
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA renewable tung oil-derived nitrile rubber and its potential use in epoxy-toughening modifiers†
Laihui Xiao ab,
Zengshe Liuc,
Fangfang Huab,
Yigang Wanga,
Jinrui Huanga,
Jie Chena and
Xiaoan Nie*a
ab,
Zengshe Liuc,
Fangfang Huab,
Yigang Wanga,
Jinrui Huanga,
Jie Chena and
Xiaoan Nie*a
aInstitute of Chemical Industry of Forestry Products, CAF, National Engineering Lab for Biomass Chemical Utilization, Key Lab on Forest Chemical Engineering, SFA, Key Lab of Biomass Energy and Material, Nanjing, Jiangsu 210042, PR China. E-mail: niexiaoan@126.com; Tel: +86-025-8548-2528
bCo-Innovation Center of Efficient Processing and Utilization of Forest Resources, Nanjing Forestry University, China
cUSDA, ARS, National Center for Agricultural Utilization Research, Bio-Oils Research Unit, 1815 N University St, Peoria, IL 61604, USA
First published on 19th August 2019
Abstract
In this study, a modifier (CTMA) prepared by emulsion copolymerization of tung oil fatty acid, methyl esters of tung oil fatty acid and acrylonitrile was used to toughen epoxy resins. The structural characterization of the copolymer was carried out by Fourier transform infrared spectroscopy, 1H NMR spectroscopy and high-temperature gel permeation chromatography. Mechanical testing, thermal characterization and scanning electron microscopy were conducted to investigate the properties of epoxy resin modified by the copolymer and further reveal its toughening mechanism. The results indicated that the newly synthesized copolymer effectively toughened the epoxy resin because the elongation-at-break was increased to 89.48%, the maximum toughness calculated by work before break was nearly 4.6 times that of the neat epoxy resin, and apparent shear yields and plastic deformations were observed in the morphology of the fractured surfaces. CTMA, which acts as a flexible cross-linker in the epoxy thermoset, may decrease the cross-linking density.
Introduction
Epoxy resins are widely used in materials science, engineering and industry because of their excellent mechanical properties, low cure shrinkage, excellent cohesiveness and good chemical resistance. However, the high cross-linking structure of epoxy resins is responsible for their poor fracture toughness.1 To overcome this drawback, modifiers are incorporated into the epoxy curing system.Many kinds of modified resins, such as rubbers, thermoplastic particles, core–shell polymers and hyperbranched polymers, have been used to improve the flexibility of epoxy resins. Acting as stress concentrators, the modifiers in epoxy matrices terminate cracks by different mechanisms such as multiple crazing mechanism, multiple shear yielding mechanism, multiple crazing with multiple shear yielding mechanism and cavitation mechanism.2–5 However, because of the reaction-induced mechanism, the generation of phase separation depends on the choice of epoxy resin, curing agent and processing conditions. Therefore, a curing system toughened by modified resins will not always induce phase separation.6–9
Among the rubbers used as modifiers, the carboxyl-terminated copolymer of acrylonitrile and butadiene (CTBN) has been extensively used. The chemical reaction between carboxyl and epoxy groups is able to improve the compatibility between CTBN and epoxy resin, and the long flexible chains may toughen epoxy matrices by developing stress concentrations under stress.10–12 Laura et al. have studied CTBN-modified epoxy resin by laser confocal microscopy characterization and found that the fracture toughness increases, whereas the glass transition temperature and the tensile modulus decrease as the size and phase volume of CTBN increase;13 Huang et al. have prepared a flexible and transparent epoxy resin with tunable mechanical properties by the reaction between hydrazine hydrate and the epoxy functional group diglycidyl ether of bisphenol A (DGEBA). The curing systems incorporated with the modified resin demonstrated excellent flexibility; however, no phase separation was observed.14 Day et al. have toughened epoxy resin with a series of multilayer core–shell particles. Compared to acrylic toughening particles (ATP) and CTBN under the same conditions, the epoxy resin modified by these multilayer core–shell particles improved the toughness, which led to a significant decrease in Young's modulus.15 Fei et al. used hyperbranched polymers to toughen the epoxy resin that were synthesized from the diglycidyl ether of bisphenol A, succinic acid (SA) and 1,3,5-benzenetricarboxylic acid (BA). After being cured by methylhexahydrophthalic anhydride (MeHHPA), the modified epoxy thermosets exhibited better toughness as the elongation-at-break of 5.25% could be achieved as compared to 3% of the neat epoxy thermoset; in addition, the glass transition temperatures of the modified epoxy thermosets increased because of their hyperbranched structure.8
However, due to the growing concerns over sustainability and environmental friendliness, the use of renewable materials is necessary; therefore, traditional methods of toughening the epoxy resin by petroleum-based modified resins are being gradually replaced by those involving the use of renewable materials. Thus, the synthesis of modifiers partly or completely from renewable feedstock is urgent.
Vegetable oils (VOs) are excellent alternatives due to their abundance, low price and biodegradability.16,17 Many studies have been devoted towards the toughening of epoxy resin by epoxidized vegetable oils.18,19 These epoxidized vegetable oils are incorporated into epoxy thermosets as a flexible cross-linker, which toughen the epoxy resin by decreasing the cross-linking density. Ratna et al. toughened DGEBA by epoxidized soybean oil using an ambient-temperature hardener. When the addition amount of the modifier was increased from 5% to 30%, the maximum elongation-at-break obtained was 7.5% (which was 3.2% for the sample without the modifier); this led to a significant improvement in toughness;20 however, it still could not satisfy the requirement for application in certain special occasions.
Moreover, the conjugated structure in tung oil obtained via the Diels–Alder reaction rather than epoxidization has received significant research interest. By the Diels–Alder reaction, a number of tung oil-derived toughened epoxy resins have been produced.21–24 As is well-known, the products obtained via the Diels–Alder reaction contain a ring structure, which may reduce the toughening effect. Hence, to avoid the formation of a ring structure, an alternative strategy is to synthesize the tung oil-based toughener via radical polymerization; however, only few studies have been reported on the synthesis of an epoxy resin toughener by the radical polymerization of tung oil or tung oil-derives.
In this study, a renewable toughener was synthesized by the emulsion polymerization of tung oil fatty acid (TOFA), methyl esters of tung oil fatty acid (METOFA) and acrylonitrile. The long flexible aliphatic chains of tung oil-derives were able to toughen the rigid epoxy matrix, and the carboxyl groups of TOFA improved the compatibility of blends. Then, to study the effect of the addition content of the synthesized resin on the properties of the modified epoxy resin, the synthesized resin in different contents was incorporated into the epoxy matrix by a pre-reaction between the carboxyl groups and epoxy groups. After curing, the mechanical properties and thermal characterizations were investigated by mechanical testing, differential scanning calorimetry (DSC), dynamic mechanical analysis (DMA) and thermogravimetric analysis (TGA). In addition, the fracture surfaces were observed by a scanning electron microscope (SEM). The results of this study show that the synthesized copolymer resin can effectively toughen the epoxy resin.
Experimental
Material
Tung oil was obtained from the Institute of Chemical Industry of Forestry Products. Diglycidyl ether of bisphenol A (epoxide value: 0.48–0.54 equiv. (100 g)−1), Baling Petrochemical Branch Company, PetroChina Co., Ltd, polyetherimide (D230, amine value: 8.1–8.7 mequiv. g−1, Huntsman Chemical Co., Ltd), triphenylphosphine (99.5%, Shanghai Lingfeng Chemical Reagent Co., Ltd), acrylonitrile (98%, Sinopec Shanghai Petrochemical Co., Ltd), potassium oleate (98%, Shanghai Macklin Biochemical Co., Ltd), ammonium persulphate (98%, Shanghai Lingfeng Chemical Reagent Co., Ltd), sodium chloride (Xilong Scientific Co., Ltd), sodium hydroxide (Xilong Scientific Co., Ltd), potassium hydroxide (Xilong Scientific Co., Ltd), methanol (99.5%, Nanjing Chemical Reagent Co., Ltd), hydrochloric acid (36.0–38.0%, Nanjing Chemical Reagent Co., Ltd) and epoxidized soybean oil (epoxide value: 0.38 equiv. (100 g)−1, Shanghai Aladdin Chemical Reagent Co., Ltd) were used as received.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) were taken in a flask, followed by refluxing for 3 h at 75 °C. After decomposing the soaps by hydrochloric acid, the water layer was removed. The crude product was rinsed with water, and the trace amount of water was removed by a rotary evaporator under vacuum at 70 °C. Then, 161 g of a light brown product was obtained, whose acid value was 200.3 mg g−1 (theoretical: 201.8 mg g−1).
