DOI:
10.1039/C9RA02161B
(Paper)
RSC Adv., 2019,
9, 34691-34698
A bromine-catalysis-synthesized poly(3,4-ethylenedioxythiophene)/graphitic carbon nitride electrochemical sensor for heavy metal ion determination†
Received
20th March 2019
, Accepted 21st October 2019
First published on 28th October 2019
Abstract
In this paper, poly(3,4-ethylenedioxythiophene)/graphitic carbon nitride (PEDOT/g-C3N4) composites were prepared by the bromine catalysed polymerization (BCP) method with varying weight ratios of monomer to g-C3N4. For comparison, solid-state polymerization (SSP) and metal oxidative polymerization (MOP) methods were also used for the synthesis of PEDOT/g-C3N4 composites. Electrochemical determination of heavy metal ions (Cd2+ and Pb2+) was carried out by differential pulse voltammetry (DPV) on composite-modified glass carbon electrodes (GCEs), which were prepared by different methods. The obtained composites were analysed by Fourier transform infrared spectroscopy (FT-IR), ultraviolet-visible absorption spectroscopy (UV-vis), X-ray diffraction (XRD), energy-dispersive X-ray spectroscopy (EDS), scanning electron microscopy (SEM), and transmission electron microscopy (TEM). The results showed that the bromine catalysed polymerization (BCP) method is an effective way to prepare the PEDOT/g-C3N4 composite, and the combination of PEDOT with g-C3N4 can improve the electrochemical activity of electrode materials. And, the composite from the BCP method modified electrode (PEDOT/10 wt% g-C3N4/GCE) exhibited the widest linear responses for Cd2+ and Pb2+, ranging from 0.06–12 μM and 0.04–11.6 μM with detection limits (S/N = 3) of 0.0014 μM and 0.00421 μM, respectively.
1. Introduction
Conducting polymers (CPs), such as polyaniline (PANI), polypyrene (PPy) and poly(3,4-ethylenedioxythiophene) (PEDOT), have been prevalent in the field of photoelectric materials for several decades since they were first discovered by Shirakawa et al.1–3 PEDOT is one of the most widely used conductive polymers due to its good electrochemical stability and high electrical conductivity.4 Since Leverkusen Bayer first synthesized this compound in 1988, much research has been done on PEDOT. Due to its unique π-conjugation system, high conductivity, easy processing and film formation, PEDOT has been used in many applications, such as energy storage,5,6 electrochromic devices,7 ion sensing8 and biosensors.9 In general, PEDOT is not used alone but is compounded with other materials such as metals,10 metal oxides,11,12 and carbon materials.13,14
Carbon-based materials typically include carbon nanotubes, single-wall carbon nanohorns, bucky papers, graphene, and fullerenes.15 New features of carbon materials increase the wide variability of multifunctional carbon-based sensing electrodes that exhibit high sensitivity and low sensing limits for a variety of chemical and biological analytes due to the extraordinary specific surface area, electrical conductivity, chemical durability, biocompatibility and mechanical strength of carbon-based materials.16–19 Graphene-like two-dimensional nanomaterials have been extensively studied for electrochemical sensing applications due to their structural properties similar to those of graphene and tuneable bandgap capability,20 such as transition metal dichalcogenides,21,22 hexagonal boron nitride nanosheets,23,24 and graphitic carbon nitride (g-C3N4) nanosheets.25–27 Among these, g-C3N4 has attracted massive attention due to its simple synthesis, satisfactory catalytic activity, excellent chemical stability and environmental friendliness. According to reports, the strong electron donor properties of nitrogen can affect the electronic structure and result in different activity-enhancing mechanisms.20 In addition, g-C3N4 ultrathin sheets demonstrate a strong affinity towards heavy metal ions due to the intrinsic –NH and –NH2 groups of g-C3N4, and this affinity is very beneficial for metal ions detection.28 However, the mediocre electroconductivity of g-C3N4 seriously affects its application in reality.27,29–31 Therefore, modifying it by compounding with other materials, such as graphene or CPs, is necessary to enable efficient electron transfer.
Recently, a number of PEDOT/g-C3N4 composites have been synthesized for studies on supercapacitors, biosensors, photocatalysis and catalytic hydrogen production. For example, a g-C3N4 nanofibre-doped PEDOT-modified electrode was used for the simultaneous determination of ascorbic acid and acetaminophen;32 a new type of carbon nitride-based polymer composite was used for enhanced photocatalytic hydrogen production.33,34 However, there is no research in the field of heavy metal ion detection. Hence, the preparation of PEDOT/g-C3N4 nanocomposites is a feasible method for determining heavy metal ions.
In general, the methods employed to prepare polymer-based composites include electrochemical polymerization,35 chemical oxidation polymerization,36 metal-catalysed coupling,37 solid-state polymerization (SSP),38 acid-assisted polymerization,39 and photoinduced polymerization.40 However, polymer-based composites synthesised by SSP often have performance fluctuations due to uneven mixing of the inorganic phase and organic phase. Furthermore, the metals used in the abovementioned chemical polymerization methods are difficult to remove and act as dopants.41 For example, FeCl3-catalysed polymerization can result in residual iron, and after several cycles of de-doping, an obvious electron paramagnetic resonance signal and sharp XRD peaks (FeCl4−-doping) are still observed.42–46 Hence, obtaining a metal-free polymer is essential for improving the performance of devices, such as OLEDs, solar cells and heavy metal ion sensors, because the metal present in the polymer occupies the active sites, which act as quenching sites for excitons.47 In summary, the selection of a suitable polymerization method is important for the preparation of polymer composites. Patra et al. reported a metal free bromine catalyzed polymerization for the synthesis of PEDOT and found this method providing metal free and virtually defect free PEDOT under mild reaction conditions.48 However, there is no report for preparation of composites with PEDOT by this method.
In this paper, PEDOT/g-C3N4 composites were prepared by the bromine catalyzed polymerization (BCP) method with varying weight ratios of monomer to g-C3N4. For comparison, the SSP and metal oxidative polymerization (MOP) methods were also used for the synthesis of PEDOT/g-C3N4 composites. And, the composites are used as electrochemical sensors for determination of heavy metal ions (Cd2+ and Pb2+). The results showed that among the composites from the bromine catalyzed polymerization method, PEDOT/10 wt% g-C3N4 exhibited the widest linear responses and excellent sensitivity for heavy metal ions (Cd2+ and Pb2+). Furthermore, compared with the SSP and MOP method composites, the reliability and linear detection range of the composite from the Br2-catalysed polymerization method were significantly improved.
2. Experimental
2.1 Reagents
3,4-Ethylenedioxythiophene (EDOT) and carbamide were obtained from J&K Scientific Ltd. N-Bromosuccinimide (NBS) and Br2 were purchased from Aladdin. Anhydrous ethanol, isopropanol, tetrahydrofuran (THF), anhydrous ferric chloride, CHCl3, and CH3COONa were purchased from Tianjin ZhiYuan Chemical Reagent Ltd. for use without further purification. The 0.1 mol L−1 acetate buffer solution (ABS) was prepared by 0.1 mol L−1 sodium acetate and acetic acid, and standard solutions of 0.001 M Cd2+ and Pb2+ were obtained by dissolving Pb(AcO)2 and Cd(AcO)2 in ultrapure water, respectively.
