
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteraction of carbohydrate binding module 20 with starch substrates†
Son Tung Ngo *ab,
Phuong Duy Tran-Lec,
Giap T. Hoc,
Loan Q. Led,
Le Minh Buic,
Bao Khanh Vuc,
Huong Thi Thu Phungc,
Hoang-Dung Nguyencd,
Thanh-Sang Voc and
Van V. Vu*c
*ab,
Phuong Duy Tran-Lec,
Giap T. Hoc,
Loan Q. Led,
Le Minh Buic,
Bao Khanh Vuc,
Huong Thi Thu Phungc,
Hoang-Dung Nguyencd,
Thanh-Sang Voc and
Van V. Vu*c
aLaboratory of Theoretical and Computational Biophysics, Ton Duc Thang University, Ho Chi Minh City, Vietnam. E-mail: ngosontung@tdtu.edu.vn
bFaculty of Applied Sciences, Ton Duc Thang University, Ho Chi Minh City, Vietnam
cNTT Hi-Tech Institute, Nguyen Tat Thanh University, Ho Chi Minh City, Vietnam. E-mail: vanvu@ntt.edu.vn
dInstitute of Tropical Biology, Vietnam Academy of Science and Technology, Ho Chi Minh City, Vietnam
First published on 9th August 2019
Abstract
CBM20s are starch-binding domains found in many amylolytic enzymes, including glucoamylase, alpha-amylase, beta-amylases, and a new family of starch-active polysaccharide monooxygenases (AA13 PMOs). Previous studies of CBM20–substrate interaction only concerned relatively small or soluble amylose molecules, while amylolytic enzymes often work on extended chains of insoluble starch molecules. In this study, we utilized molecular simulation techniques to gain further insights into the interaction of CBM20 with substrates of various sizes via its two separate binding sites, termed as BdS1 and BdS2. Results show that substrate binding at BdS1 involving two conserved tryptophan residues is about 2–4 kcal mol−1 stronger than that at BdS2. CBM20 exhibits about two-fold higher affinity for helical substrates than for the amylose random coils. The affinity for amylose individual double helices does not depend on the helices' length. At least three parallel double helices are required for optimal binding. The binding affinity for a substrate containing 3 or more double helices is ∼−15 kcal mol−1, which is 2–3 kcal mol−1 larger than that for individual double helices. 100 ns molecular dynamics simulations were carried out for the binding of CBM20 to an extended substrate containing 3 layers of 9 60-unit double helices (A3L). A stable conformation of CBM20–A3L was found at BdS1. However, when CBM20 binds A3L via BdS2, it moves across the surface of the substrate and does not form a stable complex. MD simulations show that small amylose helices are quickly disrupted upon binding to CBM20. Our results provide some important molecular insights into the interactions of CBM20 with starch substrates, which will serve as the basis for further studies of CBM20-containing enzymes, including AA13 PMOs.
Introduction
Starch is one of the most abundant natural polymers found on earth, which has been playing important roles in human society, including both food and non-food sectors.1 Starch consists of ∼20–30% of amylose and ∼70–80% amylopectin.2 Amylose contains linear polymers of several to thousands of α(1→4) linked D-glucose units. Amylopectin also contains α(1→6) linkages at about every ∼30 units along the α(1→4) linked chain. In biology, starch is metabolized via hydrolysis to oligosaccharide by amylolytic enzymes. Amylose often exists in nano/microcrystal forms of helices that are resistant to hydrolysis by amylolytic enzymes. In contrast, amylopectin is highly branched and has less ordered structures that are more amenable to hydrolysis.Amylolytic enzymes, including alpha-amylase, beta-amylase, and gamma-amylase (glucoamylases) often contain one or more carbohydrate binding modules (CBMs), such as CBM20. CBMs are ubiquitous with 84 families spreading in all kingdoms of life.3 They are often thought of as supporting modules that help carbohydrate active enzymes to bind to their target substrates. However, accumulated data suggest that CBMs's roles are more diverse. Pre-incubation of amylose with a stand-alone CBM20 was found to significantly enhance its hydrolysis by a truncated glyucoamylase without any CBM.4 It was proposed that CBM20 disrupted the helical structure of amylose, making it more amenable to hydrolysis by amylase. Two starch binding sites were revealed in CBM20.5 Binding site 1 (BdS1) contains two critical tryptophan residues. Binding site 2 (BdS2) does not contain any tryptophan residues, but contains two tyrosine residues. It was also later shown on the basis of Atomic Force Microscopy (AFM) that CBM20 disrupted the structure of soluble amylose, presumably via binding at these two binding sites.6,7 Due to the low resolution of AFM, the atomic detail of CBM20–amylose interaction was not clear.
In the last decade, extensive studies of carbohydrate activate enzymes in both academic and industrial sectors were driven by the enormous demand for next-generation biofuels. Among the large number of new enzymes discovered, polysaccharide monooxygenases (PMOs) or lytic PMOs (LPMOs) exhibit unpreceded oxidative mechanism in glycosidic bond cleavages.8–12 Some PMOs could boost the activity of glycoside hydrolases,13–16 and thus could play key role in reducing the cost of biomass conversion to fermentable sugar, the bottleneck step in cellulosic biofuel production. During this period, the interests in the role of CBMs in substrate binding and catalysis of carbohydrate-active enzymes were also reignited.17–21 CBM20 was found as a C-terminal domain in the majority of starch-active PMOs (AA13),22–24 the only PMO family that oxidatively cleaves on α-glycosidic bond in starch. CBM20 appears to have significant roles in the activity of AA13 PMOs on various type of starch.
Understanding the nature of binding between CBMs and starch substrates is of great fundamental and practical interests, which has proved to be almost infeasible to obtain experimentally at the atomic levels. Previous studies using AFM6,7 or NMR5 only revealed the interaction of CBM20 with individual molecules of soluble amylose or starch analogues, respectively. Although these studies revealed the binding sites (NMR) or how CBM20 would disrupt small amylose molecules, they did not reflect the true interaction of CBM20 with insoluble substrates relevant to the industries. In this present work, we used molecular docking, molecular dynamics simulations to obtain atomic-level insights into the interaction between CBM20 and various starch substrates, including a large bundle of extended amylose double helices. Our study provides new insights into the interactions of CBM20 with starch substrates and provide some important implications to the biochemistry of CBM20 and CBM20-containing enzymes.