:1 v/v) were taken in a flask, followed by refluxing for 3 h at 75 °C. After decomposing the soaps by hydrochloric acid, the water layer was removed. The crude product was rinsed with water, and the trace amount of water was removed by a rotary evaporator under vacuum at 70 °C. Then, 161 g of a light brown product was obtained, whose acid value was 200.3 mg g−1 (theoretical: 201.8 mg g−1).The preparation method is shown in Fig. 1.
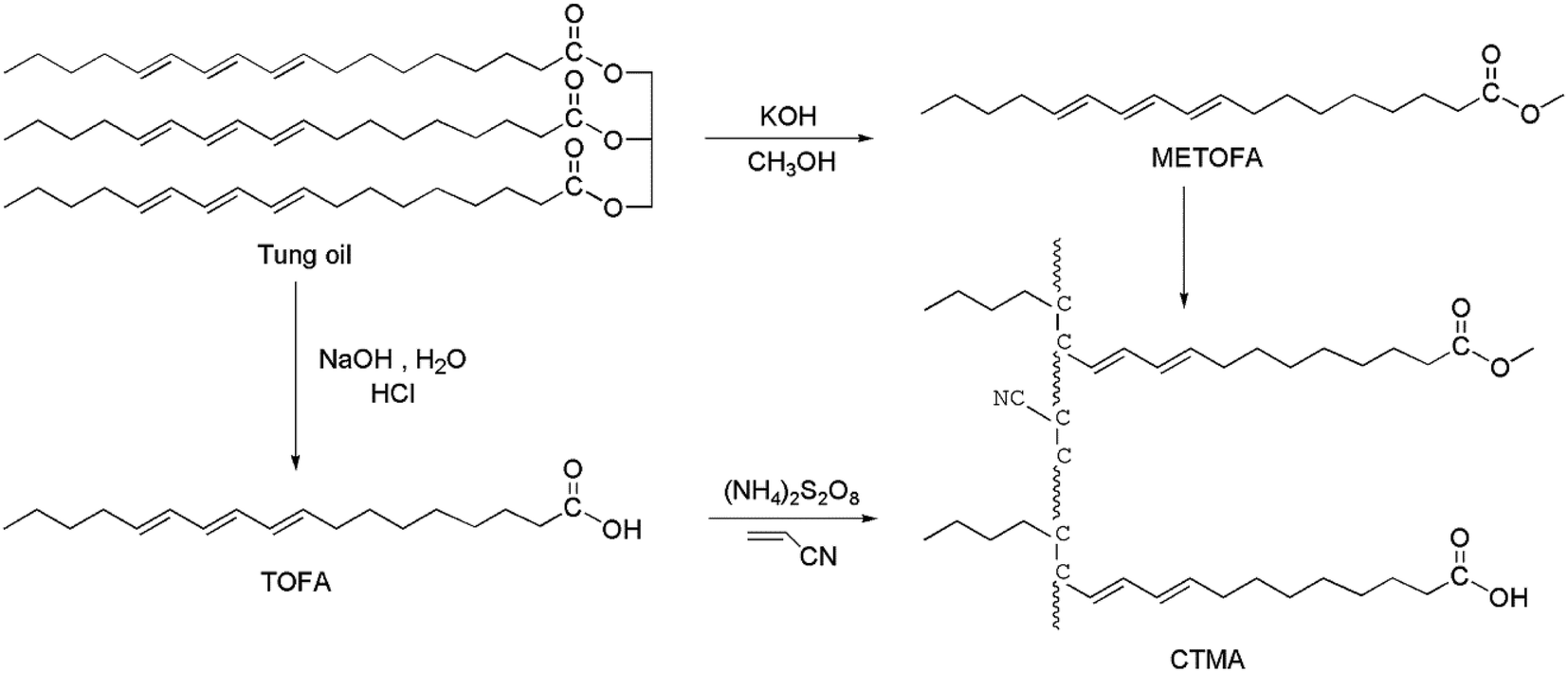 | ||
| Fig. 1 Synthesis routes of CTMA. | ||
Preparation of the CTMA-toughened epoxy resin (E-CTMA) and curing procedure
Epoxy resin and CTMA (in the mass ratios of 100:25, 100:35, and 100:45) were charged into a flask. After catalysis by triphenylphosphine (0.2 wt% of epoxy resin), the mixture was stirred at 130 °C for 3 h. After the product was cooled down to room temperature, polyetherimide was added (epoxy and curing agent were in the 1:1 equivalent ratio). The mixture was then charged into a stainless mold preheated at 80 °C. Curing was performed at 40 °C for 24 h and at 80 °C for 3 h. These samples were then coded as E-CTMA25, E-CTMA35 and E-CTMA45. Epoxidized soybean oil (ESO) was blended with epoxy resin in the mass ratio of 100:45 at room temperature, and the sample was coded as E-ESO45, which was then used for comparison purposes. Neat epoxy resin (EP) was also prepared in the same way as a control group.
Characterization
Results and discussion
Structure determinations
As shown in Fig. 2, functional group conversion can be detected by comparing the infrared spectra. In the CTMA spectrum, a weaker peak of![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H appeared at 3012 cm−1 as compared to that of tung oil-derives, which indicated the polymerization of the double bond. Furthermore, the peaks of –COOCH3 at 1739 cm−1, –COOH at 1707 cm−1 and –CN at 2228 cm−1 were observed in the CTMA spectrum, indicating that CTMA contained these three monomer units. Due to the reaction between the carboxyl groups and the epoxy groups, the peak of –COOH at 1707 cm−1 disappeared in the spectrum of E-CTMA45.
C–H appeared at 3012 cm−1 as compared to that of tung oil-derives, which indicated the polymerization of the double bond. Furthermore, the peaks of –COOCH3 at 1739 cm−1, –COOH at 1707 cm−1 and –CN at 2228 cm−1 were observed in the CTMA spectrum, indicating that CTMA contained these three monomer units. Due to the reaction between the carboxyl groups and the epoxy groups, the peak of –COOH at 1707 cm−1 disappeared in the spectrum of E-CTMA45.
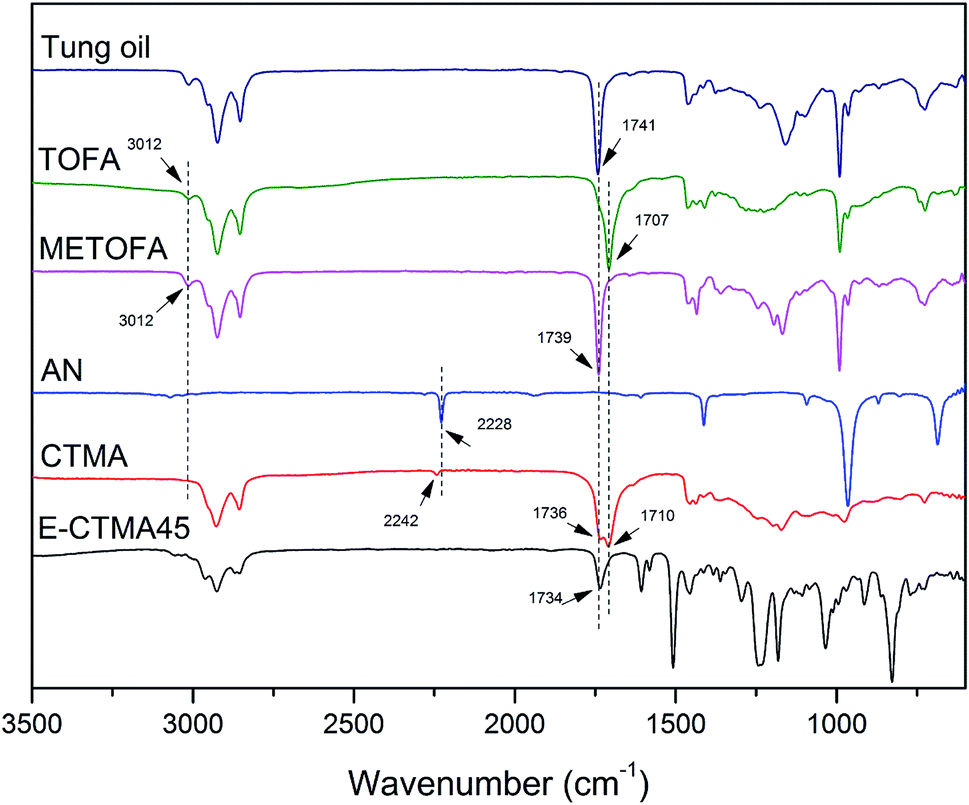 | ||
| Fig. 2 FTIR spectra of CTMA and materials. | ||
Fig. 3 shows the GPC spectrum of CTMA. Only one peak was observed in the spectrum, which confirmed that the main product was the copolymer of the three monomer units. The number-average molecular weight is about 28740 g mol−1, and the Đ value is 5.43. The high Đ value may be attributed to the different monomer activities of acrylonitrile and tung oil-derives. Furthermore, the simultaneous addition of monomers contributed to high Đ value.