2.2 Synthesis of g-C3N4
The g-C3N4 sample was synthesized according to the literature.49 First, 10 g of carbamide was added to a 40 mL crucible and placed in a muffle furnace. After that, the precipitate was heated to 500 °C for 2 h at a rate of 10 °C per minute, followed by heating to 550 °C for an additional 2 h. Finally, the crucible was cooled to ambient temperature, and a yellowish solid powder was obtained after washing with absolute ethanol and distilled water.
2.3. Synthesis of monomer DBEDOT
DBEDOT was prepared according to a method used in previous research.50,51 EDOT (3.0 g, 21.1 mmol) was dissolved in a mixture of THF and HAc (200 mL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume). NBS (8.6 g, 48.3 mmol) was added to the mixture. After stirring at room temperature and in the dark for 2 h, a certain amount of ice was added to the solution, and some silvery white crystals appeared. The crystals were then filtered, washed with iced ethanol and dried over a vacuum at room temperature to obtain the pure product (3.9 g, 61.7% yield).
:1 by volume). NBS (8.6 g, 48.3 mmol) was added to the mixture. After stirring at room temperature and in the dark for 2 h, a certain amount of ice was added to the solution, and some silvery white crystals appeared. The crystals were then filtered, washed with iced ethanol and dried over a vacuum at room temperature to obtain the pure product (3.9 g, 61.7% yield).
2.4 Preparation of PEDOT/g-C3N4 composites by BCP
To obtain composites with different contents of g-C3N4, DBEDOT (0.3 g, 1 mmol) was added to 20 mL CHCl3 that had different ratios of g-C3N4 and was ultrasonically dispersed for 20 min. Then, Br2 (0.64 g, 4 mmol) solution was added to CHCl3 (2 mL) at room temperature. Next, the reaction mixture was heat to 50 °C and stirred for 48 h. After completion of the reaction, a purple-black solid precipitate was obtained. A black solid powder was obtained after washing with acetone and CHCl3. Pure PEDOT was obtained using the same method without adding g-C3N4.
2.5 Preparation of PEDOT/g-C3N4 composites by SSP
A mixture of DBEDOT (0.3 g, 1 mmol) and a certain amount of g-C3N4 in 10 mL CHCl3 was ultrasonicated for 20 min. After ultrasonication, the mixture was placed in an agate mortar to completely evaporate the chloroform. Then, the mixture was gently ground for 60 min until the DBEDOT was evenly mixed with g-C3N4; the mixture was kept in a vacuum oven at 60 °C for 24 h. Finally, the residue was washed with CHCl3 and absolute ethanol and then dried at 60 °C for 24 h.
2.6 Preparation of PEDOT/g-C3N4 composites by MOP
First, EDOT (0.3 g, 1 mmol) and a certain amount of g-C3N4 were added to chloroform (30 mL) and ultrasonically dispersed for 30 min. After ultrasonication, anhydrous ferric chloride (0.65 g, 4 mmol) was added to the mixture by ultrasonication for an additional 30 min. Then, the reaction was magnetically stirred at room temperature for 24 h. Finally, the reaction product was filtered to give a black solid, which was washed sequentially with chloroform, distilled water and anhydrous ethanol until the filtrate was colourless and then dried under vacuum at 60 °C for 24 h.
2.7 Preparation of modified electrodes
In general, the GCEs were sequentially polished with 1.0, 0.3, and 0.05 μm aluminium oxide abrasives and then modified by a simple method. A 5 μL aliquot of the PEDOT/g-C3N4 suspension (1 mg/4 mL) was dropped onto the surface of the GCE and dried at 40 °C for 30 min. g-C3N4/GCE, PEDOT (BCP)/GCE and PEDOT/10 wt% g-C3N4/GCE (obtained by different methods) were prepared by the same procedure.
2.8 Characterization and instruments
The FTIR spectra of the g-C3N4, PEDOT and PEDOT/g-C3N4 composites were obtained using a BRUKER EQUINOX-55 Fourier transform infrared spectrometer (Bruker, Billerica, MA, USA) (frequency range 4000 to 500 cm−1). The UV-vis spectra of the samples were recorded on a UV-vis spectrophotometer (UV4802, Unico, Dayton, NJ, USA). The XRD patterns of the samples were conducted on a Bruker AXS D8 diffractometer with Cu-Kα radiation (λ = 0.154 nm); the scan range (2θ) was 10° to 80°. The morphology analysis of the g-C3N4, PEDOT and PEDOT/g-C3N4 composites was obtained on a scanning electron microscope (JSM7610F Plus, JEOL, Japan, operating voltage, 5 kV) and transmission electron microscope (JEOL model 2100, JEOL Ltd., Tokyo, Japan). The elemental compositions of the samples were measured using energy-dispersive X-ray spectroscopy (EDS), which was conducted on a Leo 1430VP microscope (Carl Zeiss Inc., Oberkochen, Germany). The real samples were verified by atomic absorption spectrometry (AAS) (Shimadzu AA6300C, lead wavelength 283.3 nm, lamp current 5.0 mA; cadmium wavelength 228.8 nm, lamp current 5.0 mA; slit 0.4 nm).
The electrochemical performances were investigated via a three-electrode system on an electrochemical workstation. (ChenHua, CHI660C, Instruments Co.). The modified GCEs were used as the working electrode, a platinum electrode was the counter electrode and a calomel electrode was the reference electrode. Cyclic voltammetry (CV) was performed in a mixture of 5.0 mM [Fe(CN)6]3−/4− and 0.1 M KCl at 50 mV s−1. Differential pulse voltammetry (DPV) was performed in 0.1 M ABS (pH = 4.5) with an incremental potential of 2 mV from −1.2 to −0.2 V, with a pulse width of 50 ms and pulse period of 100 ms.
