
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn energy-blocking nanoparticle decorated with anti-VEGF antibody to reverse chemotherapeutic drug resistance†
Liu-Qing
Gu‡
a,
Peng-Fei
Cui‡
a,
Lei
Xing
abcd,
Yu-Jing
He
a,
Xin
Chang
a,
Tian-Jiao
Zhou
a,
Yu
Liu
*e,
Ling
Li
*f and
Hu-Lin
Jiang
 *abcd
*abcd
aState Key Laboratory of Natural Medicines, Department of Pharmaceutics, China Pharmaceutical University, Nanjing 210009, China. E-mail: jianghulin3@163.com; Fax: +86-25-83271027; Tel: +86-25-83271027
bJiangsu Key Laboratory of Druggability of Biopharmaceuticals, China Pharmaceutical University, Nanjing 210009, China
cJiangsu Key Laboratory of Drug Screening, China Pharmaceutical University, Nanjing 210009, China
dJiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, China Pharmaceutical University, Nanjing 210009, China
eDepartment of Biochemistry, School of Life Science and Technology, China Pharmaceutical University, Nanjing 210009, China. E-mail: liuyuyaoda@163.com; Fax: +86-25-83271019; Tel: +86-25-83271543
fDepartment of Endocrinology, Zhongda Hospital, Southeast University, Nanjing 210009, China. E-mail: li-ling76@hotmail.com; Fax: +86-25-83272011; Tel: +86-25-83272012
First published on 16th April 2019
Abstract
Multi-drug resistance (MDR) of tumor cells has greatly hindered the therapeutic efficacy of chemotherapeutic drugs, resulting in chemotherapy failure, while overexpression of ATP-binding cassette (ABC) transporters in cell membranes is the leading cause of MDR. In this study, we reported novel self-assembled triphenylphosphine-quercetin–polyethylene glycol–monoclonal antibody nanoparticles (TQ–PEG–mAb NPs) for overcoming MDR primarily through mitochondrial damage to block ATP supply to ABC transporters both in vitro and in vivo. The doxorubicin (DOX)-loaded NPs (TQ/DOX–PEG–mAb) were composed of two drugs (TQ and DOX) and an outer shielding shell of the PEG–mAb conjugate. Besides, the outer shell could be acid-responsively detached to expose the positive charge of TQ inside the NPs to enhance cellular uptake. TQ was proved to effectively induce mitochondrial damage with increased ROS levels and depolarization of mitochondrial membrane potential (MMP), leading to prominently reduced ATP supply to ABC transporters. Moreover, the involvement of the anti-vascular endothelial growth factor (VEGF) mAb was not only for efficient targeting but also for combined therapy. Consequently, TQ/DOX–PEG–mAb showed that the internalized amount of DOX was largely improved while the efflux amount was dramatically inhibited on MCF-7/ADR cells, indicating excellent reversal of DOX resistance. Importantly, the growth of DOX-resistant breast tumors was significantly inhibited with no evident systemic toxicity. Therefore, the employment of TQ–PEG–mAb is believed to be a new approach to improve the efficacy of chemotherapeutic drugs in MDR tumors.
Introduction
Breast cancer is a leading cause of death in women globally with more than one million women annually diagnosed with this disease. Chemotherapy remains the principle choice for most patients to treat breast cancer.1,2 However, multi-drug resistance (MDR) is always the key obstacle in the current chemotherapeutics.3,4 Although the mechanisms of MDR are complex,5 overexpression of ATP-binding cassette (ABC) transporters in the plasma membrane has always played a crucial role, including P-glycoprotein (P-gp), Breast Cancer Resistance Protein (BCRP), and Multidrug Resistance-associated Protein 1 (MRP1).6,7 ABC transporters can pump chemotherapeutic drugs out of the cancer cells by consuming the energy from adenosine 5′-triphosphate (ATP) hydrolysis,8,9 leading to remarkably decreased intracellular drug concentration.10 To address this issue, many drug delivery systems based on nanoparticles (NPs) have been reported to avoid the efflux pump via endocytosis pathway.11,12 Meanwhile, together with the introduction of stimuli-responsive burst release of drugs trapped in NPs,13,14 the intracellular drug concentration can be high enough to effectively kill tumor cells.15,16 However, the intracellular drugs still can be slowly pumped out by ABC transporters over time,17 resulting in reduced therapeutic efficacy. Therefore, many studies using NPs that can inhibit the function of ABC transporters to further overcome MDR have been reported.18,19Previous work has only focused on the ABC transporter itself,20,21 but few strategies are reported through mitochondria to reverse MDR. In addition, it was well recognized that mitochondria is the energy-supplying center of cells and the regulatory center of apoptosis.22,23 Hence, causing mitochondrial specific damage not only inhibits the ATP supply to ABC transporters, but also induces mitochondria-mediated apoptosis.24,25 Our group has previously synthesized a conjugate of triphenylphosphine-quercetin (TQ), which could effectively target mitochondria and induce mitochondrial damage with increased ROS level and depolarization of mitochondrial membrane potential (MMP).24 Therefore, we assume that TQ can be combined with traditional chemotherapeutic drugs to reverse MDR of tumor cells. On one hand, TQ can block the ATP generation to prevent ABC transporters from pumping chemotherapeutic drugs out; and on the other hand, TQ can play a synergistic anti-tumor effect.
It has been demonstrated that vascular endothelial growth factor (VEGF) is overexpressed in breast cancer compared with normal tissue.26,27 Meanwhile, VEGF is a growth factor secreted by tumor cells that can promote the generation of angiogenesis through the VEGF–VEGFR signaling pathway,28,29 resulting in constant supply of oxygen and nutrients to tumor to promote its survival.30,31 Therefore, VEGF-targeting strategy has always been attractive in oncology treatment.32,33
As illustrated in Fig. 1a, we designed a novel self-assembled nanoformulation composed of drugs and PEG without redundant carrier materials, which may lead to potential systemic toxicity.34,35 TQ can be linked with phenylboronic acid (PBA)–PEG–maleimide (Mal) through boronate ester bond, and the formed complex can self-assemble into TQ–PEG NPs.24 After that, anti-VEGF mAb can be decorated on the surface of TQ–PEG to form TQ–PEG–mAb via the reaction between Mal of PBA–PEG–Mal and SH of thiolated mAb.36,37 Here doxorubicin (DOX), a substrate of ABC transporters, was selected as the model drug encapsulated in the core of TQ–PEG–mAb through hydrophobic effect to form TQ/DOX–PEG–mAb.19 Owing to the shielding effect of PEG–mAb, TQ/DOX–PEG–mAb was stable in blood circulation and normal tissues (Fig. 1b), and could accumulate at the tumor site efficaciously with the enhanced permeability and retention (EPR) effect and targeting function of anti-VEGF mAb.38,39 Upon reaching the tumor microenvironment (pH ∼ 6.5),40,41 part of the PEG–mAb conjugate shed due to the presence of pH-sensitive borate ester linkages. The detached mAb could combine with the free VEGF in extracellular matrix to block VEGF–VEGFR signaling pathway of endothelial cells to inhibit angiogenesis. Meanwhile, the attached mAb on the surface of NPs bound to the VEGF on tumor cell membrane to promote uptake, which could be further improved by the exposed positive charge of TPP.42 When entering a more acidic environment of lysosomes (pH ∼ 5),40,41 TQ and DOX could be released in large quantities due to the dissociation of NPs caused by further shedding of PEG. TQ was able to target mitochondria efficiently to cause mitochondrial damage, leading to a dramatic reduction in ATP supply to ABC transporters and mitochondria-mediated apoptosis. As a result, DOX could successfully enter the nucleus without being pumped out to trigger apoptosis. Consequently, TQ/DOX–PEG–mAb could effectively treat MDR breast cancer mainly through mitochondrial damage caused by TQ not only reversing the resistance to DOX but also inducing apoptosis, together with the synergistic therapeutic effect of anti-VEGF mAb and rational nanoparticle design.