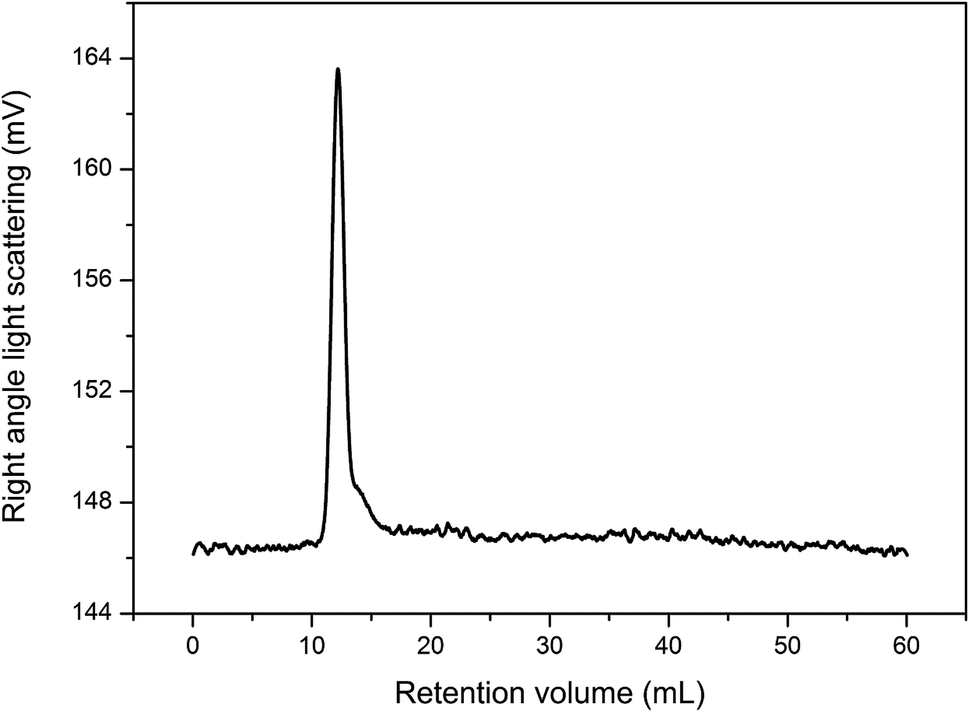 | ||
| Fig. 3 GPC spectrum of CTMA. | ||
Fig. 4 and 5 show the 1H NMR spectra of tung oil-derives and CTMA, which have been used to determine the copolymer composition. In the 1H NMR spectrum of CTMA, the proton resonances of –COOCH3 (3.69 ppm) in METOFA and –CH3 (0.91 ppm) in TOFA and METOFA are well separated, whereas those for methylene protons and methine protons (1–2.5 ppm) are overlapped. Hence, their relative peak areas can be used to calculate the mole fraction of the monomer by the following formulas,
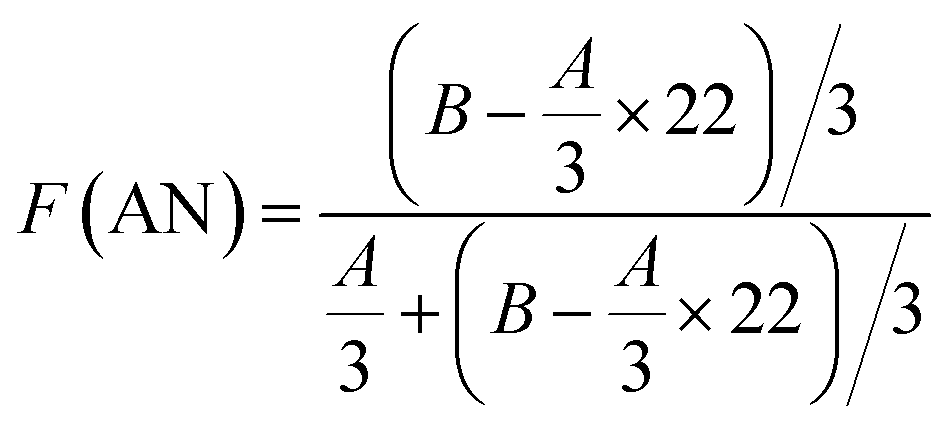 | (1) |
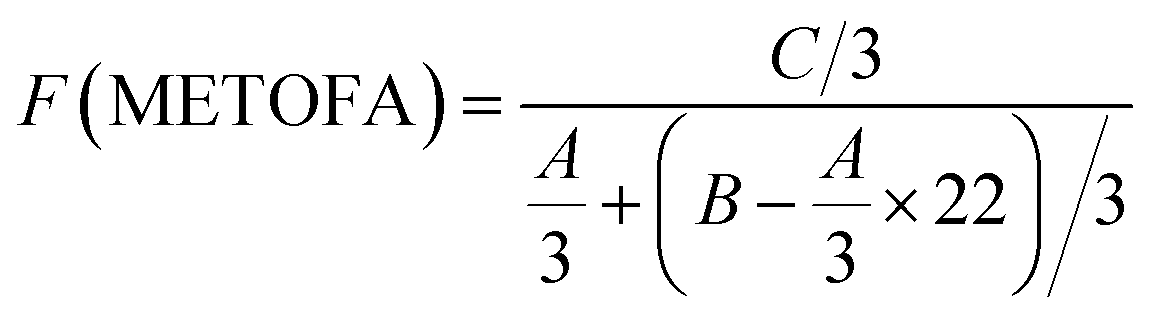 | (2) |
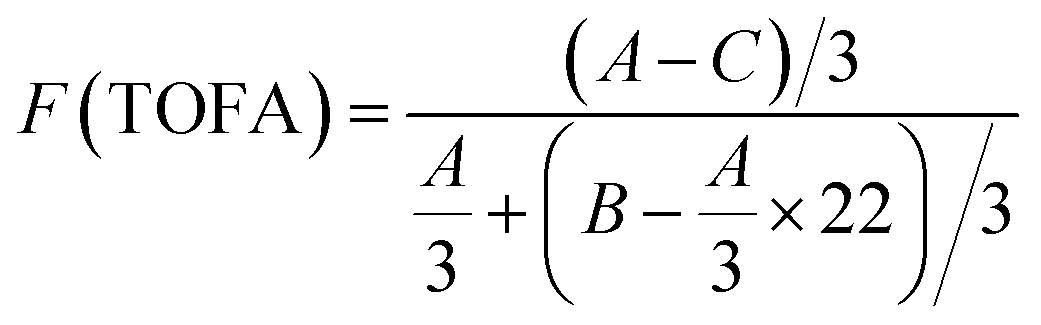 | (3) |
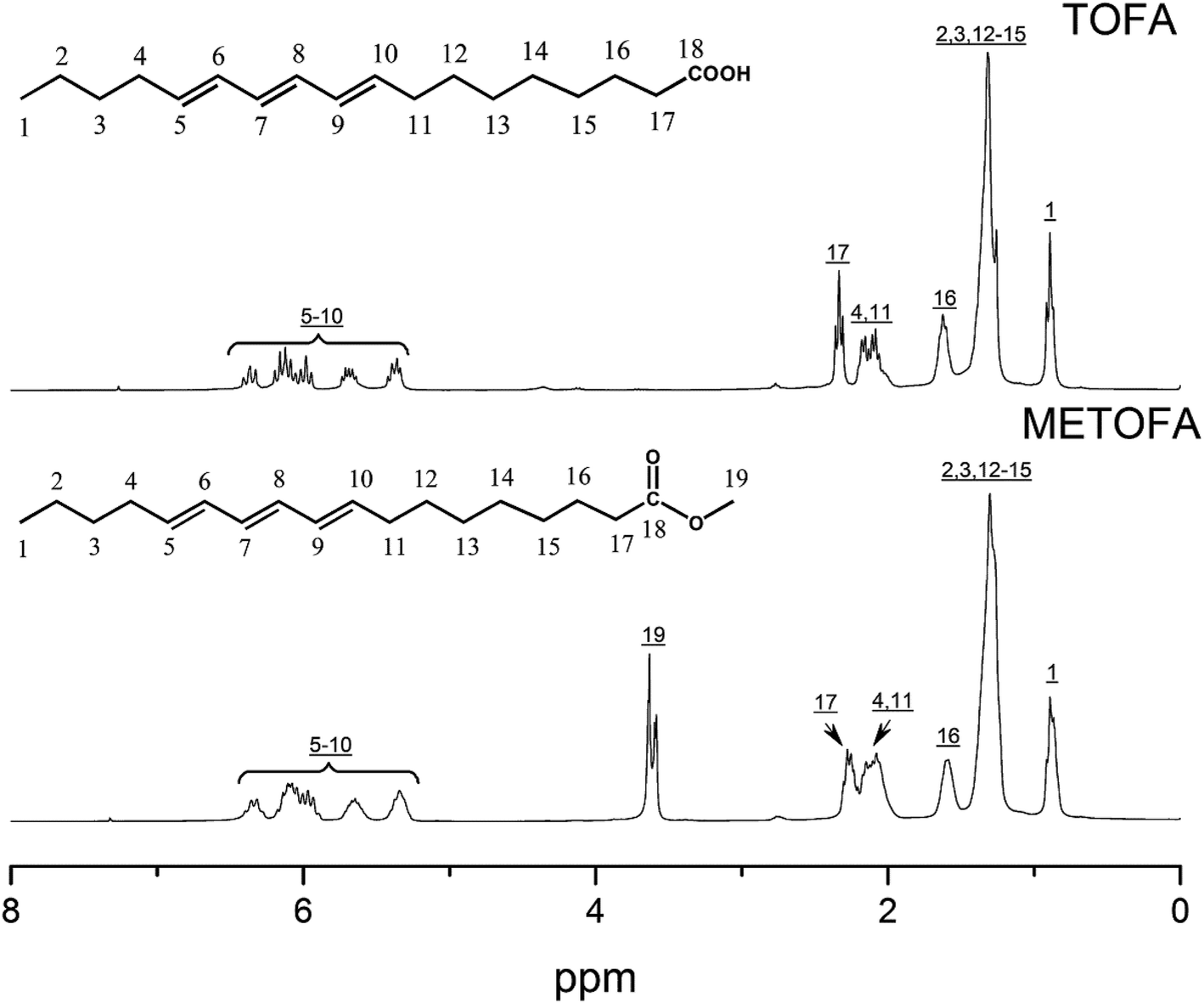 | ||
| Fig. 4 1H NMR spectra of TOFA and METOFA. | ||
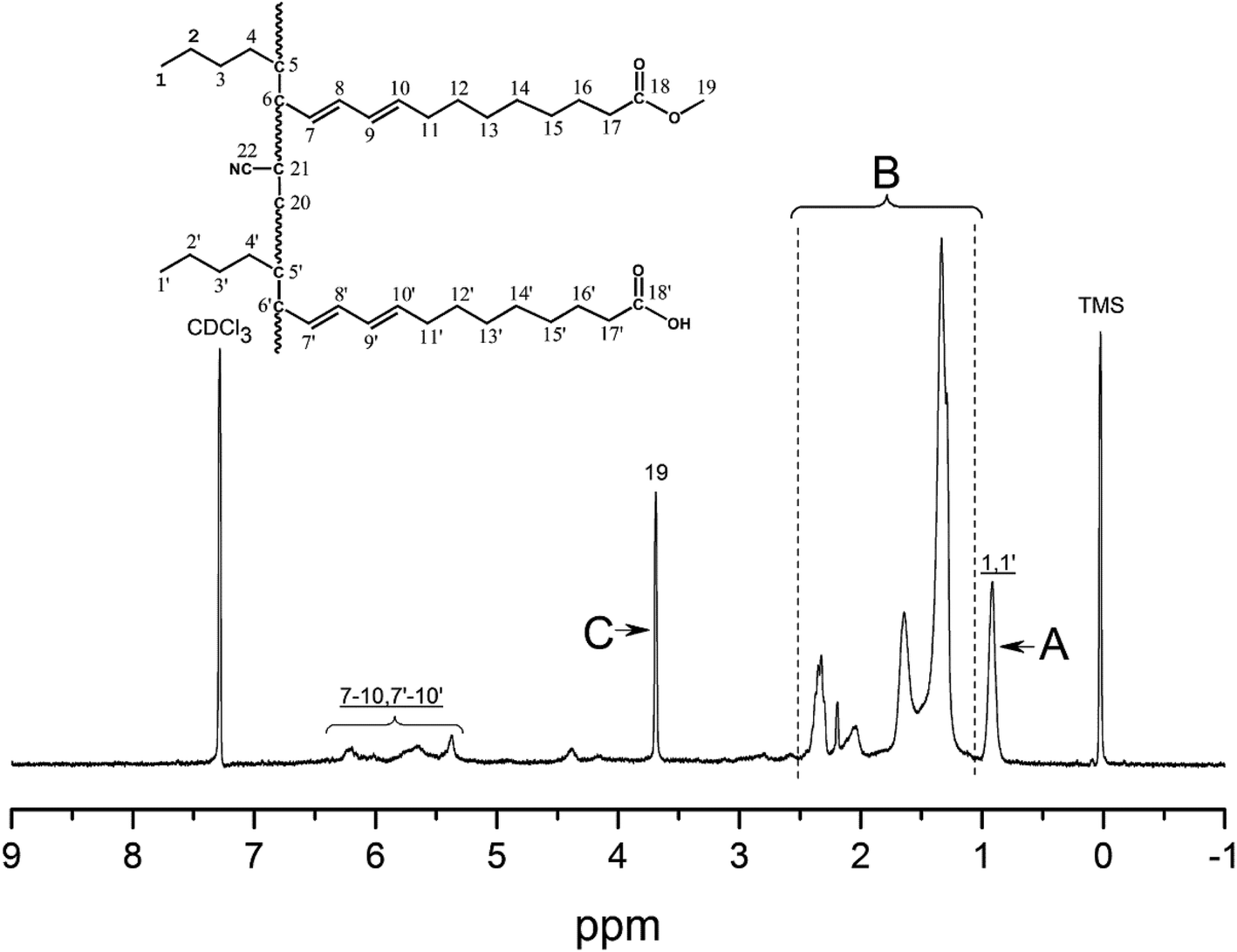 | ||
| Fig. 5 1H NMR spectrum of CTMA. | ||
| Part | A | B | C |
|---|---|---|---|
| Peak area (relative to C) | 1.75 | 14.28 | 1 |
| Monomer | AN | METOFA | TOFA |
| Mole fraction (%) | 45.26 | 31.28 | 24.06 |
Differential scanning calorimetry