3. Results and discussion
3.1 FT-IR spectra
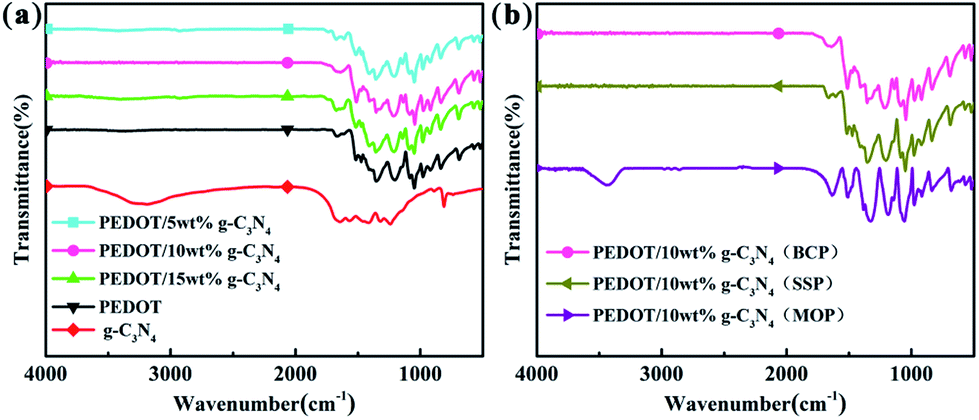
Fig. 1 shows the FTIR spectra of g-C3N4, PEDOT (BCP) and the PEDOT/g-C3N4 nanocomposites. In the spectrum of g-C3N4, the band at 813 cm−1 was identified as the bending vibration of the planar triazine ring mode. The weak peak appearing at 890 cm−1 can be attributed to the deformation mode of crosslinked heptazine.52 The stretching vibrations of C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N in the heterocyclic ring cause the characteristic peaks in the range of 1230–1650 cm−1.53 The wide vibration band appearing in the 3000–3500 cm−1 region is caused by the stretching vibration of the aromatic N–H bond and the uncondensed amino group (–NH2).54 For pure PEDOT (BCP), the characteristic peaks at 1515 and 1320 cm−1 are the typical C–C asymmetric stretching vibration and inter-ring stretching vibration, respectively. The peaks at approximately 1209, 1139 and 1088 cm−1 are the C–O–C bending vibrations in the ethylenedioxy group.55 The bands at approximately 974, 918, 833, and 693 cm−1 are the characteristic absorption bands of the C–S–C bond in the thiophene ring.56
N in the heterocyclic ring cause the characteristic peaks in the range of 1230–1650 cm−1.53 The wide vibration band appearing in the 3000–3500 cm−1 region is caused by the stretching vibration of the aromatic N–H bond and the uncondensed amino group (–NH2).54 For pure PEDOT (BCP), the characteristic peaks at 1515 and 1320 cm−1 are the typical C–C asymmetric stretching vibration and inter-ring stretching vibration, respectively. The peaks at approximately 1209, 1139 and 1088 cm−1 are the C–O–C bending vibrations in the ethylenedioxy group.55 The bands at approximately 974, 918, 833, and 693 cm−1 are the characteristic absorption bands of the C–S–C bond in the thiophene ring.56
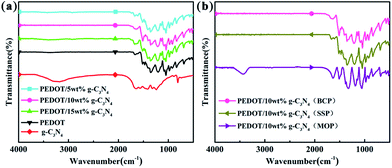 |
| | Fig. 1 FTIR spectra of (a) PEDOT (BCP) and PEDOT/g-C3N4 nanocomposites with different weight percentages of g-C3N4 (b) PEDOT/10 wt% g-C3N4 prepared by different polymerization methods. | |
For the composites, as shown in Fig. 1(a) and (b), the main vibration bands are similar to those of PEDOT indicating that the successful incorporation of PEDOT component in composites. Fig. 1(a) and (b) show that the positions of the characteristic peaks for the PEDOT/10 wt% g-C3N4 composites prepared by the SSP, MOP and BCP methods are basically identical. Furthermore, according to report,43 the intensity ratio of the vibration bands at approximately 1470/1515 cm−1 is indicative of the conjugation degree of the composites, with a smaller ratio indicating a higher degree of conjugation. Therefore, it is clear from Fig. 1(a) and (b) that PEDOT/10 wt% g-C3N4 has a longer conjugated bond length.57 However, there are no characteristic peaks corresponding to g-C3N4 in the spectra of the PEDOT/g-C3N4 composites, which may caused by overlapping of characteristic peak of PEDOT and g-C3N4. In addition, the g-C3N4 can also be enwrapped by PEDOT in some extent, consequently bring a decrease in peak intensity of g-C3N4.
3.2 UV-vis spectra
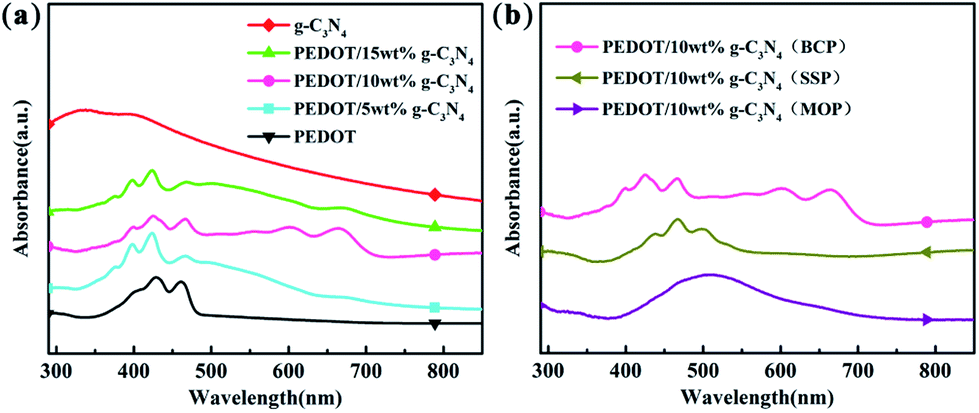
The UV-vis spectra of g-C3N4, PEDOT (BCP), and the PEDOT/g-C3N4 composites are presented in Fig. 2. It is clear from Fig. 2(a) that the UV-vis spectral peaks of g-C3N4 appears from 270 to 430 nm, which are absorption peaks characteristics of carbon nitride. The absorption bands of PEDOT (BCP) at approximately 399, 426 and 460 nm can be ascribed to different conjugation lengths, and they are assigned to the π–π transition of the thiophene ring.58 Furthermore, the absorption peaks of PEDOT/g-C3N4 (BCP) at 550, 600 and 667 nm are assigned to the polaron and/or bipolaron band,59,60 which indicates a strong interaction between PEDOT and g-C3N4. In Fig. 2(b), PEDOT/10 wt% g-C3N4 (SSP) shows characteristic peaks at 437, 465 and 498 nm, which can also be assigned to the π–π transition of the thiophene ring. Moreover, very weak absorption bands are also observed at 606 nm, which indicates that the π–π interaction between PEDOT and g-C3N4 is weak.
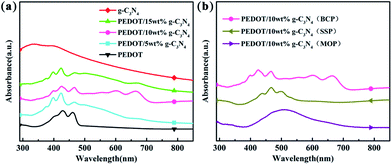 |
| | Fig. 2 UV-vis spectra of (a) PEDOT (BCP) and PEDOT/g-C3N4 nanocomposites with different weight percentages of g-C3N4 (b) PEDOT/10 wt% g-C3N4 prepared by different polymerization methods. | |
In the spectrum of PEDOT/10 wt% g-C3N4 (MOP), the absorption peak is located in the range of 400–600 nm, which is in good agreement with that previously reported.45 By comparison, among composites, PEDOT/10 wt% g-C3N4 (BCP) shows the highest π–π interaction between PEDOT and g-C3N4, while it is the lowest in the case of PEDOT/10 wt% g-C3N4 (SSP).