Materials and methods
Input structure of CBM20
NMR structure of a complex of CBM20 with beta-cyclodextrin (PDB ID: 1AC0) reveals two binding sites (Fig. 1A).5 Binding site 1 (BdS1) contains two critical tryptophan residues W543 and W590. Binding site 2 (BdS2) does not contain any tryptophan residues, but contains two tyrosine residue Y527 and Y556. The structure of this CBM20 and its binding sites will be used for molecular docking and molecular dynamics simulations in this study.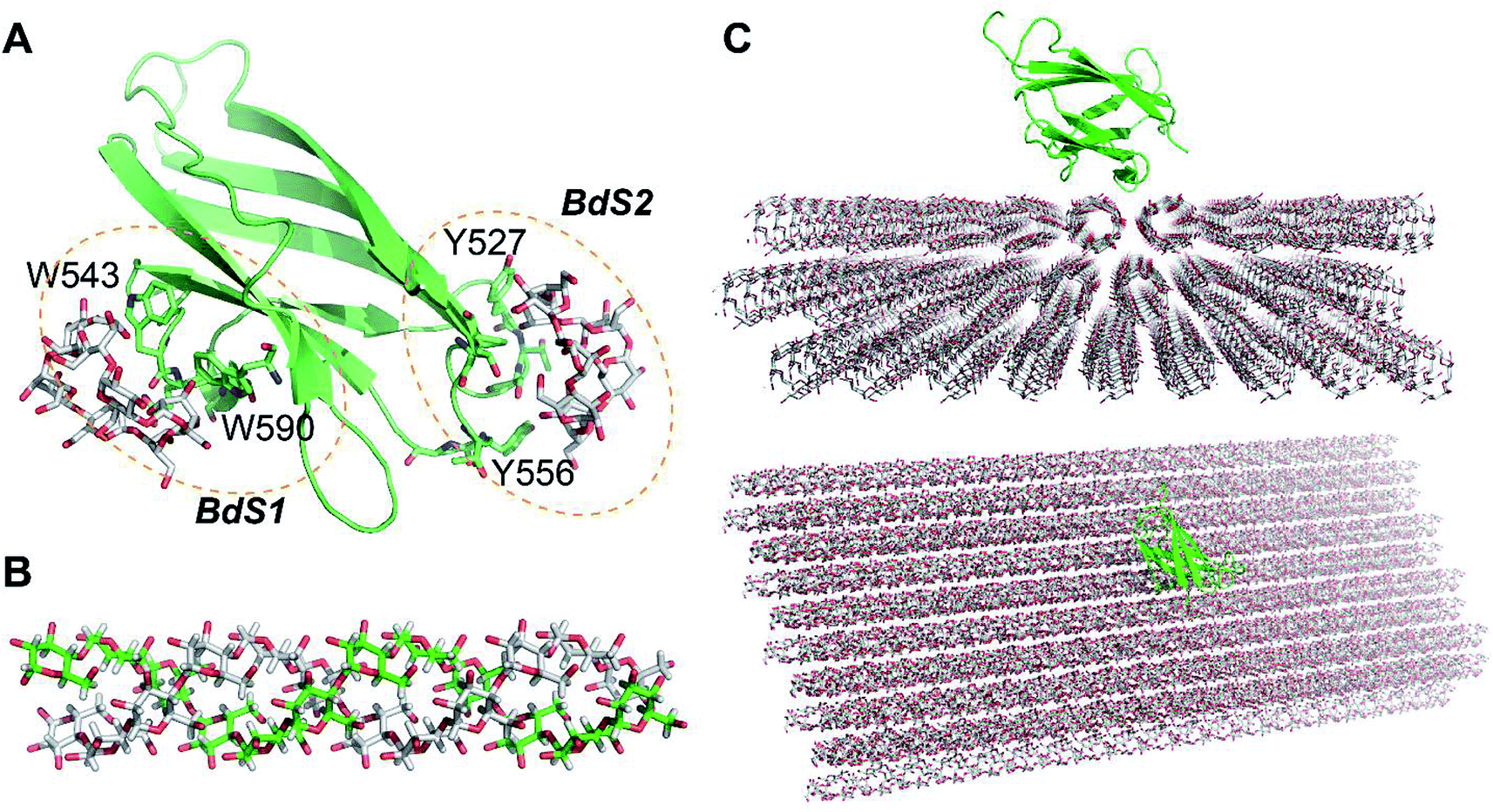 | ||
| Fig. 1 Molecules used in this study. (A) A CBM20 with β-cyclodextrin molecules bound to two separate binding sites BdS1 and BdS2 (PDB ID 1AC0). (B) A 12-unit amylose double helix.25 (C) An amylose-A cluster consisting of 3 layers of 9 60-unit amylose double helices. This cluster is large enough to ensure that the simulations of the binding of CBM20 (green) to the middle sections are not affected by the boundary parameters. | ||
Amylose substrate models
Amylose double helix (ADH10) (AmyA_double.pdb)25 containing 10 glucose units in each strand was obtained from the database of polysaccharide 3D structures (POLYSAC3DB) (Fig. 1B). Amylose single helix (ASH10) and amylose random coil were generated form 1 of the strands of ADH10.The structure of amyloses including 3 layers of 9 60-unit amylose double helix (A3L) (Fig. 1C) was generated using PyMOL 1.3 and the Carbohydrate builder at the GLYCAM server.26 A 60-unit amylose double helix (ADH60) was first generated using the Carbohydrate builder using the ψ and φ angles obtained from a crystal structure of an amylose-A (A-amylose_2009-popov_expanded.pdb)27 available at POLYSAC3DB.28 Crystal packing parameters were also obtained from this structure and used to generate A3L with PyMOL.
Molecular docking
CBM20 and substrates were parameterized using AutodockTools 1.5.6.29 The ligands were docked to receptor utilizing Autodock Vina version 1.0 (ref. 30) with the optimization using Broyden–Fletcher–Goldfarb–Shanno (BFGS) scheme.31 The exhaustiveness was chosen as 40. The CBM20 molecule was set as rigid molecule during simulation. CBM20 was used as the ligand in the docking studies with A3L and as the receptor in all other cases. In the docking experiment to A3L, a grid of 4.0 × 4.0 × 6.0 nm was used for the CBM20–A3L complex, which is centered at the center of A3L. The substrates containing 1–9 parallel ADH60 molecules (nADH60, n = 1–9) resembling fragments of one layer in A3L were docked to CBM20 using a grid of 2.0 × 30 × 30 nm. The ADH10, ASH10, and ASC10 molecules were docked to CBM20 with the grid size of 4.0 × 4.0 × 6.0 nm. During the simulation, the helical substrates were set as rigid molecules while ASC10 was fully flexible. The best docked model was designated to the binding pose with the lowest binding affinity.Molecular dynamics simulation
The CBM20 molecule was parameterized by Amber99SB-ildn force field.32 All of substrates were parameterized using GLYCAM 06j-1 force field.26 The CBM20–starch complexes structures were generated via molecular docking. These complexes were solvated using TIP3P water model.33 The CBM20 + A3L complex was put into a ∼1921 nm3 rectangular box, resulting in a system containing more than 194![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 atoms. The CBM20 + ADH10/ASH10 complexes were inserted into a 585 nm3 dodecahedron box, forming systems consisting of more than 59000 atoms.
300 atoms. The CBM20 + ADH10/ASH10 complexes were inserted into a 585 nm3 dodecahedron box, forming systems consisting of more than 59000 atoms.
The solvated systems were energy-minimized with the steepest descent scheme. After that, the CBM20–substrate complexes were relaxed in 500 ps of NVT ensemble with a harmonic positional restraint force applied on them, which were then relaxed in 500 ps of NPT ensemble. The last snapshot of NPT simulations was used as the starting conformation of MD simulation. The MD simulation length ranges from 50–100 ns. The MD simulation were performed on GPU using GROMACS 5.1.3 (ref. 34) with the MD parameters are derived from our previous works.35,36 The simulation temperature was set at 300 K. All-bonds were constrained using the LINCS method.37 The non-bond cutoff was set at 0.9 nm. The particle mesh Ewald was used to mimic the electrostatic interaction. van der Waals interaction simulated with a 0.9 nm cutoff. The regions of A3L forming contacts with CBM20 were left unrestrained, while other regions and atoms were restrained using a small harmonic potential during MD simulations (Fig. S1†).
