
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced electrocatalytic hydrodechlorination of 2,4-dichlorophenoxyacetic acid by a Pd-Co3O4/Ni foam electrode
Qiuxiang Liuab,
Yanting Shenb,
Shuang Song ab and
Zhiqiao He*b
ab and
Zhiqiao He*b
aCollaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, Hangzhou 310032, People's Republic of China
bCollege of Environment, Zhejiang University of Technology, Hangzhou 310032, People's Republic of China. E-mail: zqhe@zjut.edu.cn; Fax: +86-571-88320276; Tel: +86-571-88320726
First published on 17th April 2019
Abstract
A new Pd-Co3O4/Ni foam electrode was synthesized by a facile two-step method comprising co-electrodeposition and calcination. Compared with Ni foam-supported Pd electrodes obtained by electrodeposition or chemical deposition, the new Pd-Co3O4/Ni foam electrode exhibited greatly enhanced catalytic hydrodechlorination activity. The introduction of Co3O4 reduced the amount of Pd required. For the same degree of dechlorination of 2,4-D, only 25% of the Pd was required in the Pd-Co3O4/Ni foam electrode compared with the Ni foam electrode prepared by chemical deposition. Various characterizations indicated that Co3O4 on the surface of the Ni foam enhanced catalytic performance through accelerated generation of atomic H*. In addition, the good distribution of macropores, providing a larger specific surface area and lower electron transfer impedance, enabled more adsorption of atomic 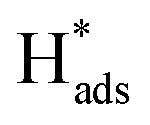 .
.
1. Introduction
The chlorinated organic compound (COC) 2,4-dichlorophenoxyacetic acid (2,4-D) is a commonly used herbicide to control a wide range of broadleaf weeds and grasses in crops because of its low cost and good selectivity.1,2 However, 2,4-D is a weakly biodegradable pollutant that is easily transported into bodies of water as a result of leaching and surface runoff.3–5 Additionally, studies on the toxicological effects of 2,4-D have shown that it is a threat to public health.6,7 Some studies have reported toxicity of 2,4-D as a consequence of endocrine disrupting activities in animals and humans, causing irreparable damage to the central nervous system. The prevalence and toxicity of 2,4-D have prompted research into the reductive dechlorination of COCs to form less dangerous products.8–11Compared with various methods of reduction, including chemical and biological methods and photochemical dechlorination,12–15 electrocatalytic reductive dechlorination is considered to be efficient because of intrinsic advantages such as mild reaction conditions, rapid reaction rate and the absence of recalcitrant secondary contaminants.9,16–18 The electrolytic dechlorination of COCs has been studied on Ag, Au, Pd, Pt, Cu, Fe, Ni, Pb, Zn.19 In particular, palladium (Pd) is generally utilized as the catalyst for hydrodechlorination reactions.20,21 The metal has the ability to retain large amounts of nascent adsorbed atomic 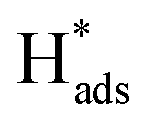 via Pd–H bonds that result from multiple σ-bonding interactions between hydrogen atoms and the d orbitals of the Pd atoms.22,23 However, Pd is expensive and it is desirable to make the electrocatalytic hydrodechlorination process more economically feasible. Transition metal oxides and some low cost metals, such as TiO2, CeO2, NiO, Co3O4, Cu and Ag,24–28 have shown potential as co-catalysts to reduce the amount of precious metal catalyst, increase catalytic activity, enhance tolerance to catalyst poisoning, and they cost less than precious metals.
via Pd–H bonds that result from multiple σ-bonding interactions between hydrogen atoms and the d orbitals of the Pd atoms.22,23 However, Pd is expensive and it is desirable to make the electrocatalytic hydrodechlorination process more economically feasible. Transition metal oxides and some low cost metals, such as TiO2, CeO2, NiO, Co3O4, Cu and Ag,24–28 have shown potential as co-catalysts to reduce the amount of precious metal catalyst, increase catalytic activity, enhance tolerance to catalyst poisoning, and they cost less than precious metals.
Recently, there has been widespread application of transition metal oxides (Fe3O4, Co3O4 and NiO) for hydrogen production via the hydrogen evolution reaction (HER) as water-splitting electrocatalysts in the field of energy research.29–32 According to the well-known theoretical “Volcano Curve”,33 the catalytic performance of Co is slightly inferior to those of the expensive platinum group metals such as Pt, Pd and Ir. It has been demonstrated by other researchers that Co3O4 enhances the performance of HER.34 Since H2 molecules would readily dissociate on the surface of Pd,35 it is expected that Co3O4 could be used to improve the activity of Pd in electrochemical hydrodechlorination of 2,4-D.
In this work, hybrids of Pd and Co3O4 supported on Ni foam (Pd-Co3O4/Ni foam) were fabricated by co-electrodeposition followed by calcination, and used as electrodes for electrochemical hydrodechlorination of 2,4-D. Ni foam was selected as the electrode substrate because of its large specific surface area, porous structure and excellent electrical conductivity.36–38 The reaction mechanism and pathway of 2,4-D dechlorination by the Pd-Co3O4/Ni foam electrode were also studied.
2. Experimental
2.1. Materials
All chemical reagents used in this study were of analytical grade and purchased from Huadong Medical Co., Ltd, China. Nickel foam with a pore density of 110 ppi was purchased from Suzhou Jiashide Materials, Ltd, China. Ultra-pure water (18.2 mΩ cm−1) was obtained from a Millipore-Q system.2.2. Electrode preparation
In a typical procedure, a piece of Ni foam (20 × 20 × 1.2 mm) was used as the substrate for electrodeposition. Before electrode preparation, the Ni foam was dipped in acetone in an ultrasonic bath for 20 min, rinsed with H2SO4 solution (0.5 mol L−1) for 5 min to remove the surface oxidized layer,39 and then rinsed with ultrapure water and dried in a stream of nitrogen. The electrodeposition device was a standard three-electrode glass cell (100 mL) consisting of the clean Ni foam working electrode, a platinum plate counter electrode and a saturated calomel electrode (SCE) in a water bath (THD-2015, Ningbo Tian Heng Instrument Factory, China) at 25 °C ± 1 °C. All of the potentials mentioned in this paper are relative to the SCE unless otherwise specified. Firstly, the Ni foam was subjected to constant potential deposition at −1.0 V for 20 min in a 40 mL aqueous solution containing 5 mmol L−1 Co(NO3)2 and 1 mmol L−1 PdCl2. The treated Ni foam electrode was carefully rinsed several times with ultrapure water and dried in N2. Subsequently, the electrode was placed in a quartz tube and calcined at 250 °C for 120 min with a ramping rate of 1 °C min−1 to transform into the hybrid Pd-Co3O4/Ni foam electrode. The Pd loading was determined to be 0.47 mg cm−2 using inductively coupled plasma optical emission spectrometry (ICP-OES) by dissolution of the electrode in nitrohydrochloric acid. Two other electrodes with the same Pd loading (0.47 mg cm−2) were prepared for comparison: constant potential deposition of Pd on Ni foam (Pd(CPD)/Ni foam) was performed at −1.0 V for 25 min in a 40 mL aqueous solution containing 1 mmol L−1 PdCl2; chemical deposition of Pd on Ni foam (Pd(CD)/Ni foam) was carried out according to a published method.40 In addition, the Pd(CD)/Ni foam electrode was prepared with 4 times the Pd loading (4Pd(CD)/Ni foam). The Co3O4 supported on the Ni foam (Co3O4/Ni foam) electrode was prepared using the same method used for Pd-Co3O4/Ni foam in the absence of PdCl2.2.3. Batch experiments
Electrochemical dechlorination of 2,4-D was conducted in a conventional two-compartment glass H-cell in a water bath at 25 °C ± 1 °C. The initial concentration of 2,4-D (50 mg L−1) was selected to be within the usual range for chlorinated organic compounds found in agricultural wastewater.41 The cathode (80 mL) and anode (50 mL) compartments were separated by Nafion-117 membranes, and the catholyte was magnetically stirred during electrolysis. In each experiment, aqueous Na2SO4 solution (0.017 mol L−1, 30 mL) containing 2,4-D (50 mg L−1) was added into the cathode compartment and aqueous Na2SO4 (0.017 mol L−1, 25 mL) was added into the anode compartment. The cathode (modified Ni foam) and anode (platinum foil) were placed vertically at a separation of about 80 mm. The surface of the working electrode exposed to electrolytes was a square film with a geometric surface area of 4 cm2. Platinum foil with a surface area of 4 cm2 was used as the counter electrode and a SCE was used as the reference electrode. A constant current of 0.006 A was supplied by the electrochemical workstation (CHI760E Shanghai Chen Hua Instrument Co., China) for 120 min. Samples (0.4 mL) were withdrawn from the catholyte at predetermined time intervals for analysis.The concentrations of 2,4-D, 2-chlorophenoxyacetic acid (2-CPA), 4-chlorophenoxyacetic acid (4-CPA) and phenoxyacetic acid (PA), were analyzed on an Agilent 1200 series high-performance liquid chromatograph (HPLC) equipped with an Agilent XDB-C18 column (150 × 4.6 mm). The mobile phase was methanol/water (0.2% H3PO4) (60/40 by volume) at a flow rate of 0.8 mL min−1. The chosen determination wavelength was 210 nm and the volume of the injected sample was 5 μL.