3.3 XRD analysis
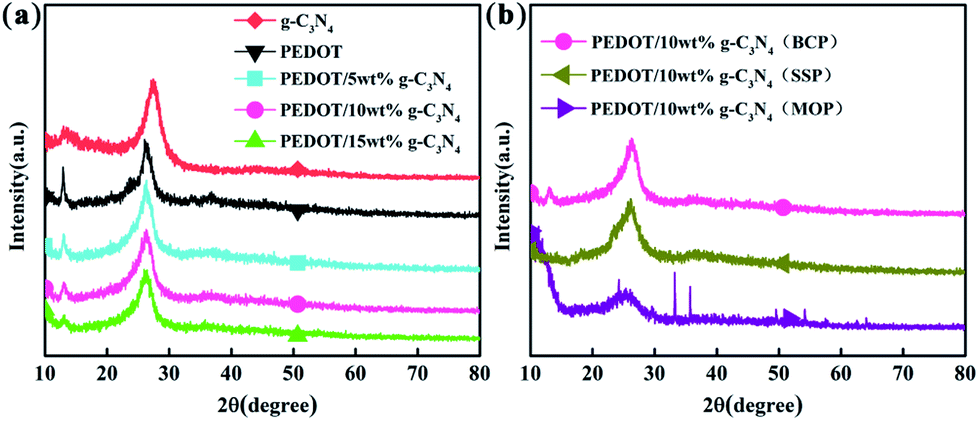
Fig. 3 shows the XRD patterns of g-C3N4, PEDOT (BCP) and the PEDOT/g-C3N4 nanocomposites. In Fig. 3(a), the diffraction peaks of g-C3N4 can be seen at 27.3° and 13.1°, which can be attributed to the (002) and (100) diffraction planes, respectively, of the graphite material. Among them, the stronger diffraction peak is assigned to the typical planes of graphitic structures, and the weaker peak at 13.1° is attributed to the hole-to-hole arrays of the tri-s-triazine units. The XRD pattern of PEDOT (BCP) also shows two characteristic peaks at approximately 2θ = 26.2° and 12.8°, which are due to intermolecular π–π stacking. In the patterns of the PEDOT/g-C3N4 composites (BCP), as the g-C3N4 content increases, the intensity of the diffraction peak at 12.8° decreases, which is consistent with the increasing g-C3N4. In Fig. 3(b), PEDOT/g-C3N4 (SSP) shows the same characteristic peaks as PEDOT/g-C3N4 (BCP). However, PEDOT/g-C3N4 (MOP) shows broader diffraction peaks at 26.2° and sharp diffraction peaks at 2θ ≈ 33°, 35°, 49° and 54° due to FeCl4− doping in the composite.
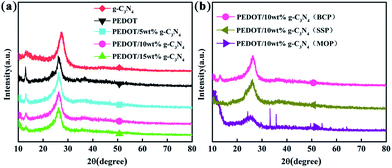 |
| | Fig. 3 The XRD patterns of FTIR spectra of (a) PEDOT (BCP) and PEDOT/g-C3N4 nanocomposites with different weight percentages of g-C3N4 (b) PEDOT/10 wt% g-C3N4 prepared by different polymerization methods. | |
3.4 EDS analysis
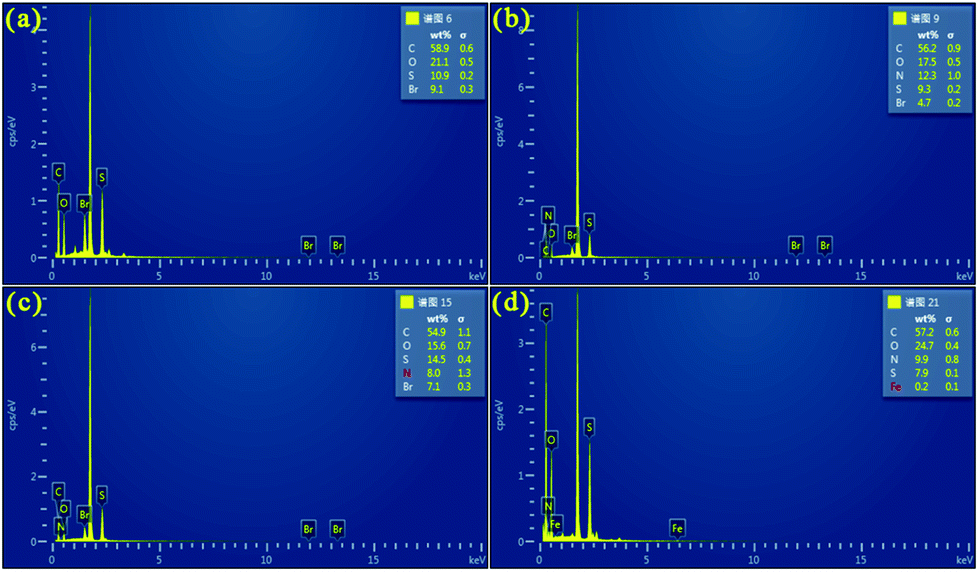
Fig. 4 shows the EDS analysis of PEDOT (BCP) and the PEDOT/g-C3N4 composites on a silicon wafer. As shown in Fig. 4(a), the EDS spectrum of the pure PEDOT sample indicates the presence of C, S, O, and Br. After the addition of g-C3N4, some N was detected in the PEDOT/g-C3N4 composites prepared by the three different methods. In Fig. 4(d), anhydrous ferric chloride was added as a catalyst in the reaction, which introduced Fe in the composites. Compared with PEDOT, the weight percentage of N in the composites increased with the addition of g-C3N4.
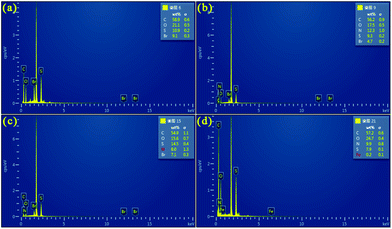 |
| | Fig. 4 EDS of (a) PEDOT (BCP), (b) PEDOT/10 wt% g-C3N4 (BCP), (c) PEDOT/10 wt% g-C3N4 (SSP), (d) PEDOT/10 wt% g-C3N4 (MOP). | |
3.5 Morphology studies
Fig. 5 and S1† show the SEM and TEM images of g-C3N4, PEDOT (BCP), PEDOT/10 wt% g-C3N4 (BCP), PEDOT/10 wt% g-C3N4 (SSP) and PEDOT/10 wt% g-C3N4 (MOP), respectively. The pure PEDOT prepared by SSP presents a rule and plate-like structure, as shown in Fig. 5(b) and 6(b). After sonication for a period of time, due to π–π stacking and aromatic electrostatic attraction, the DBEDOT monomer will be adsorbed onto the surface of flake g-C3N4, which will allow the polymer to be uniform on the surface of g-C3N4.
 |
| | Fig. 5 SEM images of (a) g-C3N4 (b) PEDOT (BCP), (c) PEDOT/10 wt% g-C3N4 (BCP), (d) PEDOT/10 wt% g-C3N4 (SSP), (e) PEDOT/10 wt% g-C3N4 (MOP). | |
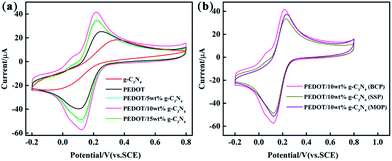 |
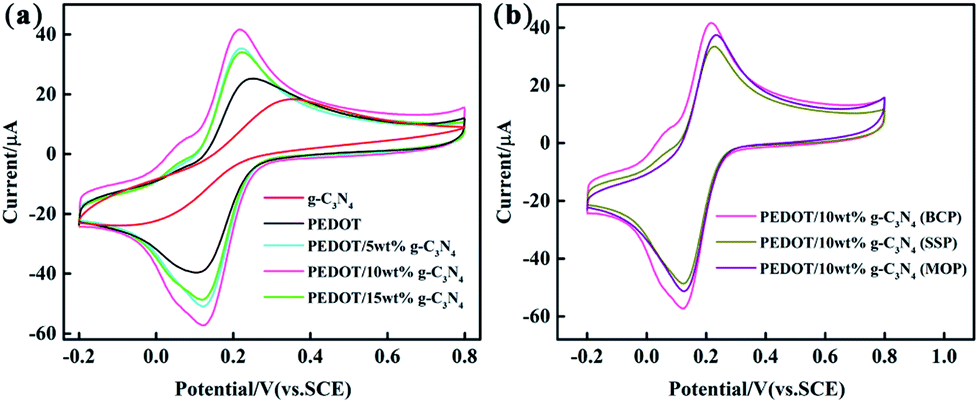
| | Fig. 6 CV measured with g-C3N4, PEDOT (BCP), PEDOT/g-C3N4 composite-modified glassy carbon electrode (GCE) in a solution of 5 mM Fe(CN)63−/4− containing 0.1 M KCl. (a) PEDOT/g-C3N4 nanocomposites with different weight percentages of g-C3N4 (b) PEDOT/10 wt% g-C3N4 prepared by different polymerization methods. | |
Therefore, the PEDOT/10 wt% g-C3N4 composite prepared by BCP has a layered morphology, as shown in Fig. 5(c) and S1(c).† Furthermore, as shown in Fig. 5(d), (e) and S1(d), (e),† the PEDOT/10 wt% g-C3N4 composite prepared by SSP and MOP showed fragmented and irregular groups, respectively.