Data analysis
Pull-down assays
NCU08746 were expressed and purified as previously described.23 Cornstarch (S4126) and corn amylopectin (10120) were purchased from Sigma-Aldrich. 100 mg of each substrate was washed three times with 1 mL of 10 mM sodium acetate buffer pH 5.0 (buffer A) using centrifugation. The final pellet was re-suspended in 1 mL buffer A. NCU08746 was added to each substrate suspension to 5 μM final concentration. The assays were incubated at room temperature with gentle rotation for 30 minutes, which were then centrifuged to separate the pellets from the supernatant. The pellets were then washed three times with 1 mL buffer A. The final pellet was re-suspended in 1 mL buffer A. 50 uL aliquots of the supernatants collected after the initial incubation step and 3 washing steps, as well as the final suspension were mixed with 10 μL 6× SDS-PAGE sample buffer. SDS-PAGE was then carried out using Protean TGX precast gels (Bio-Rad) as instructed by the manufacturer. The gels were analyzed using a ChemiDoc Imaging system (Bio-Rad).Results and discussion
Binding affinity of CBM20 to starch substrates derived from molecular docking
Because the CBM20–A3L complex is very large, we chose a 4.0 × 4.0 × 6.0 nm docking grid that is large enough to provide the space for CBM20 to alternate between various binding positions to A3L. Two docking poses of CBM20 to A3L substrate were observed (Fig. 2). These poses are consistent with the observed binding sites of CBM20 with β-cyclodextrin reported previously.43
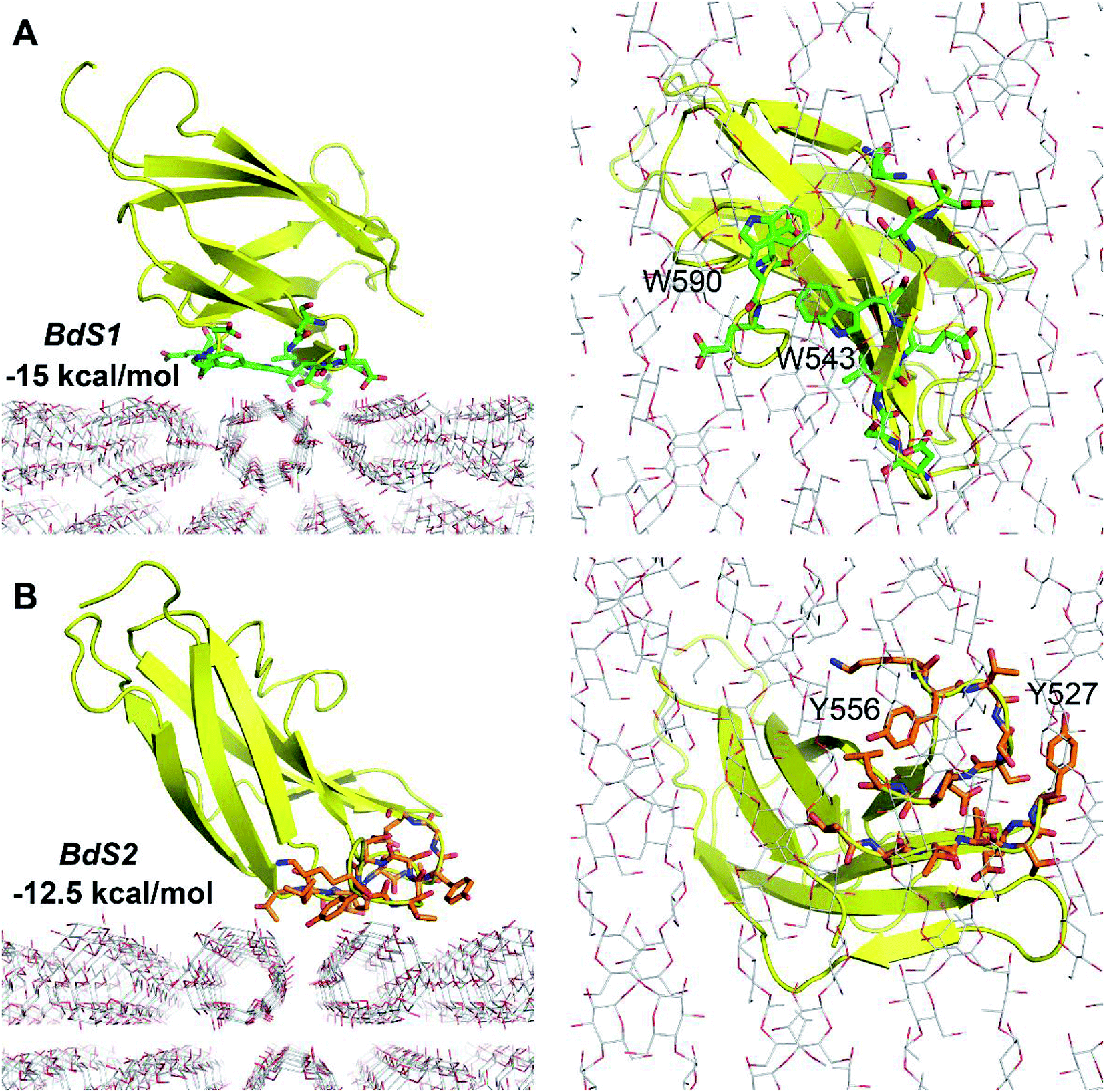 | ||
| Fig. 2 Molecular docking of CBM20 to A3L at BdS1 (A) and BdS2 (B). | ||
The binding affinity at BdS1 (−15 kcal mol−1) is significantly larger than that at BdS2 (−12.5 kcal mol−1), which is in good agreement with the numbers of H-bonds between CBM20 and A3L found at the two binding sites. At BdS1 K578, S592, and E589 residues forms 4 H-bonds to A3L, while at BdS2, only two H-bonds are formed by T557 and R616. Moreover, the critical aromatic residues W543 and W590 at BdS1 appear to contribute to the binding of CBM20 to A3L significantly more that by Y527 and Y556 at BdS2. While the aromatic rings of Y527 and Y556 at BdS2 do not align well on a surface, W543 and W590 side chains are positioned close to one another in BdS1 and form a relatively flat surface as found in many other CBMs.44
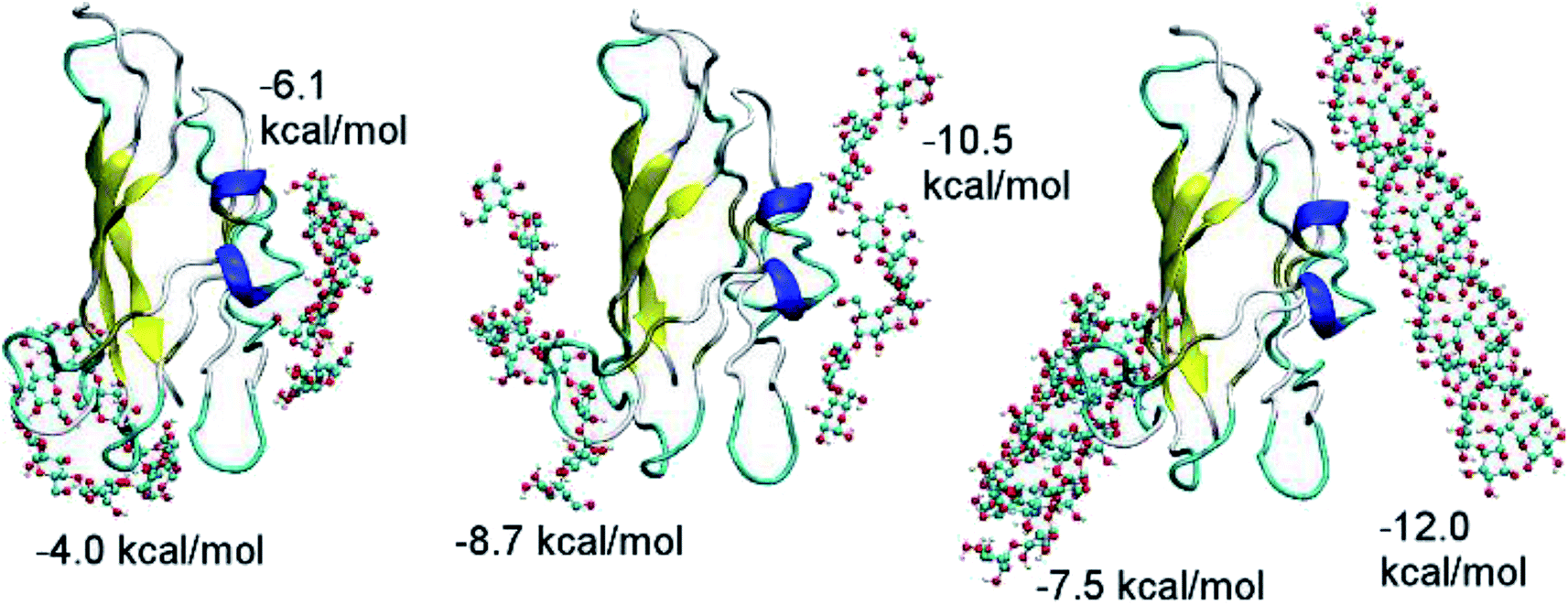 | ||
| Fig. 3 Molecular docking of ADH10, ASH10, and ASC10 to CBM20. | ||
 | ||
| Fig. 4 The binding affinity of the CBM20 to helical amylose substrates as the function of the number of the double helices. Each chain of the double helices contain 60 glucose units. | ||
Molecular dynamics of the CBM20–A3L complex
The CBM20–A3L complex at BdS2 did not reach the equilibrium states after 100 ns of MD simulations. The protein moved on the surface of A3L substrate during the entire MD trajectory (Fig. S2 and S3†). Analysis of several snapshots of the CBM20–A3L complex at BdS2 reveals a few polar contacts involving mainly T522, T614, Y527, and Y566. Further analyses of the CBM20–A3L binding process at BdS2 were not performed.
