cancer cells, which could be counteracted by HGFR overexpression. Conversely, HGFR upregulation by miR-3681 promoted activation of the PI3K/AKT pathway and cell proliferation/migration in cervical cancer cells, which could be antagonized by HGFR knockdown.
5. Conclusions
In conclusion, miR-3681 expression was sharply downregulated in human cervical cancer tissues and cell lines. MiR-3681 had a negative effect on the proliferation and invasion of cervical cancer cells through directly targeting and post-transcriptionally downregulating the oncogene HGFR.Conflicts of interest
The authors declare that they have no conflicts of interest.References
- P. Sasieni and A. Castanon, J. Med. Screen., 2012, 19, 127–132 CrossRef.
- D. W. Cramer, Cancer, 2015, 34, 2018–2027 CrossRef.
- A. D. Mwaka, C. G. Orach, E. M. Were, L. Georgios, W. Henry and R. Martin, Health Expectations, 2016, 19, 854–867 CrossRef.
- S. Rubina and C. M. Krishna, J. Cancer Res. Ther., 2015, 11, 10 CrossRef CAS.
- S. Giglio and A. Vecchione, J. Nucleic Acids Invest., 2010, 1, e4 CrossRef.
- B. Wang, J. Nucleic Acids, 2013, 2013, 951570 Search PubMed.
- X. Hu, J. K. Schwarz, J. S. Lewis, P. C. Huettner, J. S. Rader, J. O. Deasy, P. W. Grigsby and X. Wang, Cancer Res., 2010, 70, 1441 CrossRef CAS.
- V. Laengsri, U. Kerdpin, C. Plabplueng, L. Treeratanapiboon and P. Nuchnoi, Lab. Med., 2018, 49, 97–111 CrossRef.
- L. Wang, L. Chang, Z. Li, Q. Gao, D. Cai, Y. Tian, L. Zeng and M. Li, Med. Oncol., 2014, 31, 934–941 CrossRef.
- L. Wang, Q. Wang, H. L. Li and L. Y. Han, Asian Pac. J. Cancer Prev., 2013, 14, 2113–2118 CrossRef.
- H. Ping, X. Jie and S. Liu, Biomed. Pharmacother., 2016, 83, 850–856 CrossRef.
- J. Cong, R. Liu, X. Wang, H. Jiang and Y. Zhang, Artif. Cells, Nanomed., Biotechnol., 2015, 1–8 CAS.
- Z. Fan, H. Cui, H. Yu, Q. Ji, L. Kang, B. Han, J. Wang, Q. Dong, Y. Li, Z. Yan, X. Yan, X. Zhang, Z. Lin, Y. Hu and S. Jiao, Oncogenesis, 2016, 5, e223 CrossRef CAS.
- X. Yu, W. Zhao, X. Yang, Z. Wang and M. Hao, Int. J. Gynecol. Cancer, 2016, 26, 851–858 CrossRef.
- C. Vaz, H. M. Ahmad, P. Sharma, R. Gupta, L. Kumar, R. Kulshreshtha and A. Bhattacharya, BMC Genomics, 2010, 11, 288 CrossRef.
- V. Vaira, L. Roncoroni, D. Barisani, G. Gaudioso, S. Bosari, G. Bulfamante, L. Doneda, D. Conte, C. Tomba, M. T. Bardella, S. Ferrero, M. Locatelli and L. Elli, Clin. Sci., 2014, 126, 417–423 CrossRef CAS PubMed.
- Y. Xu, W. Xia, D. Baker, J. Zhou, H. C. Cha, J. J. Voorhees and G. J. Fisher, J. Biol. Chem., 2011, 286, 15980 CrossRef CAS.
- M. Anum, A. H. Muhammad, S. Asfandyar, K. A. M. Hammad and S. Shafaq, Infect. Agents Cancer, 2013, 8, 13 CrossRef.
- G. Fan and N. Nicholas, Small GTPases, 2017, 3, 1–5 CrossRef PubMed.
- Y. Sagara, Y. Miyata, K. Nomata, K. Abe, J. Eguchi, T. Hayashi, H. Sakai, S. Kanda and H. Kanetake, Pathol., Res. Pract., 2009, 205, 57–61 CrossRef PubMed.
- M. Jücker, A. Günther, G. Gradl, C. Fonatsch, G. Krueger, V. Diehl and H. Tesch, Leuk. Res., 1994, 18, 7 CrossRef.
- K. Helou, V. Wallenius, Y. Qiu, F. Ohman, F. Ståhl, K. Klinga-Levan, L. G. Kindblom, N. Mandahl, J. O. Jansson and G. Levan, Oncogene, 1999, 18, 3226 CrossRef CAS.
- C. Comunoğlu, K. Boynukalin, M. G. Uğur, R. Al, G. M. Kuzey and C. Baykal, Eur. J. Gynaecol. Oncol., 2012, 33, 625–628 Search PubMed.
- H. W. Tsai, N. H. Chow, C. P. Lin, S. H. Chan, C. Y. Chou and C. L. Ho, Hum. Pathol., 2006, 37, 198–204 CrossRef CAS.
- Y. Zhang, M. Xia, K. Jin, S. Wang, H. Wei, C. Fan, Y. Wu, X. Li, X. Li and G. Li, Mol. Cancer, 2018, 17, 45 CrossRef PubMed.
- K. Wang, Y. Zhuang, C. Liu and Y. Li, Arch. Biochem. Biophys., 2012, 526, 38–43 CrossRef CAS PubMed.
- F. Wang, W. H. Tan, W. Liu, Y. X. Jin, D. D. Dong, X. J. Zhao and Q. Liu, Eur. Rev. Med. Pharmacol. Sci., 2018, 22, 1891 CAS.
- A. Li, Y. Gu, X. Li, H. Sun, H. Zha, J. Xie, J. Zhao, M. Huang, L. Chen, Q. Peng, Y. Zhang, Y. Weng and L. Zhou, Oncol. Lett., 2018, 15, 5685–5693 Search PubMed.
| This journal is © The Royal Society of Chemistry 2019 |