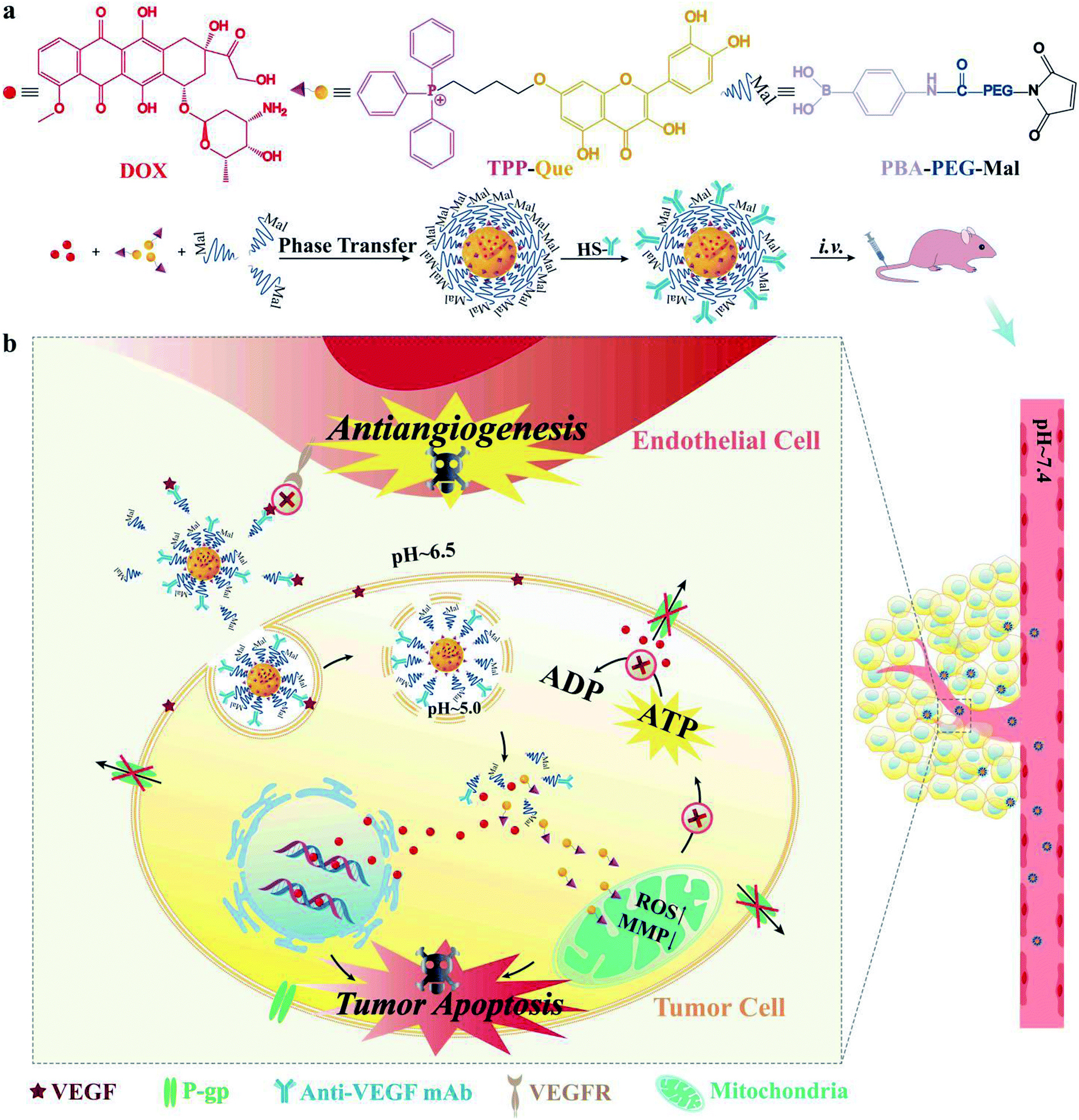 | ||
| Fig. 1 (a) Structures of DOX, TQ and PBA–PEG–Mal as well as the preparation method of TQ/DOX–PEG–mAb. (b) Schematic diagram of the behavior of TQ/DOX–PEG–mAb in blood circulation and tumor tissues. | ||
Experimental section
Materials
Quercetin (Que), imidazole and Traut's Reagent were all purchased from Sigma-Aldrich (St. Louis, MO, USA). Triphenylphosphine (TPP) was purchased from Alfa Aesar Co., Inc. (Ward Hill, MA, USA). Sodium iodide, cesium carbonate and 1-bromo-4-chlorobutane were all purchased from J&K Co., Ltd. (Beijing, China). Alpha-maleinimido-omega-carboxy succinimidyl ester poly (ethylene glycol) (NHS–PEG–Mal, Mw = 2000 Da) was obtained from Shanghai yarebio Co., Ltd. (Shanghai, China). 4-Aminophenylboronic acid was obtained from Shanghai aladdin Co., Ltd. (Shanghai, China). Doxorubicin hydrochloride (DOX) was purchased from China Langchem Inc. (Shanghai, China). Anti-VEGF mAb was kindly provided by Prof. Yu Liu.43 RPMI 1640 and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were both purchased from KeyGEN Biotech (Nanjing, China). Fetal bovine serum (FBS) was purchased from Gibco (Burlington, ON, Canada). All other chemicals and reagents in this study were analytical grade.Cell culture
MCF-7 cells and MCF-7/ADR cells were cultured in RPMI 1640 medium supplemented with 10 and 20% (v/v) FBS at 37 °C and 5% CO2, respectively. One mM of DOX was added to maintain the drug resistance of MCF-7/ADR cells.Xenograft model
Female nude balb/c mice (6 weeks, 20–22 g) were purchased from Qinglongshan breading farm (Nanjing, China). All animal experiments were carried out in compliance with the Guidelines for Care and Use of Laboratory Animals of China Pharmaceutical University, and were approved by the Animal Ethics Committee of China Pharmaceutical University. MCF-7/ADR cells (1 × 107 cells per animal) were subcutaneously injected into the right axilla of mice to obtain the tumor model.Synthesis and characterization of triphenylphosphine-quercetin
Triphenylphosphine-quercetin (TQ) was synthesized as outlined in Fig. S1† according to a published procedure. Briefly, (i) quercetin (1, 1.0 g, 3.0 mmol, 1.0 equiv.), acetic anhydride (6.13 g, 60.0 mmol, 20 equiv.) and pyridine (15 mL) were mixed and refluxed with stirring (140 °C, 5 h). Ice-water (50 g) was added to the warm mixture. The resulting precipitate was filtered, washed and recrystallized with cold ethyl acetate to afford 2; (ii) cesium carbonate (1.43 g, 4.4 mmol, 1.5 equiv.) and 1-bromo-4-chlorobutane (1.51 g, 8.8 mmol, 3.0 equiv.) were added to the solution of 2 (1.50 g, 2.9 mmol, 1.0 equiv.) in DMF (30 mL). Imidazole (0.03 g, 2% w/w) was then added and the mixture was stirred at room temperature (N2, 48 h). The reaction mixture was diluted in EtOAc (200 mL) and washed with 3 M aq. HCl (3 × 100 mL). The organic layer was dried with Na2SO4 and the solvent was evaporated under reduced pressure; (iii) without further purification, the crude product was added to a mixture of acetonitrile (60 mL) and 3 M aq. HCl (30 mL). The resulting solution was refluxed and stirred for 1 h, and then EtOAc (200 mL) and water (200 mL) were added. The organic layer was washed with 3 M aq. HCl (3 × 200 mL) and dried with Na2SO4. The solvent was evaporated under reduced pressure and the resulting residue was purified by silica gel flash chromatography (CHCl3/MeOH, 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to obtain 3 as a bright yellow solid; (iv) 3 was added to a saturated solution of sodium iodide in dry acetone (20 mL), which was refluxed for 20 h. After cooling, the resulting mixture was diluted in EtOAc (100 mL), filtered and washed with water (3 × 30 mL). The organic layer was dried with Na2SO4 and evaporated under reduced pressure to afford 4; (v) 4 and triphenylphosphine (0.825 g, 3.15 mmol, 5.0 equiv.) were dissolved in toluene (15 mL) and heated at 95 °C under argon. After 3 h, the solvent was removed under reduced pressure and the resulting solid was dissolved in the minimum volume of CH2Cl2 and precipitated with diethyl ether (5 × 50 mL). The solvents were decanted to achieve the final product (yellow solid). The successful synthesis of TQ was confirmed by mass spectrometry, 1H NMR spectra and UV-Vis spectrum. Besides, the purity was determined by HPLC (chromatographic conditions: C18 reversed-phase column (250 mm × 4.6 mm) packed with 5 μm diameter particles, mobile phase: acetonitrile–water (35:65, v/v) containing 0.12% acetic acid, detection wavelength: 360 nm, flow rate: 1.0 mL min−1, Analytical Liquid Phase-DAD, Shimadzu LC-20A).
:1) to obtain 3 as a bright yellow solid; (iv) 3 was added to a saturated solution of sodium iodide in dry acetone (20 mL), which was refluxed for 20 h. After cooling, the resulting mixture was diluted in EtOAc (100 mL), filtered and washed with water (3 × 30 mL). The organic layer was dried with Na2SO4 and evaporated under reduced pressure to afford 4; (v) 4 and triphenylphosphine (0.825 g, 3.15 mmol, 5.0 equiv.) were dissolved in toluene (15 mL) and heated at 95 °C under argon. After 3 h, the solvent was removed under reduced pressure and the resulting solid was dissolved in the minimum volume of CH2Cl2 and precipitated with diethyl ether (5 × 50 mL). The solvents were decanted to achieve the final product (yellow solid). The successful synthesis of TQ was confirmed by mass spectrometry, 1H NMR spectra and UV-Vis spectrum. Besides, the purity was determined by HPLC (chromatographic conditions: C18 reversed-phase column (250 mm × 4.6 mm) packed with 5 μm diameter particles, mobile phase: acetonitrile–water (35:65, v/v) containing 0.12% acetic acid, detection wavelength: 360 nm, flow rate: 1.0 mL min−1, Analytical Liquid Phase-DAD, Shimadzu LC-20A).
Synthesis and characterization of PBA–PEG–Mal
4-Aminophenylboronic acid (4-NH2-PBA) was reacted with NHS–PEG–Mal at the molar ratio of 1.8:1 in PBS (pH 8.0, 50 mM) for 3 h at room temperature as shown in Fig. S6.† The resulting conjugate was purified by dialysis using a dialysis bag (MWCO = 500 Da) against distilled water for 48 h, and then lyophilized to obtain white solid. The successful synthesis of PBA–PEG–Mal was characterized by mass spectrometry and 1H NMR spectrum.
Preparation of TQ/DOX–PEG
Briefly, TQ, DOX and PBA–PEG–Mal at a molar ratio of 1:0.5:0.4 were completely dissolved in 100 μL DMSO. Then the solution was added into 3 mL deionized water (pH = 10.5) with continuous stirring for 20 min. After dialyzed using a dialysis bag (MWCO = 14000 Da) against distilled water for 8 h, TQ/DOX–PEG were obtained.
Conjugation of anti-VEGF mAb to TQ/DOX–PEG
Firstly, anti-VEGF mAb was thiolated with Traut's reagent. Traut's reagent and mAb at a molar ratio 100:1 were incubated in PBS (pH = 8.0, 50 mM) at room temperature for 1 h (Fig. S9†), and then unreacted Traut's reagent was removed by a Sephadex G-25 column to afford purified SH-mAb. Finally, TQ/DOX–PEG–mAb was obtained by mixing TQ/DOX–PEG and SH-mAb at 4 °C overnight. Unbound mAb was removed by centrifuging the suspension at 12000 rpm for 5 min. SDS-PAGE (12%) was used to determine whether the mAb was connected to the surface of TQ/DOX–PEG and confirm the integrity of anti-VEGF mAb. The amount of anti-VEGF mAb conjugated to TQ/DOX–PEG was quantified using a bicinchoninic acid (BCA) protein assay.41
Characterization of TQ/DOX–PEG and TQ/DOX–PEG–mAb
Particle size, polydispersity index (PDI), and zeta potential of TQ/DOX–PEG and TQ/DOX–PEG–mAb were measured via ZetaPlus particle size analyzer (Brookhaven Instruments, USA). Transmission electron microscopy (TEM) was used to observe the morphology of micelles. In addition, to confirm whether both two drugs were successfully loaded through UV-Vis spectrum (Thermo Scientific Multiskan Go, USA), the lyophilized NPs were dissolved in a mixed solvent (DMSO:1 M HCl:H2O = 5:1:4, v/v/v), where DMSO was to depolymerize NPs, 1 M HCl was to break the boronate ester bond and H2O was to blend previous two systems homogeneously. Meanwhile, the content of TQ and DOX in NPs were determined by UV-Vis spectrum as well. The drug loading and encapsulation efficiency were calculated as previously reported.44,45
Stability study of TQ/DOX–PEG–mAb
The prepared TQ/DOX–PEG–mAb were diluted to 3 mL with distilled water, 5% glucose solution and 1640 medium containing 10% FBS, respectively. Then the particle size and PDI of each sample were measured at 37 °C over one week period using ZetaPlus particle size analyzer.In vitro release of TQ and DOX from TQ/DOX–PEG–mAb
The prepared TQ/DOX–PEG–mAb were dissolved in 3 mL PBS buffer (10 mM, pH 5.0, pH 6.5 and pH 7.4) and transferred into a dialysis bag (MWCO = 14000 Da). The dialysis bag was then immerged into the same pH release medium (50 mL) and incubated at 37 °C. Two mL of medium solution was taken out and 2 mL of fresh blank PBS buffer was supplemented into the release medium periodically. The released TQ and DOX were measured via UV-Vis spectrum.