The binding at BdS1 reached equilibrium states after 40 ns (Fig. S4†), and analyses were performed for this system in the last 60 ns of MD simulation. Overall, CBM20 forms 14.78 ± 1.46 side chain contacts (SCs) and ∼6.09 ± 1.19 hydrogen bonds (HBs) to the substrate (Fig. S5†). The probabilities of intermolecular contacts between individual residues of CBM20 to A3L substrate are shown in Fig. 5. Critical residues involved in the binding are D542, W543, E544, E576, K578, D585, D586, S587 and W590. Previous NMR study revealed that W543, K578, and W590 are key residues in the binding of CBM20 with beta-cyclodextrin.46 The residues 541–545, 576, 578, 585–590, 592, and 595 form intermolecular SC contacts to substrate in at least half of MD simulation time (Fig. 5). Especially, residues 542, 543, 578, 585–588, and 590 adopt SC contacts to A3L in all equilibrium snapshots (Fig. 5). Moreover, residues 542–544, 576, 578, 585–587, and 590 form HB to the substrate in more than 35% of the equilibrium snapshots. In addition, four independent trajectories with the same starting initial structure but different generated starting-velocity were also carried out (Fig. S6†). The superposition of the CBM20–A3L complex at BdS1 in different independent MD trajectories help validate our results (Fig. S7†).
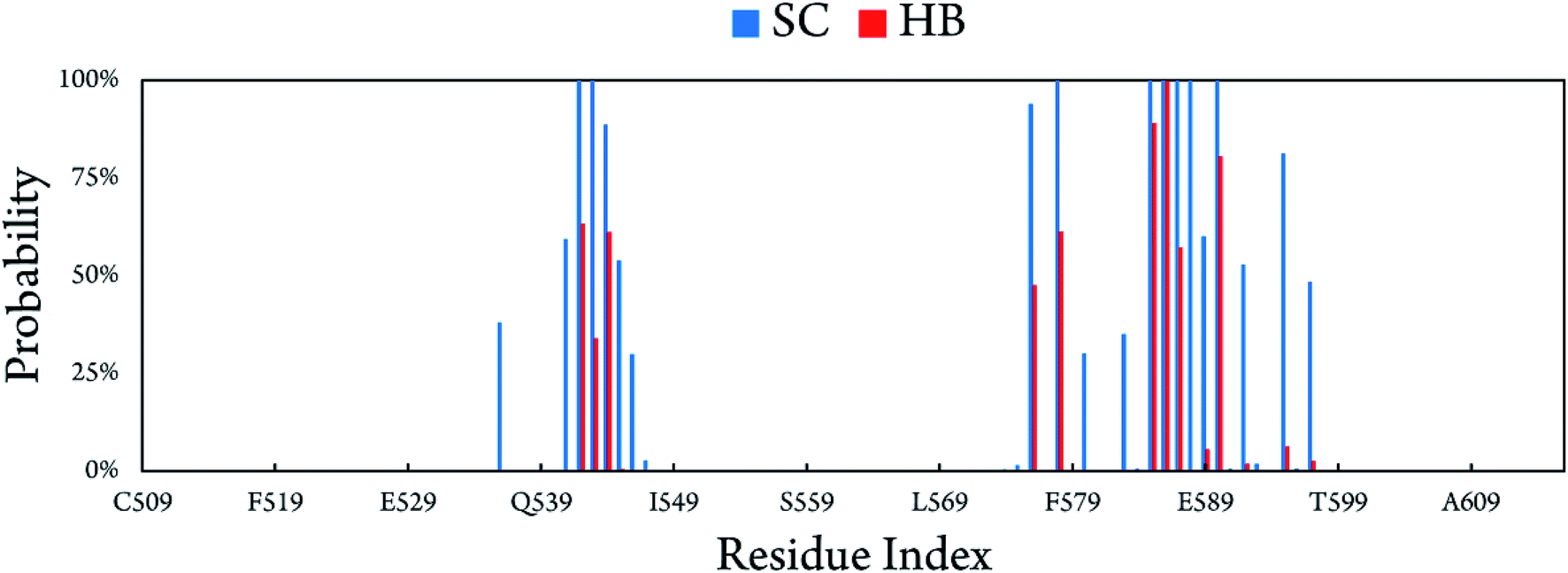 | ||
| Fig. 5 Intermolecular contacts between CBM20 and A3L at BdS1 derived as the average those in of all equilibrium snapshots of MD simulations. | ||
CBM20 undergoes small structural changes upon binding to A3L at BdS1s. There are slight changes in the β-content (from 39.44 ± 1.92% to 38.90 ± 2.40%) and coil content (from 48.68 ± 3.11% to 46.82 ± 2.71%) of CBM20 when it binds to A3L (Fig. S8†). Lager changes are observed for the helical structure content (from 13.75 ± 1.92% to 1.33 ± 1.76%) and turn content (from 0 to 11.09 ± 2.59%). The collision cross section, which represents the overall size of CBM20, decreased from 14.89 ± 0.16 to 14.70 ± 0.15 nm2 (Fig. S9†). In addition, the region of A3L that binds CBM20 appears to be slightly stabilized compared to that in free A3L as indicated by slightly lower RMSD throughout 100 ns MD simulation (Fig. S10†).
The relative binding free energy between the CBM20 protein to A3L substrate was evaluated by using the free energy perturbation method. The obtained results indicate that the electrostatics free interaction energy dominates over the van der Waals free interaction energy during the binding process of the CBM20 protein to the A3L substrate (Fig. S11†).
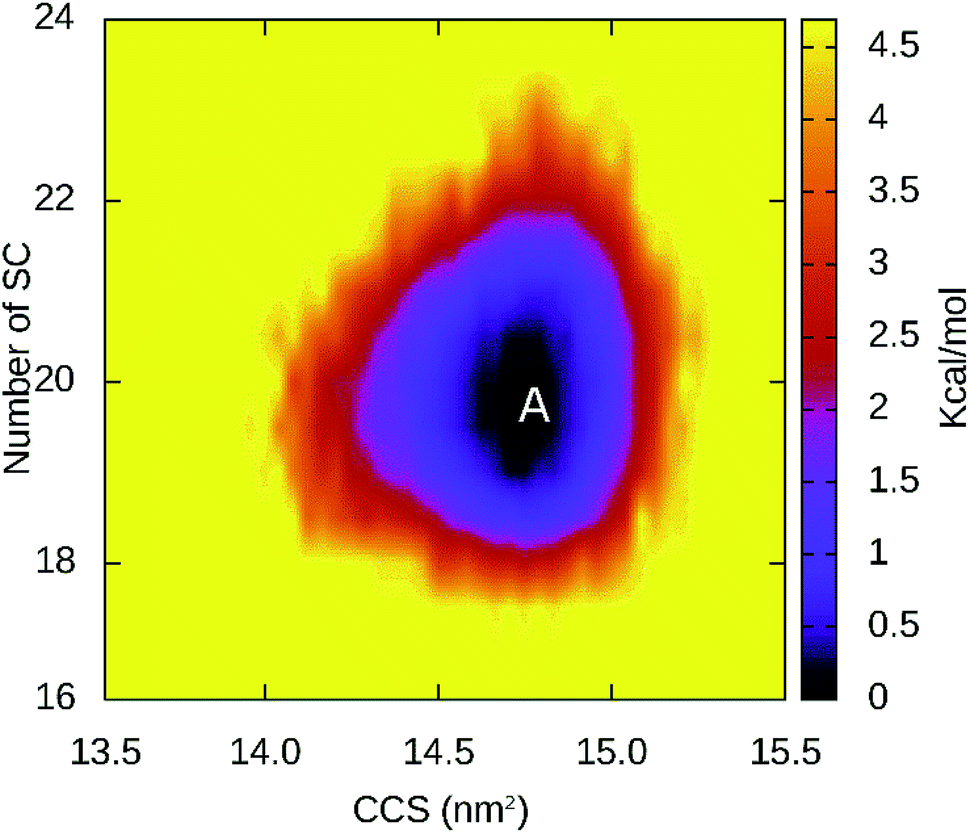 | ||
| Fig. 6 Free energy landscape of CBM20–A3L complex at BdS1 constructed for all equilibrium snapshots using the number of SC between two molecules and CCS of CBM20 as the coordinates. The minimum A is found at (∼14.73 nm2; ∼19.5) coordinate. | ||
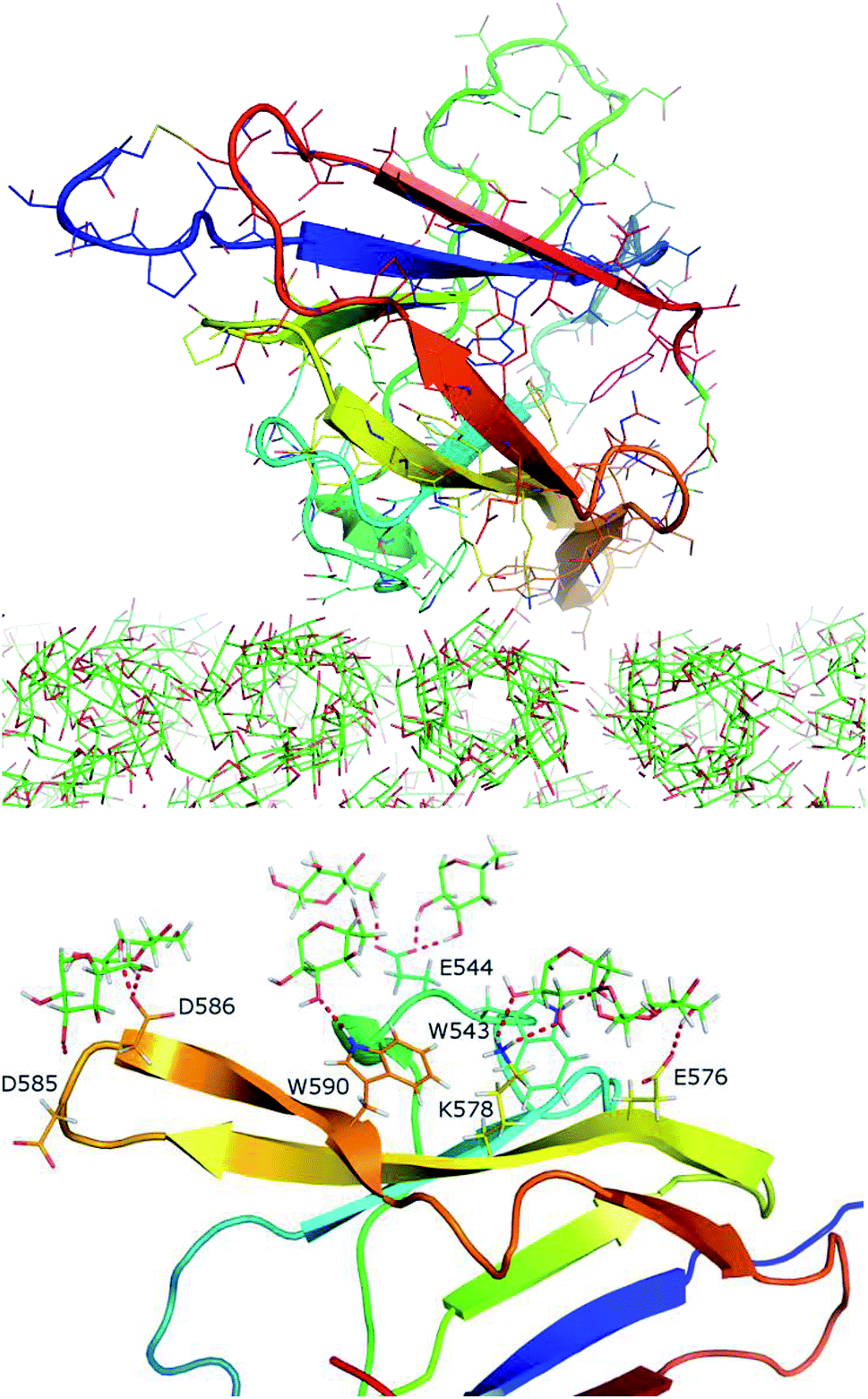 | ||
| Fig. 7 Optimized structure of CBM20–A3L complex at BdS1 found in the minimum A using clustering method with a resolution of 0.15 nm. (Top) Overall structure. (Bottom) Close-up view of the interaction region. | ||
CBM20 disrupt the helical structure of small substrates
MD simulations were carried out without any restraint to gain insights the interactions of CBM20 with small amylose substrates. MD simulations were carried out for the CBM20–ADH10 and CBM20–ASH10 complexes obtained with dockings. Structural change of soluble complexes was monitored over MD simulation intervals. Interestingly, we observed that the amylose helices were rapidly disrupted (Fig. 8). Thus simulations were only carried out for 50 ns for both complexes.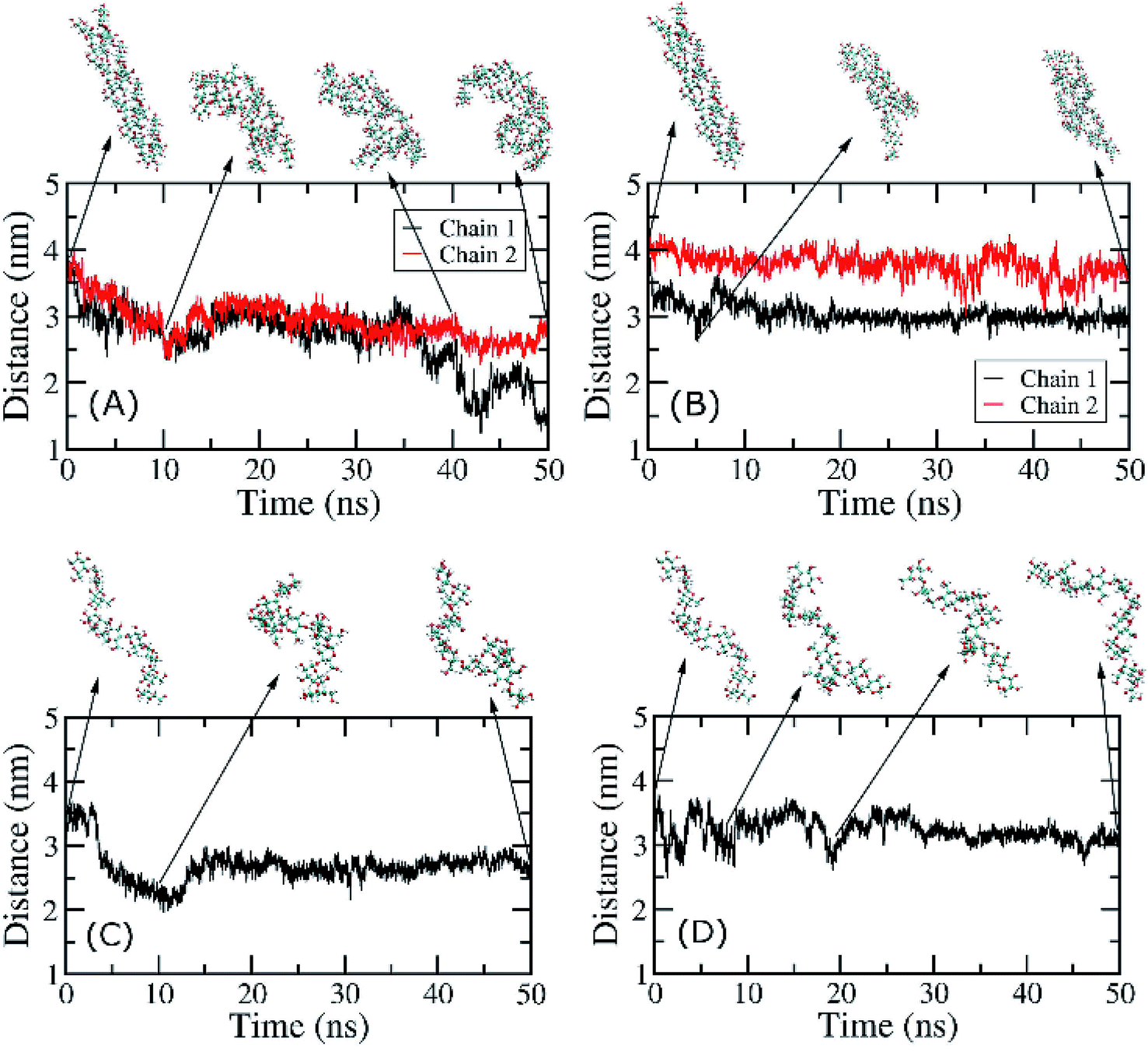 | ||
| Fig. 8 End-to-end distances of individual chains of ADH10 and ASH10 during MD simulations. (A) CBM20–ADH10 at BdS1. (B) CBM20–ADH10 at BdS2. (C) CBM20–ASH10 at BdS1. (D) CBM20–ASH10 at BdS2. | ||
The structural changes of ADH10 during simulation under effects of CBM20 were measured through the end-to-end distance of amylose chains. At BdS1, the end-to-end distances of the two chains in ADH10 decreased from 4.1 nm to ∼3 nm and ∼1.5 nm after 50 ns of MD simulations (Fig. 8A). At BdS2, the end-to-end distances also decreases to ∼3.5 and 3.0 nm (Fig. 8B). The curves representing these distances over time are clearly different from one another. Thus, it is evident that the double helix was rapidly disrupted in the presence of CBM20 at both binding sites, which is consistent with the structures of the double helix taken at various snapshots (Fig. 8A and B). Likewise, the single helix was also disrupted at both binding sites of CBM20 within 50 ns of MD simulations (Fig. 8C and D). It is worth noting that the isolated ADH10 and ASH10 in solution was stable during 50 ns of MD simulations (Fig. S12 and S13†). This result is consistent with previous experimental results with small soluble amylose.4,6