Fig. 6 shows the results of DSC. In Fig. 6(a), we can observe a melting peak and a crystallization peak, which illustrates the crystallizability of CTMA. Moreover, the approximate enthalpy values confirm the corresponding melt crystallization process. As observed in the CTMA structure shown in Fig. 1, the flexibility of the long aliphatic chain causes the molecules to align; this leads to crystallization. Furthermore, the induction forces induced by the cyano groups were able to stabilize the crystallization. Note that via both the heating curve and the cooling curve, the glass transition temperature of CTMA can be detected, which is around −26.76 °C.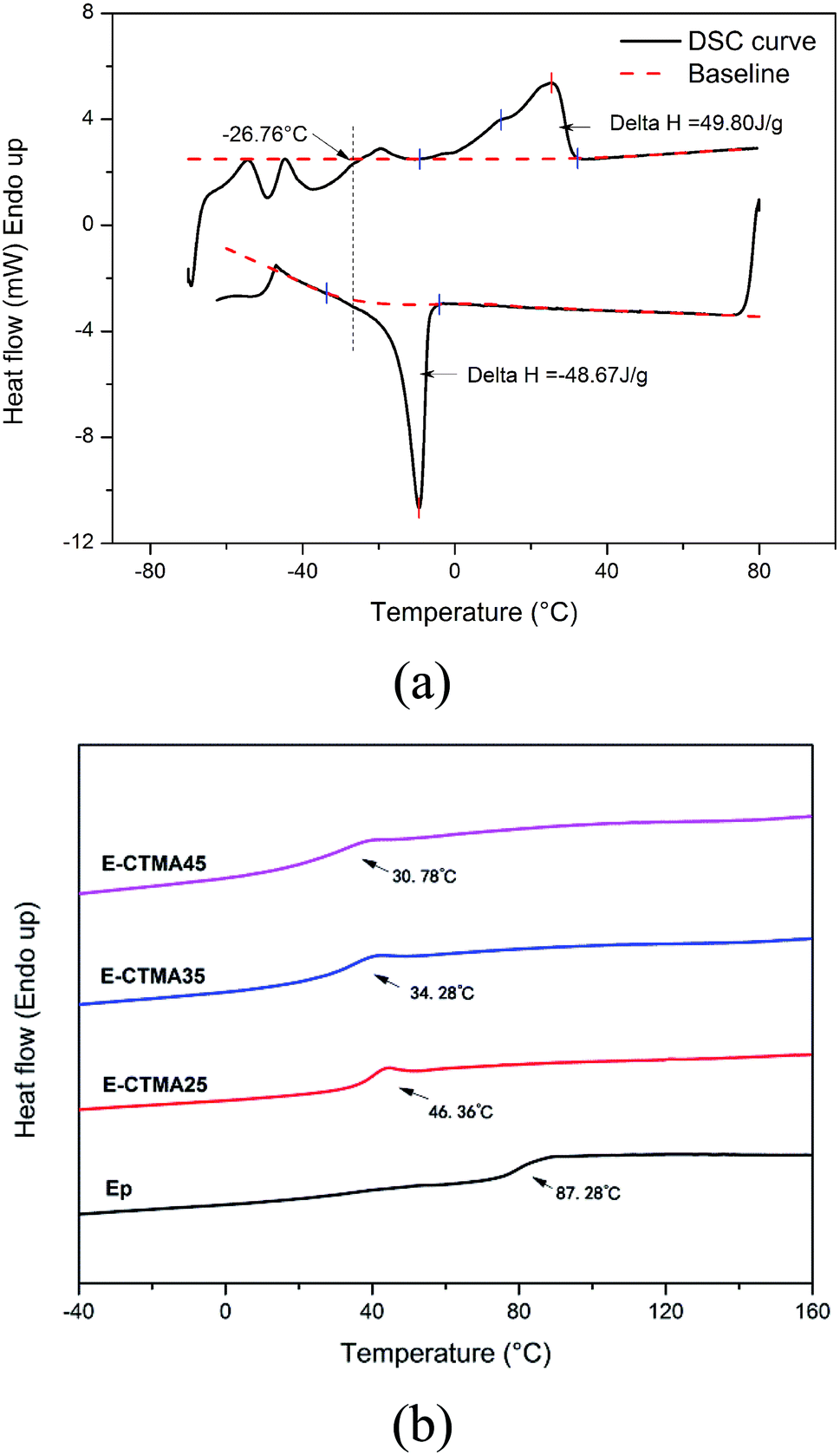 | ||
| Fig. 6 DSC curves of CTMA (a) and cured thermosets (b). | ||
However, no melting or crystallization peaks were observed in the curves of E-CTMA, and only one glass transition temperature could be detected, which indicated that no phase separation occurred. This was because the epoxy resin incorporated CTMA by the reaction between the carboxyl groups and the epoxy groups; this contributed to the molecular-level compatibility.
As flexible cross-linkers, many long flexible aliphatic chains were incorporated into E-CTMA thermosets. Segment movement in flexible aliphatic chains can be easily induced by increasing the temperature, which will contribute to a lower glass transition temperature; furthermore, the high cross-linking network system will be destroyed, and the less restriction from covalent bonds contributes to segment movement, which will also contribute to a lower glass transition temperature. All these results are in accordance with the decreasing Tg value shown in Fig. 6(b); moreover, further explanation about this point has been provided in the discussion of DMA.
Furthermore, the curing behavior was studied by DSC. In Fig. 7(a), all samples show a similar curing behavior, and only one curing process has been determined. The accurate parameters of the curing process are presented in Table 2. Obviously, the To and Tp values of E-CTMAs are significantly lower than that of the neat epoxy resin, and a decreasing tendency is observed as the content of CTMA is increased. Previous studies have demonstrated that the existence of strong hydrogen bonding species, such as acids and alcohols, is able to accelerate the epoxy–amine reaction.25 In the blending process, the carboxyl groups in CTMA react with the epoxy groups, and the same amount of hydroxyls are simultaneously produced. Therefore, the curing process is accelerated, which is in agreement with the decreased values of To and Tp. Furthermore, this positive effect on the curing process is shown in Fig. 7(b) in which it is observed that CTMA enhances the conversion at a specific temperature; however, higher content of CTMA results in lower enthalpy (ΔH). This is because the addition of CTMA reduces the content of epoxy groups and curing agent; this leads to fewer reactions. Moreover, the incorporation of CTMA increases the viscosity of epoxy resin (Table 2), which is attributed to the much higher molecular weight of CTMA. Furthermore, the pre-reaction between the carboxyl and the epoxy groups may produce hydroxyls, which may form hydrogen bonds with other polar groups and further increase the viscosity. Similar results have been obtained in previous studies, in which rubber and other oil-based polymers have been used to toughen the epoxy resin.12,21,26 The increased viscosity may restrict the movement of reaction groups and then reduce the enthalpy.
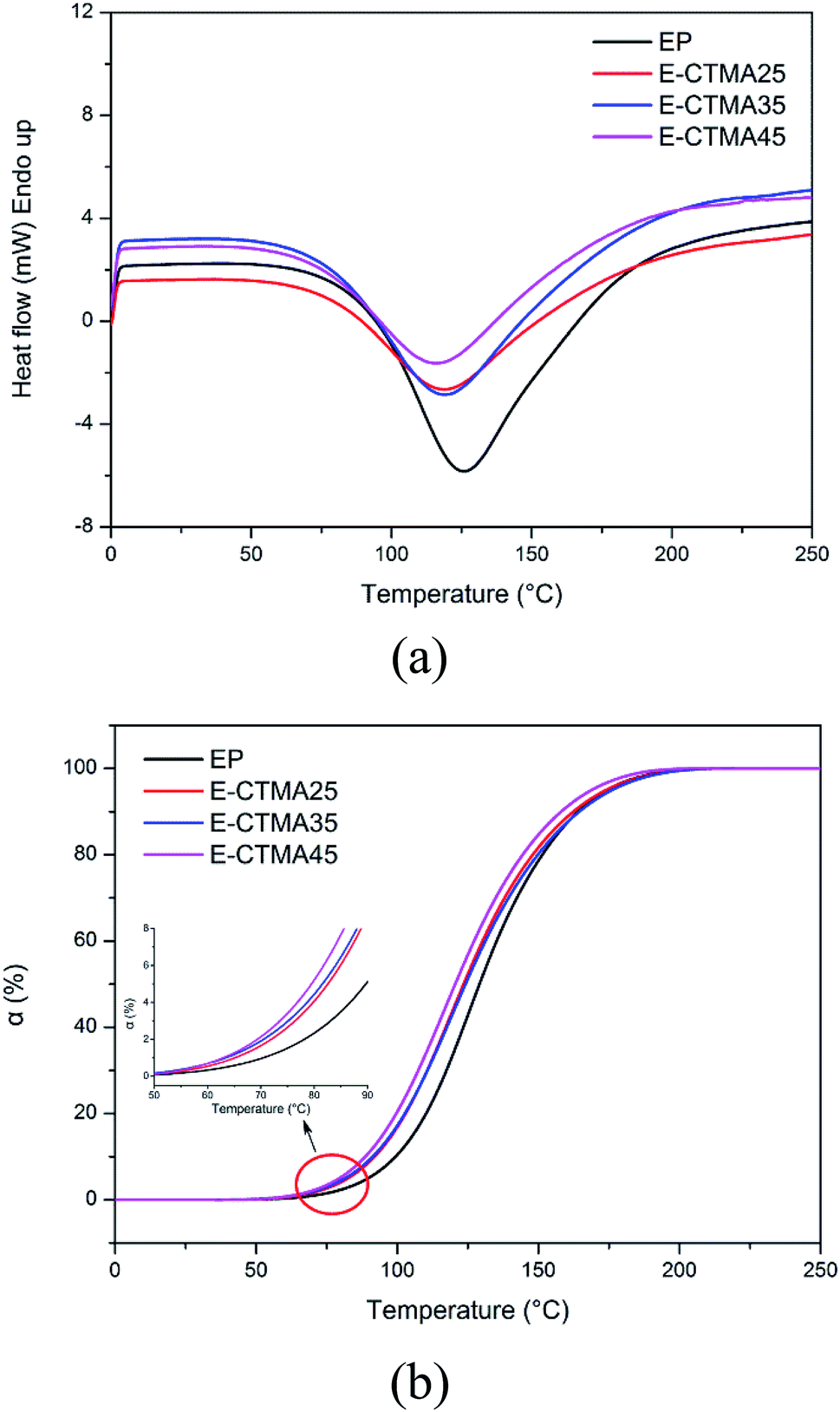 | ||
| Fig. 7 DSC curves of the curing process: heat flow against temperature (a) and conversion against temperature (b). | ||
| Sample | Toa (°C) | Tpb (°C) | ΔH (J g−1) | Viscosityc (kPa s) |
|---|---|---|---|---|
| a To is the initial curing temperature.b Tp is the temperature at which the curing rate is maximum.c Viscosity is measured at room temperature. | ||||
| EP | 89.17 | 126.18 | 379.65 | 12.36 |
| E-CTMA25 | 78.37 | 119.43 | 297.89 | 19.16 |
| E-CTMA35 | 77.89 | 119.25 | 292.83 | 27.08 |
| E-CTMA45 | 74.43 | 116.78 | 244.34 | 51.22 |
Dynamic mechanical analysis
Fig. 8 shows the temperature dependence of storage modulus and tanδ of the neat epoxy resin and E-CTMA thermosets. The accurate statistics of thermal properties are presented in Table 3.