3.6 Electrochemical characterization of PEDOT (BCP), g-C3N4 and PEDOT/g-C3N4 composites
Cyclic voltammetry was performed with different modified GCEs in a mixture solution of 5 mM [Fe(CN)6]3−/4− and 0.1 M KCl (50 mL, 1:1 by volume). As shown in Fig. 6, the anodic peak potentials are 0.38 V and 0.25 V for pure PEDOT (BCP) and g-C3N4 modified GCEs, respectively. And, the anodic peak current intensity is lower in g-C3N4-modified GCE than that of PEDOT (BCP) modified GCE. The main reason for this is that the lower electrical conductivity of g-C3N4 than that of PEDOT (BCP) can lead a low peak potential and current occurred in g-C3N4-modified GCE than that of PEDOT (BCP) modified GCE.27 After combination of g-C3N4 with PEDOT, the peak potential of samples shifted to low potential at about 0.22 V with high current intensity than that of PEDOT (BCP). All these results showed that the of PEDOT (BCP) and g-C3N4 cannot be used alone as a high sensitive materials for ions with their lower electrochemical activity than that of composites. However, the heterojunction of PEDOT (BCP) and g-C3N4 can bring an enhancement in electrochemical activity of electrode materials. According to previous report, although g-C3N4 is poor in electrical conductivity, it has fast charge transfer ability and large surface area with π–π conjugation structure.61,62 The enhancement in electrochemical activity composites should be assigned to the strong π–π interaction between PEDOT and g-C3N4 by π–π stacking effect. And, the fast charge transfer ability of g-C3N4 as well as the high electrical conductivity of PEDOT can cause a rapid electron transfer occurring at the interface of composite electrodes, which can promote the electrochemical performance of composite electrodes. As shown in Fig. 6(b), the PEDOT/10 wt% g-C3N4 (BCP) composite-modified GCE exhibits the highest peak current than that of other composite/GCEs, which can be explained by the strong π–π interaction between PEDOT and g-C3N4 in the composite prepared by BCP, while PEDOT/10 wt% g-C3N4 (SSP) composite modified GCE exhibits the lowest peak current resulting from the low π–π interaction between PEDOT and g-C3N4 in composite from SSP method.
Fig. S2† shows the DPV curves for 2.0 μM Cd2+ and Pb2+ in a 0.1 M ABS (pH = 4.5) solution with the different modified GCEs. As shown in Fig. S2,† the distance between the characteristic peaks of Cd2+ and Pb2+ is sufficiently wide, and the peak positions of Cd2+ and Pb2+ are at approximately −0.81 V and −0.58 V, respectively. As shown in Fig. S2,† the g-C3N4 presents a similar electrocatalytic performance with that of PEDOT and PEDOT/10 wt% g-C3N4 from SSP method. This can understood by the large number of nitrogen-containing groups in g-C3N4, which can coordinate with metal ions to enhance the electrocatalytic activity.28,63 The comparison also indicates that the composite from BCP method displayed a higher peak current than others. The reason for this can be that the plate-like morphology of PEDOT from the BCP method, which is beneficial for a uniform combination of PEDOT and g-C3N4 flakes, and the strong π–π interaction between PEDOT and g-C3N4, coordination effect of g-C3N4 with metal ions as well as the fast charge transfer ability and large surface of g-C3N4 can increase the synergetic effect of the components, which can also enhance the electrochemical properties of the composite. In the case of PEDOT/10 wt% g-C3N4 from SSP and MOP methods, the inventible mixing of the inorganic and organic phase as well as low π–π interaction between PEDOT and g-C3N4 in SSP method is the negative effect for the electrocatalytic oxidation of metal ions, while iron ion doping MOP method can play negative for the electrocatalytic oxidation of metal ions. In summary, PEDOT/g-C3N4 prepared by BCP not only effectively increases the electron transfer kinetics of the electrode surface but also avoids doping with iron ions. In addition, the interaction between the conjugated structure of PEDOT and flakes, such as g-C3N4, through π–π stacking allows strong adsorption of heavy metal ions.63
3.7 Individual determination of Cd2+ and Pb2+
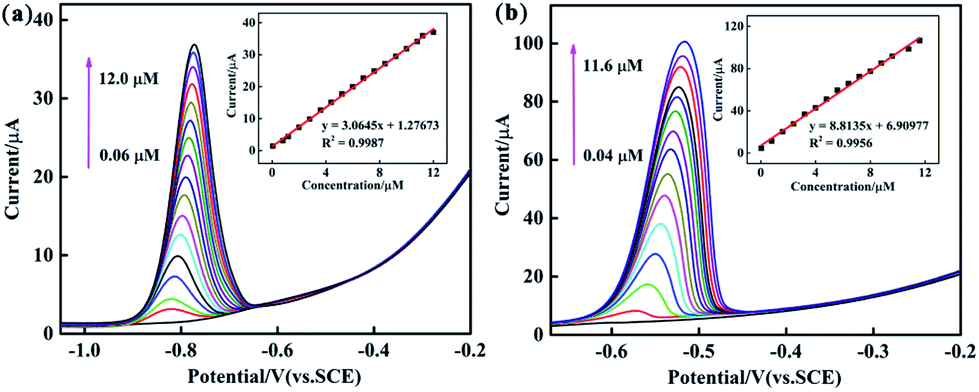
To systematically study the performance difference among the composites prepared by three different methods, under optimized conditions, various modified GCEs were used for electrochemical determination of Cd2+ and Pb2+ in 0.1 M ABS (pH = 4.5) by DPV. Fig. 7 and S3, S4† show the DPV responses of PEDOT/10 wt% g-C3N4 (BCP), PEDOT/10 wt% g-C3N4 (SSP), and PEDOT/10 wt% g-C3N4 (MOP) composite-modified GCEs towards Cd2+ and Pb2+, and the inset shows their linear equations and correlation coefficients (Fig. 7 and S3, S4† inset). As from the above results, PEDOT/10 wt% g-C3N4 (BCP) exhibited detection limits of 0.0014 μM and 0.0042 μM with linear ranges of 0.06–12.0 μM and 0.04–11.6 μM for Cd2+ and Pb2+, respectively. (Other detailed results are shown in Table 1). It is clear that the PEDOT/10 wt% g-C3N4 (BCP)/GCE shows a wide linear range and low detection limit, and the detection limit was lower than that of the WHO standard, illustrating that the PEDOT/10 wt% g-C3N4 prepared by BCP provides a large number of active sites, thereby increasing the adsorption of heavy metal ions.