The current efficiency (CE) for the 2,4-D dechlorination was calculated using eqn (1):
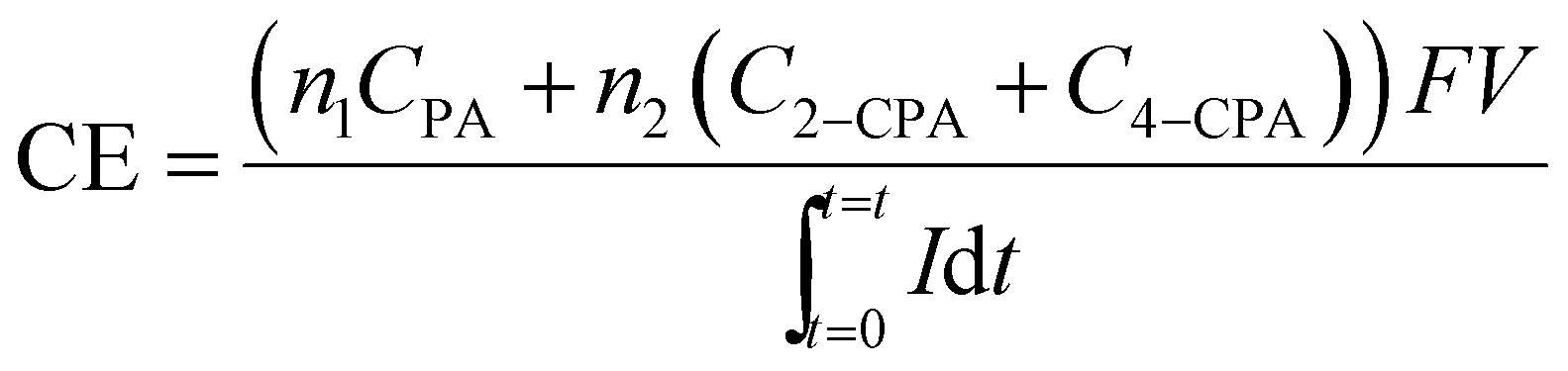 | (1) |
2.4. Electrode characterization
The Pd loading on the electrodes was analyzed by ICP-OES (Optima 7000DV, PerkinElmer Co. Ltd, USA). The morphology of the electrodes was analyzed by scanning electron microscopy (SEM, ΣIGMA, Carl Zeiss, Germany) coupled with energy dispersive X-ray spectroscopy (EDS, XFlash Detector 5010, Bruker Optics, Germany) operating at an accelerating voltage of 5 kV. The X-ray diffraction (XRD, D/max 2550/PC, Rigaku, Japan) patterns were recorded using Cu Kα radiation (λ = 1.5418 Å), operated at 40 kV and 30 mA and a scan rate of 10° min−1. X-ray photoelectron spectroscopy (XPS, 5000C ECSA system, PHI Co., USA) was conducted using a Mg-Kα source (hν = 1253.6 eV). The Brunauer–Emmett–Teller (BET) method was used to determine the surface area of the electrodes (ASAP 2020, McMeretik Instruments Co., Ltd, China).To study the electrocatalytic activity of Pd-Co3O4/Ni foam, linear sweep voltammetry (LSV), cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) analyses were performed on the prepared electrodes (20 × 20 × 1.2 mm) in a conventional three-electrode system using an electrochemical workstation. Platinum foil and SCE were used as the counter electrode and reference electrode, respectively. The electrolyte was 0.1 mol L−1 aqueous Na2SO4, which was purged with high purity N2 for 10 min to remove dissolved O2. The LSV curves were determined from −1.5 to 0 V at 20 mV s−1 and CV scans were recorded between −1.0 and 0.0 V at 50 mV s−1. The EIS spectra were recorded over the frequency range of 0.1–1000 kHz with a 5 mV amplitude at an open circuit voltage. For comparison, the electrocatalytic activities of Ni, Co3O4/Ni, and Pd(CPD)/Ni foam samples were measured under similar conditions.
3. Results and discussion
3.1. Characterization
Fig. 1 shows the XRD patterns of Pd-Co3O4/Ni, Pd(CPD)/Ni and Pd(CD)/Ni foams. As can be seen, diffraction peaks assigned to (111), (200) and (220) planes of Ni (JCPDS card no. 04-0850),42 and the (111) plane of Pd (JCPDS card no. 46-1043)43 were observed on all three electrodes. One differing feature was the presence of three weak typical diffraction peaks at 36.8°, 59.3° and 65.2° corresponding to the (311), (511), and (440) planes of Co3O4 (JCPDS card no. 042-1467)44 on the Pd-Co3O4/Ni foam. This confirmed that Co3O4 and Pd were successfully deposited onto the surface of the Ni foam.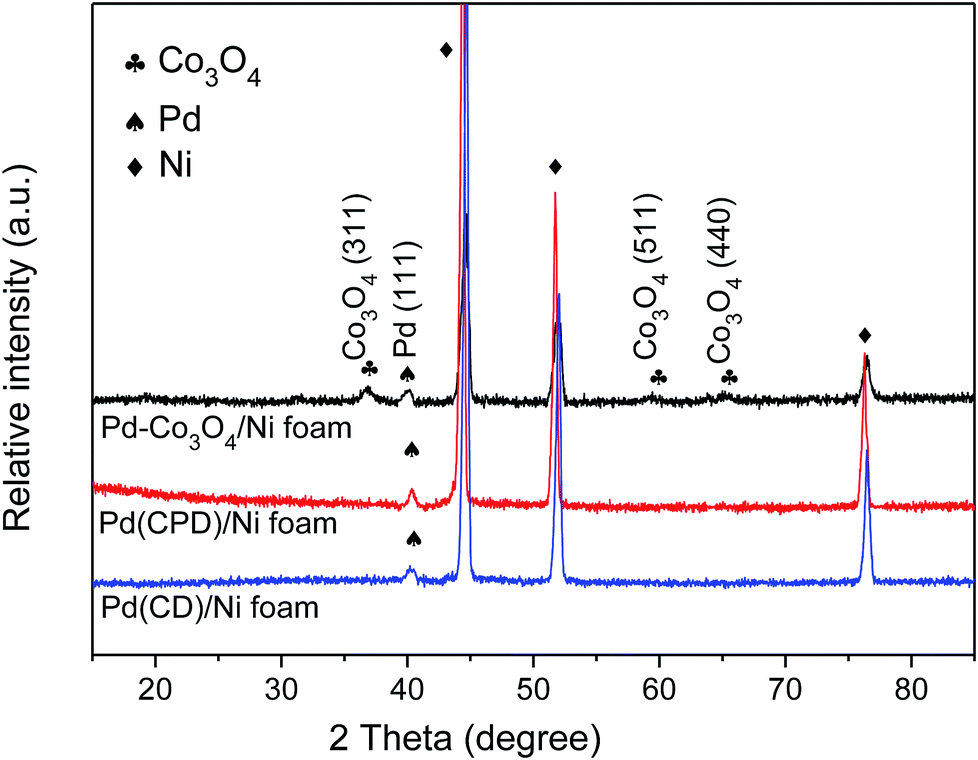 | ||
| Fig. 1 The XRD patterns of Pd-Co3O4/Ni, Pd(CPD)/Ni and Pd(CD)/Ni foams. | ||