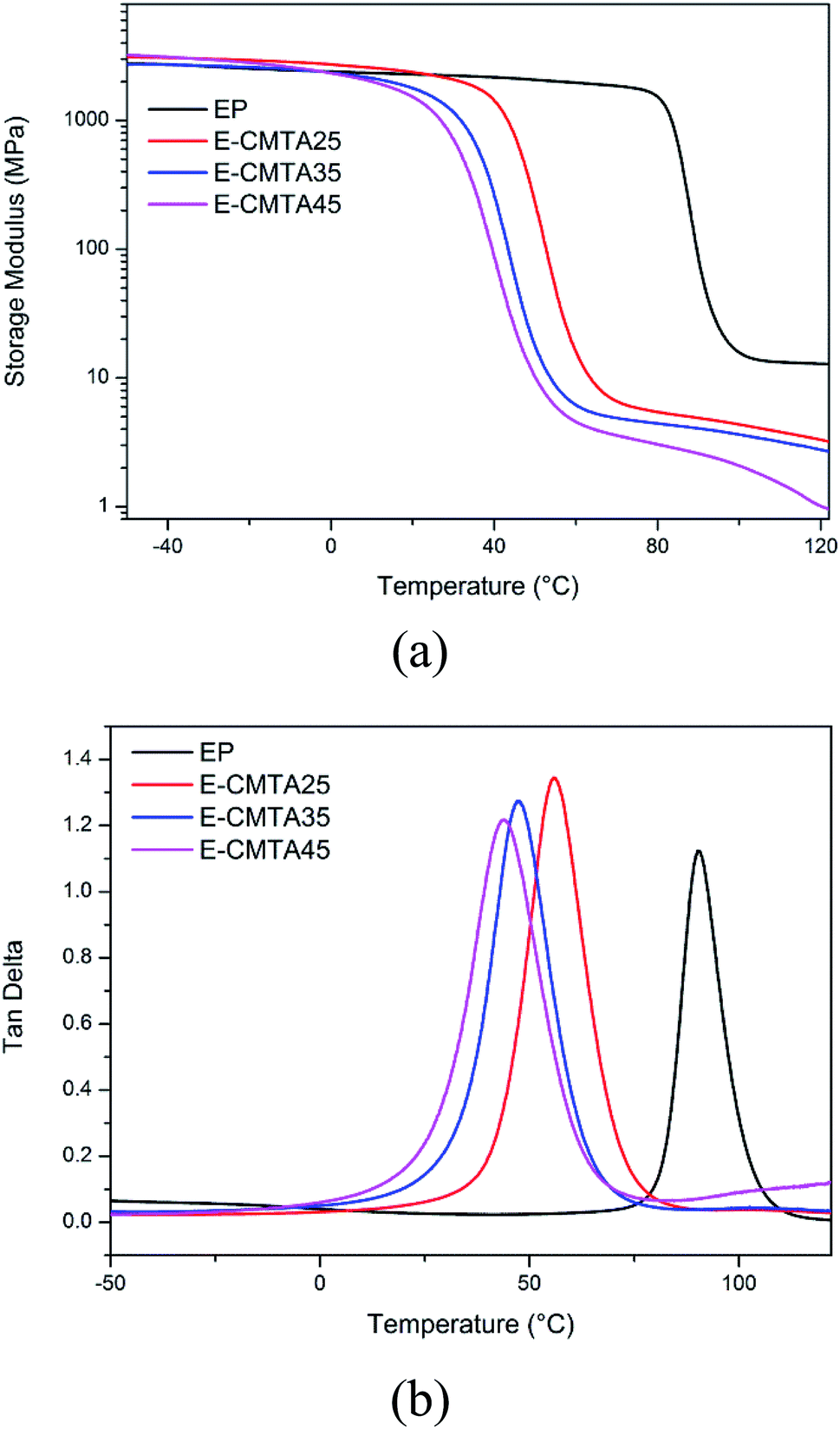 | ||
| Fig. 8 Storage modulus (a) and tanδ (b) of the neat epoxy resin and E-CTMA thermosets. | ||
Tg decreases with an increase in the CTMA content; this is in accordance with the abovementioned DSC results provided in the DSC section. Furthermore, the explanation of the decreasing Tg can be confirmed by the results of half-peak width (HPW) in tanδ curves, which is related to structural uniformity.27 The neat epoxy resin has the lowest HPW value, whereas E-CTMA45 has the highest HPW value of E-CTMA. This is because CTMA acts as a flexible crosslinker, which increases the molecular weight between cross-linking points. Furthermore, the random carboxyl distribution of CTMA extends the dispersity of molecular weight between cross-linking points. However, every tanδ curve of E-CTMAs and neat epoxy resin has only one peak, which illustrates that the uniform network structure is unable to lead to phase separation.28,29
Based on the theory of rubber elasticity, the cross-linking density ve can be calculated from the storage modulus in the rubbery region:
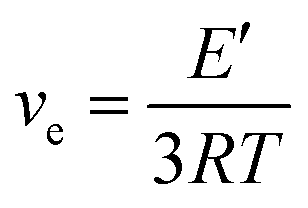 | (4) |
The value of ve decreases with the increasing content of CTMA; this indicates lower cross-linking density. With fewer cross-linking points, chain segments move more easily; this contributes to a lower glass transition temperature. Thus, the decreasing glass transition temperatures are presented as Tg value in Table 3.
Morphology of the fractured surfaces
In the SEM images shown in Fig. 9, we can observe that the fracture surface of neat epoxy samples is very smooth, whereas that of the CTMA-modified epoxy samples is rather rough. No cavity was observed on the fracture surfaces; this indicated that no phase separation occurred.14,30 This result is in line with the results of DSC and DMA. Furthermore, massive shear yields and plastic deformations observed on the E-CTMA fracture surfaces indicate a typical toughness character. | ||
| Fig. 9 SEM images of the frozen fracture morphology for the thermosets: neat epoxy, E-CTMA25, E-CTMA35 and E-CTMA45. | ||
When CTMA was added as a modifier, the carboxyl groups reacted with the epoxy groups; this led to molecular level incorporation instead of phase separation. This phenomenon has also been observed by other researchers.31–34 Because of the long aliphatic chains of CTMA, external stresses can be redistributed, and plastic deformations will be induced by the flexible cross-linker. This is the reason why the fracture surfaces become rougher.