Several isothermal calorimetry studies revealed 5–8 kcal mol−1 binding affinities for oligosaccharides of the CBM20s of the glucoamylase from Aspergillus niger45 and starch-active polysaccharide monooxygenases (AA13) from Magnaporthe oryzae and Aspergillus terreus.18 Moderate affinity for starch granules were also demonstrated for these CBM20s. Given the moderate affinity of CBM20 for starch, we have been able to use CBM20 as an affinity tag for facile purification of NCU08746, an AA13 polysaccharide monooxygenase from Neurospora crassa (NcAA13), using an amylose resin column.
We performed pull-down assays of NCU08746 with starch granules and amylopectin from corn to compare the binding affinity between these substrates. SDS-PAGE analysis indicate that, for both starch granules and amylopectin, there is a significant amount of protein remained in the supernatant (Fig. 9). A fraction of the protein was found to bind to the pellet, which was gradually released to the solutions in each washing steps. This result is consistent with previous studies that CBM20 has moderate affinity for starch substrates. Moreover, the density of the bands corresponding to NCU08746 on the gels indicate that NCU08746 has higher affinity for amylopectin than for starch granules. Both amylopectin and starch granules used in this study are from corn. It is likely that the separation of amylopectin from starch granules made the helical regions more accessible to CBM20s, resulting in higher affinity. This result is consistent with our docking studies that CBM20 has higher affinity for helical amylose than for flexible coil amylose.
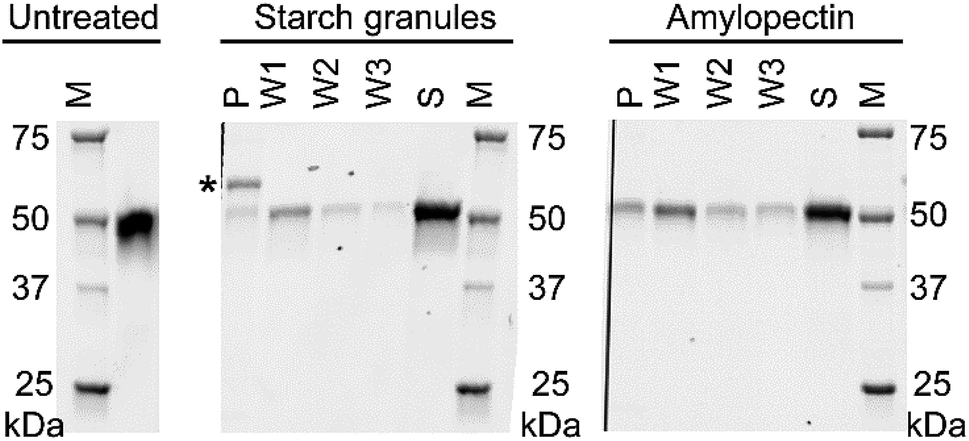 | ||
| Fig. 9 SDS-PAGE analysis of the pull-down assays of NCU08746, a CBM20-containing enzyme, using corn starch granules and corn amylopectin as the substrates. P: final pellet. S: the supernatant obtained after the first incubation step. W1, W2, and W3: the supernatants obtained at the washing steps 1, 2, and 3, respectively. M: protein ladder. *Unknown protein(s) in starch granules. | ||
Previous activity4 and AFM6,7 studies provided evidences that CBM20 disrupted starch structure. These studies carried out on individual relatively short chain amylose molecules that are soluble. Our computational studies on short chain amylose is consistent with these experimental studies and provide a dynamics picture of the amylose disruption process. However, for extended insoluble amylose chains in a large bundle, our MD simulations suggest that the disrupting effect of CBM20 might not be as strong as for short chain amylose.
Moreover, our computational study indicates that CBM20 has higher affinity for ADH10 and ASH10 than for ASC10. It might be possible that CBM20 first binds to the helical region of starch then disrupts this structure, resulting in weaker affinity. This could explain the moderate affinity of CBM20 for starch, which allows for subsequent dissociation of CBM20 to make the substrate available for hydrolysis by catalytic domains of amylases.
Conclusions
Our molecular docking, molecular dynamics simulations, and pull-down assays provide new insights into the interactions of CBM20 with various starch substrates. First, CBM20 has two binding sites, namely BdS1 and BdS2, that exhibit different binding affinity to starch substrates. Binding at BdS1 involving two conserved tryptophan residues is 2–4 kcal mol−1 stronger than that at BdS2 with two conserved tyrosine residues. Second, CBM20 has higher affinity for helical amylose molecules than for random coil starch molecules. The binding affinity for the double helices does not depend on the length of the helices. CBM20 requires 3 parallel helices for optimal binding and binding affinity does not change when the substrate has more than 3 helices. Finally, CBM20 quickly disrupts the helical structure of short substrates, but does not disrupt the helices in extended substrate during 100 ns MD simulations. On extended substrate, CBM20 from stable complex at BdS1, but moves along the substrate surface when interacting via BdS2.These insights, which were not observed in previous studies with small soluble amylose and cyclodextrins, are helpful for future studies on the interactions of CBM20-containing starch-active enzymes with industrially relevant starch substrates. Recently, we found that CBM20-containing AA13 polysaccharide monooxygenases (AA13 PMOs) has about two folds higher activity than that of the corresponding catalytic domains.47 In addition, AA13 PMOs is likely the first PMO family, and the first oxidative enzyme family, to have processivity-like activity. AA13 PMOs may slide on the amylose helices and cleave the glycosidic linkages separated by multiples of a helical turns, generating major products with degree of polymerization (DP) of 6n (n = 1, 2, 3…). CBM20 appears to help the enzymes retain longer on extended amylose helices and generate more products at higher DP. The understanding of the interaction of CBM20 with starch substrates obtained in this work will serve as the basis for computational studies of CBM20 containing AA13 PMOs with starch substrates, which will provide further inside into the processivity-like activity of these enzymes.
Author contributions
The manuscript was written through contributions of all authors.Funding sources
Vietnam National Foundation for Science and Technology Development (NAFOSTED) grant # 106-NN.02-2016.33.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant # 106-NN.02-2016.33.References
- S. C. Zeeman, J. Kossmann and A. M. Smith, Starch: Its Metabolism, Evolution, and Biotechnological Modification in Plants, Annu. Rev. Plant Biol., 2010, 61(1), 209–234 CrossRef CAS PubMed.
- S. Pérez and E. Bertoft, The molecular structures of starch components and their contribution to the architecture of starch granules: a comprehensive review, Starch/Staerke, 2010, 62(8), 389–420 CrossRef.
- Carbohydrate-Active enZYmes Database, http://www.cazy.org/, accessed on March 13, 2019 Search PubMed.
- S. M. Southall, P. J. Simpson, H. J. Gilbert, G. Williamson and M. P. Williamson, The starch-binding domain from glucoamylase disrupts the structure of starch, FEBS Lett., 1999, 447(1), 58–60 CrossRef CAS PubMed.
- K. Sorimachi, M.-F. L. Gal-Coëffet, G. Williamson, D. B. Archer and M. P. Williamson, Solution structure of the granular starch binding domain of Aspergillus niger glucoamylase bound to β-cyclodextrin, Structure, 1997, 5(5), 647–661 CrossRef CAS PubMed.
- V. J. Morris, A. P. Gunning, C. B. Faulds, G. Williamson and B. Svensson, AFM Images of Complexes between Amylose and Aspergillus niger Glucoamylase Mutants, Native and Mutant Starch Binding Domains: A Model for the Action of Glucoamylase, Starch/Staerke, 2005, 57(1), 1–7 CrossRef CAS.
- T.-Y. Jiang, Y.-P. Ci, W.-I. Chou, Y.-C. Lee, Y.-J. Sun, W.-Y. Chou, K.-M. Li and M. D.-T. Chang, Two Unique Ligand-Binding Clamps of Rhizopus oryzae Starch Binding Domain for Helical Structure Disruption of Amylose, PLoS One, 2012, 7, e41131 CrossRef CAS PubMed.
- W. T. Beeson, V. V. Vu, E. A. Span, C. M. Phillips and M. A. Marletta, Cellulose Degradation by Polysaccharide Monooxygenases, Annu. Rev. Biochem., 2015, 84(1), 923–946 CrossRef CAS PubMed.
- W. T. Beeson, C. M. Phillips, J. H. D. Cate and M. A. Marletta, Oxidative Cleavage of Cellulose by Fungal Copper-Dependent Polysaccharide Monooxygenases, J. Am. Chem. Soc., 2012, 134(2), 890–892 CrossRef CAS PubMed.
- R. J. Quinlan, M. D. Sweeney, L. Lo Leggio, H. Otten, J.-C. N. Poulsen, K. S. Johansen, K. B. R. M. Krogh, C. I. Joergensen, M. Tovborg, A. Anthonsen, T. Tryfona, C. P. Walter, P. Dupree, F. Xu, G. J. Davies and P. H. Walton, Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15079–15084 CrossRef CAS PubMed.
- C. M. Phillips, W. T. Beeson, J. H. D. Cate and M. A. Marletta, Cellobiose Dehydrogenase and a Copper-Dependent Polysaccharide Monooxygenase Potentiate Cellulose Degradation by Neurospora crassa, ACS Chem. Biol., 2011, 6(12), 1399–1406 CrossRef CAS PubMed.
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. Liu, H. Zhai, M. Sørlie and V. G. H. Eijsink, An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides, Science, 2010, 330(6001), 219–222 CrossRef CAS PubMed.
- P. V. Harris, D. Welner, K. C. McFarland, E. Re, J.-C. Navarro Poulsen, K. Brown, R. Salbo, H. Ding, E. Vlasenko, S. Merino, F. Xu, J. Cherry, S. Larsen and L. Lo Leggio, Stimulation of Lignocellulosic Biomass Hydrolysis by Proteins of Glycoside Hydrolase Family 61: Structure and Function of a Large, Enigmatic Family, Biochemistry, 2010, 49, 3305–3316 CrossRef CAS PubMed.
- http://www.bioenergy.novozymes.com/en/cellulosic-ethanol/Cellic-HTec3/Documents/CE_APP_Cellic_Ctec3.pdf, accessed on July 7, 2016.
- http://bioenergy.novozymes.com/en/cellulosic-ethanol/CellicCTec3/Documents/AS_2010-01668-03.pdf, accessed on July 7, 2016.
- M. V. Semenova, A. V. Gusakov, P. V. Volkov, V. Y. Matys, V. A. Nemashkalov, V. D. Telitsin, A. M. Rozhkova and A. P. Sinitsyn, Enhancement of the enzymatic cellulose saccharification by Penicillium verruculosum multienzyme cocktails containing homologously overexpressed lytic polysaccharide monooxygenase, Mol. Biol. Rep., 2019 Search PubMed.
- L. I. Crouch, A. Labourel, P. H. Walton, G. J. Davies and H. J. Gilbert, The Contribution of Non-catalytic Carbohydrate Binding Modules to the Activity of Lytic Polysaccharide Monooxygenases, J. Biol. Chem., 2016, 291(14), 7439–7449 CrossRef CAS PubMed.
- L. Nekiunaite, T. Isaksen, G. Vaaje-Kolstad and M. Abou Hachem, Fungal lytic polysaccharide monooxygenases bind starch and β-cyclodextrin similarly to amylolytic hydrolases, FEBS Lett., 2016, 590(16), 2737–2747 CrossRef CAS PubMed.
- A. Várnai, M. R. Mäkelä, D. T. Djajadi, J. Rahikainen, A. Hatakka and L. Viikari, Chapter Four – Carbohydrate-Binding Modules of Fungal Cellulases: Occurrence in Nature, Function, and Relevance in Industrial Biomass Conversion, in Advances in Applied Microbiology, ed. S. Sariaslani, G. M. Gadd, Academic Press, 2014, vol. 88, pp. 103–165 Search PubMed.
- H. J. Gilbert, J. P. Knox and A. B. Boraston, Advances in understanding the molecular basis of plant cell wall polysaccharide recognition by carbohydrate-binding modules, Curr. Opin. Struct. Biol., 2013, 23(5), 669–677 CrossRef CAS PubMed.
- C. M. Payne, B. C. Knott, H. B. Mayes, H. Hansson, M. E. Himmel, M. Sandgren, J. Ståhlberg and G. T. Beckham, Fungal Cellulases, Chem. Rev., 2015, 115(3), 1308–1448 CrossRef CAS PubMed.
- L. Lo Leggio, T. J. Simmons, J.-C. N. Poulsen, K. E. H. Frandsen, G. R. Hemsworth, M. A. Stringer, P. von Freiesleben, M. Tovborg, K. S. Johansen, L. De Maria, P. V. Harris, C.-L. Soong, P. Dupree, T. Tryfona, N. Lenfant, B. Henrissat, G. J. Davies and P. H. Walton, Structure and boosting activity of a starch-degrading lytic polysaccharide monooxygenase, Nat. Commun., 2015, 6 CAS.
- V. V. Vu, W. T. Beeson, E. A. Span, E. R. Farquhar and M. A. Marletta, A family of starch-active polysaccharide monooxygenases, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(38), 13822–13827 CrossRef CAS PubMed.
- V. V. Vu and M. A. Marletta, Starch-degrading polysaccharide monooxygenases, Cell. Mol. Life Sci., 2016, 73(14), 2809–2819 CrossRef CAS PubMed.
- A. Imberty, H. Chanzy, S. Pérez, A. Bulèon and V. Tran, The double-helical nature of the crystalline part of A-starch, J. Mol. Biol., 1988, 201(2), 365–378 CrossRef CAS PubMed.
- K. N. Kirschner, A. B. Yongye, S. M. Tschampel, J. González-Outeiriño, C. R. Daniels, B. L. Foley and R. J. Woods, GLYCAM06: A generalizable biomolecular force field. Carbohydrates, J. Comput. Chem., 2008, 29(4), 622–655 CrossRef CAS PubMed.
- D. Popov, A. Buléon, M. Burghammer, H. Chanzy, N. Montesanti, J. L. Putaux, G. Potocki-Véronèse and C. Riekel, Crystal Structure of A-amylose: A Revisit from Synchrotron Microdiffraction Analysis of Single Crystals, Macromolecules, 2009, 42(4), 1167–1174 CrossRef CAS.
- http://polysac3db.cermav.cnrs.fr/db-connect.php?number=10, accessed on February 22.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30(16), 2785–2791 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 CAS.
- D. F. Shanno, Conditioning of Quasi-Newton Methods for Function Minimization, Mathematics of Computation, 1970, 24(111), 647–656 CrossRef.
- A. E. Aliev, M. Kulke, H. S. Khaneja, V. Chudasama, T. D. Sheppard and R. M. Lanigan, Motional timescale predictions by molecular dynamics simulations: case study using proline and hydroxyproline sidechain dynamics, Proteins: Struct., Funct., Bioinf., 2014, 82(2), 195–215 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79(2), 926–935 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- S. T. Ngo, H. M. Hung, D. T. Truong and M. T. Nguyen, Replica exchange molecular dynamics Study of the Truncated Amyloid Beta (11-40) Trimer in Solution, Phys. Chem. Chem. Phys., 2017, 19(3), 1909–1919 RSC.
- S. T. Ngo, X.-C. Luu, M. T. Nguyen, C. N. Le and V. V. Vu, In silico studies of solvated F19W amyloid β (11-40) trimer, RSC Adv., 2017, 7(67), 42379–42386 RSC.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, LINCS: A Linear Constraint Solver for Molecular Simulations, J. Comput. Chem., 1997, 18(12), 1463–1472 CrossRef CAS.
- E. G. Marklund, M. T. Degiacomi, C. V. Robinson, A. J. Baldwin and J. L. P. Benesch, Collision cross sections for structural proteomics, Structure, 2015, 23(4), 791–799 CrossRef CAS PubMed.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren and A. Mark, Peptide folding: When simulation meets experiment, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. G. Touw, C. Baakman, J. Black, T. A. H. te Beek, E. Krieger, R. P. Joosten and G. Vriend, A Series of PDB-related databanks for everyday needs, Nucleic Acids Res., 2015, 43(D1), D364–D368 CrossRef CAS PubMed.
- J. Madine, M. J. Pandya, M. R. Hicks, A. Rodger, E. A. Yates, S. E. Radford and D. A. Middleton, Site-Specific Identification of an Aβ Fibril–Heparin Interaction Site by Using Solid-State NMR Spectroscopy, Angew. Chem., Int. Ed., 2012, 51(52), 13140–13143 CrossRef CAS PubMed.
- K. Sorimachi, M. F. Le Gal-Coëffet, G. Williamson, D. B. Archer and M. P. Williamson, Solution structure of the granular starch binding domain of Aspergillus niger glucoamylase bound to beta-cyclodextrin, Structure, 2017, 5(5), 647–661 CrossRef.
- A. B. Boraston, D. N. Bolam, H. J. Gilbert and G. J. Davies, Carbohydrate-binding modules: fine-tuning polysaccharide recognition, Biochem. J., 2004, 382(Pt 3), 769–781 CrossRef CAS PubMed.
- B. W. Sigurskjold, B. Svensson, G. Williamson and H. Driguez, Thermodynamics of Ligand Binding to the Starch-Binding Domain of Glucoamylase from Aspergillus Niger, Eur. J. Biochem., 1994, 225(1), 133–141 CrossRef CAS PubMed.
- L. Nekiunaite, T. Isaksen, G. Vaaje-Kolstad and M. Abou Hachem, Fungal lytic polysaccharide monooxygenases bind starch and β-cyclodextrin similarly to amylolytic hydrolases, FEBS Lett., 2016, 590(16), 2737–2747 CrossRef CAS PubMed.
- V. V. Vu, J. A. Hangasky, T. C. Detomasi, S. J. W. Henry, S. T. Ngo, E. A. Span and M. A. Marletta, Substrate selectivity in starch polysaccharide monooxygenases, J. Biol. Chem., 2019 DOI:10.1074/jbc.RA119.009509.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01981b |
| This journal is © The Royal Society of Chemistry 2019 |