XPS was used to characterize the elemental composition and understand the chemical states of Pd and Co on the surface of the Ni foam electrode. The overall XPS spectrum (Fig. 2(a)) of the Pd-Co3O4/Ni foam exhibited peaks corresponding to Co 2p, O 1s, Pd 3d and C 1s. High resolution XPS spectra of Co 2p, Pd 3d and O 1s are shown in Fig. 2(b–d). The high resolution XPS spectrum of Co 2p showed two peaks at binding energies of 779.8 and 794.8 eV with a spin-energy separation of 15 eV. These two peaks are ascribed to Co 2p3/2 and Co 2p1/2, respectively, which is in good agreement with reports in the literature on the characterization of Co3O4.45,46 The Pd 3d spectrum contained two spin–orbit peaks that corresponded to signals for Pd 3d5/2 and Pd 3d3/2 at binding energies of 336.6 and 342.0 eV, respectively. The value of 336.6 eV was higher than the binding energy for metallic Pd 3d5/2 (335.4 eV) due to superficial oxidation of Pd as a consequence of the nano-scale nature of the Co3O4 and Pd co-deposition on the Ni foam.47,48 The high resolution XPS spectrum of O 1s was fitted into two peaks at binding energies of 529.8 and 530.8 eV, corresponding to the Co–O bond and surface absorbed water, respectively.49–51
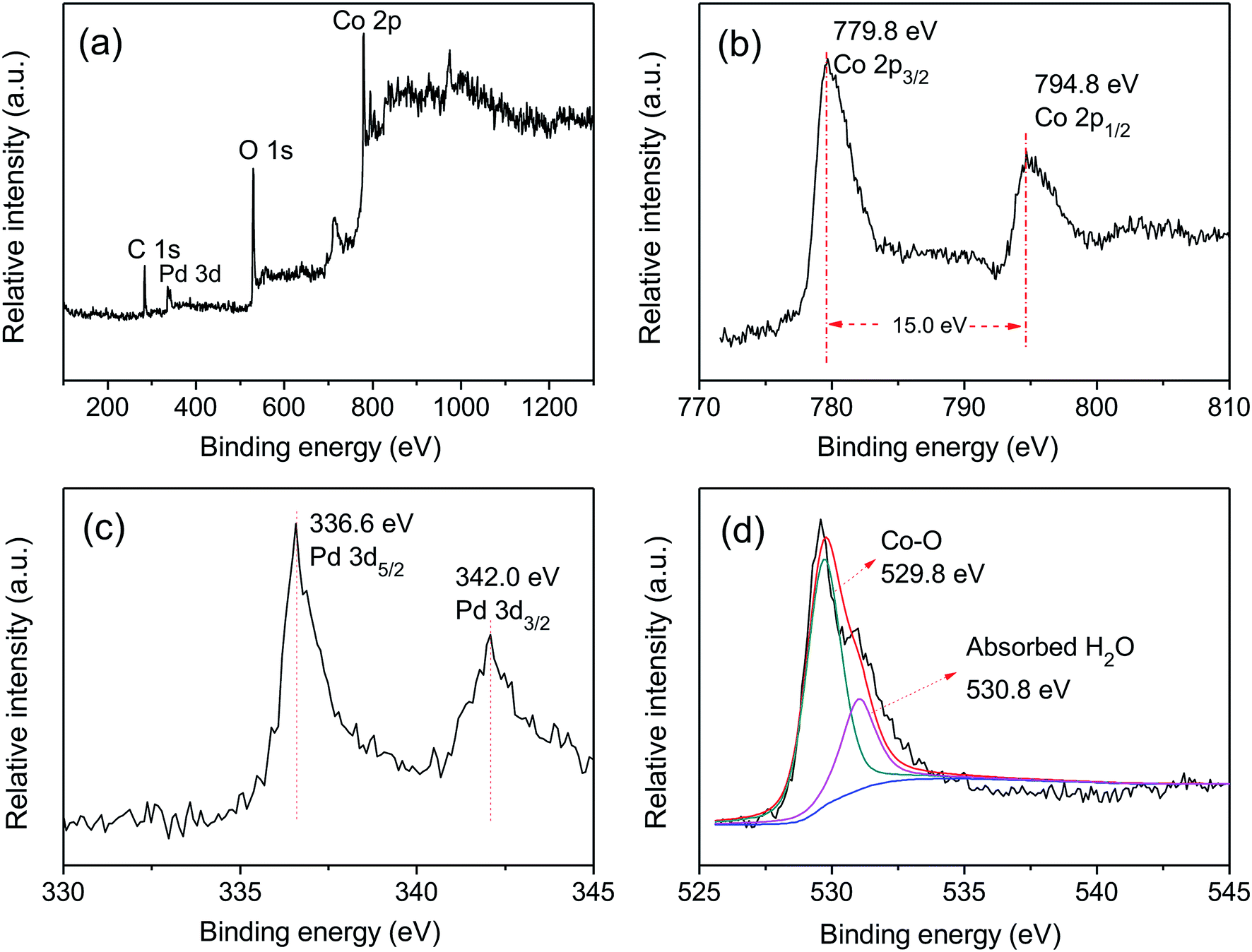 | ||
| Fig. 2 The XPS spectra of (a) overall spectra, (b) Co 2p, (c) Pd 3d and (d) O 1s for the Pd-Co3O4/Ni foam. | ||
Fig. 3 shows the determination of elemental composition of the electrode surface by EDS analysis. As can be seen, Ni, Co, O and Pd were present on the Pd-Co3O4/Ni foam, while only Ni, Pd and a small amount of oxygen, caused by surface oxidation of the metals, were present on the Pd(CD)/Ni and Pd(CPD)/Ni foam electrodes.
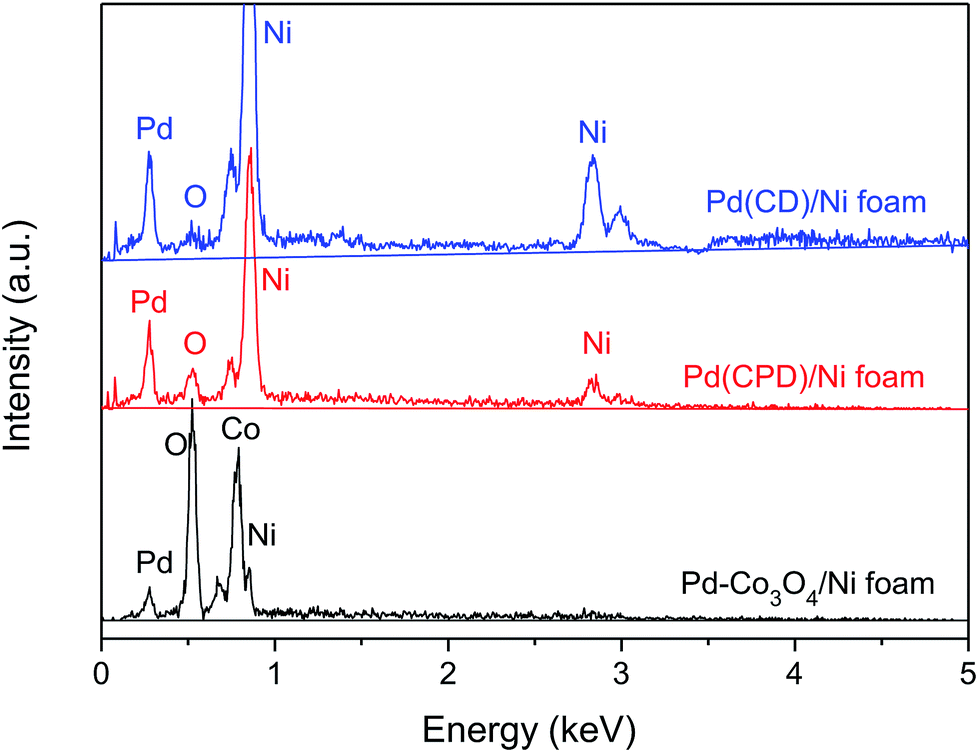 | ||
| Fig. 3 The EDS spectra of Pd-Co3O4/Ni, Pd(CPD)/Ni and Pd(CD)/Ni foams. | ||
The XRD and XPS characterizations confirmed the presence of Pd and Co3O4 on the surface of the Ni foam after constant potential deposition at a potential of −1.0 V and calcining at 250 °C. The EDS spectrum further confirmed the result.
The preparation of the Pd-Co3O4/Ni foam electrode was achieved in two steps. In the first step, a green Co(OH)2 precursor and Pd were co-electrodeposited onto the Ni foam at a potential of −1.0 V. The origin of Co(OH)2 generation has been explained previously.37 Electrodeposition of Pd on the Ni foam surface at a potential of −1.0 V has also been reported in the literature.52 In the second step, Co(OH)2 was calcined in air at 250 °C and transformed to black Co3O4 in a simple oxidation reaction. ICP-OES results showed a Pd loading of 0.47 mg cm−2 and a cobalt loading of 0.4 mg cm−2.