Thermogravimetric analysis
The thermal stability of CTMA and the neat epoxy resin and E-CTMA thermosets was examined, as shown in Fig. 10. We can observe that E-CTMAs tend to decompose at lower temperatures, T5% and T10% presented in Table 4; this is attributed to the decomposition of small molecules (carbonyl and ester) in CTMA. Consequently, a subtle peak at 250 °C appears in the derivative curves with an increase in CTMA content, which has been magnified in the inset of Fig. 10(b). The peak around 395 °C in the derivative curves demonstrates the main decomposition process, which is considered to be caused by the degradation of the cross-linked epoxy resin networks.35,36 Hence, the maximum degradation rate temperatures of E-CTMA and neat epoxy resin thermosets are approximate. Similar results have been reported by other researches.35,37–39 Kumar et al. toughened the epoxy resin using ESO in different contents, and the results show that T5% decreases from 341 °C to 283 °C when the mass ratio between epoxy resin and ESO is 80/20;38 however, CTMA leads to a greater weight loss; this may be explained in terms of the cross-linking density. As is well-known, higher cross-linking density contributes to higher thermal stability.38 The incorporation of CTMA results in a great loss in the cross-linking density, which may account for the loss in thermal stability. However, the epoxide equivalent weight of commercial ESO is about 260 g mol−1. In other words, each ESO molecule contains more than 3 epoxy groups on an average, which may react with the curing agent and avoid a sharp decrement in the cross-linking density. Therefore, the reduction of thermal stability is less.35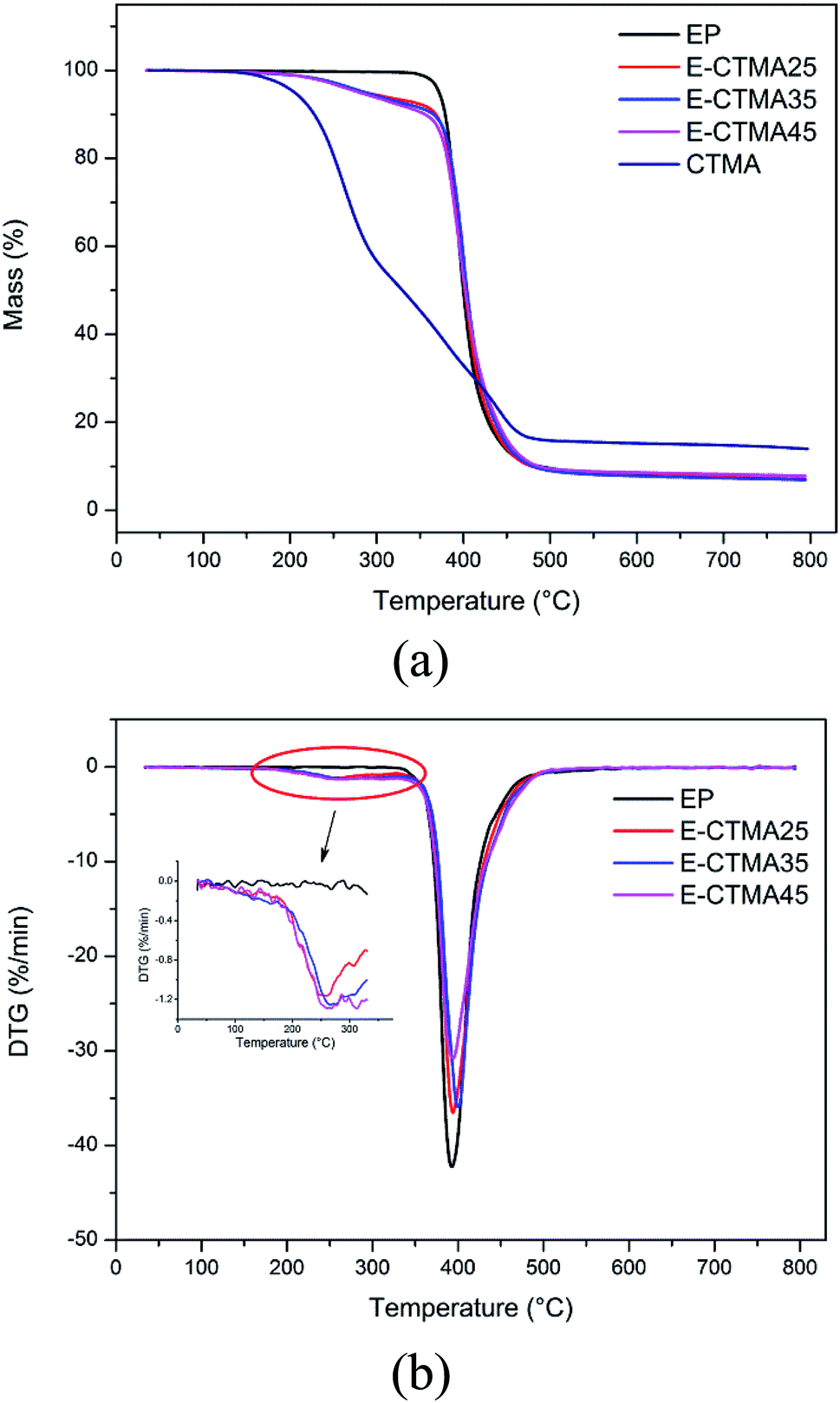 | ||
| Fig. 10 Thermogravimetric plots of the curing system: TG curves (a) and DTG curves (b). | ||
| Sample code | T5%a (°C) | T10%a (°C) | Tmb (°C) |
|---|---|---|---|
| a T5% and T10% are the temperatures of 5% and 10% weight loss, respectively.b Tm is the temperature at which the degradation rate is maximum. | |||
| EP | 372.06 | 378.92 | 392.7 |
| E-CTMA25 | 286.11 | 368.04 | 394.0 |
| E-CTMA35 | 285.91 | 365.29 | 400.2 |
| E-CTMA45 | 279.49 | 355.32 | 393.8 |
| CTMA | 204.94 | 227.84 | 263.9 |
Mechanical properties
The mechanical properties of all thermosets are shown in Fig. 11. As can be seen, the elongation-at-break, which indicates the toughness of the material, has increased dramatically from 5.6% to 104.5%; this suggests that CTMA can effectively toughen the epoxy curing system. This is owing to the flexibility of the modifier, which may restrict crack extension and induce crazes.40 Compared with other tung oil-based products synthesized by the Diels–Alder reaction, whose elongation-at-break is less than 5%, E-CTMAs show higher toughness.21,23,24 However, an increase in the content of CTMA decreases the tensile strength and Young's modulus. Because the addition of CTMA reduces the cross-linking density and enhances ductility, the modified samples are less sensitive to deformation; this leads to lower Young's modulus.41,42 In addition, due to the presence of less covalent bonds, the samples are unable to withstand high stress; this accounts for the decrement of tensile strength. Furthermore, toughness can be indicated in terms of fracture energy. The increasing toughness, calculated by integrating the area under stress–strain curves,43 demonstrates that CTMA can effectively toughen the epoxy curing system as well.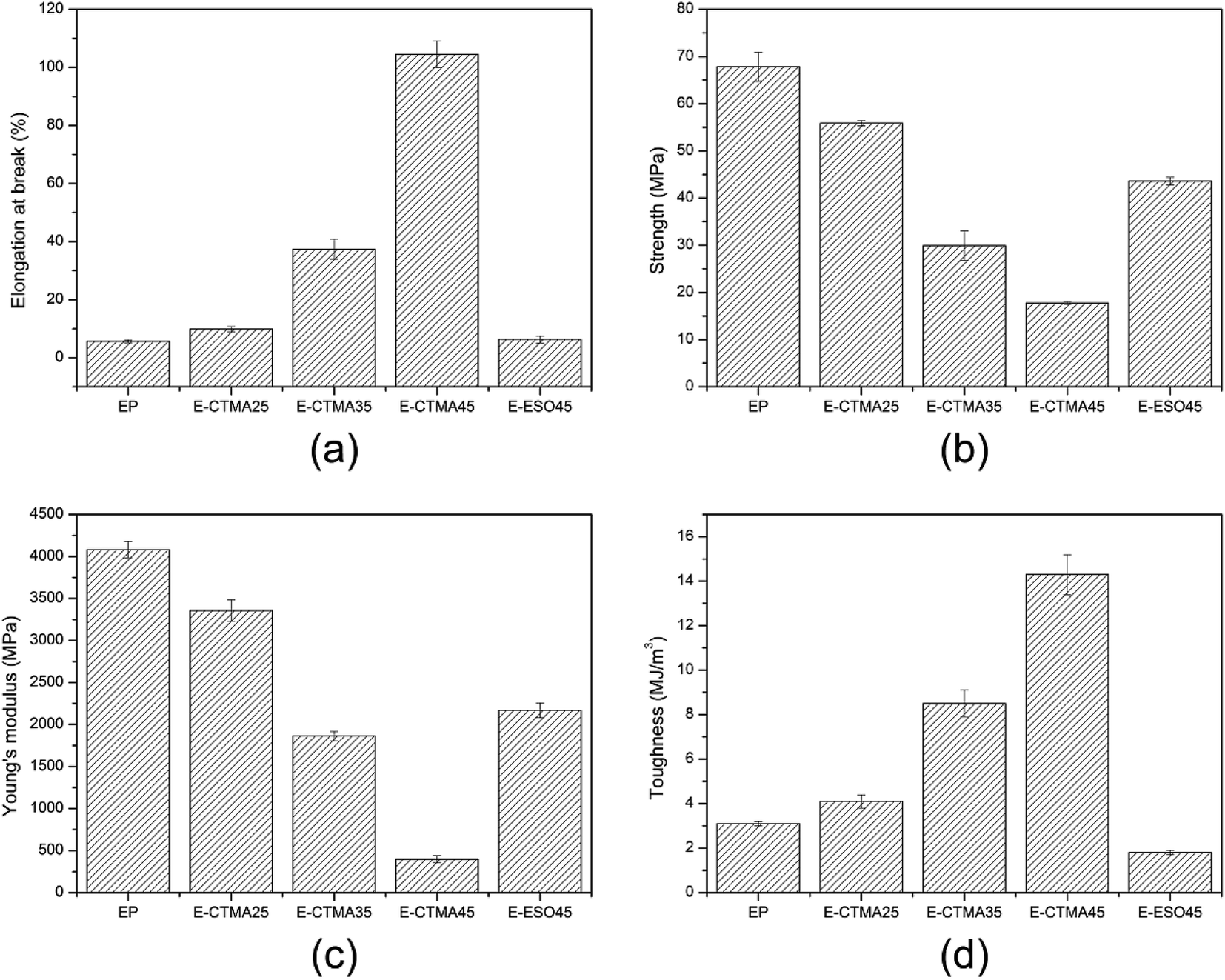 | ||
| Fig. 11 Mechanical properties of the neat epoxy resin and E-CTMA thermosets. | ||
The sample E-ESO45 was used for comparison. Obviously, although E-ESO45 has higher tensile strength and Young's modulus, its toughness value is much lower than that of E-CTMA45; this is mainly attributed to its low elongation-at-break. Moreover, many studies on the toughening of epoxy resin with ESO have been reported, which indicate that ESO shows limited improvement in the elongation-at-break.20,35,38,39 Hence, in terms of elongation-at-break and toughness value, CTMA exhibits excellent toughening ability, which is better than ESO. Moreover, the mechanical properties suggest that the epoxy resin modified by CTMA is a potential material that can be used as a coating, pouring sealant and other casting material and has higher requirement for toughness but less demand for strength.44 However, owing to the higher viscosity of some formulations, some solvents should be used to reduce the viscosity for applications.
Conclusions
Epoxy resin modified by CTMA shows a great improvement in toughness when compared with the neat epoxy resin and the sample modified by ESO. By acting as a flexible cross-linker, CTMA reduces the cross-linking density of epoxy resin, simultaneously increasing the elongation-at-break and toughness value. However, the incorporation of flexible aliphatic chains decreases the tensile strength, Young's modulus and Tg value; this is similar to the results of rubber-modified epoxy resin. Due to the reaction between the carboxyl groups and the epoxy groups, no phase separation has been detected by thermal characterization and SEM. Based on these characteristics, the epoxy resin modified by CTMA is a potential material for application as a coating, pouring sealant and casting material; moreover, the results demonstrate that the modified resins obtained from renewable feedstock, such as tung oil, will be potential alternatives to petroleum-based modified resins in the future.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This work was financially supported by the National “Thirteenth Five-Year” Key Research and Development Plan (2017YFD0601304) and the Science Foundation of Key Laboratory of Biomass Energy and Materials of Jiangsu Province (JSBEM-S-201703).References
- Y. Huang, Y. Tian, Y. Li, X. Tan, Q. Li, J. Cheng and J. Zhang, RSC Adv., 2017, 7, 49074–49082 RSC.
- R. Pearson and A. Yee, J. Mater. Sci., 1989, 24, 2571–2580 CrossRef CAS.
- A. F. Yee and R. A. Pearson, J. Mater. Sci., 1986, 21, 2462–2474 CrossRef CAS.
- H. Ren, X. Zhan, Q. Zhang and F. Chen, RSC Adv., 2016, 6, 35621–35627 RSC.
- H. Ren, X. Zhan, Q. Zhang and F. Chen, J. Appl. Polym. Sci., 2016, 133, 42826 Search PubMed.
- X. Fernández-Francos, D. Foix, À. Serra, J. M. Salla and X. Ramis, React. Funct. Polym., 2010, 70, 798–806 CrossRef.
- M. Flores, X. Fernández-Francos, F. Ferrando, X. Ramis and À. Serra, Polymer, 2012, 53, 5232–5241 CrossRef CAS.
- X. Fei, W. Wei, Y. Tang, Y. Zhu, J. Luo, M. Chen and X. Liu, Eur. Polym. J., 2017, 90, 431–441 CrossRef CAS.
- D. Foix, A. Serra, L. Amparore and M. Sangermano, Polymer, 2012, 53, 3084–3088 CrossRef CAS.
- L. Calabrese and A. Valenza, Eur. Polym. J., 2003, 39, 1355–1363 CrossRef CAS.
- K. Srivastava, A. K. Rathore and D. Srivastava, Spectrochim. Acta, Part A, 2018, 188, 99–105 CrossRef CAS PubMed.
- J. Xi and Z. Yu, J. Appl. Polym. Sci., 2018, 135, 45767 CrossRef.
- L. Hader-Kregl, G. M. Wallner, R. W. Lang and B. Strauß, J. Appl. Polym. Sci., 2018, 135, 46094 CrossRef.
- J. Huang and X. Nie, Polym. Int., 2016, 65, 835–840 CrossRef CAS.
- R. J. Day, P. A. Lovell and A. A. Wazzan, Polym. Int., 2001, 50, 849–857 CrossRef CAS.
- G. M. Roudsari, A. K. Mohanty and M. Misra, ACS Sustainable Chem. Eng., 2017, 5, 9528–9541 CrossRef.
- S. Miao, P. Wang, Z. Su and S. Zhang, Acta Biomater., 2014, 10, 1692–1704 CrossRef CAS PubMed.
- S. Qureshi, J. A. Manson, J. C. Michel, R. W. Hertzberg and L. H. Sperling, in Characterization of Highly Cross-linked Polymers, American Chemical Society, 1984, vol. 243, ch. 8, pp. 109–124 Search PubMed.
- H. Miyagawa, A. K. Mohanty, M. Misra and L. T. Drzal, Macromol. Mater. Eng., 2004, 289, 636–641 CrossRef CAS.
- D. Ratna, Polym. Int., 2015, 50, 179–184 CrossRef.
- K. Huang, J. Xia, X. Yang, M. Li and H. Ding, Polym. J., 2010, 42, 51–57 CrossRef CAS.
- L. Hou and Y. Liu, J. Appl. Polym. Sci., 2012, 126, 1572–1579 CrossRef CAS.
- K. Huang, Z. Liu, J. Zhang, S. Li, M. Li, J. Xia and Y. Zhou, Biomacromolecules, 2014, 15, 837–843 CrossRef CAS PubMed.
- K. Huang, P. Zhang, J. Zhang, S. Li, M. Li, J. Xia and Y. Zhou, Green Chem., 2013, 15, 2466–2475 RSC.
- J. M. Barton, The application of differential scanning calorimetry (DSC) to the study of epoxy resin curing reactions, 1985 Search PubMed.
- A. Saikia and N. Karak, Polym. Degrad. Stab., 2017, 135, 8–17 CrossRef CAS.
- A. R. Kannurpatti, J. W. Anseth and C. N. Bowman, Polymer, 1998, 39, 2507–2513 CrossRef CAS.
- L. Wang, Y. Liang, Q. Yu, S. Chen, J. Zhang, M. Miao and D. Zhang, Polym. Compos., 2018, 39, 2046–2055 CrossRef.
- X. Miao, Y. Meng and X. Li, Polymer, 2015, 60, 88–95 CrossRef CAS.
- L. Lijuan, M. Yan, Q. Teng, L. Zhuoxin, Y. Jing, C. Xingzhong and L. Xiaoyu, Polym. Compos., 2013, 34, 1051–1060 CrossRef.
- T. Li, H. Qin, Y. Liu, X. Zhong, Y. Yu and A. Serra, Polymer, 2012, 53, 5864–5872 CrossRef CAS.
- J.-P. Yang, Z.-K. Chen, G. Yang, S.-Y. Fu and L. Ye, Polymer, 2008, 49, 3168–3175 CrossRef CAS.
- M. Morell, M. Erber, X. Ramis, F. Ferrando, B. Voit and A. Serra, Eur. Polym. J., 2010, 46, 1498–1509 CrossRef CAS.
- L. Luo, T. Qiu, Y. Meng, L. Guo, J. Yang, Z. Li, X. Cao and X. Li, RSC Adv., 2013, 3, 14509–14520 RSC.
- S. K. Sahoo, S. Mohanty and S. K. Nayak, RSC Adv., 2015, 5, 13674–13691 RSC.
- P. Chand, C. V. Reddy, J. G. Verkade, T. Wang and D. Grewell, Energy Fuels, 2009, 23, 989–992 CrossRef CAS.
- C. Yan, L. Yang, L. Ma, D. E. Finlow, S. Lin and K. Song, J. Therm. Anal. Calorim., 2013, 113, 939–945 CrossRef.
- S. Kumar, S. K. Samal, S. Mohanty and S. K. Nayak, Ind. Eng. Chem. Res., 2017, 56, 687–698 CrossRef CAS.
- S. K. Sahoo, S. Mohanty and S. K. Nayak, Chin. J. Polym. Sci., 2015, 33, 137–152 CrossRef CAS.
- J. M. Misasi, Q. Jin, K. M. Knauer, S. E. Morgan and J. S. Wiggins, Polymer, 2017, 117, 54–63 CrossRef CAS.
- H. Ren, X. Zhan, Q. Zhang, G. Zhang and F. Chen, Polymer, 2016, 92, 222–230 CrossRef.
- H. Ren, X. Zhan, Q. Zhang and F. Chen, J. Appl. Polym. Sci., 2017, 134, 44849 Search PubMed.
- J. Song, C. Chen, S. Zhu, M. Zhu, J. Dai, U. Ray, Y. Li, Y. Kuang, Y. Li and N. Quispe, Nature, 2018, 554, 224–239 CrossRef CAS PubMed.
- L. Ying and S. Mao, J. Appl. Polym. Sci., 1996, 61, 2059–2063 CrossRef.
Footnote |
| † Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer. |
| This journal is © The Royal Society of Chemistry 2019 |