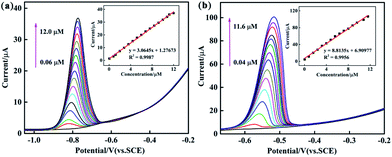 |
| | Fig. 7 DPV response of the PEDOT/10 wt% g-C3N4 (BCP) composite-modified GCE for the individual analysis of (a) Cd2+ (b) Pb2+. The inset shows their linear equations as well as correlation coefficient. | |
Table 1 Performance of the PEDOT/10 wt% g-C3N4/GCE for individual determination of Cd2+ and Pb2+
| Methods |
Analytes |
Linear range (μM) |
Linear regression equation |
R2 |
Detection limit (μM) |
| BCP |
Cd2+ |
0.06–12.0 |
y = 3.0645x + 1.27673 |
0.9987 |
0.0014 |
| Pb2+ |
0.04–11.6 |
y = 8.8135x + 6.90977 |
0.9956 |
0.00421 |
| SSP |
Cd2+ |
0.1–6.8 |
y = 0.63559x − 0.3114 |
0.9821 |
0.0521 |
| Pb2+ |
0.1–6.0 |
y = 10.6259x − 1.4879 |
0.9934 |
0.0263 |
| MOP |
Cd2+ |
0.1–3.6 |
y = 1.6021x + 0.2147 |
0.9923 |
0.0065 |
| Pb2+ |
0.1–2.8 |
y = 27.3213x + 5.1612 |
0.9844 |
0.0637 |
3.8 Simultaneous determination of Cd2+ and Pb2+
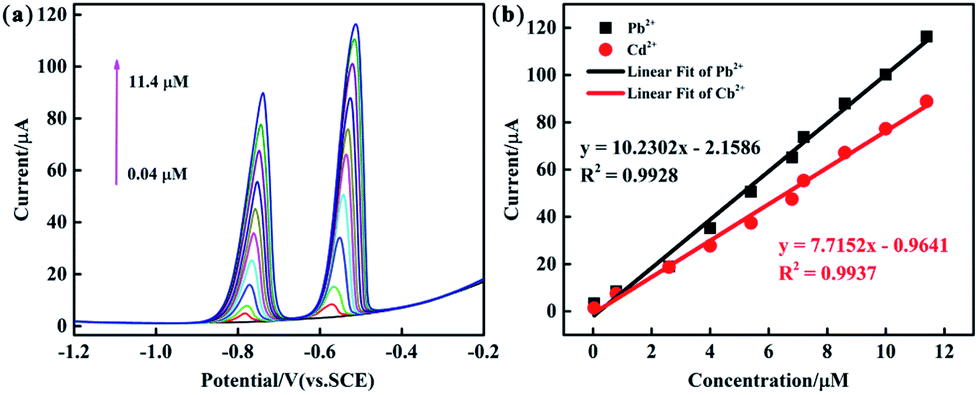
The analytical performance of the PEDOT/10 wt% g-C3N4/GCE (BCP), PEDOT/10 wt% g-C3N4/GCE (SSP), and PEDOT/10 wt% g-C3N4/GCE (MOP) was investigated through simultaneous determination of Cd2+ and Pb2+ in 0.1 M ABS (pH = 4.5). Fig. 8 and S5, S6† show the linear equations and correlation coefficients for each modified electrode. For simultaneous detection, there may be two factors affecting the performance results: the active sites on the modified electrode and the competitive effect between different ions. The PEDOT/10 wt% g-C3N4 (BCP) exhibited the widest linear ranges of those tested with a linear range of 0.04–11.4 μM for Cd2+ and Pb2+ and detection limits of 0.00547 μM and 0.00694 μM for Cd2+ and Pb2+, respectively. (Other detailed results are shown in Table 2). This result indicates that the composite prepared by the BCP method has a larger number of active sites than the other two composites and no iron bonding.
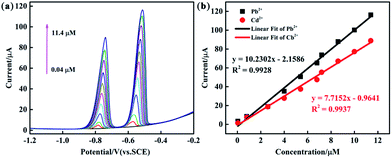 |
| | Fig. 8 (a) DPV response of the PEDOT/10 wt% g-C3N4 (BCP) composite-modified GCE for the simultaneous analysis of Cd2+ and Pb2+ (b) the respective calibration curves of Cd2+ and Pb2+. | |
Table 2 Performance of the PEDOT/10 wt% g-C3N4/GCE for simultaneous determination of Cd2+ and Pb2+
| Methods |
Analytes |
Linear range (μM) |
Linear regression equation |
R2 |
Detection limit (μM) |
| BCP |
Cd2+ |
0.04–11.4 |
y = 7.7152x − 0.9641 |
0.9937 |
0.0068 |
| Pb2+ |
y = 10.2302x − 2.1586 |
0.9928 |
0.0079 |
| SSP |
Cd2+ |
0.1–4.0 |
y = 1.5714x + 1.3715 |
0.9927 |
0.0054 |
| Pb2+ |
y = 17.0702x + 2.4552 |
0.9887 |
0.064 |
| MOP |
Cd2+ |
0.1–2.8 |
y = 2.3403x + 1.4897 |
0.9994 |
0.0011 |
| Pb2+ |
y = 21.9224x + 3.0524 |
0.9911 |
0.0059 |
3.9 Real sample analysis
To quantify Cd2+ and Pb2+ in real samples and estimate the applicability of PEDOT/10 wt% g-C3N4 (BCP), a standard addition method was carried out with a tap water sample. First, 0.1 M ABS (pH = 4.5), which included certain amounts of tap water, was prepared. Subsequently, different amounts of Cd2+ and Pb2+ standard solutions were added to the testing system. The test sample was found by DPV and the recoveries of Cd2+ and Pb2+ were 96–103.5% and 96–101.7%, respectively. The ion concentration of the test sample was verified by AAS and the recoveries of Cd2+ and Pb2+ were 89–98.5% and 92–106.6%, respectively (Table 3). Based on the above results, the PEDOT/10 wt% g-C3N4 (BCP)/GCE has practical application for the determination of Cd2+ and Pb2+ in tap water samples with excellent sensitivity and recovery.