Fig. 4 shows the surface morphological properties of the fresh Ni foam, as-prepared Pd(CD)/Ni foam, Pd(CPD)/Ni foam and Pd-Co3O4/Ni foam electrodes. As observed in Fig. 4(a), the fresh Ni foam exhibited an irregular block structure with grooves on the surface. An irregular dense agglomeration of Pd particles was formed on the Pd(CD)/Ni foam electrode after chemical deposition (Fig. 4(b)), which was warped in many places because of weak contact with the Ni foam substrate. In contrast, Fig. 4(c) shows that the Pd particles were more uniformly dispersed on the surface of Pd(CPD)/Ni and tightly bound to the Ni foam. Comparison of Fig. 4(c) and (d) reveals that Pd particles and Co3O4 nanosheets were evenly deposited on the surface of the Pd-Co3O4/Ni foam electrode. The Co3O4 nanosheets lay aslant or perpendicular, were evenly deposited on the surface of the Ni foam and interconnected with Pd particles, providing a highly open and porous structure. The open porous channels between the Co3O4 nanosheets and Pd particles facilitated fast penetration of the electrode.37 In other words, these hierarchical multi-porous structures ensured efficient contact between surface active sites and the electrolyte.53,54 Furthermore, the ultrathin macroporous surface facilitated electron transfer across the electrode surface. Therefore, the introduction of Co3O4 provided a particular surface morphology that the Pd(CD)/Ni and Pd(CPD)/Ni foams cannot.
 | ||
| Fig. 4 The SEM images of (a) Ni, (b) Pd(CD)/Ni, (c) Pd(CPD)/Ni and (d) Pd-Co3O4/Ni foams. | ||
According to the results of the BET test, the surface areas of the Pd(CD)/Ni, Pd(CPD)/Ni and Pd-Co3O4/Ni foam electrodes were 3.7, 4.3 and 8.5 m2 g−1, respectively, which also demonstrated that the Pd and Co3O4 co-deposition electrodes had larger surface areas. The larger surface area of the electrode provided more active sites for dechlorination reaction.
3.2. Comparative experiments with different electrodes
In general, the current density is a significant factor affecting the removal rate of 2,4-D by catalytic electrodes. As shown in Fig. 5(f), an increase of the applied current density from 0.5 to 1.5 mA cm−2 resulted in increased removal efficiency of 2,4-D from 22.5% to 94.2% during 60 min electrolysis. A further increase of current density to 2.5 mA cm−2 led to a slight reduction of dechlorination efficiency. This is because the high current density provides sufficient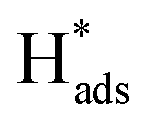 for the electrocatalytic hydrodechlorination, but an excessive current density accelerates H2 generation, retarding mass transfer of the chlorinated organic material to the reaction active site. Thus, the optimal current density for removal of 2,4-D is 1.5 mA cm−2.
for the electrocatalytic hydrodechlorination, but an excessive current density accelerates H2 generation, retarding mass transfer of the chlorinated organic material to the reaction active site. Thus, the optimal current density for removal of 2,4-D is 1.5 mA cm−2.
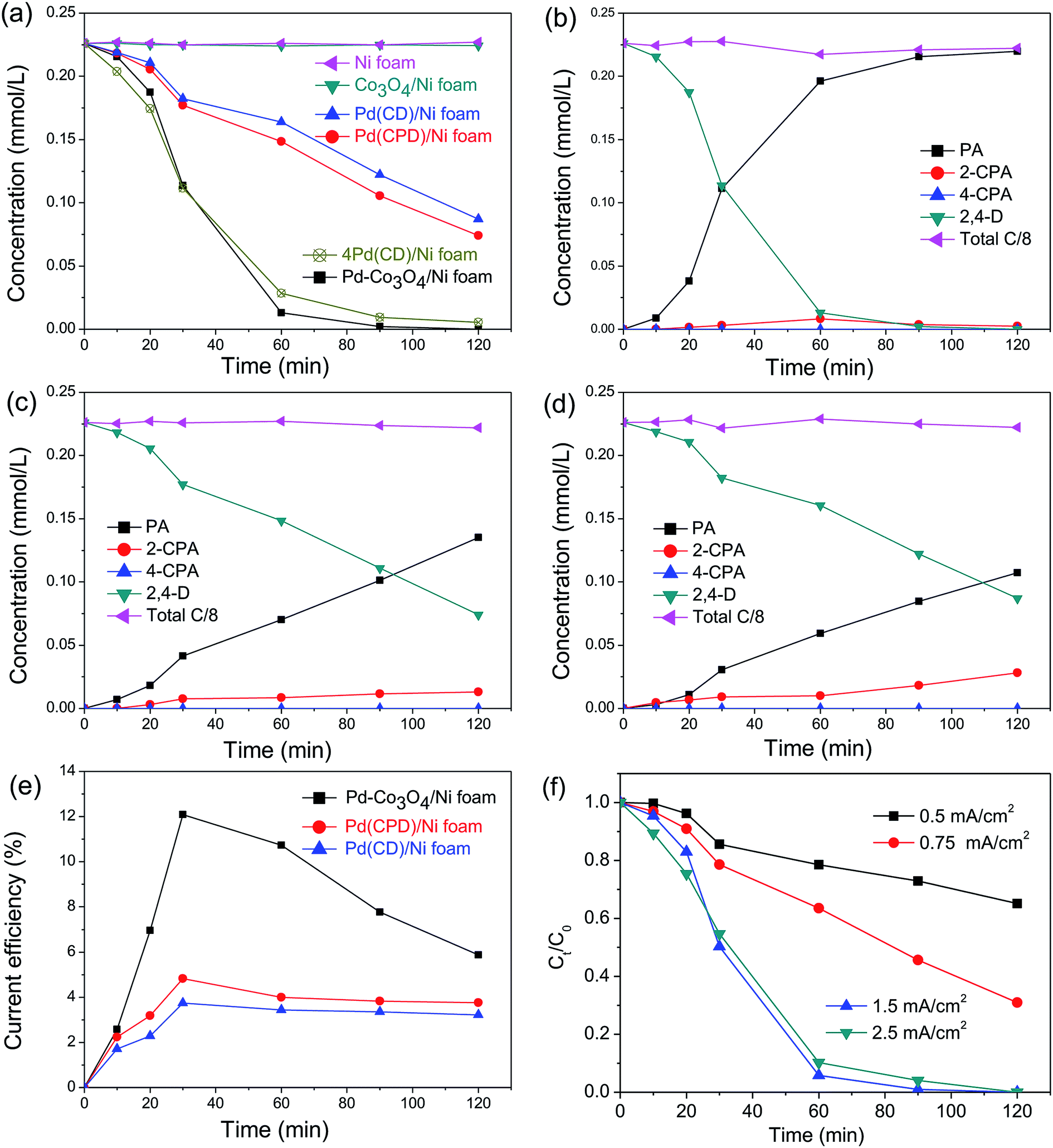 | ||
| Fig. 5 (a) The time course of 2,4-D concentration with as-prepared electrodes of Ni, Co3O4/Ni, Pd(CD)/Ni, Pd(CPD)/Ni, 4Pd(CD)/Ni and Pd-Co3O4/Ni foams; the time course of PA, 2-CPA, 4-CPA and 2,4-D concentrations with the (b) Pd-Co3O4/Ni foam electrode, (c) Pd(CPD)/Ni foam electrode and (d) Pd(CD)/Ni foam electrode; (e) the current efficiencies as functions of dechlorination time on Pd-Co3O4/Ni, Pd(CPD)/Ni and Pd(CD)/Ni foam electrodes at a current density of 1.5 mA cm−2; (f) the dechlorination efficiencies as functions of time on the Pd-Co3O4/Ni foam electrode at current densities of 0.5, 0.75, 1.5 and 2.5 mA cm−2. | ||
To compare electrocatalytic activity, removal of 2,4-D by electrochemical hydrodechlorination was conducted with as-prepared Ni, Co3O4/Ni, Pd(CD)/Ni, Pd(CPD)/Ni, 4Pd(CD)/Ni and Pd-Co3O4/Ni foams. Fig. 5(a) shows the time course for reaction of 2,4-D. After 120 min of electrolysis, hardly any 2,4-D was removed by the bare Ni foam, while 67.2% and 61.5% were removed by the Pd(CPD)/Ni foam and Pd(CD)/Ni foam, respectively. This was because Pd is a well-known catalyst that promotes dechlorination.55,56 The dechlorination of 2,4-D was remarkably enhanced by the Pd-Co3O4/Ni foam, and 94.2% of 2,4-D was removed after 60 min, indicating that the presence of Co3O4 played an important role in dechlorination. However, the rate of removal of 2,4-D by the Co3O4/Ni foam in the absence of Pd was similar to that using the bare Ni foam, which implied that Co3O4 acted as a co-catalyst for dechlorination. It is worthwhile noting that four times the amount of Pd (1.88 mg cm−2) in the 4Pd(CD)/Ni foam was required to achieve similar removal efficiency to the Pd-Co3O4/Ni foam (Fig. 5(a)). The results demonstrated that the Pd-Co3O4/Ni foam electrode significantly reduced the amount of Pd required while maintaining good dechlorination performance.
The molar concentrations of intermediates and the final product as a function of time for various electrodes at a current density of 1.5 mA cm−2 are shown in Fig. 5(a–d). As can be seen in Fig. 5(b–d), PA was the main product. Only a small amount of 2-CPA was generated, and 4-CPA was undetectable by HPLC. The rate of dechlorination of 2-CPA was slower than that of 4-CPA, since the attached –OCH2COOH group hindered cleavage of the C–Cl bond at the 2-position. The selectivity of the dechlorination was similar to that reported in the literature,38,55 in which electrocatalytic dechlorination of 2,4-D first yielded the mono-chlorinated intermediates, 2-CPA or 4-CPA, which were subsequently converted to PA, the ultimate dechlorinated product. The sum concentration of all products and the remaining reactants translated to about 95.9–101.0% of the initial total carbon mass, suggesting that the adsorption of these pollutants on the electrode was negligible during the reaction. The finding that the total amount of carbon was unchanged is consistent with results reported in the literature.40,57
Fig. 5(e) shows the variation of CEs on Pd-Co3O4/Ni, Pd(CPD)/Ni and Pd(CD)/Ni foam electrodes over time at a current density of 1.5 mA cm−2. All of the CEs increased first and then declined as the reaction time was extended. This is likely due to establishment of an equilibrium between the hydrogen atoms in the solid solution phase and those in the metal hydride phase in the preliminary stage.57 Subsequently, the 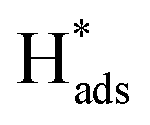 in solid solution is consumed for dechlorination of 2,4-D. Consequently, an initial rise of the CE was observed. The subsequent reduction of CE might be due to limited mass transfer of the reactants.11,58
in solid solution is consumed for dechlorination of 2,4-D. Consequently, an initial rise of the CE was observed. The subsequent reduction of CE might be due to limited mass transfer of the reactants.11,58
The maximal CEs of the Pd-Co3O4/Ni, Pd(CPD)/Ni and Pd(CD)/Ni foam electrodes at a current density of 1.5 mA cm−2 were determined to be 12.1%, 4.8% and 3.7%, respectively. Clearly, the maximum CE of the Pd-Co3O4/Ni foam electrode was ∼2.5-fold that of the Pd(CPD)/Ni electrode and ∼3.3-fold that of the Pd(CD)/Ni electrode. Therefore, the Pd-Co3O4/Ni foam electrode has superior dechlorination activity compared with the Pd(CPD)/Ni and Pd(CD)/Ni foam electrodes.
Regarding the electrochemical dechlorinated process, two mechanisms that have been proposed involve direct electron transfer and indirect reduction via atomic H*.59–61 As shown in Fig. 5(a) where no Pd catalyst was used, the Ni and Co3O4/Ni foams exhibited almost no decrease of 2,4-D at a current density of 1.5 mA cm−2 over 120 min of electrolysis, suggesting an indirect electrochemical dechlorination process in the reaction of 2,4-D.62 In this work, the indirect reduction mechanism may involve the following pathways,63 as shown in eqn (2)–(5):
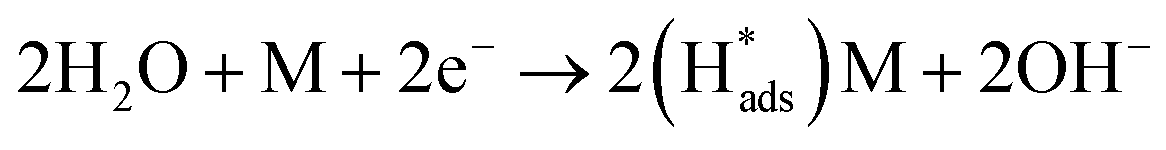 | (2) |
| R–Cl + AS ↔ (R–Cl)adsAS | (3) |
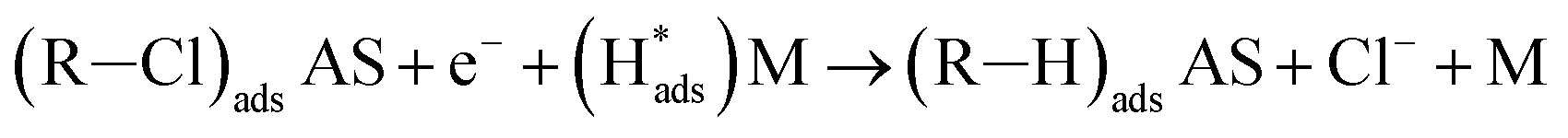 | (4) |
| (R–H)adsAS ↔ R–H + AS | (5) |
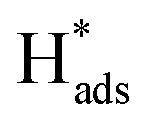 is adsorbed atomic hydrogen, and M is the Pd catalyst.
is adsorbed atomic hydrogen, and M is the Pd catalyst.
3.3. Enhanced mechanism
It was reasonable to speculate that the introduction of Co3O4 could accelerate the hydrogen evolution reaction. To confirm this hypothesis, LSV measurements of Ni, Pd(CPD)/Ni, Pd-Co3O4/Ni, and Co3O4/Ni foams were recorded in the catholyte in the absence of 2,4-D. As shown in Fig. 6(a), the rates of HER were in the following order: Ni foam < Co3O4/Ni foam < Pd(CPD)/Ni foam < Pd-Co3O4/Ni foam. The introduction of Co3O4 on the Ni foam resulted in an improvement of potential of hydrogen evolution from −1.0 V (bare Ni foam) to −0.9 V (Co3O4/Ni foam). However, the Co3O4/Ni foam electrode does not enable dechlorination because Pd is the key catalyst to store atomic H*, which is the major reductant for catalytic hydrodechlorination.40,56,59 The potential of Pd-Co3O4/Ni foam (−0.50 V) was relatively positive compared to that of Pd(CPD)/Ni foam (−0.6 V), further indicating that HER was promoted in the presence of Co3O4. Although it would be inappropriate to draw conclusions on the effectiveness of dechlorination based simply on the LSV data for the HER as a side reaction in the dechlorination of 2,4-D, it could offer some insights into the process.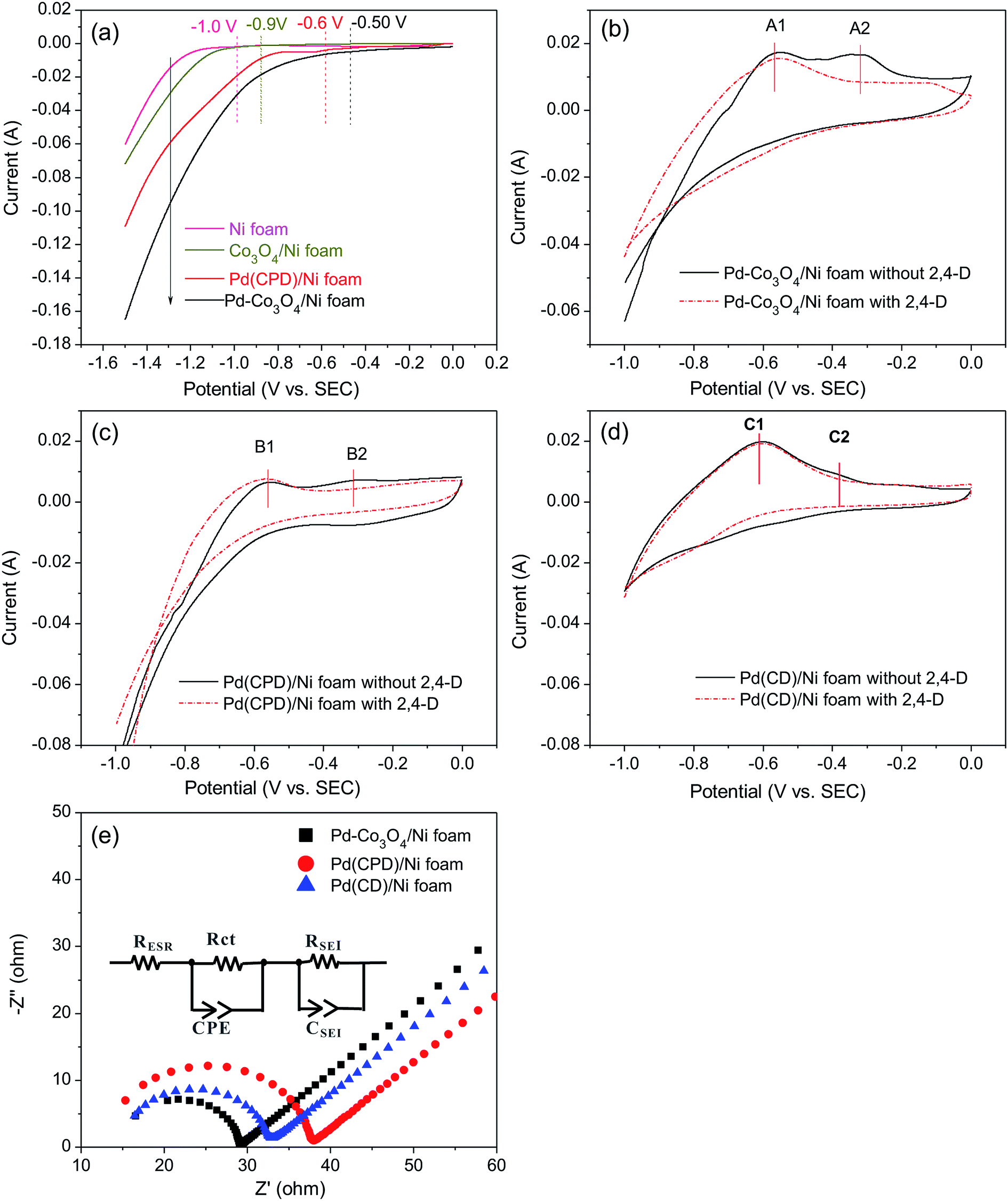 | ||