In vitro pH-responsive shedding of PEG
The prepared TQ/DOX–PEG–mAb were divided into three parts. And then the pH of NPs solution was adjusted to pH 5.0, 6.5 and 7.4, respectively. After standing for a while, the zeta potential of each was determined.In vitro cytotoxicity
The cytotoxicity against MCF-7, MCF-7/ADR and MDA-MB-231 cells was evaluated by the MTT assay. Briefly, the cells were seeded into 96-well plates (1 × 104/well) and cultured for 24 h. When the confluence of the culture cells used in our study was around 70–80%, the culture medium was replaced with different concentrations of drug samples. After incubated for 48 h, the drug was removed and the wells were washed with PBS, followed by adding the fresh medium containing MTT (5 mg mL−1). The medium was taken away after 4 h of incubation, and 150 μL of DMSO was added to dissolve the formazan. The absorbance was determined at 490 nm by using multiskan (Thermo Scientific Multiskan Go, USA). Besides, synergic index (SI) of different molar ratios between TQ and DOX (1:1, 2:1, 3:1 and 4:1, respectively) was calculated by CompuSyn software.
Intracellular ROS detection
The intracellular ROS detection was measured using an oxidation-sensitive probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA). Briefly, MCF-7/ADR cells were cultured on 12-well plates (1 × 105/well) for 24 h, and then treated with DOX, TQ, TQ + DOX, TQ/DOX–PEG and TQ/DOX–PEG–mAb, respectively (TQ: 60 μM). After 12 h of culture, cells were washed with PBS twice and incubated with 1640 containing DCFH-DA (10 μM) for 20 min. After washed with PBS twice again to remove residual DCFHDA, cells were observed via an Inverted Fluorescence Microscope (Nikon ti-s, Japan).Determination of mitochondrial membrane potential
Mitochondrial membrane potential (MMP) detection kit (JC-1) was used to determine the change of MMP. MCF-7/ADR cells were cultured on 12-well plates (1 × 105/well) for 24 h. Then the culture medium was removed and replaced with DOX, TQ, TQ + DOX, TQ/DOX–PEG and TQ/DOX–PEG–mAb, respectively (TQ: 60 μM). Twelve h later, drug was extracted and cells were washed twice with PBS. After that, cells was added with 2.5 mg L−1 of JC-1 and incubated for 20 min at 37 °C, followed by rinsed with JC-1 buffer twice. Finally, the fluorescence was observed via Inverted Fluorescence Microscope (Nikon ti-s, Japan) at 510–540 nm (monomeric JC-1) and at 570–600 nm (aggregated JC-1).Drug internalization and efflux measurement
Firstly, MCF-7/ADR cells and MCF-7 cells were cultured on 6-well plates (5 × 105/well) for 24 h. For DOX internalization determination, cells were incubated with free DOX, TQ + DOX, TQ/DOX–PEG and TQ/DOX–PEG–mAb (DOX: 20 μM) for 4 h, respectively. After that, cells were washed thoroughly and treated with 0.5 mL of trypsin. And then the cells were collected and counted. The next step was to add the cell lysis buffer to both cell lines and kept vortexing for 5 min, followed by freeze-thawing three times. Then ethanol was added and keep sonicating for 30 min. Finally the cells were centrifuged (12000 rpm min−1, 10 min) to take the supernatant, and the DOX amount was determined by fluorometric spectrophotometry (RF-5302, Japan). As for DOX efflux measurement, both cells were exposed to various drug formulations for 4 h, and then half of them were treated to determine the internalized DOX amount; the other half were replaced with culture medium and incubated for another 20 h. The processing method of cells was the same as above. The efflux amount of DOX was calculated by subtracting the final intracellular DOX amount from the initial internalized amount of DOX.
In vivo biodistribution study
MCF-7/ADR cells (1 × 107 cells per animal) were subcutaneously injected into the right axilla of female nude balb/c mice, whose age were around 6 weeks and body weight were 20–22 g. When tumor volume grew up to 150–200 mm3, the mice were randomly divided into four groups (three mice per group). After that, the tumor-bearing mice were treated with PBS, DOX, TQ/DOX–PEG and TQ/DOX–PEG–mAb, respectively via tail vein injection (DOX: 1.2 mg kg−1, TQ: 4.6 mg kg−1, anti-VEGF mAb: 2 mg kg−1). After 24 h of administration, the mice were sacrificed, and the tumor, heart, liver, spleen, lungs and kidneys were collected for fluorescence imaging. The fluorescence intensity of tumor and other organs was detected by using an In Vivo Imaging System (IVIS®spectrum, PerkinElmer).In vivo anti-tumor effect
The establishment of tumor-bearing mice model was the same as above. When the tumor size reached around 100 mm3, mice were randomly assigned to six groups (eight mice per group). PBS, DOX, TQ, TQ + DOX, TQ/DOX–PEG and TQ/DOX–PEG–mAb (DOX: 1.2 mg kg−1, TQ: 4.6 mg kg−1, anti-VEGF mAb: 2 mg kg−1) were injected to the tumor-bearing mice by tail vein every three days (days 0, 3, 6, 9, 12, 15, 18 and 21). Tumor volume and body weight were measured every 3 days as well. On day 21, mice were sacrificed, and the blood and major tissues (tumor, heart, liver, spleen, lungs and kidneys) were harvested. Among them, tumor tissues were examined by means of hematoxylin and eosin (H&E), terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), Ki-67 and CD31 staining to investigate the effect of drug resistance reversal and combined therapy. Besides, TUNEL staining was quantified by ImageJ software.In vivo safety evaluation
Furthermore, to assess the safety of drug formulations, the main organs (heart, liver, spleen, lung and kidneys) were analyzed by H&E staining. Meanwhile, blood samples were centrifuged for 15 min at 3000 rpm to collect serum, which were used to detect biochemical indices, including the level of alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase (ALP); blood urea nitrogen (BUN) and creatinine (CR); lactate dehydrogenase (LDH) and creatine kinase (CK), which indicate the damage of liver, kidneys and heart, respectively.Statistical analysis
Student's t test or one-way ANOVA test was used for statistical analysis. Differences were assessed for significance at two thresholds: *P < 0.05, **P < 0.01, ***P < 0.001.Results and discussion
Characterization of TQ and PBA–PEG–Mal
As shown in Fig. S2,† the [M]+ peak of TQ at 619.3 of mass spectrum indicated that TPP was successfully connected with Que. Moreover, the shift of absorption band of Que from 374 to 357 nm in UV-Vis spectrum (Fig. S5†) also proved the covalent connection between TPP and Que. Meanwhile, according to the 1H NMR spectrum of TQ (Fig. S3†), the peak of 7-OH (10.74 ppm) disappeared and TPP (7.65–7.98 ppm) appeared, manifesting that TPP took place of the 7-OH position of Que. The purity of TQ was up to 98% through the analysis by HPLC (Fig. S4†).As for PBA–PEG–Mal, through the analysis of mass spectrum (Fig. S8†), we found that the average molecular weight of NHS–PEG–Mal and PBA–PEG–Mal were at 2174.3 and 2305.3, respectively, whose molecular weight difference was exactly the molecular weight of PBA (173.41). It meant that PBA was successfully grafted on PEG by covalent bond. Besides, the signal of 1H NMR spectrum at 7.5–8.5 ppm (–PhB(OH)2) further implied the successful connection (Fig. S7†).
Identification of anti-VEGF mAb decorated NPs
SDS-PAGE was used to verify whether anti-VEGF mAb was conjugated to the surface of TQ/DOX–PEG. The molecular weight of anti-VEGF mAb was originally 150 kD, which was increased after conjugation to TQ/DOX–PEG through the reaction between Mal–PEG–PBA and SH-mAb. Just as shown in Fig. S10,† the position of the band for conjugated mAb was higher than the non-conjugated one, which indicated the successful coupling between anti-VEGF mAb and TQ/DOX-PEG. The change in position was not so apparent owing to the large molecular weight of anti-VEGF mAb. Besides, the decrease in zeta potential of TQ/DOX–PEG–mAb compared to TQ/DOX–PEG also indicated successful coupling of anti-VEGF mAb (Table S1†).The characterization of TQ/DOX–PEG–mAb
Dynamic light scattering (DLS) determination revealed that the hydrodynamic size of TQ/DOX–PEG–mAb was around 109.6 ± 3.3 nm (Fig. 2a), a little increase compared to TQ/DOX–PEG with the particle size of 89.3 ± 2.5 nm (Table S1†). Besides, PDI below 0.2 (Table S1†) implied the homogenous size distribution. The TEM image further indicated that TQ/DOX–PEG–mAb were uniform, well-dispersed and spherical in aqueous solution (Fig. 2a). Furthermore, as shown in Fig. 2b, the characteristic peaks of TQ and DOX appeared in TQ/DOX–PEG–mAb, implying the successful entrapment of both two drugs. The encapsulation efficiency of TQ and DOX were 89.3 ± 0.23 and 94.7 ± 0.19%, respectively (Table S2†), and the molar ratio of the two drugs loaded was around 3:1. In vitro stability was investigated in distilled water, 5% glucose solution and 1640 medium containing 10% FBS at 37 °C. There were no significant change in hydrodynamic diameter during one week (Fig. 2f), demonstrating the good stability of TQ/DOX–PEG–mAb and its potential for in vivo applications. A slight increase in the particle size of TQ/DOX–PEG–mAb in the FBS containing medium may be due to a small amount of serum protein adsorbed on the surface of NPs.
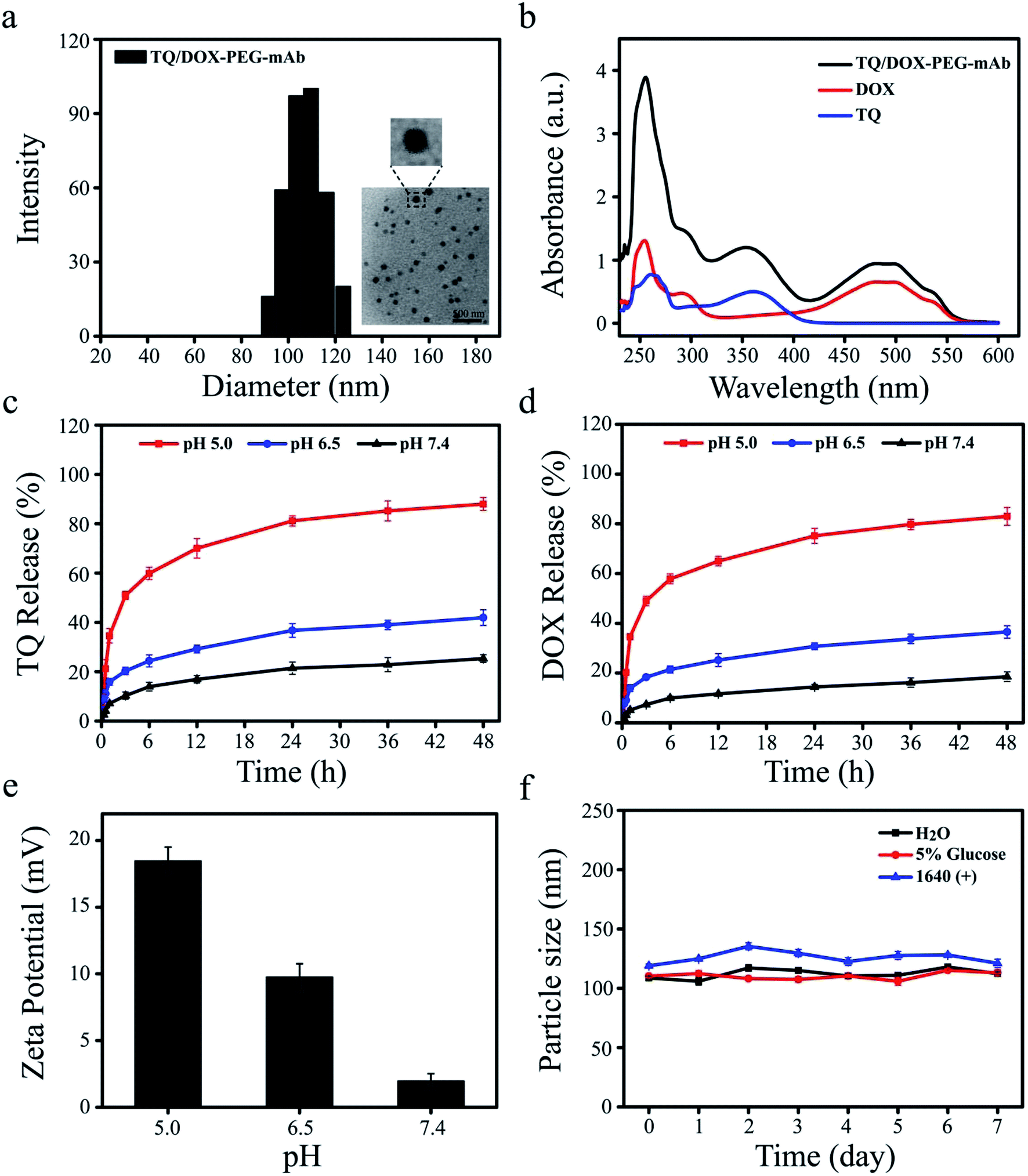 | ||
| Fig. 2 (a) Particle size distribution and TEM image of TQ/DOX–PEG–mAb. Scare bar = 500 nm. (b) UV-Vis spectrum of TQ/DOX–PEG, TQ and DOX. Cumulative release of (c) TQ and (d) DOX from TQ/DOX–PEG–mAb under different pH conditions (pH 5.0, 6.5 and 7.4) in 48 h. (e) Zeta potential changes of TQ/DOX–PEG–mAb after incubation with different pHs (pH 5.0, 6.5 and 7.4). (f) In vitro stability of TQ/DOX–PEG–mAb in H2O, 5% glucose and RPMI 1640 containing 10% FBS during 7 days. | ||
In vitro pH-responsive study of TQ/DOX–PEG–mAb
To explore the pH-responsive behavior of TQ/DOX–PEG–mAb, the release study of TQ and DOX from TQ/DOX–PEG–mAb was carried out in pH 7.4, 6.5, and 5.0 PBS media, simulating the pH of physiological environment, tumor extracellular matrix and endosome, respectively. Fig. 2c and d showed that only a small amount of TQ and DOX were released from NPs at pH 7.4 after 48 h, and a slight increase to about 40% at pH 6.5. However, both the release amount and release rate of TQ and DOX at pH 5.0 were much higher than that at pH 7.4 and 6.5. These results were ascribed to the pH-responsive breakage of boronate ester bond, which is stable at neutral or alkaline pH and dissociated in a low-pH environment.46 Therefore, as the pH decreased, the gradually detached PEG–mAb could lead to the dissociation of NPs and subsequently rapid release of TQ and DOX from TQ/DOX–PEG–mAb. Besides, to further verify the pH-sensitive characteristic, the determination of zeta potential of TQ/DOX–PEG–mAb after incubation with different pHs (pH 5.0, 6.5 and 7.4) was also conducted. The positive electricity of TQ/DOX–PEG–mAb would be exposed gradually with the shedding of PEG–mAb due to the positive charge of TPP. Hence, the determination of the zeta potential changes of NPs under different pH environments could also reflect pH-responsiveness. As shown in Fig. 2e, the zeta potential of NPs was 1.95 mV under normal physiological pH environment, and increased up to 9.75 mV under pH 6.5. When pH was further lowered to 5.0, there was a sharp increase in zeta potential to 18.46 mV. This demonstrated that PEG could gradually fall off under reduced pH to expose the positive charge of NPs, resulting in enhanced cell uptake to increase cytotoxicity owing to the negatively charged cell membrane. As shown in Fig. S11,† when the pH of drug-containing medium was lowered to 6.5, cytotoxicity was increased in both MCF-7 and MCF-7/ADR cells.Prescription screening of nanoparticles
It was essential to investigate what the ratio of TQ to DOX trapped into TQ/DOX–PEG–mAb could reverse the drug resistance to a large extent. As shown in Fig. 3a–d, the cell viability of MCF-7/ADR cells declined with the increase of TQ ratio, and the synergic index (SI) between TQ and DOX decreased correspondingly (Fig. S12†), indicating more effective reversal of DOX resistance. When the ratio of TQ to DOX was 1:1 and 2:1, SI (DOX: 20 μM) were relatively high, 0.627 and 0.572, respectively. However, as the ratio was raised to 3:1, SI (DOX: 20 μM) dropped a lot to 0.181. No obvious changes have been made in further improvements in the ratio to 4:1. SI between 0.1 and 0.3 means that the two drugs have a strong cooperative effect,47 hence the ratio of 3:1 of TQ to DOX was chosen for the next experiment.
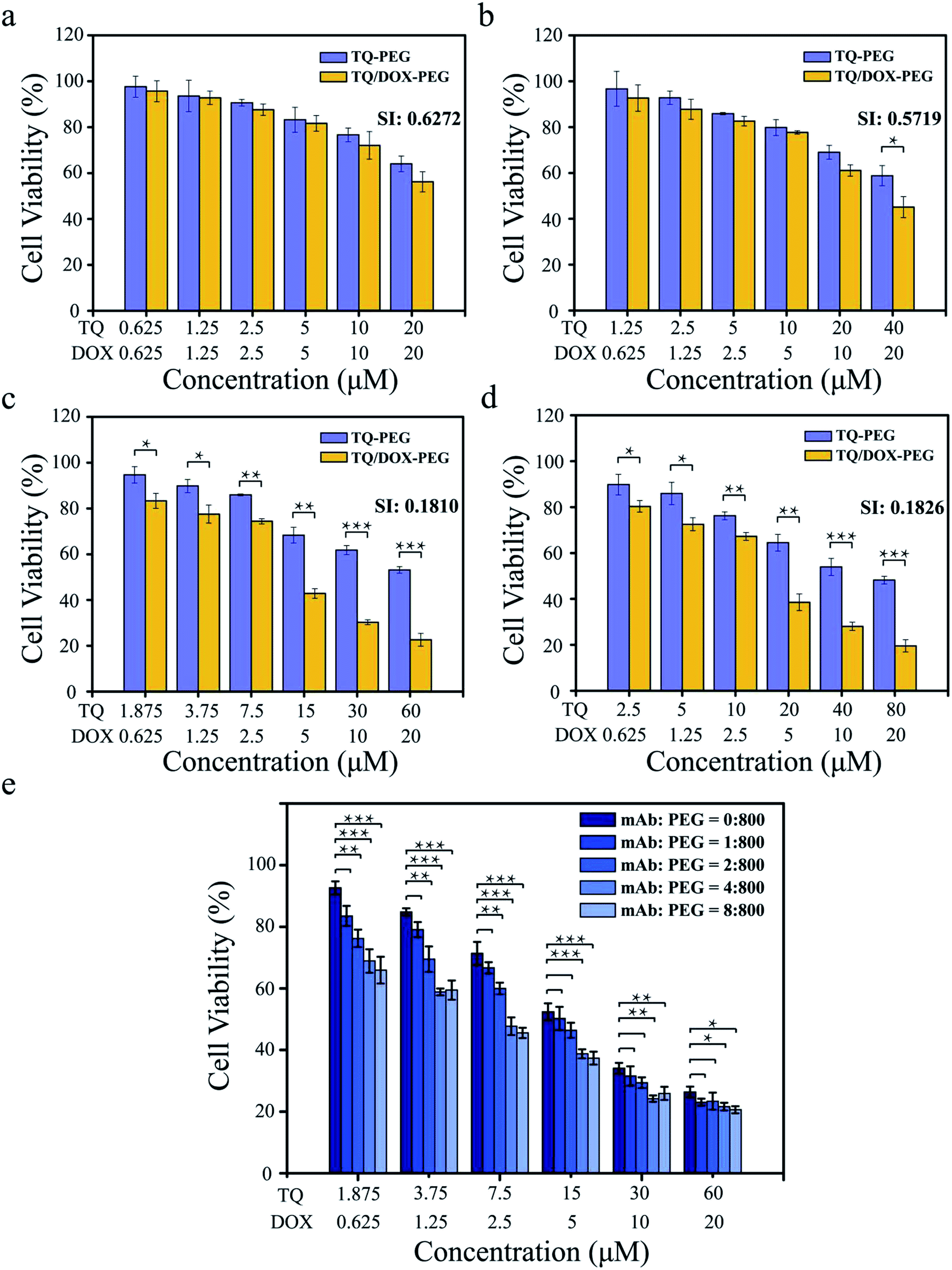 | ||
| Fig. 3 Cell viability of MCF-7/ADR cells after 48 h incubation with TQ–PEG and TQ/DOX–PEG. The molar ratio of TQ to DOX was (a) 1:1, (b) 2:1, (c) 3:1 and (d) 4:1. *p < 0.05, **p < 0.01, ***p < 0.001 were measured by independent-samples t test. (e) Cell viability of MCF-7/ADR cells after 48 h incubation with TQ/DOX–PEG–mAb with the molar ratio of mAb to PEG 0:800, 1:800, 2:800, 4:800 and 8:800, respectively. *p < 0.05, **p < 0.01, ***p < 0.001 were measured by One-Way ANOVA. | ||
Furthermore, it was also necessary to study the coupling amount of anti-VEGF mAb on the surface of TQ/DOX–PEG so as to achieve desirable synergistic treatment (Fig. 3e). With the proportion of mAb rising, the cell viability declined gradually. As the ratio of PBA–PEG–Mal to mAb was up to 800:4, a significant improvement was made in the cytotoxicity of MCF-7/ADR cells compared to the group without mAb. And there was no more noticeable effect with the further increase of mAb. Hence, the ratio of 800:4 of PBA–PEG–MAl to mAb was applied to the subsequent experiments.
In vitro cytotoxicity
The cytotoxicity of free DOX, free TQ, physical mixture (TQ + DOX), TQ/DOX–PEG and TQ/DOX–PEG–mAb were evaluated on MCF-7 cells and MCF-7/ADR cells (Fig. 4a and b). Free DOX showed concentration-dependent cytotoxicity in MCF-7 cells, whose IC50 was 4.05 μM (Table S3†). However, as for MCF-7/ADR cells, free DOX had little effect, and cell viability could still reach 80% at the concentration of 20 μM when integrating it with TQ into TQ/DOX–PEG NPs, there was significant improvement in cytotoxicity in both MCF-7 and MCF-7/ADR cells. Especially the sensitivity of MCF-7/ADR cells to DOX was heightened a lot with the IC50 of merely 5.47 μM (Table S3†), which meant that the combination of TQ could effectively reverse the resistance to DOX. Meanwhile, the cytotoxicity of TQ/DOX–PEG was much higher than that of the TQ + DOX because the formation of NPs could enhance drug internalization of cells. After the modification of anti-VEGF mAb, TQ/DOX–PEG–mAb exhibited higher cytotoxicity in both cells owing to the targeting function of mAb. These results suggested that TQ/DOX–PEG–mAb could kill drug-resistant tumor cells effectively through combined therapy. Interestingly, through the further investigation on MDA-MB-231 cells (Fig. S13†), TQ/DOX–PEG–mAb also showed obvious synergistic therapeutic effects.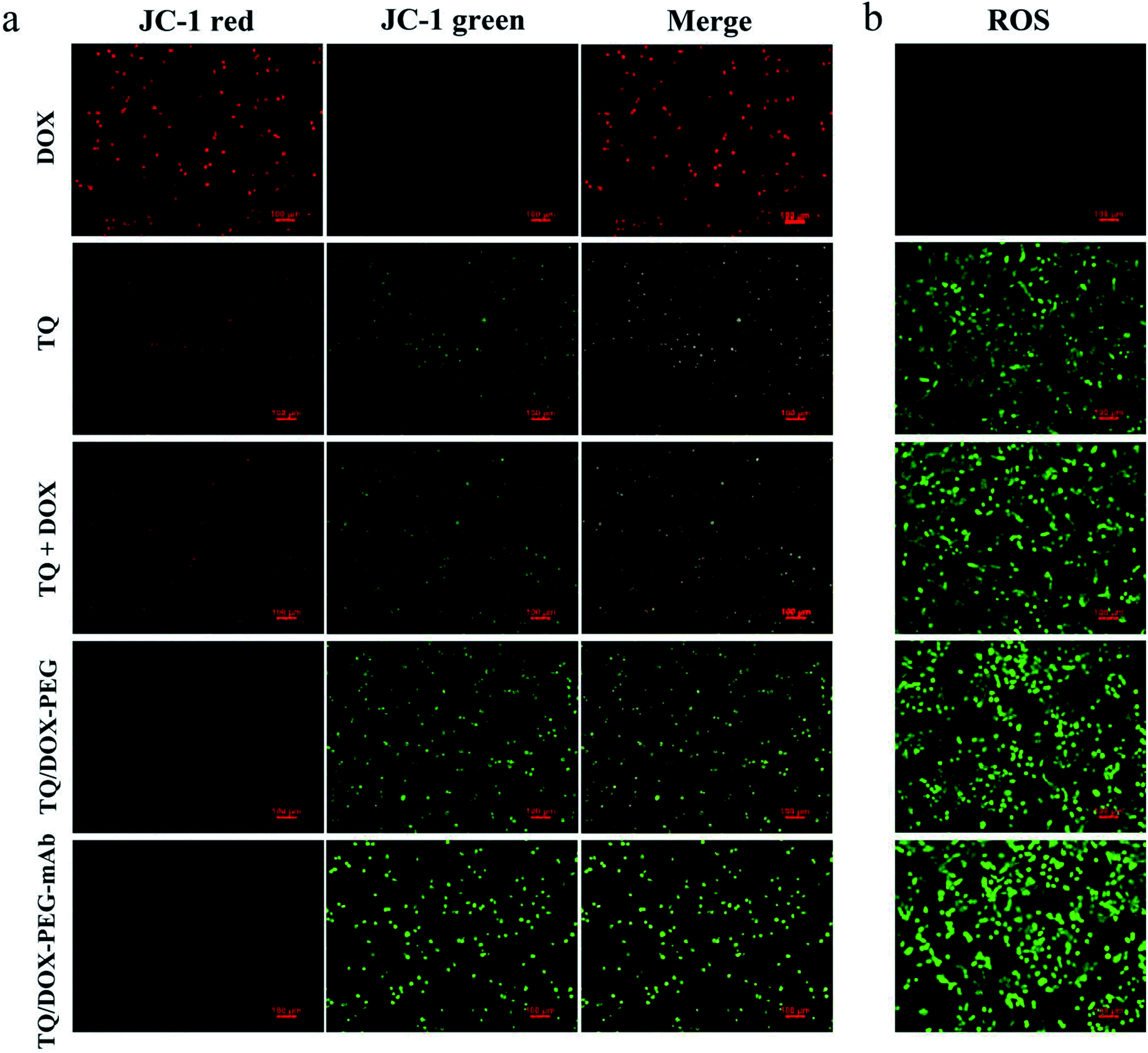 | ||
| Fig. 4 (a) Fluorescence images of mitochondrial membrane potential change of MCF-7/ADR cells after incubation with DOX, TQ, TQ + DOX, TQ/DOX–PEG and TQ/DOX–PEG–mAb for 12 h (TQ: 60 μM). JC-1 was the indicator. Red, JC-1 aggregate; green, monomeric JC-1. Scale bar = 100 μm. (b) Intracellular ROS generation in MCF-7/ADR cells treated with DOX, TQ, TQ + DOX, TQ/DOX–PEG and TQ/DOX–PEG–mAb respectively for 12 h (TQ: 60 μM). Green, DCF. Scale bar = 100 μm. | ||
The effect on mitochondrial membrane potential
In order to study the destruction of mitochondria, MMP was evaluated. Here, we chose JC-1 as the fluorescent probe, which was widely used to detect MMP. The MMP is high in normal mitochondria, and JC-1 can aggregates into the matrix of mitochondria to form a polymer (JC-1-aggregates), which produces red fluorescence; when the MMP in dysfunctional mitochondria, JC-1 keeps monomeric and cannot aggregate into mitochondria, resulting in green fluorescence. This makes it very convenient to detect changes in MMP by a shift in fluorescent color. As shown in Fig. 5a, almost no green fluorescence was observed in the MCF-7/ADR cells treated with DOX. However, free TQ caused noticeable green fluorescence with reduced red fluorescence. When integrating TQ and DOX into NPs, green fluorescence was significantly enhanced. With the involvement of anti-VEGF mAb, green fluorescence was further heightened, and almost no red fluorescence was in view at this time. Obviously, TQ had the ability to target mitochondria and cause damage, leading to the decreased MMP. Besides, the formation of NPs and modification of mAb could promote the uptake of TQ, together with the pH-responsive release, resulting in more destructive damage to mitochondria.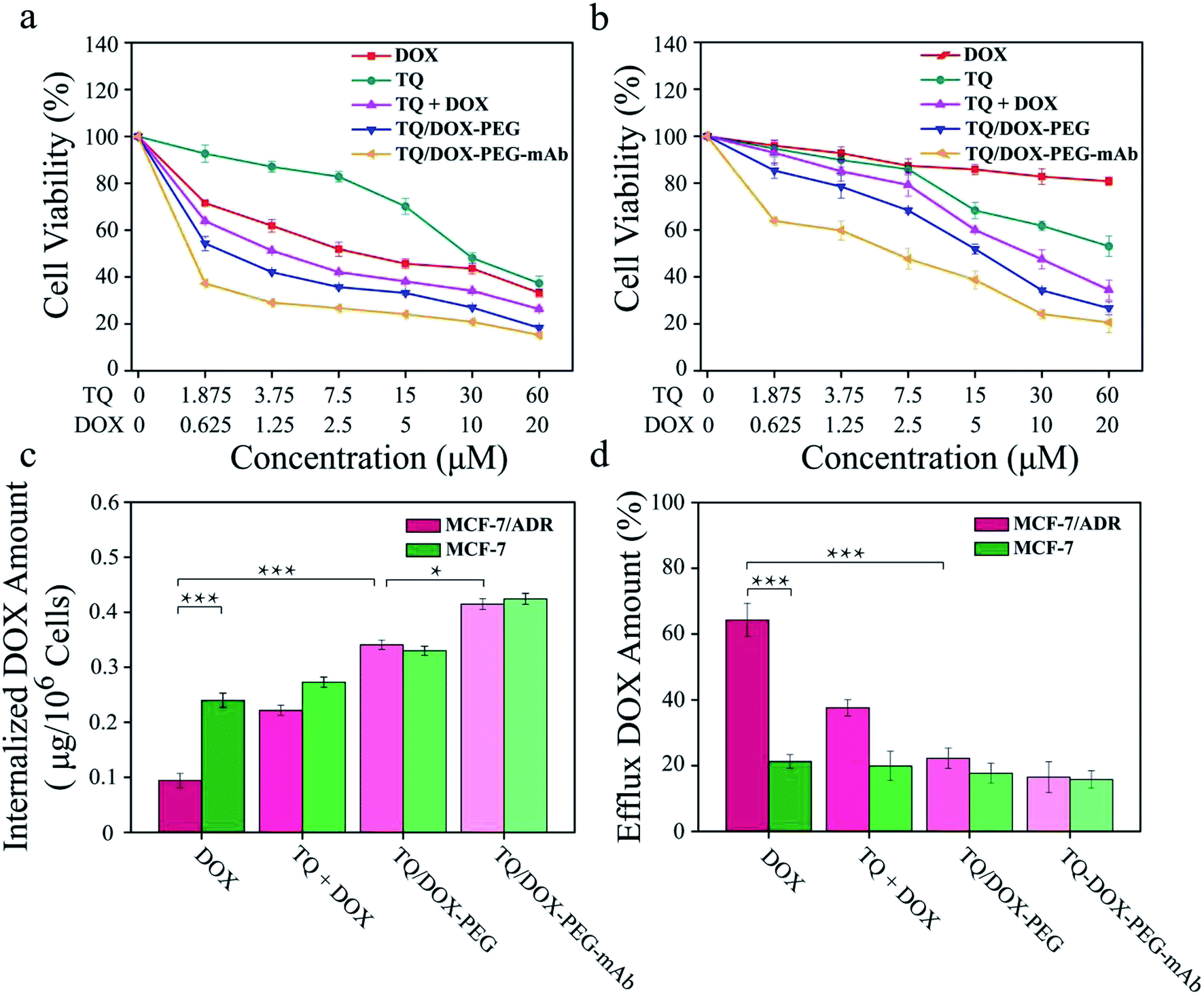 | ||
| Fig. 5 Cell viability of (a) MCF-7 cells and (b) MCF-7/ADR cells after 48 h incubation with DOX, TQ, TQ + DOX, TQ/DOX–PEG and TQ/DOX–PEG–mAb. (c) Internalization and (d) efflux amount of DOX in free DOX, TQ + DOX, TQ/DOX–PEG and TQ/DOX–PEG–mAb against MCF-7 and MCF-7/ADR cells. *p < 0.05, ***p < 0.001 were measured by independent-samples t test. | ||
Intracellular ROS level assay
To investigate the effect of TQ/DOX–PEG–mAb on oxidative damage of mitochondria functions, the intracellular ROS was assessed by DCFH-DA, which once met ROS would form DCF with green fluorescence. Significant green fluorescence appeared when MCF-7/ADR cells were incubated with free TQ while almost no fluorescence was detected in cells treated with free DOX (Fig. 5b), indicating that TQ could effectively induce ROS generation. Meanwhile, remarkable increase in intracellular ROS level was observed in cells treated with TQ/DOX–PEG, and more ROS was generated after processed by TQ/DOX–PEG–mAb. All of these results manifested that the effective accumulation of TQ in mitochondria led to the ROS generation, which could be enhanced by integration into NPs and coordination with mAb. Besides, it was reported that the increased ROS levels could induce cell apoptosis.48 Hence on one hand TQ could destroy the mitochondria to reduce the level of ATP, and on the other hand cause cell apoptosis to exert synergistic anti-tumor effect with DOX.Cellular uptake and efflux of DOX
ABC transporters was overexpressed on resistant cell membrane, which required ATP supply when pumping the drug out of the cell. TQ was mitochondria-targeting and could cause mitochondrial damage, resulting in the dysfunction of mitochondrial energy supply. To confirm the effect of mitochondrial damage on drug efflux, the internalization and efflux amount of DOX were determined. As shown in Fig. 4c, the internalized DOX amount of free DOX-treated MCF-7/ADR cells was far less than that in MCF-7 cells. Through the combination with TQ, the internalization of DOX was noticeably increased in both cell lines. Especially in MCF-7/ADR cells, the uptake of DOX was effectively reversed and almost same as that in MCF-7 cells. Besides, due to the targeting characteristic of anti-VEGF mAb, the uptake of TQ/DOX–PEG–mAb was further improved. All of these results suggested that the combination of TQ and DOX together with anti-VEGF mAb to form NPs could dramatically enhance the internalization of DOX in drug-resistant tumor cells.Meanwhile, the efflux behavior of DOX was also investigated (Fig. 4d). The efflux amount of free DOX in MCF-7/ADR cells was much higher than that in MCF-7 cells due to the overexpression of ABC transporters on drug-resistant cell membrane. However, the pumping amount of DOX was significantly reduced by uniting DOX with TQ in MCF-7/ADR cells compared with that in MCF-7 cells. In conclusion, TQ played a crucial role in inhibiting the efflux of DOX in drug-resistant cells. Consequently, the internalization of DOX was prominently increased resulting in high efficacy.
In vivo biodistribution study
In order to further study the tumor targeting capability of TQ/DOX–PEG–mAb in vivo, free DOX, TQ/DOX–PEG and TQ/DOX–PEG–mAb were investigated through the tail intravenous injection. As shown in Fig. 6a, drugs were mainly distributed in liver and tumor tissues after 24 h of administration. And the group of TQ/DOX–PEG showed remarkable fluorescence in tumor tissue while very weak fluorescence signal was observed in that of free DOX. Meanwhile, TQ/DOX–PEG–mAb displayed higher fluorescence intensity compared to TQ/DOX–PEG. Moreover, the accumulation amount of DOX in main organs was quantified in Fig. 6b. We could find that the fluorescence intensity of DOX loaded into TQ/DOX–PEG at the tumor site was around 3.8 times higher than that of free DOX. The accumulative DOX in tumor tissue of mice treated with TQ/DOX–PEG–mAb was further improved, which was 6.1 fold greater than that of free DOX, while less accumulation in liver was detected. As a supplement, the ratio of tumor to liver concerning the fluorescence intensity of DOX was calculated in Fig. 6c. It showed that the accumulative DOX was higher in tumor and lower in liver of TQ/DOX–PEG–mAb compared with the other two. These results all suggested the efficient tumor targeting effect of TQ/DOX–PEG–mAb resulting from the EPR effect of proper particle size, prolonged circulation time of the PEG–mAb shield and active targeting effect of the modified anti-VEGF mAb.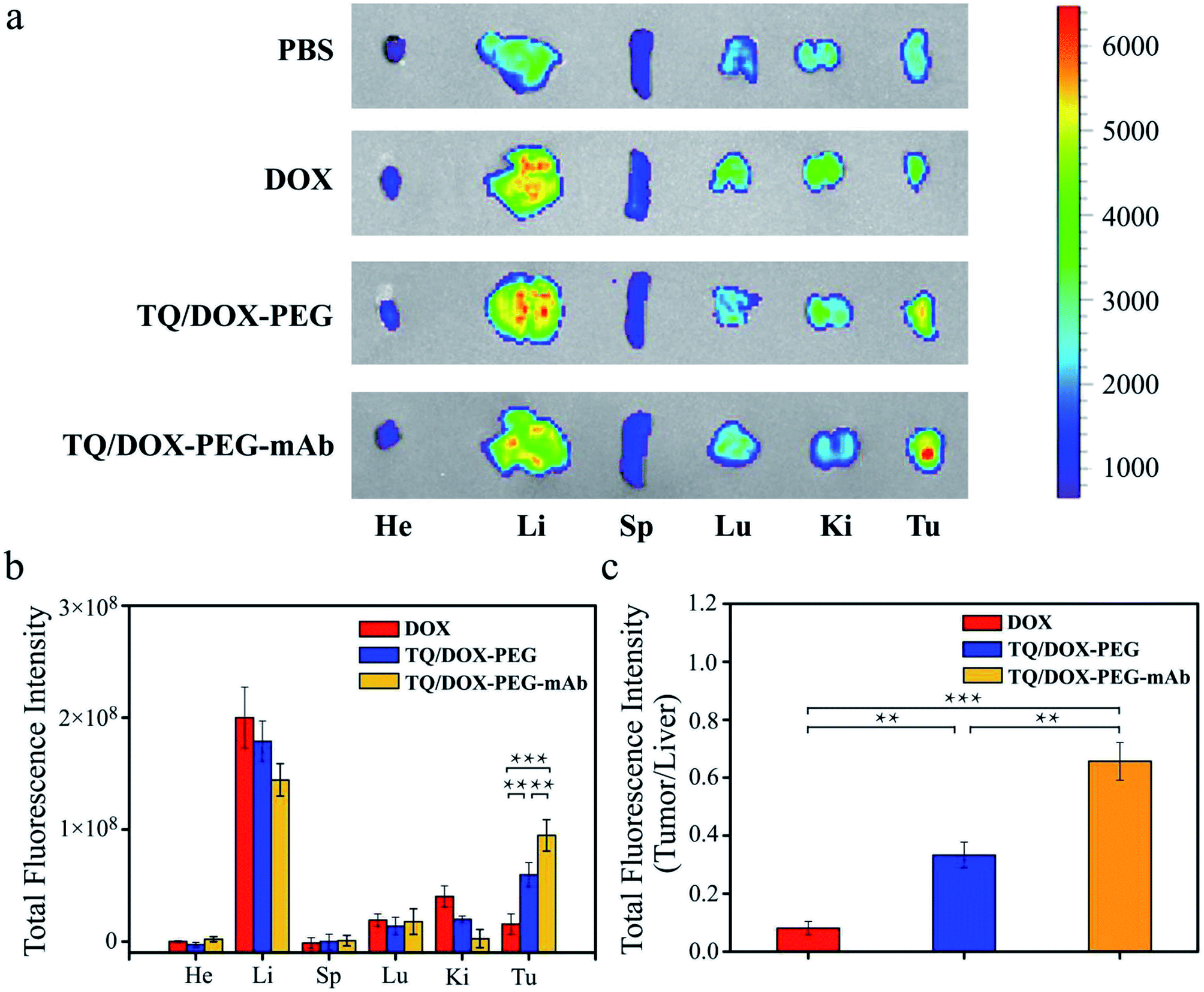 | ||
| Fig. 6 (a) Ex vivo fluorescence imaging of tumors and major organs at 24 h post injection of DOX, TQ/DOX–PEG and TQ/DOX–PEG–mAb. He: heart, Li: liver, Sp: spleen, Lu: lungs, Ki: kidneys, Tu: tumor. (b) Total fluorescence intensity of tumors and major organs (n = 3). (c) Total fluorescence intensity ratio of tumor to liver (n = 3). **p < 0.01, ***p < 0.001 were measured by One-Way ANOVA. | ||
In vivo antitumor effects and safety assay
In order to evaluate the therapeutic effect in vivo, the MCF-7/ADR tumor-bearing mice were injected intravenously with DOX, TQ, TQ + DOX, TQ/DOX–PEG and TQ/DOX–PEG–mAb every three days, lasting for 21 days. As shown in Fig. 7a, free DOX hardly had the effect on the inhibition of DOX-resistant tumor growth, which was almost as the same with the mice treated with PBS. Meanwhile, free TQ also had poor anti-tumor effect. Physical mixture group could only retard the tumor growth in the moderate manner due to the poor accumulation of drugs in tumor. However, treatment with TQ/DOX–PEG significantly hampered tumor growth. When cooperated with anti-VEGF mAb, TQ/DOX–PEG–mAb further reversed the drug resistance of MCF-7/ADR tumors, with the hardly growth of tumor volume. The same tendency could also be observed in the visual images and average weights of excised tumor (Fig. 7b). Moreover, the body weight of each treatment group in Fig. 7c implied that the physical mixture of free drugs had the obvious toxicity with the loss of weight, while TQ/DOX–PEG–mAb had the minimal toxicity in vivo. The safety for in vivo applications could be assessed more clearly from the survival curve in Fig. 7d. The mice treated with free drugs all had a high mortality during the treatment, whereas the survival rate was significantly improved when integrating the free drugs into TQ/DOX–PEG owing to the PEG shield and the EPR effect of NPs. When further modified into TQ/DOX–PEG–mAb, no mice was dead, implying that mAb decoration could further reduce the systemic toxicity in vivo through the reduced zeta potential and improved stability. The blood biochemistry assessment (Fig. S14†) further confirmed that TQ/DOX–PEG–mAb could reduce the cardiotoxicity of DOX with the normal level of LDH and CK. The results of H&E staining of normal tissues (Fig. S15†) also showed that no damage or inflammation was observed in the major organs after treatment of TQ/DOX–PEG–mAb. All these indicated that this nanoplatform could effectively treat drug-resistant tumors and was extremely secure applied in vivo.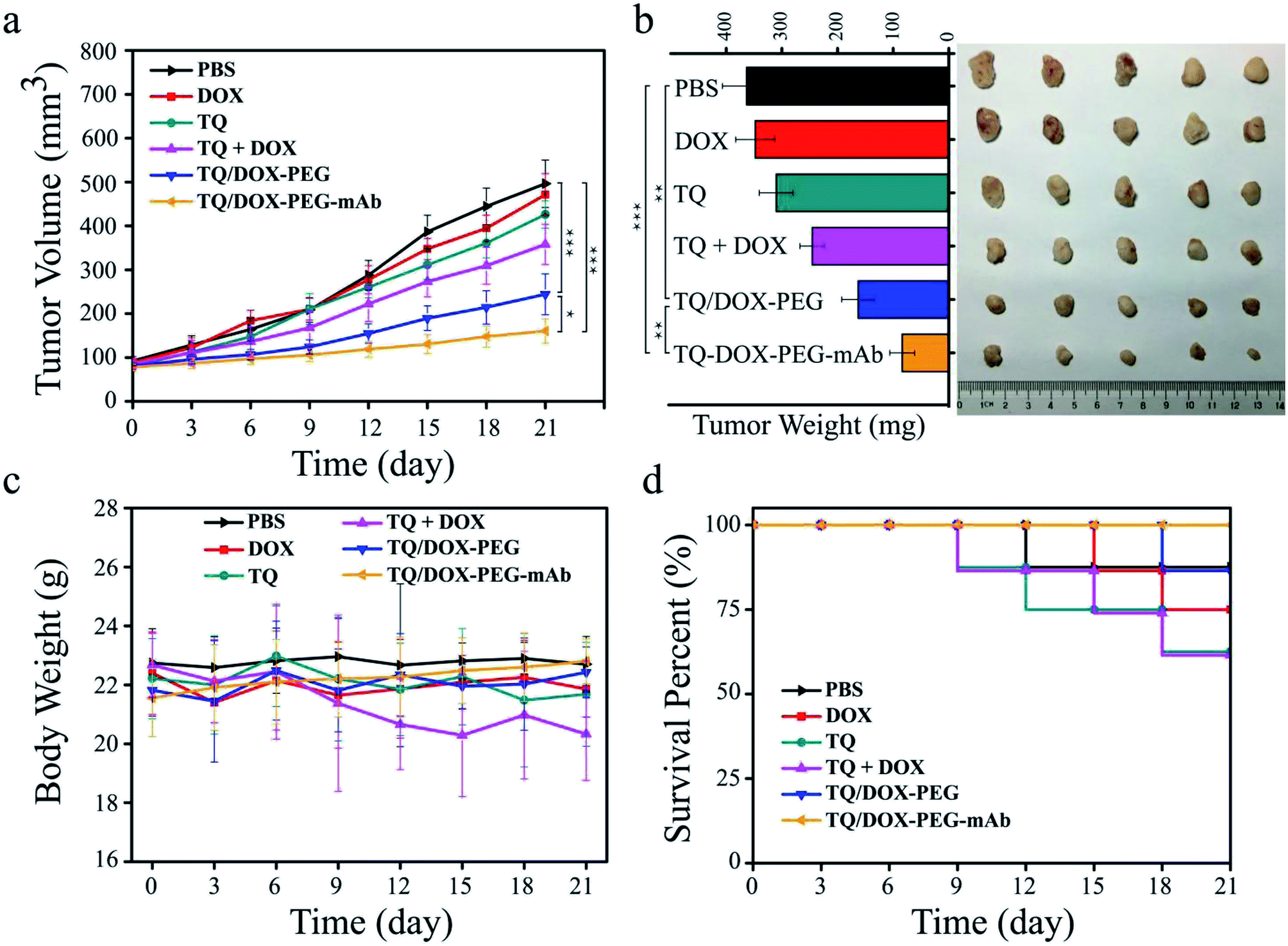 | ||
| Fig. 7 (a) The tumor volume changes, (b) tumor weight and photograph of excised tumors at the 21st day, (c) body weight changes, (d) survival curves of nude mice bearing MCF-7/ADR tumors after vail intravenous injection of different formulations for 21 days (n = 9, DOX: 1.2 mg kg−1, TQ: 4.6 mg kg−1, anti-VEGF mAb: 2 mg kg−1). *p < 0.05, **p < 0.01, ***p < 0.001 were measured by One-Way ANOVA. | ||
To further investigate the effect of TQ/DOX–PEG–mAb on tumor suppression. CD31, TUNEL, H&E and Ki67 staining were performed. To evaluate the ability of anti-VEGF mAb to inhibit neovascularization, blood vessels on tumor sections were labeled with CD31, which is one of the classical targets to identify endothelial cells. As shown in Fig. 8, the group of TQ/DOX–PEG–mAb significantly suppressed angiogenesis compared to other groups with no apparent red fluorescence. Besides, TUNEL, H&E and Ki67 staining were executed to explore the apoptosis in tumor tissues after 21 day treatment. Green fluorescence was most noticeable in TUNEL staining of TQ/DOX–PEG–mAb and almost no cell nucleus was observed in H&E staining of that, implying the strong effect to induce tumor apoptosis. Meanwhile, the quantitative analysis of TUNEL staining (Fig. S16†) also showed that TQ/DOX–PEG–mAb had a strong role in triggering apoptosis. In addition, Ki67-positive proliferating cells were prominently reduced in the group of TQ/DOX–PEG–mAb in contrast to other groups. Taken together, these results manifested that TQ/DOX–PEG–mAb could improve therapeutic effect through inducing apoptosis and inhibiting proliferation of tumor cells, as well as suppressing neovascularization effectively.
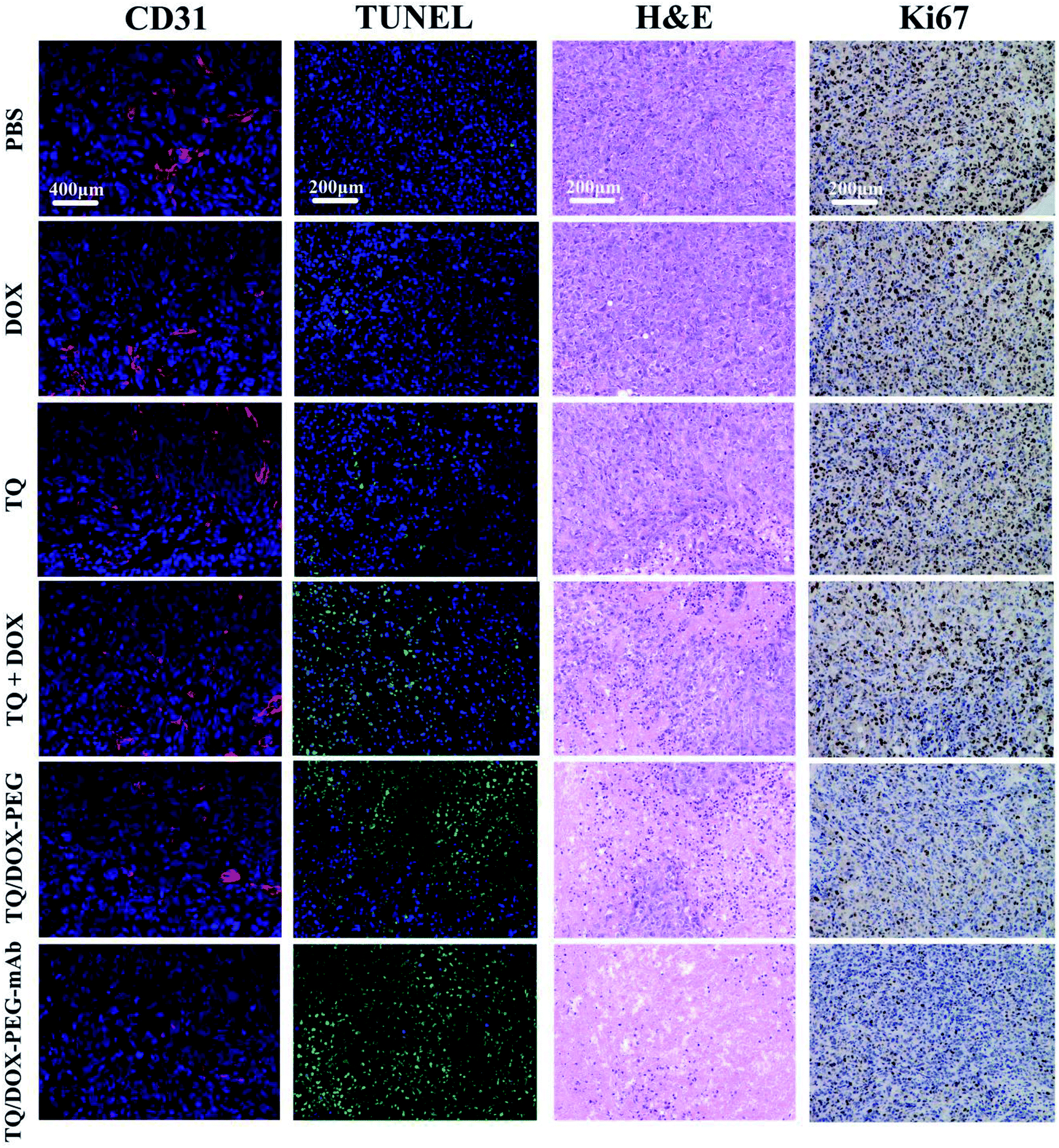 | ||
| Fig. 8 CD31, TUNEL, H&E and Ki67 staining of tumor tissues after 21 day treatment. In CD31 staining, blue: nuclei, red: endothelial cells, scare bar = 400 μm; in TUNEL staining, blue: normal cells, green: apoptotic cells, scare bar = 200 μm; in H&E staining, blue: nuclei, pink: extracellular matrix and cytoplasm, scale bar = 200 μm; in Ki67 staining, brown: proliferating cells, scare bar = 200 μm. | ||
In summary, we constructed a self-assembled TQ/DOX–PEG–mAb for reversing DOX-resistant breast cancer. The existence of boronate ester bond endowed NPs a pH-responsive release and increased surface charge density to enhance cellular uptake. Meanwhile, TQ could effectively target mitochondria and cause damage with increased ROS level and decreased MMP. Hence mitochondria was unable to afford ATP to ABC transporters, as a result, the efflux amount of DOX was significantly reduced and the cytotoxicity was correspondingly enhanced in drug-resistant tumor cells. Besides, the participation of anti-VEGF mAb could not only promote the accumulation of TQ/DOX–PEG–mAb in tumor tissues, but also exhibit a superior synergistic treatment in DOX-resistant breast cancer. This strategy showed great potential in treatment of drug-resistant tumor mainly through mitochondrial damage to reduce ATP and induce synergistic apoptosis, together with rational nanoparticle design and combined therapeutic effects of anti-VEGF mAb.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science and Technology Major Project (2017YFA0205400) and the National Natural Science Foundation of China (81773667, 81573369), and NSFC Projects of International Cooperation and Exchanges (81811540416) and the Outstanding Youth Fund of Jiangsu Province of China (Grant No. BK20160031). This work was also supported by the Fundamental Research Funds for the Central Universities (2632018PT01 and 2632018ZD12) and First-class Project (CPU2018GY06) and the “111” Project from the Ministry of Education of China and the State Administration of Foreign Experts Affairs of China (B16046). We thank the Cellular and Molecular Biology Center of China Pharmaceutical University for assistance with confocal microscopy work.References
- R. A. Smith, K. Andrews, D. Brooks, C. E. Desantis, S. A. Fedewa, J. L. Tieulent, D. M. Baptiste, O. W. Brawley and R. C. Wender, Ca-Cancer J. Clin., 2016, 66, 95–114 CrossRef PubMed.
- M. Akram, M. Iqbal, M. Daniyal and A. U. Khan, Biol. Res., 2017, 50, 33 CrossRef PubMed.
- S. T. Pan, Z. L. Li, Z. X. He, J. X. Qiu and S. F. Zhou, Clin. Exp. Pharmacol. Physiol., 2016, 43, 723–737 CrossRef CAS PubMed.
- P. Ranji, Z. Heydari and A. M. Alizadeh, Curr. Cancer Drug Targets, 2015, 15, 7 Search PubMed.
- Y. J. He, L. Xing, P. F. Cui, J. L. Zhang, Y. Zhu, J. B. Qiao, J. Y. Lyu, M. Zhang, C. Q. Luo and H. L. Jiang, Biomaterials, 2017, 113, 266–278 CrossRef CAS PubMed.
- A. H. Schinkel and J. W. Jonker, Adv. Drug Delivery Rev., 2012, 64, 138–153 CrossRef.
- E. Arrigoni, S. Galimberti, M. Petrini, R. Danesi and A. Di Paolo, Expert Opin. Drug Metab. Toxicol., 2016, 12, 1419–1432 CrossRef CAS PubMed.
- K. P. Hopfner, Biopolymers, 2016, 105, 492–504 CrossRef CAS PubMed.
- M. Zhang, L. Xing, H. Ke, Y. J. He, P. F. Cui, Y. Zhu, G. Jiang, J. B. Qiao, N. Lu and H. L. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 11337–11344 CrossRef CAS PubMed.
- H. Caitriona, V. S. Sandra, D. B. Longley and P. G. Johnston, Nat. Rev. Cancer, 2013, 13, 714–726 CrossRef PubMed.
- J. L. Markman, R. Arthur, H. Eggehard and J. Y. Ljubimova, Adv. Drug Delivery Rev., 2013, 65, 1866–1879 CrossRef CAS PubMed.
- A. R. Kirtane, S. M. Kalscheuer and J. Panyam, Adv. Drug Delivery Rev., 2013, 65, 1731–1747 CrossRef CAS PubMed.
- R. Cheng, F. Meng, C. Deng, H. A. Klok and Z. Zhong, Biomaterials, 2013, 34, 3647–3657 CrossRef CAS PubMed.
- L. L. Tayo, Biophys. Rev., 2017, 9, 1–10 CrossRef PubMed.
- C. Kwangjae, W. Xu, N. Shuming and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef PubMed.
- M. E. Davis, C. Zhuo and D. M. Shin, Nanoscience and Technology: a Collection of Reviews from Nature Journals, 2008, vol. 7, pp. 771–782 Search PubMed.
- L. Wang, Q. Sun, X. Wang, T. Wen, J. J. Yin, P. Y. Wang, R. Bai, X. Q. Zhang, L. H. Hua, A. H. Lu and C. Y. Chen, J. Am. Chem. Soc., 2015, 137, 1947–1955 CrossRef CAS PubMed.
- Y. Li, R. Li, Q. Liu, W. Li, T. Zhang, M. Zou, H. Li, T. Wu, S. Cheng, Z. Su, Z. Zhang and J. He, Mol. Pharm., 2016, 13, 3934 CrossRef CAS PubMed.
- C. G. D. Silva, G. J. Peters, F. Ossendorp and L. J. Cruz, Cancer Chemother. Pharmacol., 2017, 80, 881–894 CrossRef PubMed.
- H. G. Cui, A. J. Zhang, M. W. Cheng and J. J. Liu, Curr. Drug Targets, 2015, 16, 1356–1371 CrossRef CAS PubMed.
- K. Stefan, S. M. Schmitt and M. Wiese, J. Med. Chem., 2017, 60, 8758–8780 CrossRef CAS PubMed.
- Y. Pan, M. Cao, J. Liu, Q. Yang, X. Miao, V. L. W. Go, P. W. N. Lee and G. G. Xiao, Mitochondrial DNA and Diseases, 2017, vol. 1, pp. 149–171 Search PubMed.
- M. J. Runswick, J. V. Bason, M. G. Montgomery, G. C. Robinson, I. M. Fearnley and J. E. Walker, Open Biol., 2013, 3, 120–160 Search PubMed.
- L. Xing, J. Y. Lyu, Y. Yang, P. F. Cui, L. Q. Gu, J. B. Qiao, Y. J. He, T. Q. Zhang, M. J. Sun and H. L. Jiang, Chem. Commun., 2017, 53, 8790–8793 RSC.
- H. Wang, Z. Gao, X. Liu, P. Agarwal, S. Zhao, D. W. Conroy, G. Ji, J. Yu, C. P. Jaroniec, Z. G. Liu, X. D. Li and X. M. He, Nat. Commun., 2018, 9, 562 CrossRef PubMed.
- Y. Lu, T. Qin, J. Li, L. Wang, Q. Zhang, Z. Jiang and J. Mao, Cancer Gene Ther., 2017, 24, 386–392 CrossRef CAS PubMed.
- L. Liang, Z. Yue, W. Du, Y. Li, H. Tao, D. Wang, R. Wang, Z. Huang, N. He, X. Xie, Z. Han, N. Liu and Z. Li, Cell. Physiol. Biochem., 2017, 42, 407–415 CrossRef CAS PubMed.
- M. Shibuya, J. Biochem., 2013, 153, 13–19 CrossRef CAS PubMed.
- P. Carmeliet, Oncology, 2005, 69, 4–10 CrossRef CAS PubMed.
- Z. Zhou, H. Yao and H. Hu, Translational Research in Breast Cancer, 2017, vol. 1026, pp. 171–195 Search PubMed.
- P. S. Krishna, R. P. Nagare, V. S. Sneha, C. Sidhanth, S. Bindhya, P. Manasa and T. S. Ganesan, Int. J. Cancer, 2016, 139, 729–735 CrossRef PubMed.
- T. Simon, T. Gagliano and G. Giamas, Trends Mol. Med., 2017, 23, 282–292 CrossRef CAS PubMed.
- C. Viallard and B. Larrivée, Angiogenesis, 2017, 20, 409–426 CrossRef CAS PubMed.
- S. Shen, Y. Wu, Y. Liu and D. Wu, Int. J. Nanomed., 2017, 12, 4085–4109 CrossRef CAS PubMed.
- J. E. Chung, S. Tan, S. J. Gao, Y. Nunnarpas, S. H. Kim, J. H. Lee, H. S. Choi, Y. Hirohisa, L. Zhuo, K. Motoichi and J. Y. Ying, Nat. Nanotechnol., 2014, 9, 907–912 CrossRef CAS PubMed.
- P. Yan, K. J. Chen, J. Wu, L. Sun, H. W. Sung, R. D. Weisel, J. Xie and R. K. Li, Biomaterials, 2014, 35, 1063–1073 CrossRef CAS PubMed.
- M. C. Shin, J. Zhang, K. A. Min, K. Lee, C. Moon, J. P. Balthasar and V. C. Yang, J. Controlled Release, 2014, 194, 197–210 CrossRef CAS PubMed.
- H. Y. Liang, B. Peng, C. Dong, L. X. Liu, J. J. Mao, S. Wei, X. L. Wang, H. S. Xu, J. Shen, H. Q. Mao, X. H. Gao, K. W. Leong and Y. M. Chen, Nat. Commun., 2018, 9, 4291 CrossRef PubMed.
- M. A. Abakumov, N. V. Nukolova, M. S. Papkov, S. A. Shein, T. O. Sandalova, H. M. Vishwasrao, N. F. Grinenko, I. L. Gubsky, A. M. Abakumov, A. V. Kabanov and V. P. Chekhonin, Nanomedicine, 2015, 11, 825–833 CrossRef CAS PubMed.
- M. Hu and K. Polyak, Eur. J. Cancer, 2008, 44, 2760–2765 CrossRef CAS PubMed.
- S. H. Crayton and A. Tsourkas, ACS Nano, 2011, 5, 9592–9601 CrossRef CAS PubMed.
- X. Liang, S. M. Xu, J. Zhang, J. Li and Q. Shen, ACS Appl. Mater. Interfaces, 2018, 10, 38749–38759 CrossRef CAS PubMed.
- X. Q. Liang, C. Y. Shi, K. Wang, H. E. Xin, S. G. Liu, X. F. Huang and Y. Liu, J. China Pharm. Univ., 2012, 43, 560–566 CAS.
- C. J. Mable, R. R. Gibson, S. Prevost, B. E. Mckenzie, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 16098–16108 CrossRef CAS PubMed.
- Y. T. Fan, T. J. Zhou, P. F. Cui, Y. J. He, X. Chang, L. Xing and H. L. Jiang, Adv. Funct. Mater., 2019, 1806708 CrossRef.
- S. J. Rowan, S. J. Cantrill, G. R. Cousins, J. K. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2010, 41, 898–952 CrossRef.
- N. Fu, N. Zhang, J. H. Chou, H. J. Dong, S. F. Lin, G. S. U. Merzenich and T. C. Chou, Synergy, 2016, 3, 15–30 CrossRef.
- N. Sassi, L. Biasutto, A. Mattarei, M. Carraro, V. Giorgio, A. Citta, P. Bernardi, S. Garbisa, I. Szabò, C. Paradisi and M. Zoratti, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1095–1106 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01356c |
| ‡ Joint first authors with equal contribution. |
| This journal is © The Royal Society of Chemistry 2019 |