Table 3 Determination of Cd2+ and Pb2+ in tap water by PEDOT/10 wt% g-C3N4 (BCP)
| No. |
Added (μM) |
DPV |
AAS |
| Found (μM) |
Recovery (%) |
Found (μM) |
Recovery (%) |
| Cd2+ |
Pb2+ |
Cd2+ |
Pb2+ |
Cd2+ |
Pb2+ |
Cd2+ |
Pb2+ |
Cd2+ |
Pb2+ |
| 1 |
1 |
1 |
0.97 |
0.96 |
97 |
96 |
0.89 |
0.94 |
89 |
94 |
| 2 |
2 |
2.07 |
1.93 |
103.5 |
96.5 |
1.90 |
2.11 |
95 |
105.5 |
| 3 |
3 |
2.95 |
3.02 |
98.3 |
100.7 |
2.87 |
2.94 |
95.6 |
98 |
| 2 |
1 |
1 |
0.96 |
1.08 |
96 |
108 |
0.94 |
0.92 |
94 |
92 |
| 2 |
2 |
2.02 |
1.98 |
101 |
99 |
1.97 |
2.16 |
98.5 |
108 |
| 3 |
3 |
2.99 |
3.05 |
99.7 |
101.7 |
2.78 |
3.20 |
92.6 |
106.6 |
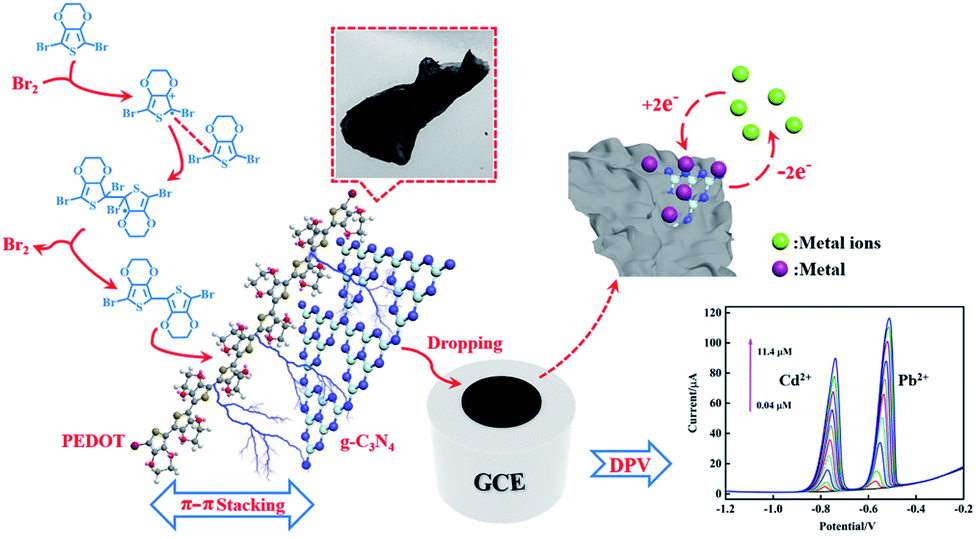
To compare with the SSP and MOP method composites, the results from electrochemical tests showed that the reliability and linear detection range of the composite from the bromine catalysed polymerization method were significantly improved, which can be related to the absence of iron bonding enhancing the absorption of Cd2+ and Pb2+. Moreover, the plate-like morphology of PEDOT from the Br2-catalysed polymerization method is beneficial for a uniform composition of PEDOT and flakes, such as g-C3N4, which can increase the synergetic effect of the beneficial for a uniform composition of PEDOT and flakes, such as g-C3N4, which can increase the synergetic effect of the components and enhance the electrochemical properties of the composite by π–π stacking interactions between PEDOT and g-C3N4 (Fig. 9).
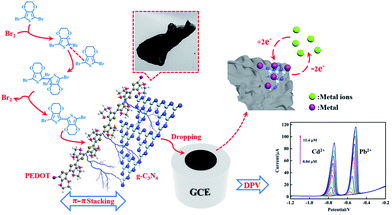 |
| | Fig. 9 Schematic illustration for the electrochemical determination of Cd2+ and Pb2+ by PEDOT/g-C3N4 composite (BCP). | |
4. Conclusion
In this paper, PEDOT/g-C3N4 composites were prepared by different methods and applied for the electrochemical determination of Cd2+ and Pb2+. The results showed that the bromine catalysed polymerization (BCP) method is an effective way for preparation of PEDOT/g-C3N4 composite. Among composites from different methods, PEDOT/10 wt% g-C3N4 (BCP) shows a highest π–π interaction between PEDOT and g-C3N4, while it is the lowest in the case of PEDOT/10 wt% g-C3N4 (SSP). In comparison with the SSP and MOP methods, the reliability and linear detection range of the composite from the Br2-catalysed polymerization method were significantly improved. All these results are originated from the plate-like morphology of PEDOT from the BCP method was beneficial for a uniform combination of PEDOT and g-C3N4 flakes, and the strong π–π interaction between PEDOT and g-C3N4, coordination effect of g-C3N4 with metal ions as well as the fast charge transfer ability and large surface of g-C3N4, which could enhance the electrochemical properties of the composite from BCP by strong synergetic effect of the components. In the case of PEDOT/10 wt% g-C3N4 from SSP and MOP methods, the inventible mixing of the inorganic and organic phase as well as low π–π interaction between PEDOT and g-C3N4 in SSP method is the negative effect for the electrocatalytic oxidation of metal ions, while iron ion doping MOP method can play negative for the electrocatalytic oxidation of metal ions. Furthermore, the PEDOT/10 wt% g-C3N4 (BCP) composite possesses a high sensitivity and low detection limit for Cd2+ and Pb2+, this will have potential applications in areas such as wastewater treatment and environmental protection.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the research grant provided by National Natural Science Foundation of China (No. 21564014, No. 21865034).
References
- H. Shirakawa, E. J. Louis, A. G. Macdiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., 1977, 16, 578–580 Search PubMed.
- Q. Zhou and G. Shi, J. Am. Chem. Soc., 2016, 138, 2868–2876 CrossRef CAS PubMed.
- S. C. Rasmussen, S. J. Evenson and C. B. McCausland, Chem. Commun., 2015, 51, 4528–4543 RSC.
- M. Sassi, L. Mascheroni, R. Ruffo, M. M. Salamone, G. A. Pagani, C. M. Mari, G. D'Orazio, F. B. La and L. Beverina, Org. Lett., 2013, 15, 3502–3505 CrossRef CAS.
- J. E. Yoo and A. J. Bard, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5712–5717 CrossRef CAS.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef CAS.
- G. A. Sotzing, J. R. Reynolds and P. J. Steel, Adv. Mater., 1997, 9, 795–798 CrossRef CAS.
- I. F. Perepichka, B. Mohamed, E. Levillain, A. Marc Sallé and R. Jean, Chem. Mater., 2002, 14, 449–457 CrossRef CAS.
- F. Mouffouk and S. J. Higgins, Electrochem. Commun., 2006, 8, 15–20 CrossRef CAS.
- L. Yang, J. Zhang, F. Zhao and B. Zeng, J. Chromatogr. A, 2016, 1471, 80–86 CrossRef CAS PubMed.
- A. Tursun, A. Ahmat, J. Ruxangul, O. Yakupjan and Z. Yu, Nanoscale Res. Lett., 2014, 9, 89 CrossRef.
- L. Tong, L. Jian, S. M. Boyer, L. A. Sonnenberg, M. T. Fox, D. Ji, J. Feng, W. E. Bernier and W. E. J. Jr, Electrochim. Acta, 2017, 224, 133–141 CrossRef CAS.
- W. Si, W. Lei, Z. Han, Q. Hao, Y. Zhang and M. Xia, Sens. Actuators, B, 2014, 199, 154–160 CrossRef CAS.
- K. Zhang, J. Xu, X. Duan, L. Lu, D. Hu, L. Zhang, T. Nie and K. B. Brown, Electrochim. Acta, 2014, 137, 518–525 CrossRef CAS.
- G. Maduraiveeran, M. Sasidharan and V. Ganesan, Biosens. Bioelectron., 2018, 103, 113 CrossRef CAS.
- S. Duan and Y. Huang, J. Electroanal. Chem., 2017, 807, 253–260 CrossRef CAS.
- J. Deep, V. K. Sangwan, L. J. Lauhon, T. J. Marks and M. C. Hersam, Chem. Soc. Rev., 2013, 42, 2824–2860 RSC.
- W. A. Marmisollé and O. Azzaroni, Nanoscale, 2016, 8, 9890–9918 RSC.