| Fig. 6 (a) LSV of Pd-Co3O4/Ni, Pd(CPD)/Ni, Co3O4/Ni and Ni foams; (b) CV of Pd-Co3O4/Ni foam; (c) CV of Pd(CPD)/Ni foam; (d) CV of Pd(CD)/Ni foam; (e) Nyquist diagrams of Pd-Co3O4/Ni, Pd(CPD)/Ni and Pd(CD)/Ni foams. The inset is the equivalent electrical circuit used to fit the EIS data (RESR is the electrolyte resistance, Rct denotes the charge transfer resistance, CPE represents the constant phase-angle element, RSEI and CSEI are the pseudo capacitance and resistance of the solid/electrolyte interface, respectively). Experimental conditions: 2,4-D concentration = 50 mg L−1 and support electrolyte (Na2SO4) concentration = 0.017 mol L−1. | ||
Generation of adsorbed atomic 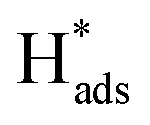 was confirmed on the Pd-Co3O4/Ni foam electrode by the CV curves shown in Fig. 6(b–d). Adsorbed atomic
was confirmed on the Pd-Co3O4/Ni foam electrode by the CV curves shown in Fig. 6(b–d). Adsorbed atomic 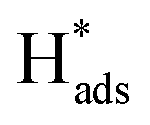 oxidation peaks were observed within potential ranges of −0.60 to −0.50 V and −0.40 to −0.30 V without addition of 2,4-D. According to previous studies,16,64 the two oxidation peaks could be associated with desorption of atomic
oxidation peaks were observed within potential ranges of −0.60 to −0.50 V and −0.40 to −0.30 V without addition of 2,4-D. According to previous studies,16,64 the two oxidation peaks could be associated with desorption of atomic 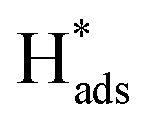 adsorbed on {111} and {100} planes of polycrystalline Pd. Since the adsorption energy of hydrogen on Pd {111} facets is stronger than that on {100} facets, the oxidation peak at 1-site (more negative potential) and 2-site (more positive potential) should be assigned to the
adsorbed on {111} and {100} planes of polycrystalline Pd. Since the adsorption energy of hydrogen on Pd {111} facets is stronger than that on {100} facets, the oxidation peak at 1-site (more negative potential) and 2-site (more positive potential) should be assigned to the 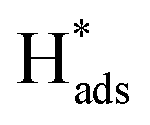 desorption on {111} and {100} planes of Pd. Interestingly, upon addition of 2,4-D, the peaks at A2, B2 and C2 totally disappeared, while peaks at A1, B1 and C1 were mostly unchanged. It can be concluded that active adsorbed atomic
desorption on {111} and {100} planes of Pd. Interestingly, upon addition of 2,4-D, the peaks at A2, B2 and C2 totally disappeared, while peaks at A1, B1 and C1 were mostly unchanged. It can be concluded that active adsorbed atomic 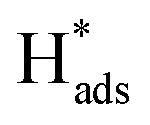 located at {111} planes were the key active hydrogen species for dechlorination. Furthermore, the current density of the A2 peak (4.2 mA cm−2) was higher than those of the B2 peak (1.93 mA cm−2) and C2 peak (1.83 mA cm−2), suggesting that more active adsorbed atomic
located at {111} planes were the key active hydrogen species for dechlorination. Furthermore, the current density of the A2 peak (4.2 mA cm−2) was higher than those of the B2 peak (1.93 mA cm−2) and C2 peak (1.83 mA cm−2), suggesting that more active adsorbed atomic 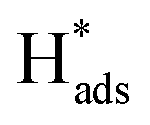 was present on the Pd-Co3O4/Ni foam.
was present on the Pd-Co3O4/Ni foam.
In addition, on the basis of the results and according to the literature,37 it was speculated that Co3O4 in the hybrid materials may have set up a hierarchical multi-porous structure,53,65 which can facilitate charge- and mass-transfer processes. To confirm this hypothesis, EIS analyses of the Pd-Co3O4/Ni foam, Pd(CPD)/Ni foam and Pd(CD)/Ni foam electrodes were conducted. In Fig. 6(e), the EIS curves show an equivalent series resistance RESR in the high frequency region, one depressed arc radius corresponding to the electron transfer resistance (Rct), a double layer capacitor represents the constant phase-angle element (CPE) region and an inclined spike reveals an electrode/electrolyte interface effect in the low frequency region.10,63,66 The size of the arc radius was reduced, revealing that the presence of Co3O4 decreased the interface impedance and accelerated electron transfer. The arc radius of the Pd(CD)/Ni foam was smaller than that of the Pd(CPD)/Ni foam, which can be explained by the resistivity of the conductive material being substantially increased as the Pd particle size decreased, as shown in other studies.67,68
In summary, the Pd-Co3O4/Ni foam enhances dechlorination because of the key role played by Co3O4, which accelerates the dissociation of H2O and electron transfer. This then produces more atomic H* intermediates, and more atomic H* is adsorbed by the Pd catalyst to facilitate dechlorination. Hence, the presence of Co3O4 greatly enhances dechlorination activity.
3.4. Reusability of the Pd-Co3O4/Ni foam electrode
Electrode stability is always a crucial concern for the practical application of contaminant degradation. The Pd-Co3O4/Ni foam electrode was evaluated to verify electrode stability via five cycles of 2,4-D dechlorination at a current density of 1.5 mA cm−2 as shown in Fig. 7. The 2,4-D removal efficiencies were 94.2%, 93.9%, 93.7%, 93.5%, and 93.2% after 60 min in each cycle, respectively. The 2,4-D removal efficiency was 100% after 120 min in each cycle. There was no significant change in the 2,4-D removal efficiency after five reaction cycles, which indicated that the Pd-Co3O4/Ni foam had good reusability. Furthermore, it was found that about 1.09% Pd was lost, as analyzed by ICP-OES during the dechlorination of 2,4-D using the Pd-Co3O4/Ni foam electrode over five reaction cycles, indicating that the electrode was stable.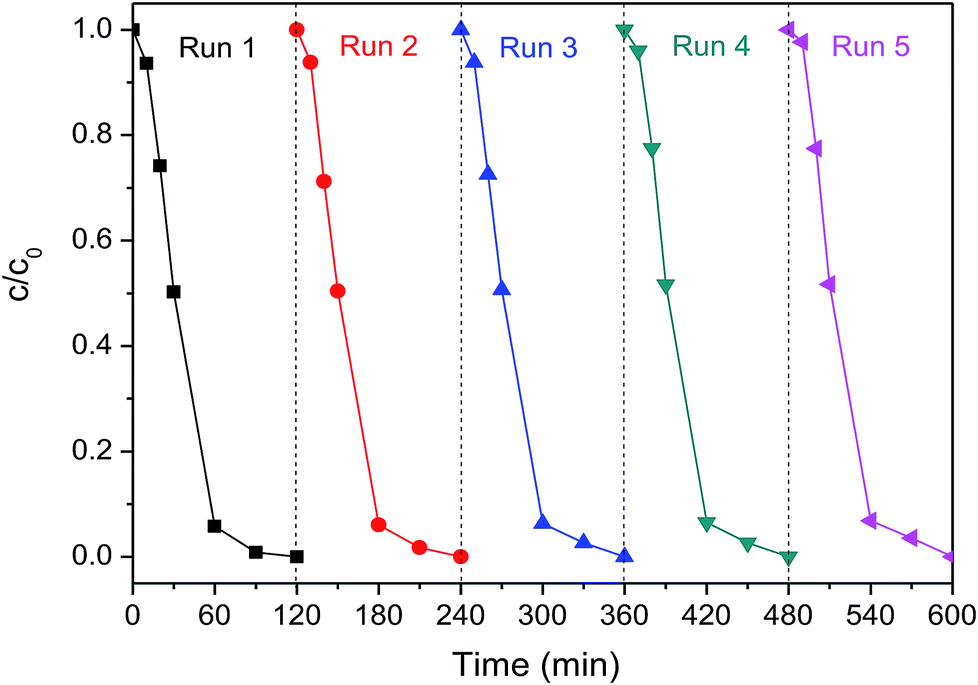 | ||
| Fig. 7 The 2,4-D removal efficiency by dechlorination using the Pd-Co3O4/Ni foam for five cycles at a current density of 1.5 mA cm−2. | ||
4. Conclusions
A well-designed Pd-Co3O4/Ni foam with high catalytic activity and excellent long-term durability for electrochemical dechlorination of 2,4-D was produced by a facile and cost-effective two-step synthesis. The Pd-Co3O4/Ni foam exhibited higher dechlorination efficiency and CE than Pd(CPD)/Ni and Pd(CD)/Ni foam electrodes. After 60 min of reaction, 94.2% of 2,4-D was dechlorinated with a maximum CE of 12.1%. The Co3O4 layer with hierarchical multi-porous structures covering the surface of the Ni foam promoted the production of atomic H* and accelerated charge- and mass-transfer. This provided the Pd catalyst with more active adsorbed atomic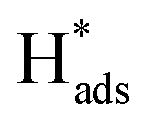 , which has been proven to play a dominant role in dechlorination of 2,4-D.
, which has been proven to play a dominant role in dechlorination of 2,4-D.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant 21477117) and the Zhejiang Provincial Natural Science Foundation of China (Grants LZ18B070001, LGF18E080017 and LR14E080001).References
- Y. Xi, M. Mallavarapu and R. Naidu, Appl. Clay Sci., 2010, 49, 255–261 CrossRef CAS.
- N. Maleki, A. Safavi and H. R. Shahbaazi, Anal. Chim. Acta, 2005, 530, 69–74 CrossRef CAS.
- R. A. Rebich, R. H. Coupe and E. M. Thurman, Sci. Total Environ., 2004, 321, 189–199 CrossRef CAS PubMed.
- J. R. de Lipthay, S. R. Sorensen and J. Aamand, Environ. Pollut., 2007, 148, 83–93 CrossRef CAS PubMed.
- K. Shareef and G. Shaw, Chemosphere, 2008, 72, 8–15 CrossRef CAS PubMed.
- S. Cenkci, M. Yildiz, I. H. Cigerci, A. Bozdag, H. Terzi and E. S. A. Terzi, Ecotoxicol. Environ. Saf., 2010, 73, 1558–1564 CrossRef CAS PubMed.
- B. Bukowska and K. Hutnik, Pestic. Biochem. Physiol., 2006, 85, 174–180 CrossRef CAS.
- B. Yang, S. Wang, G. Yu and Y. Zhou, Sep. Purif. Technol., 2008, 63, 353–359 CrossRef CAS.
- Z. Q. He, L. Y. Zhan, Q. Wang, S. Song, J. M. Chen, K. R. Zhu, X. H. Xu and W. P. Liu, Sep. Purif. Technol., 2011, 80, 526–532 CrossRef CAS.
- R. Mao, N. Li, H. C. Lan, X. Zhao, H. J. Liu, J. H. Qu and M. Sun, Environ. Sci. Technol., 2016, 50, 3829–3837 CrossRef CAS PubMed.
- Y. H. Xu, Q. Q. Cai, H. X. Ma, Y. He, H. Zhang and C. A. Ma, Electrochim. Acta, 2013, 96, 90–96 CrossRef CAS.
- H. Zilouei, B. Guieysse and B. Mattiasson, Process Biochem., 2006, 41, 1083–1089 CrossRef CAS.
- S. Eker and F. Kargi, J. Hazard. Mater., 2006, 135, 365–371 CrossRef CAS PubMed.
- H. Shimakoshi, M. Tokunaga, T. Baba and Y. Hisaeda, Chem. Commun., 2004, 1806–1807, 10.1039/b406400c.
- M. Ratti, S. Canonica, K. McNeill, J. Bolotin and T. B. Hofstetter, Environ. Sci. Technol., 2015, 49, 9797–9806 CrossRef CAS PubMed.
- G. M. Jiang, M. N. Lan, Z. Y. Zhang, X. S. Lv, Z. M. Lou, X. H. Xu, F. Dong and S. Zhang, Environ. Sci. Technol., 2017, 51, 7599–7605 CrossRef CAS PubMed.
- R. Mao, H. C. Lan, L. Yan, X. Zhao, H. J. Liu and J. H. Qu, Environ. Sci.: Nano, 2018, 5, 2282–2292 RSC.
- J. S. Zhou, Z. M. Lou, J. Xu, X. X. Zhou, K. L. Yang, X. Y. Gao, Y. L. Zhang and X. H. Xu, Chem. Eng. J., 2019, 358, 1176–1185 CrossRef CAS.
- B. B. Huang, A. A. Isse, C. Durante, C. H. Wei and A. Gennaro, Electrochim. Acta, 2012, 70, 50–61 CrossRef CAS.
- Q. Shi, H. Wang, S. L. Liu and Z. Y. Bian, RSC Adv., 2014, 4, 56263–56272 RSC.
- J. S. Zhou, Z. M. Lou, K. L. Yang, J. Xu, Y. Z. Li, Y. L. Liu, S. A. Baig and X. H. Xu, Appl. Catal., B, 2019, 244, 215–224 CrossRef CAS.
- B. P. Chaplin, M. Reinhard, W. F. Schneider, C. Schuth, J. R. Shapley, T. J. Strathmann and C. J. Werth, Environ. Sci. Technol., 2012, 46, 3655–3670 CrossRef CAS PubMed.
- I. F. Cheng, Q. Fernando and N. Korte, Environ. Sci. Technol., 1997, 31, 2443 CrossRef CAS.
- Q. Qu, G.-L. Pan, Y.-T. Lin and C.-W. Xu, Int. J. Hydrogen Energy, 2018, 43, 14252–14264 CrossRef CAS.
- C. X. Zhao, H. Luo, F. Chen, P. Zhang, L. H. Yi and K. Y. You, Energy Environ. Sci., 2014, 7, 1700–1707 RSC.
- Z. Q. He, J. J. Sun, J. Wei, Q. Wang, C. X. Huang, J. M. Chen and S. Song, J. Hazard. Mater., 2013, 250, 181–189 CrossRef PubMed.
- H. Husin, W.-N. Su, C.-J. Pan, J.-Y. Liu, J. Rick, S.-C. Yang, W.-T. Chuang, H.-S. Sheu and B.-J. Hwang, Int. J. Hydrogen Energy, 2013, 38, 13529–13540 CrossRef CAS.
- F. Li, H. Huang, G. Li and D. Y. C. Leung, J. Nanopart. Res., 2019, 21 Search PubMed.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- J. H. Wang, W. Cui, Q. Liu, Z. C. Xing, A. M. Asiri and X. P. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- G. Yanalak, A. Aljabour, E. Aslan, F. Ozel, I. H. Patir, M. Kus and M. Ersoz, Electrochim. Acta, 2018, 291, 311–318 CrossRef CAS.
- L. J. Zhang, Y. Y. Zhang, S. L. Huang, Y. L. Yuan, H. Li, Z. Y. Jin, J. H. Wu, Q. F. Liao, L. Hu, J. G. Lu, S. C. Ruan and Y. J. Zeng, Electrochim. Acta, 2018, 281, 189–197 CrossRef CAS.
- E. Skulason, V. Tripkovic, M. E. Bjoerketun, S. Gudmundsdottir, G. Karlberg, J. Rossmeisl, T. Bligaard, H. Jonsson and J. K. Norskov, J. Phys. Chem. C, 2010, 114, 22374 CrossRef CAS.
- X. Sun, J. Huo, Y. D. Yang, L. Xu and S. Y. Wang, J. Energy Chem., 2017, 26, 1136–1139 CrossRef.
- J. Lee, W. Shim, J. S. Noh and W. Lee, ChemPhysChem, 2012, 13, 1395–1403 CrossRef CAS PubMed.
- J. L. Lado, X. Wang, E. Paz, E. Carbo-Argibay, N. Guldris, C. Rodriguez-Abreu, L. Liu, K. Kovnir and Y. V. Kolen'ko, ACS Catal., 2015, 5, 6503–6508 CrossRef CAS.
- C. Yuan, L. Yang, L. Hou, L. Shen, X. Zhang and X. W. Lou, Energy Environ. Sci., 2012, 5, 7883–7887 RSC.
- J. J. Li, H. L. Liu, X. W. Cheng, Q. H. Chen, Y. J. Xin, Z. P. Ma, W. X. Xu, J. Ma and N. Q. Ren, Chem. Eng. J., 2013, 225, 489–498 CrossRef CAS.
- Y. Zhang, J. H. Li, J. Bai, Z. X. Shen, L. S. Li, L. G. Xia, S. Chen and B. X. Zhou, Environ. Sci. Technol., 2018, 52, 1413–1420 CrossRef CAS PubMed.
- K. R. Zhu, S. A. Baig, J. Xu, T. T. Sheng and X. H. Xu, Electrochim. Acta, 2012, 69, 389–396 CrossRef CAS.
- L. Tang, S. Zhang, G. M. Zeng, Y. Zhang, G. D. Yang, J. Chen, J. J. Wang, J. J. Wang, Y. Y. Zhou and Y. C. Deng, J. Colloid Interface Sci., 2015, 445, 1–8 CrossRef CAS PubMed.
- J. J. Li, H. L. Liu, X. W. Cheng, Y. J. Xin, W. X. Xu, Z. P. Ma, J. Ma, N. Q. Ren and Q. Li, Ind. Eng. Chem. Res., 2012, 51, 15557–15563 CrossRef CAS.
- J. J. Li, C. Luan, Y. Q. Cui, H. X. Zhang, L. Wang, H. Wang, Z. H. Zhang, B. Zhao, H. W. Zhang, X. Y. Zhang and X. W. Cheng, Sep. Purif. Technol., 2019, 211, 198–206 CrossRef CAS.
- S. Pocovi-Martinez, I. Zumeta-Dube and D. Diaz, J. Nanomater., 2019, 2019, 6461493 Search PubMed.
- S. L. Xiong, C. Z. Yuan, M. F. Zhang, B. J. Xi and Y. T. Qian, Chem.–Eur. J., 2009, 15, 5320–5326 CrossRef CAS PubMed.
- G. Yanalak, A. Aijabour, E. Aslan, F. Ozel and I. H. Patir, Int. J. Hydrogen Energy, 2018, 43, 17185–17194 CrossRef CAS.
- G. B. Hoflund and Z. H. Li, Appl. Surf. Sci., 2006, 253, 2830–2834 CrossRef CAS.
- J. Y. Luo, M. Meng, X. Li, X. G. Li, Y. Q. Zha, T. D. Hu, Y. N. Xie and J. Zhang, J. Catal., 2008, 254, 310–324 CrossRef CAS.
- X. D. Zhang, J. L. Xiao, X. Y. Zhang, Y. Meng and D. Xiao, Electrochim. Acta, 2016, 191, 758–766 CrossRef CAS.
- Z. Q. Liu, K. Xiao, Q. Z. Xu, N. Li, Y. Z. Su, H. J. Wang and S. Chen, RSC Adv., 2013, 3, 4372–4380 RSC.
- Z. Q. Liu, Q. Z. Xu, J. Y. Wang, N. Li, S. H. Guo, Y. Z. Su, H. J. Wang, J. H. Zhang and S. Chen, Int. J. Hydrogen Energy, 2013, 38, 6657–6662 CrossRef CAS.
- J. A. Mwanda and A. Cuesta, Electrochim. Acta, 2018, 292, 419–424 CrossRef CAS.
- B. Liu, J. Cheng, H.-Q. Peng, D. Chen, X. Cui, D. Shen, K. Zhang, T. Jiao, M. Li, C.-S. Lee and W. Zhang, J. Mater. Chem. A, 2019, 7, 775–782 RSC.
- Q. M. Sun, N. Wang, Q. M. Bing, R. Si, J. Y. Liu, R. S. Bai, P. Zhang, M. J. Jia and J. H. Yu, Chem, 2017, 3, 477–493 CAS.
- Z. R. Sun, G. Song, R. Du and X. Hu, RSC Adv., 2017, 7, 22054–22062 RSC.
- C. Sun, Z. M. Lou, Y. Liu, R. Q. Fu, X. X. Zhou, Z. Zhang, S. A. Baig and X. H. Xu, Chem. Eng. J., 2015, 281, 183–191 CrossRef CAS.
- C. Sun, S. A. Baig, Z. M. Lou, J. Zhu, Z. X. Wang, X. Li, J. H. Wu, Y. F. Zhang and X. H. Xu, Appl. Catal., B, 2014, 158, 38–47 CrossRef.
- A. I. Tsyganok and K. Otsuka, Appl. Catal., B, 1999, 22, 15–26 CrossRef CAS.
- W. J. Xie, S. H. Yuan, X. H. Mao, W. Hu, P. Liao, M. Tong and A. N. Alshaulabkeh, Water Res., 2013, 47, 3573–3582 CrossRef CAS PubMed.
- A. Z. Li, X. Zhao, Y. N. Hou, H. J. Liu, L. Y. Wu and J. H. Qu, Appl. Catal., B, 2012, 111, 628–635 CrossRef.
- T. Li and J. Farrell, Environ. Sci. Technol., 2000, 34, 173–179 CrossRef CAS.
- Z. Q. He, Q. W. Jian, J. T. Tang, T. Xu, J. L. Xu, Z. S. Yu, J. M. Chen and S. Song, Electrochim. Acta, 2016, 222, 488–498 CrossRef CAS.
- Z. Q. He, Y. W. Tong, S. L. Ni, X. C. Ye, C. P. Makwarimba, X. W. Huang, S. H. Zhang and S. Song, Electrochim. Acta, 2018, 292, 685–696 CrossRef CAS.
- Z. M. Lou, J. S. Zhou, M. Sun, J. Xu, K. L. Yang, D. Lv, Y. P. Zhao and X. H. Xu, Chem. Eng. J., 2018, 352, 549–557 CrossRef CAS.
- L. Yang, H. Zhou, X. Qin, X. Guo, G. Cui, A. M. Asiri and X. Sun, Chem. Commun., 2018, 54, 2150–2153 RSC.
- J. Yesuraj, S. A. Suthanthiraraj and O. Padmaraj, Mater. Sci. Semicond. Process., 2019, 90, 225–235 CrossRef CAS.
- R. Mahesh, R. Mahendiran, A. K. Raychaudhuri and C. N. R. Rao, Appl. Phys. Lett., 1996, 68, 2291–2293 CrossRef CAS.
- T. H. Tsai, S. C. Chiou and S. M. Chen, Int. J. Electrochem. Sci., 2011, 6, 3333–3343 CAS.
| This journal is © The Royal Society of Chemistry 2019 |