- S. Li, L. Wang, X. Zhang, H. Chai and Y. Huang, Sens. Actuators, B, 2018, 264, 312–319 CrossRef CAS.
- N. Wongkaew, M. Simsek, C. Griesche and A. J. Baeumner, Chem. Rev., 2019, 119, 120–194 CrossRef CAS.
- Y. Hu, Y. Huang, C. Tan, X. Zhang, Q. Lu, M. Sindoro, X. Huang, W. Huang, L. Wang and H. Zhang, Mater. Chem. Front., 2017, 1, 24–36 RSC.
- Y. H. Wang, K. J. Huang and X. Wu, Biosens. Bioelectron., 2017, 97, 305–316 CrossRef CAS.
- A. F. Khan, B. Dac, C. W. Foster, G. C. Smith and C. E. Banks, Analyst, 2017, 142, 1756–1764 RSC.
- G. H. Yang, J. J. Shi, S. Wang, W. W. Xiong, L. P. Jiang, C. Burda and J. J. Zhu, Chem. Commun., 2013, 49, 10757–10759 RSC.
- J. Tian, Q. Liu, A. M. Asiri, A. H. Qusti, A. O. Al-Youbi and X. Sun, Nanoscale, 2013, 5, 11604–11609 RSC.
- Y. Zhang, X. Bo, A. Nsabimana, C. Luhana, G. Wang, H. Wang, M. Li and L. Guo, Biosens. Bioelectron., 2014, 53, 250–256 CrossRef CAS.
- J. Tian, Q. Liu, C. Ge, Z. Xing, A. M. Asiri, A. O. Al-Youbi and X. Sun, Nanoscale, 2013, 5, 8921–8924 RSC.
- M. Sadhukhan and S. Barman, J. Mater. Chem. A, 2013, 1, 2752 RSC.
- Y. Xu, W. Lei, J. Su, J. Hu, X. Yu, T. Zhou, Y. Yang, D. Mandler and Q. Hao, Electrochim. Acta, 2018, 259, 994–1003 CrossRef CAS.
- J. Zhang, Z. Zhu, J. Di, Y. Long, W. Li and Y. Tu, Electrochim. Acta, 2015, 186, 192–200 CrossRef CAS.
- H. Gu, T. Zhou and G. Shi, Talanta, 2015, 132, 871–876 CrossRef CAS.
- C. Li, J. Xu, Y. Wu, Y. Zhang, C. Zhang, W. Lei and Q. Hao, J. Electroanal. Chem., 2018, 824, 52–59 CrossRef CAS.
- X. Chen, X. Zhu, Y. Xiao and X. Yang, J. Electroanal. Chem., 2015, 743, 99–104 CrossRef CAS.
- Z. Xing, Z. Chen, X. Zong and L. Wang, Chem. Commun., 2014, 50, 6762–6764 RSC.
- A. F. Diaz and K. K. Kanazawa, J. Chem. Soc., Chem. Commun., 1979, 14, 635 RSC.
- K. Yoshino, S. Hayashi and R. Sugimoto, Jpn. J. Appl. Phys., 2014, 23, L899–L900 CrossRef.
- J. Roncali, Chem. Rev., 1992, 92, 711–738 CrossRef CAS.
- M. Hong, D. F. Perepichka and W. Fred, Angew. Chem., 2010, 42, 658–661 Search PubMed.
- B. Baltasar and T. M. Swager, J. Am. Chem. Soc., 2012, 134, 18916–18919 CrossRef.
- B. Aydogan, G. E. Gunbas, A. Durmus, L. Toppare and Y. Yagci, Macromolecules, 2010, 43, 101–106 CrossRef CAS.
- A. Patra, M. Bendikov and S. Chand, Acc. Chem. Res., 2014, 47, 1465–1474 CrossRef CAS.
- M. Pomerantz, B. Chaloner-Gill, L. O. Harding, J. J. Tseng and W. J. Pomerantz, ChemInform, 1993, 24, 1672–1673 Search PubMed.
- C. Wang, M. E. Benz, E. Legoff, J. L. Schindler, J. Allbritton-Thomas, C. R. Kannewurf and M. G. Kanatzidis, Chem. Mater., 1994, 6, 40–49 Search PubMed.
- T. Yamamoto, K. Shiraishi, Y. Mahmut, I. Yamaguchi and L. Bert Groenendaal, Polymer, 2002, 43, 711–719 CrossRef CAS.
- J. M. Xu, H. S. O. Chan, S. C. Ng and T. S. Chung, Synth. Met., 2002, 132, 63–69 CrossRef CAS.
- M. H. Chahma, Synth. Met., 2005, 155, 474–479 CrossRef CAS.
- I. F. Perepichka, D. F. Perepichka and D. F. Perepichka, Mater. Today, 2009, 12, 54 Search PubMed.
- A. Patra, V. Agrawal, R. Bhargav, Shahjad, D. Bhardwaj, S. Chand, Y. Sheynin and M. Bendikov, Macromolecules, 2015, 48, 8760–8764 CrossRef CAS.
- N. Chidhambaram and K. Ravichandran, Mater. Lett., 2017, 207, 44–48 CrossRef CAS.
- F. Tranvan, S. Garreau, G. Louarn, G. Froyer and C. Chevrot, J. Mater. Chem., 2001, 11, 1378–1382 RSC.
- A. Tursun, J. Ruxangul, C. Zhao, A. Tunsagul and N. Ismayil, Synth. Met., 2010, 160, 325–332 CrossRef.
- M. J. Bojdys, M. Jens-Oliver, A. Markus and T. Arne, Chem.–Eur. J., 2010, 14, 8177–8182 CrossRef.
- S. Martha, A. Nashim and K. M. Parida, J. Mater. Chem. A, 2013, 1, 7816–7824 RSC.
- X. Zhou, J. Bei, L. Li, P. Feng, H. Wang, Y. Hao and Y. Fang, J. Mater. Chem., 2012, 22, 17900–17905 RSC.
- E. Eren, G. Celik, A. Uygun, J. Tabačiarová and M. Omastová, Synth. Met., 2012, 162, 1451–1458 CrossRef CAS.
- Y. Yang, Y. Jiang, J. Xu and J. Yu, Polymer, 2007, 48, 4459–4465 CrossRef CAS.
- Y. Furukawa, M. Akimoto and I. Harada, Synth. Met., 1987, 18, 151–156 CrossRef CAS.
- L. Yang, R. Jamal, F. Liu, Y. Wang and T. Abdiryim, RSC Adv., 2017, 7, 8625–8626 RSC.
- T. Yamamoto, T. Shimizu and E. Kurokawa, React. Funct. Polym., 2000, 43, 79–84 CrossRef CAS.
- J. J. Apperloo, R. A. J. Janssen, M. M. Nielsen and K. Bechgaard, Adv. Mater., 2000, 12, 1594–1597 CrossRef CAS.
- X. Yuan, W. Qin, X. Lei, L. Sun, Q. Li, D. Li, H. Xu and D. Xia, Chemosphere, 2018, 205, 369–379 CrossRef CAS.
- Y. Zhang, Q. Li, Y. Long, J. Zou, Z. Song, C. Liu, L. Liu, F. Qi, B. Xu and Z. Chen, Appl. Catal., B, 2019, 254, 569–579 CrossRef CAS.
- Z. Guo, Y. Yuan, W. Hui, L. Gang and Z. Wang, Electrochim. Acta, 2016, 220, 267–275 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02161b |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab