
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA supported manganese complex with amine-bis(phenol) ligand for catalytic benzylic C(sp3)–H bond oxidation†
Touraj Karimpour a,
Elham Safaei*b and
Babak Karimia
a,
Elham Safaei*b and
Babak Karimia
aDepartment of Chemistry, Institute for Advanced Studies in Basic Sciences (IASBS), P. O. Box 45137-66731, Gava Zang, Zanjan, Iran
bDepartment of Chemistry, College of Sciences, Shiraz University Shiraz, 71454, Iran. E-mail: e.safaei@shirazu.ac.ir
First published on 7th May 2019
Abstract
With regards to the importance of direct and selective activation of C–H bonds in oxidation processes, we develop a supported manganese amine bis(phenol) ligand complex as a novel catalyst with the aim of obtaining valuable products such as carboxylic acids and ketones that have an important role in life, industry and academic laboratories. We further analyzed and characterized the catalyst using the HRTEM, SEM, FTIR, TGA, VSM, XPS, XRD, AAS, and elemental analysis (CHN) techniques. Also, the catalytic evaluation of our system for direct oxidation of benzylic C–H bonds under solvent-free condition demonstrated that the heterogeneous form of our catalyst has high efficiency in comparison with homogeneous ones due to more stability of the supported complex. Furthermore, the structural and morphological stability of our efficient recyclable catalytic system has been investigated and all of the data proved that the complex was firmly anchored to the magnetite nanoparticles.
Introduction
From the environmental and economic points of view, the functionalization of the organic substrates is one of the best strategic approaches for the preparation of broad ranges of new chemicals as a novel and attractive way in organic synthesis.1 Because of the accessibility of carbon–hydrogen bonds in organic substrates, the activation of these bonds is an attractive and challenging method for selective synthesis of products with different functional groups. It is noteworthy that the methane monooxygenases (MMOs) and cytochrome P-450 are two important classes of enzymes that are applied by nature to attain high selectivity in the oxidation of alkanes at elevated rates.2 Two identified forms of the MMOs which contain iron or copper ions in their active sites are able to produce methanol from methane.3 The cytochrome P-450 uses its iron porphyrin core to catalyze the oxidation of hydrocarbons to alcohols through the carbon–hydrogen activation method with high turnover numbers.4 Several interesting homogenous catalytic systems involving transition-metals (Cu,5 Fe,6 Mn,7 Co,8 Ni,9 Pd,10 Rh,11 Ru12 and Ti13) have been reported for this important process. These systems have limited applications in comparison with the heterogeneous catalysis, due to product separation and catalyst recycling problems.In the past decade, immobilization of metal complexes on various supports has been introduced and investigated to overcome the problems pertaining to unsupported complexes. So this catalytic system has the privileges of both homogeneous and heterogeneous catalysts. From a sustainable point of view, the core–shell magnetic nanoparticles (MNPs) are quite favourable supports.14 These magnetized catalysts can be easily isolated by applying an appropriate magnetic field. This unique characteristic of MNPs eliminates the necessity of procedures such as centrifugation or filtration for the recycling of the catalysts. Despite of several interesting reports15 for activation of the inert C–H bonds, there are limited publications that address the issue of heterogeneous C–H oxidation catalysis using Earth-abundant iron16 and manganese catalysts.17
Due to the special importance of C–H bond oxidation in the pharmaceutical and chemical industries, and ideal properties of both manganese complexes and magnetic nanoparticles, our main aim is designing a novel, sustainable and selective heterogeneous system for benzylic C–H bond oxidation. Regarding the fact that amine bis(phenol) ligands consist of N and O atoms, and different substituents on the phenol groups, they are able to tune the Lewis acidity of the metal centres which plays a crucial role in the catalytic activity of metal complexes. Consequently, an amine bis(phenol) ligand was applied to design our catalyst. The immobilized amine bis(phenolate) ligand on the magnetite nanoparticles (Fe3O4@SiO2-APTES-H2LGDC) was prepared according to the previous report in our group.18 Manganese as an inexpensive, Earth-abundant and non-toxic metal was applied for metalation of these nanoparticles. It is interesting to note that we found the high cooperation between silica-coated Fe3O4 nanoparticles and manganese complex for the synthesis of a sustainable catalyst. In our designed system the stabilized manganese complex exhibits excellent efficiency in selective benzylic C–H bond oxidation for a wide range of substrates as well as toluene. Furthermore, five successful recoveries of the catalyst confirmed the stability of the manganese complex attached to the magnetic nanoparticles.
Results and discussion
Synthesis and characterization of Fe3O4@SiO2-APTES-MnLGDC
Using the presented route in Scheme 1, immobilized amine bis(phenol) ligand on the magnetite nanoparticles (Fe3O4@SiO2-APTES-H2LGDC) was prepared according to the previously report in our group,18 afterward for metalation of these nanoparticles MnCl2·4H2O was applied (Scheme 1).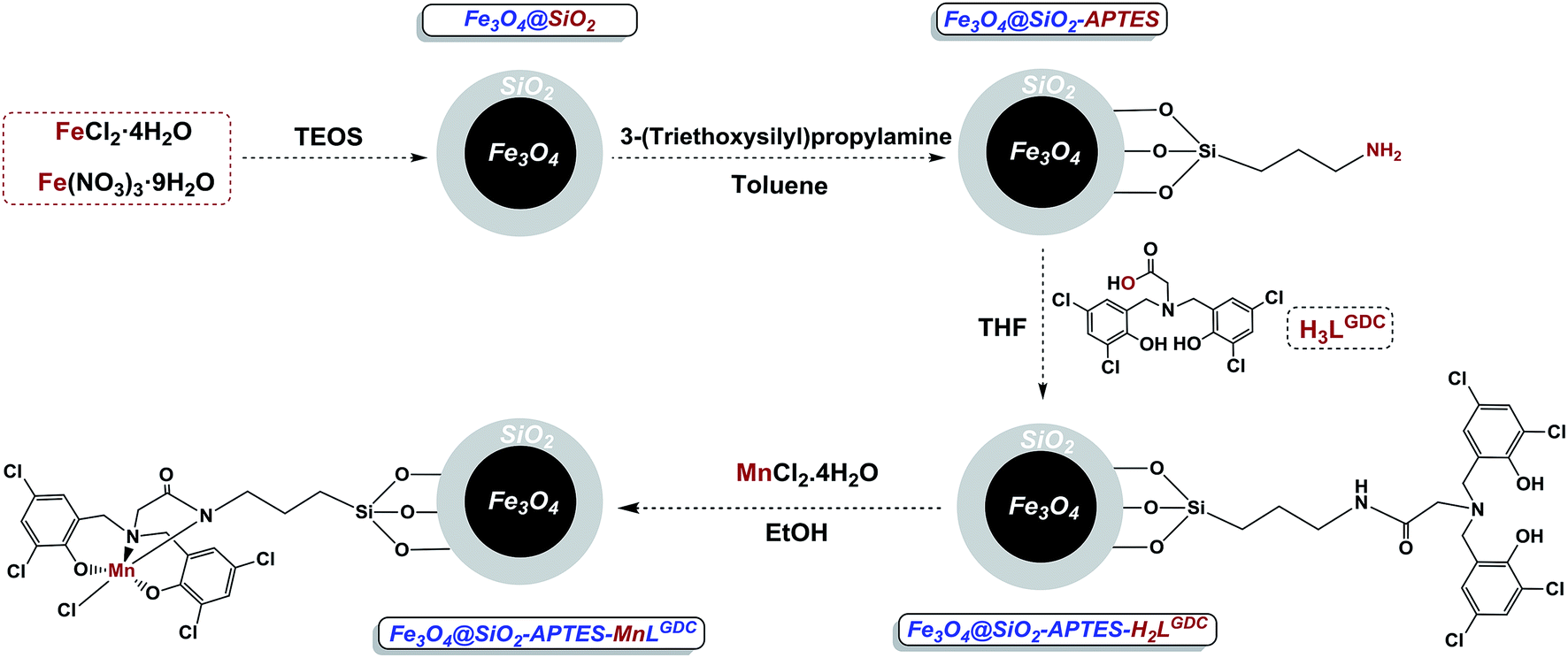 | ||
| Scheme 1 A schematic illustration of catalyst (Fe3O4@SiO2-APTES-MnLGDC). | ||
Both chemical nature and structure of our catalyst have been investigated via the common methods. The powder X-ray diffraction (XRD) analysis was performed in order to obtain information about the phase purity and crystalline structure of Fe3O4@SiO2, Fe3O4@SiO2-APTES, Fe3O4@SiO2-APTES-H2LGDC and Fe3O4@SiO2-APTES-MnLGDC (Fig. 1A). Our results are in line with previous reports of Fe3O4@SiO2 nanoparticles XRD pattern.19 XRD pattern for all nanoparticles show the cubic inverse spinel of Fe3O4 nanoparticles and a broad diffraction peak in the 2θ = 20–30°, demonstrating the presence of silica shell. Diffraction peaks of [111], [220], [311], [400], [422], [511], and [440] planes found in 18, 30, 35, 43, 54, 57 and 63 degree, respectively.
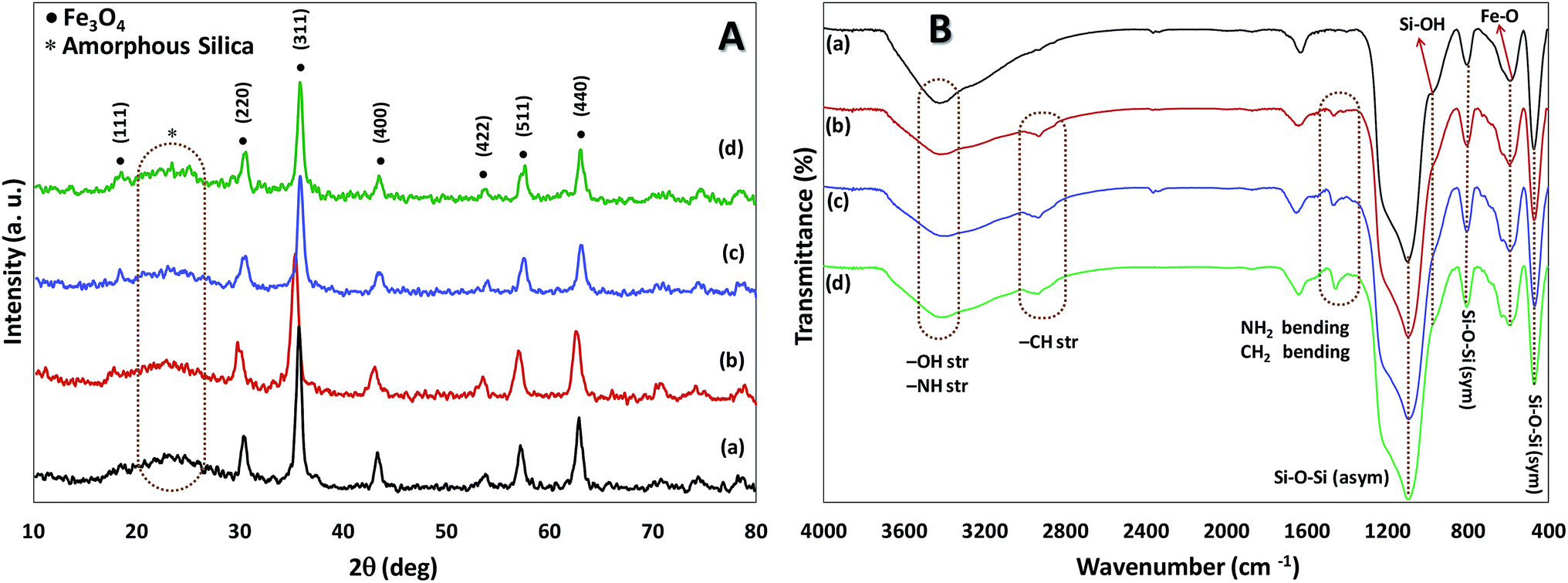 | ||
| Fig. 1 (A) The XRD pattern, (B) the FTIR spectra of Fe3O4@SiO2 (a), Fe3O4@SiO2-APTES (b), Fe3O4@SiO2-APTES-H2LGDC (c) and Fe3O4@SiO2-APTES-MnLGDC (d). | ||
FT-IR analysis was used to investigate the successful functionalization and modification of particles. As can be seen in Fig. 1B, broad peak at 3423 cm−1 is related to the O–H and N–H vibrations and the presence of stretching vibration of Fe–O in all samples was found (at around 544 cm−1). Moreover, symmetric stretching (at about 400 and 800 cm−1, respectively), asymmetric stretching of Si–O–Si (at about 1200 cm−1) and stretching vibrations of Si–OH (at around 900 cm−1) are evidence to support the existence of silica shell in all magnetic nanoparticles.20 Bending peak of –CH2 at 1400 cm−1 and stretching vibrations peaks related to –CH2 and N–H groups at about 2840–3000 cm−1 and 3400 cm−1, respectively, confirmed stabilization of amino propyl groups on the particles.
The HRTEM and FESEM analyses played a crucial role in the investigation of size, structure, and morphology of nanomagnetic particles. Our results show that nanoparticles are smaller than 45 nm and core size is about 15 nm that surrounded via 15 nm uniform grey silica shell. HRTEM images of obtained catalyst (Fig. 2A and B) confirmed the stability of the core–shell system in Fe3O4@SiO2 nanoparticles after modification. Moreover, FESEM analysis indicated our mentioned catalyst has a spherical uniform morphology and smooth surface (Fig. 2C and D).
 | ||
| Fig. 2 HRTEM (A and B) and FESEM (C and D) images of Fe3O4@SiO2-APTES-MnLGDC. | ||
We also explored the magnetization of magnetic nanoparticles by vibrating sample magnetometer (VSM) at room temperature (Fig. 3A). The superparamagnetic behavior for our catalyst was confirmed with the field-dependent magnetization curves without any coercivity field (Hc) and remanent magnetism (Mr). The resulted data in Fig. 3A shows the high saturation magnetization (Ms) of about 22 emu g−1 for the catalyst that enables it to be easily separated via an external magnet from the reaction mixture.21 According to TGA curve of catalyst initial weight at 0–200 °C can be related to trapped solvent on the surface of the catalyst (Fig. S3B†). As can be seen in Fig. 3B, main weight loss for the catalyst (Fe3O4@SiO2-APTES-MnLGDC) is observed at high temperature of about 300–600 °C which proved thermal stability of the final catalyst. Thermal behaviour of Fe3O4@SiO2-APTES-H2LGDC and Fe3O4@SiO2-APTES-MnLGDC samples under N2 atmosphere showed same decomposition pattern indicating that under metalation with Mn the total structure was preserved. A noticeable point in this matter is that after metalation with Mn the temperature range of main weight loss has been shifted to the lower temperatures (compare diagrams c and d in Fig. 3B). This issue can be attributed to the catalytic role of Mn on the decomposition process of organic moieties in the circumstance of TG experiment. Furthermore, the loading of aminopropyl in Fe3O4@SiO2-APTES and ligand in Fe3O4@SiO2-APTES-H2LGDC achieved about 1.43 and 0.5 mmol g−1, respectively. Further tests such as CHN analysis corroborated these results (1.35 mmol g−1 of APTES in Fe3O4@SiO2-APTES and 0.56 mmol g−1 of H3LGDC in Fe3O4@SiO2-APTES-H2LGDC). In an attempt to do the estimation of manganese loading, we used the AAS technique (was found to be 0.49 mmol g−1).
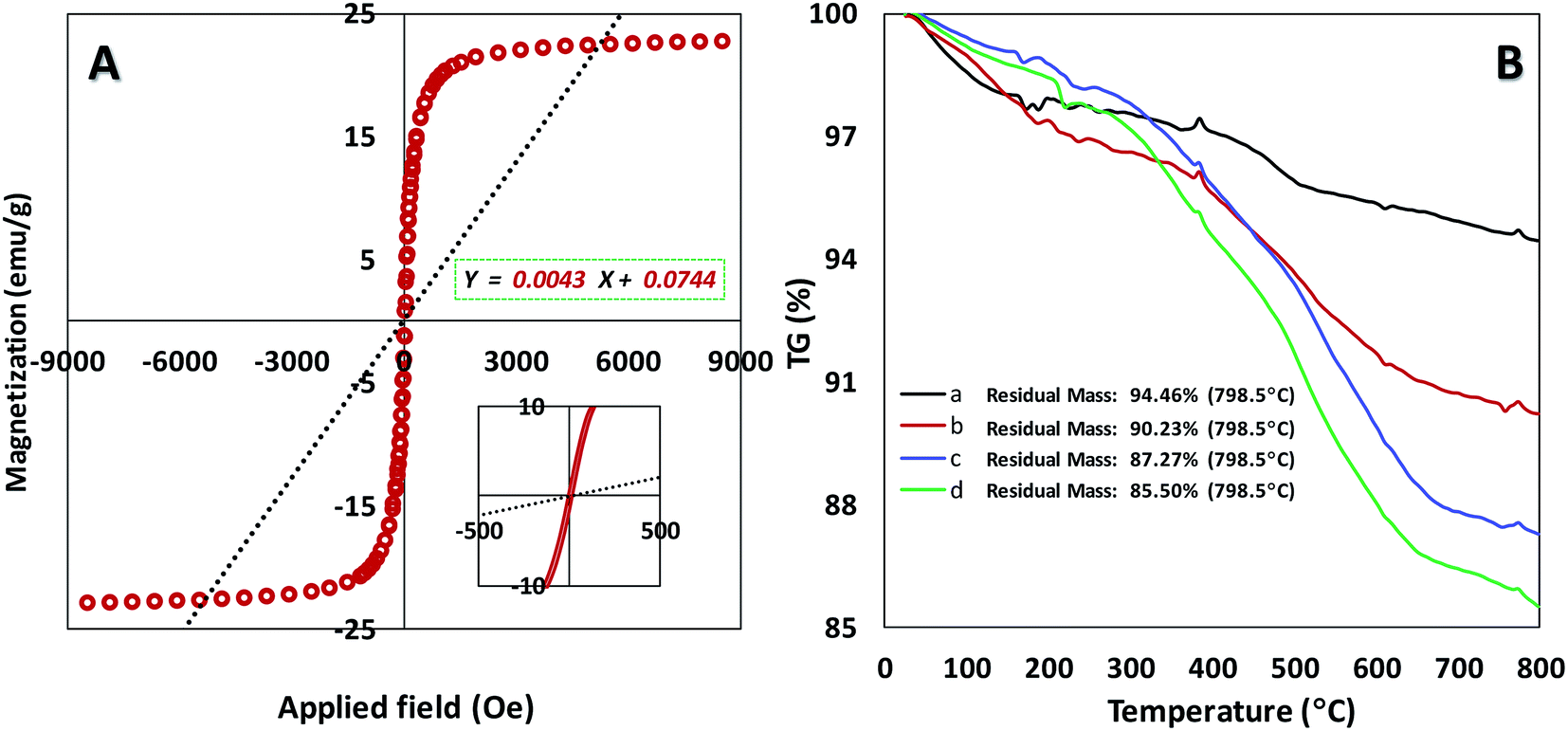 | ||
| Fig. 3 (A) Magnetization curves of Fe3O4@SiO2-APTES-MnLGDC. Inset: the enlarged image near the coercive field. (B) Thermo-gravimetric analysis (TGA) of Fe3O4@SiO2 (a), Fe3O4@SiO2-APTES (b), Fe3O4@SiO2-APTES-H2LGDC (c), Fe3O4@SiO2-APTES-MnLGDC (d). | ||
Chemical state and composition of Mn, Si, C, Cl and O of the mentioned synthesis catalyst were investigated via X-ray Photoelectron Spectroscopy (XPS), as well as Fe in the core (Fe3O4) (Fig. 4). Achieved data suggest Mn has +3 oxidation state and at 641, and 653 eV we can see lines regarding Mn 2p3/2 and Mn 2p1/2, respectively. The presence of iron(III) and (II) in the catalyst was confirmed according to the peaks at 55 (Fe 3p), 710 (Fe 2p3/2), and 724 eV (Fe 2p1/2) in Fig. 4D. In addition, appeared peak at 200 eV (Cl 2p) truly proves successful attachment of mentioned ligand (H3LGDC) on the magnetic nanoparticles (Fig. 4B). As we can see in Fig. 4, peaks related to C 1s, O 1s, Si 2s, Si 2p, and N 1s prove the presence of these elements in our catalyst.22
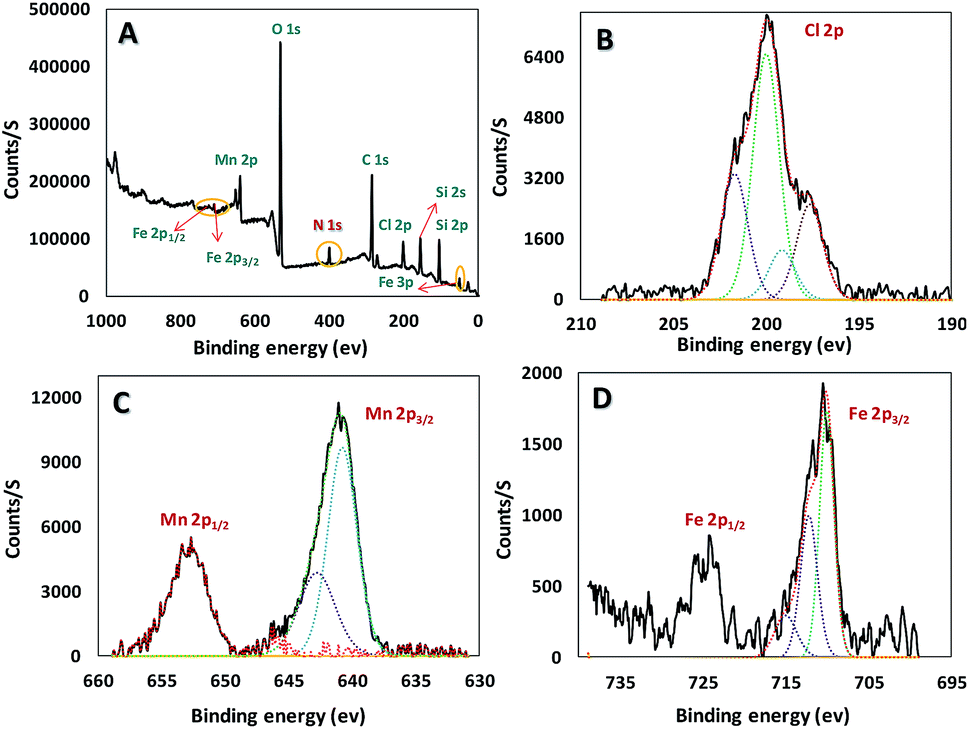 | ||
| Fig. 4 The X-ray photoelectron spectroscopy (XPS) spectrum of catalyst (Fe3O4@SiO2-APTES-MnLGDC). | ||
Catalytic activity evaluation
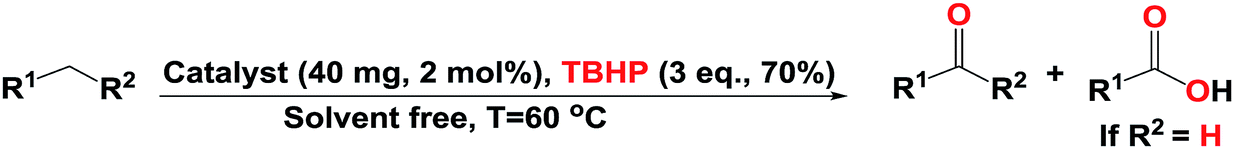
On the basis of the main goal of our studies, we have investigated the efficiency of our designed system for activation of a broad range of substrates with different ability to activation. In an attempt to achieve an optimized condition, we selected oxidation of ethylbenzene to corresponding products as a model reaction (The results are summarized in Table 1, and Fig. S3–S27†). We began our investigation into the effect of solvents such as dioxan, EtOH, THF, and CH3CN during 5 h; as we can see in Table 1 (entries 1–4) CH3CN acts as a suitable solvent and the activity of the mentioned catalyst was increased in the smaller amount of CH3CN (Table 1, entries 4–7). Given the high activity of the catalyst in the absence of solvent, so we evaluated the eligibility of our proposed system in the solvent free condition (Table 1, entry 8). The model reaction in the presence of H2O2 cannot progress (Table 1, entry 15); therefore other green oxidants such as O2 and TBHP were selected.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | TBHP (eq.) | Solvent (mL) | Time (h) | Conv.o (%) | Selectivityp (%) | TONq | ||
| A | B | C | ||||||
| a TBHP (2 eq., 70%), O2 balloon.b T = RT.c T = 70 °C.d Cat. (20, 1 mol%).e H2O2 (3 eq., 30%).f Fe3O4, 40 mg.g Fe3O4@SiO2, 40 mg.h Fe3O4@SiO2-APTES, 40 mg.i Fe3O4@SiO2-APTES-H2LGDC, 40 mg.j MnLGDC (10 mg, 2 mol%).k MnCl2·4H2O (4 mg, 2 mol%).l In the absence of catalyst.m O2 balloon, in the absence of TBHP.n Solvent free.o Conversions were determined by GC (anisole as internal standard (1 mmol, 1/1, substrate/anisole)).p Selectivity to product = [product%/(products%)] × 100.q TON = (substrate/catalyst) × conversion. | ||||||||
| 1 | 16 | 87 | 13 | 16 | ||||
| 2 | 3 | THF, 3 | 5 | Trace | ||||
| 3 | 3 | EtOH, 3 | 5 | Trace | ||||
| 4 | 3 | CH3CN, 3 | 5 | 39 | 97 | 3 | 39 | |
| 5 | 3 | CH3CN, 2 | 5 | 41 | 95 | 5 | 41 | |
| 6 | 3 | CH3CN, 1 | 5 | 47 | 96 | 4 | 47 | |
| 7 | 3 | CH3CN, 0.5 | 5 | 55 | 96 | 4 | 55 | |
| 8 | 3 | —n | 5 | 62 | 3 | 95 | 2 | 61 |
| 9 | 2 | — | 5 | 49 | 94 | 6 | 49 | |
| 10a | 2 | — | 5 | 71 | 2 | 97 | 1 | 70 |
| 11 | 4 | — | 5 | 65 | 2 | 97 | 1 | 65 |
| 12b | 3 | — | 5 | 13 | 92 | 8 | 13 | |
| 13c | 3 | — | 5 | 65 | 97 | 3 | 65 | |
| 14d | 3 | — | 5 | 50 | 2 | 92 | 6 | 49 |
| 15e | — | — | 5 | Trace | ||||
| 16f | 3 | — | 10 | Trace | ||||
| 17g | 3 | — | 10 | Trace | ||||
| 18h | 3 | — | 10 | Trace | ||||
| 19i | 3 | — | 10 | Trace | ||||
| 20j | 3 | — | 10 | 19 | 89 | 11 | 19 | |
| 21k | 3 | — | 10 | 30 | 90 | 10 | 30 | |
| 22l | 4 | — | 10 | Trace | ||||
| 23l | 4 | CH3CN, 0.5 | 10 | Trace | ||||
| 24m | — | CH3CN, 0.5 | 10 | Trace | ||||
| 25 | 2 | — | 10 | 70 | 99 | 1 | 70 | |
| 26 | 3 | — | 10 | 96 | 98 | 2 | 96 | |
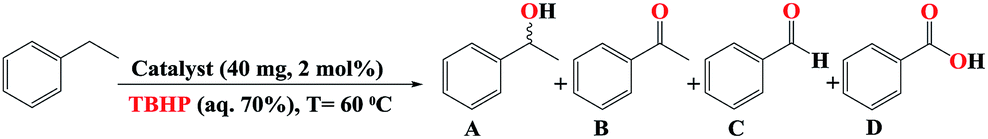
According to the Table 1 (entries 8–11), our catalyst has the best activity in the presence of 3 eq. TBHP. In order to find the optimized temperature at room temperature, and 70 °C about 13 and 65% conversion was achieved, respectively. Hence, 60 °C was selected as optimized temperature (Table 1 entries 12 and 13). By reducing the amount of catalyst to 1 mol% (20 mg) catalytic activity was decreased (Table 1, entry 14) so 2 mol% catalyst chosen as the optimum amount of catalyst. The experiment was continued by investigation the effect of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2-APTES, and Fe3O4@SiO2-APTES-H3LGDC (Table 1, entries 16–19). Slightly progress in the model reaction in the presence of them proves that these particles do not have any catalytic activity in this reaction. Moreover, by synthesizing the homogeneous catalyst (MnLGDC), we explored the catalytic activity of this form of catalyst and just about 19% conversion was achieved (Table 1 entry 20). As shown in Fig. S41,† the absorption spectra for the fresh reaction mixture in the presence of MnLGDC after an hour has been changed, this can be duo to the decomposition of homogeneous form of the catalyst (MnLGDC) during the oxidation process. This result clearly confirms the positive effect of nanoparticles on the stability of manganese complex and proves our complex has a good structural stability on the magnetic nanoparticles in the heterogeneous form of our catalyst. It is important to note that, the mentioned reaction in the presence of manganese(II) chloride salt as a catalyst has not a good conversion (Table 1, entry 21). Consequently, the last results strongly proved the dramatically influence of magnetic nanoparticles supported ligand on the activity of catalyst system. It is more notable, in the absence of the catalyst but in the presence of 4 eq. of TBHP, slightly progress in the modal reaction truly confirms the eligibility of our system (Table 1, entries 22 and 23).
In the process of investigation, the high efficiency of mentioned catalyst in the aerobic oxidation of the ethylbenzene in the presence of air and O2 was checked. In the presence of 2 eq. of TBHP and O2 our catalyst showed same activity similar to 3 mmol of TBHP without O2 (Table 1 entries 8 and 10) but at 60 °C and in the absence of TBHP trace conversion was achieved (Table 1, entry 24). Our results show that this oxidant (O2) can act at a higher temperature and longer time. This point of view is supported by good resulted data in Scheme 2 (Scheme 2b and c, Fig. S29 and 30†). These data confirm the eligibility of our catalyst not only by TBHP (Scheme 2a, Fig. S28†) but also by O2.
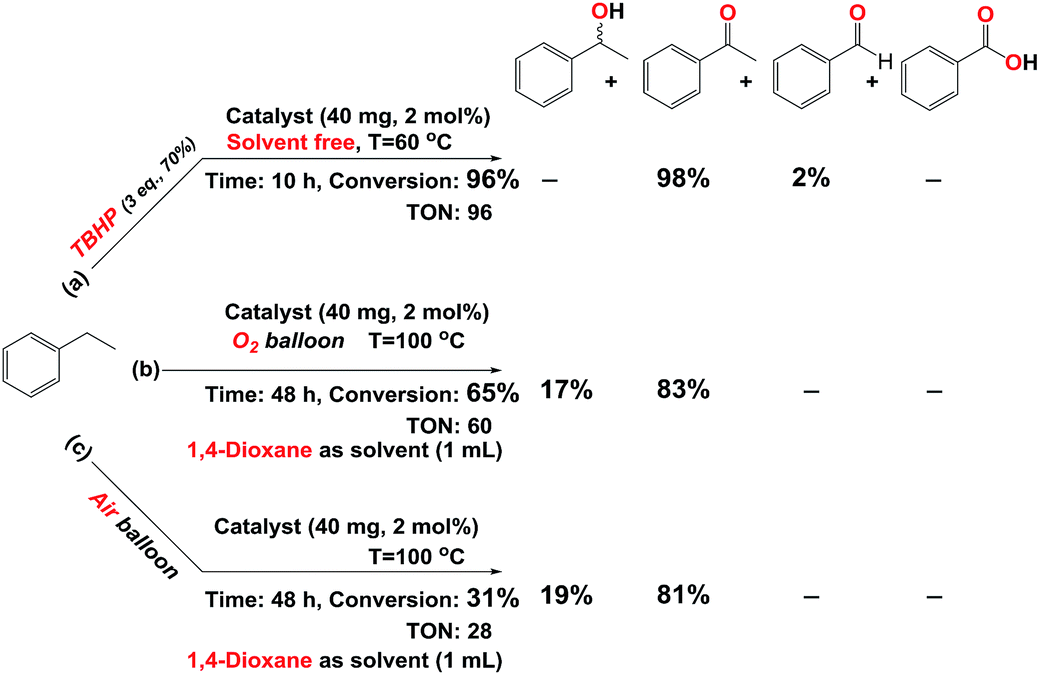 | ||
| Scheme 2 Oxidation of ethylbenzene catalyzed by catalyst (Fe3O4@SiO2-APTES-MnLGDC) in the presence of (a) TBHP (aq. 70%). (b) O2. (c) Air. | ||
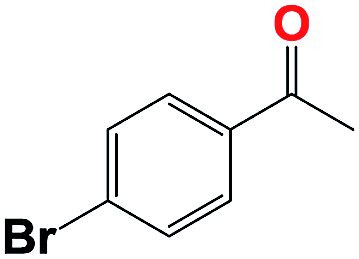
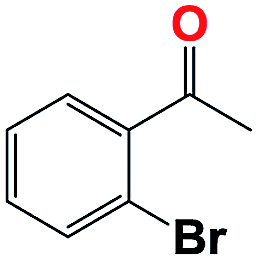
Into confirming the efficiency of the proposed system as an ideal candidate for C–H bond activation, further investigations were continued for a broad range of substrates (Table 2, Fig. S31–S39† and 1H NMR spectrum of products Fig. S42–S49†). 1-Ethyl-4-methoxybenzene (Table 2 entry 1) and 1-ethyl-4-bromobenzene (Table 2, entry 2) were chosen to peruse the effect of electron donating (MeO−) and electron withdrawing (Br−) groups in comparison with ethylbenzene. Based on Table 2, the substrate with the electron donating group, converted to products easier than substrates contain electron withdrawing groups. The harder reaction of C–H bond in 1-ethyl-2-bromobenzene (Table 2 entry 3) in comparison with 1-ethyl-4-bromobenzene, causes by the steric effect of bromo group. For bulky molecule such as diphenylmethane, our catalyst showed high activity and selectivity (Table 2 entry 4). It should be noted that high selectivity of the catalyst in a catalytic reaction is as important as high activity. This point of view was confirmed in the oxidation of 1,2,3,4-tetrahydronaphthalene to α-tetralone with high conversion and selectivity in 10 h in the presence of 2 eq. of TBHP (Table 2, entry 5). Furthermore, the designed catalyst showed a good efficiency for oxidation of 1-indanone without any C–C bond cleavage (Table 2 entry 6).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Major product | Time (h) | Conv.c (%) | Selectivityd (%) | TONe | |
| Major product | Other product | ||||||
| a TBHP (2 eq., 70%).b TBHP (4 eq., 70%).c Conversions were determined by GC.d Selectivity to product = [product%/(products%)] × 100.e TON = (substrate/catalyst) × conversion. | |||||||
| 1 |  |
 |
91 | 9 | 92 | ||
| 2 | 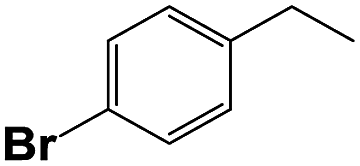 |
 |
10 | 96 | 98 | 2 | 95 |
| 3 | 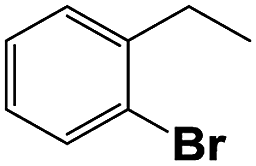 |
 |
10 | 64 | 48 | 52 | 47 |
| 4 | 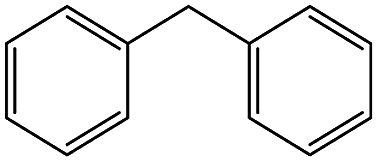 |
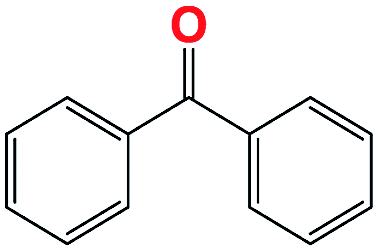 |
10 | 93 | 100 | — | 93 |
| 5a | 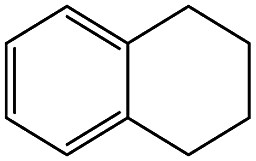 |
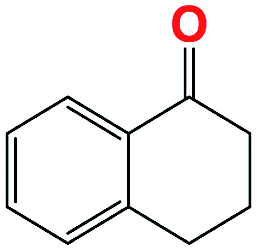 |
10 | 91 | 91 | 9 | 89 |
| 6 | 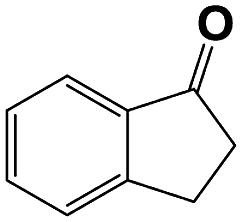 |
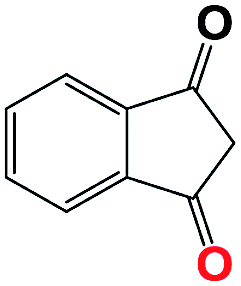 |
10 | 80 | 100 | — | 80 |
| 7b | 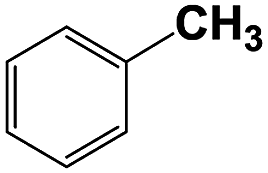 |
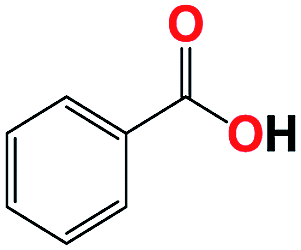 |
10 | 57 | 100 | — | 86 |
| 8b | 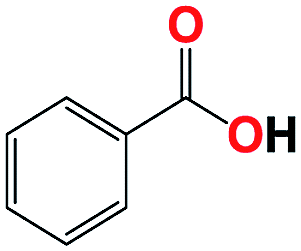 |
20 | 71 | 100 | — | 107 | |
| 9b | 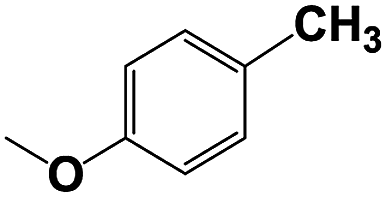 |
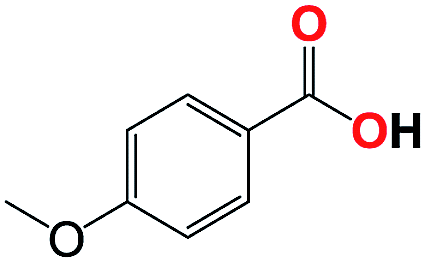 |
20 | 87 | 92 | 8 | 131 |
With respect to the industrial application of caprolactam, so direct oxidation of toluene to benzoic acid as one of the important steps for synthesis of caprolactam is so remarkable.23 Therefore we investigated the competency of our introduced system for direct oxidation of toluene to benzoic acid as interesting transformation. As we can see in Table 2 (entries 7 and 8) our catalyst demonstrated high activity in mentioned reaction and 71% benzoic acid was achieved with 100% selectivity at 20 h. Moreover, to further investigation direct oxidation of 4-methylanisole was performed (Table 2 entry 9) and 83% 4-methoxybenzoic acid as the main product was obtained and this percentage of conversion proves the positive effect of electron donating group (MeO−).
Monitoring, hot filtration test and recovery of the catalyst
As we can see in Fig. 5A the oxidation reaction of ethylbenzene was monitored as a function of time and 97% of conversion was achieved after 10 h. Moreover, after removal of the catalyst at the half of the completion reaction time (5 h) less than 10% progress in conversion was observed. Also, the amount leaching of manganese into the final aqueous phase was checked and negligible leaching based on the ICP-AES analysis (less than the detection limit) was detected. These results truly confirmed the heterogeneous nature of our catalytic system.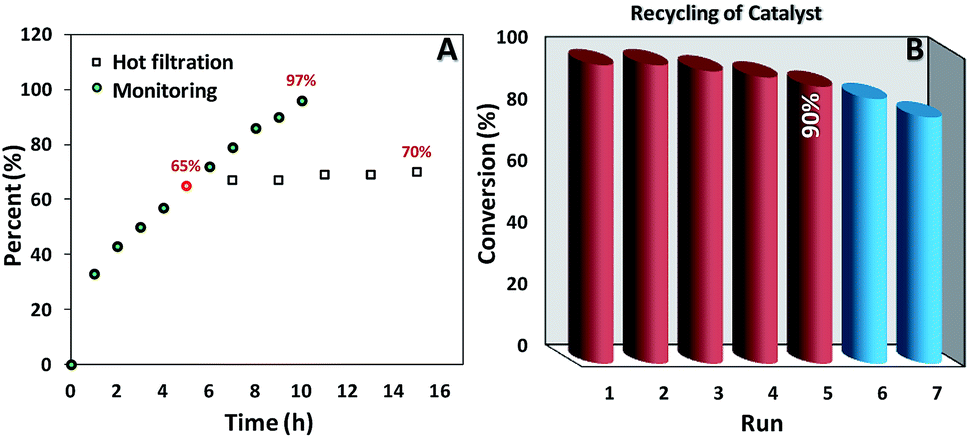 | ||
| Fig. 5 (A) Monitoring and hot filtration test, (B) recycling for oxidation of ethylbenzene. | ||
It is noteworthy that simple separation and good recovery of the catalyst is one of the important branches of green chemistry, accordingly, to further validation of our catalytic system the feasibility of catalyst recovery was investigated for oxidation of ethylbenzene under optimal condition (Fig. 5B). Five successive recycling of catalyst is a good validation on the durability of our catalyst. In addition, XRD patterns (Fig. 1A and S1B†), FESEM image (Fig. 2C and D and S1A†), VSM data (Fig. 3A and S2A†) as well as TGA analysis (Fig. 3B and S2B†) of pristine and recovered catalysts clearly show the high chemical and physical stability of our catalysts. The Mn content of the recovered catalyst was determined by AAS technique. The result indicated a small leaching in the Mn content at the end of recovery process (0.49 mmol g−1 for fresh catalyst vs. 0.47 mmol g−1 for recovered catalyst). It may be concluded that the negligible decreasing of catalytic activity after the 5th run could be attributed to this minimal leaching of Mn species and/or small losing of catalyst mass during the recovery process.
Proposed mechanism
According to the previous studies, the oxidation mechanism of C–H bond in the presence of a non-heme manganese complex involves a high-valent manganese-oxo species.Therefore, we think that our system containing a non-heme manganese complex can produce metal-oxo species which can generate short-lived alkyl radicals in this reaction (Scheme 3, cycle A). As can be seen in following Scheme (cycle B), the formed alcohol converts to the aldehyde or ketone form. Also, further oxidation of prepared benzaldehydes by using of oxidant (TBHP) or H2O can be done (Scheme 3, cycle C).24
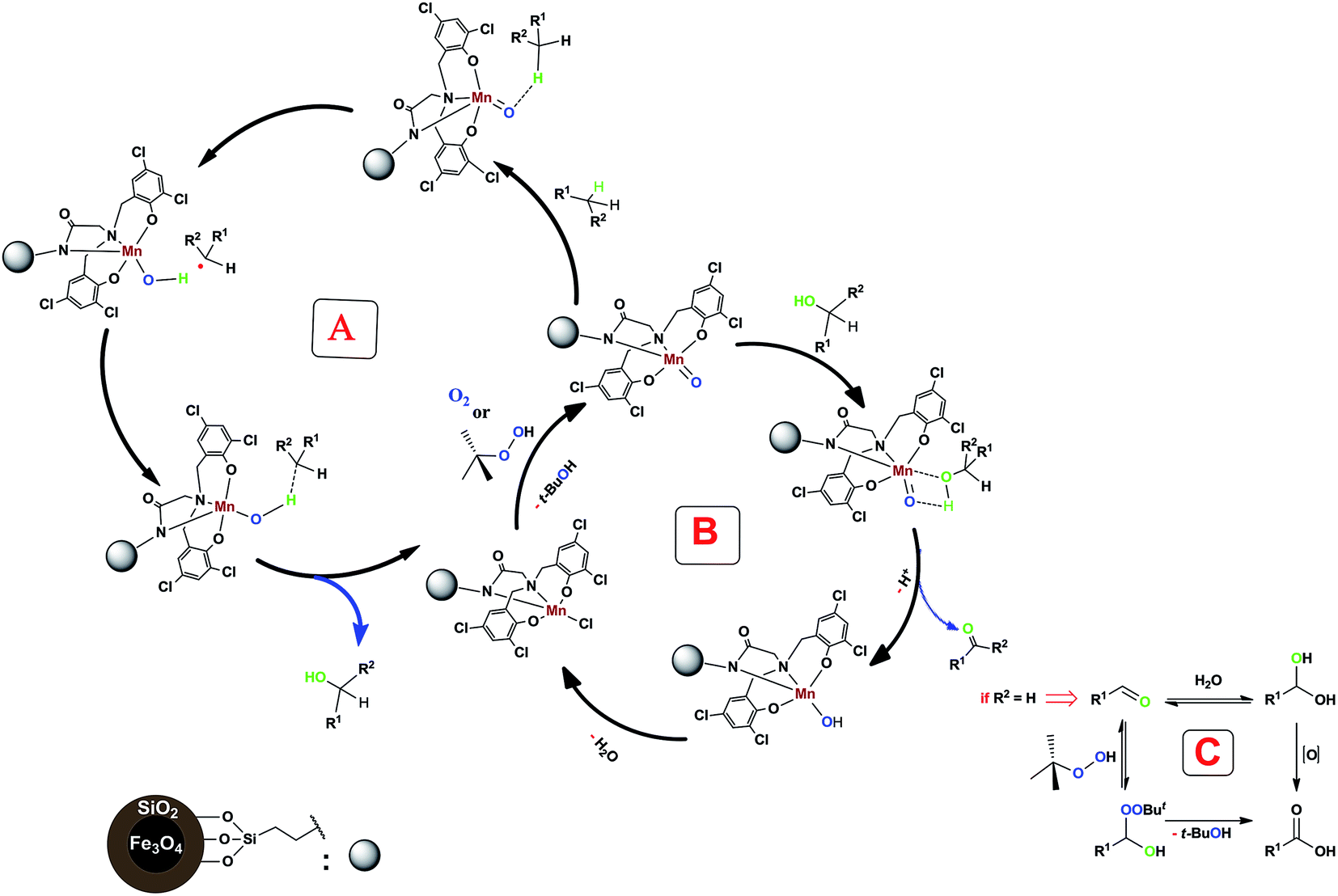 | ||
| Scheme 3 Proposed mechanism for our non-heme manganese complex-catalyzed benzylic C–H bond oxidation reaction. | ||
Conclusions
In conclusion, our reported system consisting of three parts, Mn(III) ion, amine bis(phenol) ligand (H3LGDC) and magnetic nanoparticles. These components showed a good cooperation effect acting as an efficient, recyclable and environmentally friendly catalyst under solvent-free and mild conditions in benzylic C–H bond oxidation reaction for a variety of substrates as well as toluene. The chemical nature and structural stability of the catalyst were confirmed by various techniques. Furthermore, both the leaching experiment and five successive recycling of the catalyst, proved the strong attachment of the manganese complex onto the magnetite nanoparticles.Experimental section
Materials and methods
All reagents and solvents were purchased from commercial sources and used without further purification. Fourier transform infrared spectroscopy on KBr pellets of the compounds was recorded on a Bruker Vector 22 in the range of 400 and 4000 cm−1. UV-vis absorbance digitized spectra were collected using a CARY 100 spectrophotometer. The morphological features of particles were characterized by using field emission transmission electron microscopy (JEOL, JEM-2100F, 200 kV TEM) and Field emission scanning electron microscopy (JEOL, JSM-7610F). Elemental analyses (C, H and N) were performed by the Elementar, Vario EL III. The X-ray powder patterns were recorded using a PHILIPS PW1730 (step size: 0.05, time per step: 1 s). The content of manganese in the catalyst was determined using an Atomic Absorption Spectrometer Varian Spectre AA 110. The organic composition of the modified MNPs base materials was determined by thermogravimetric analysis (TGA) and differential thermoanalysis (DTG), heating from room temperature to 800 °C under nitrogen flow using a STA 409 PC/PG analyser (Netzsch). The magnetic properties of the prepared materials were measured using a homemade vibrating sample magnetometer (Meghnatis Daghigh Kavir Company, Iran) at room temperature from −10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to +10000 Oe. The electronic states of powders were evaluated using X-ray photoelectron spectroscopy (was performed using a Thermo Scientific, ESCALAB 250Xi with Mg X-ray resource). The content of Mn in final aqueous phase was determined by applying an inductively coupled plasma-optical emission spectrometer (ICP-OES). Furthermore, the products were determined and analysed using a VARIAN CP-3800 gas chromatograph equipped with a capillary column and a flame-ionization detector.
000 to +10000 Oe. The electronic states of powders were evaluated using X-ray photoelectron spectroscopy (was performed using a Thermo Scientific, ESCALAB 250Xi with Mg X-ray resource). The content of Mn in final aqueous phase was determined by applying an inductively coupled plasma-optical emission spectrometer (ICP-OES). Furthermore, the products were determined and analysed using a VARIAN CP-3800 gas chromatograph equipped with a capillary column and a flame-ionization detector.
Synthesis of Mn(III) complex of Fe3O4@SiO2-APTES-H2LGDC (Fe3O4@SiO2-APTES-MnLGDC)
For synthesis of our catalyst, triethylamine (2 mmol, 0.20 g) was added to a stirred mixture of Fe3O4@SiO2-APTES-H2LGDC (1.00 g) and MnCl2·4H2O (1 mmol, 0.20 g) in ethanol (80 mL) under continuous stirring. The mixture reaction was stirred at room temperature for 4 days. Final product was separated from the reaction mixture by applying an external magnetic field and washed with ethanol and acetone. The resulting solid was dried at 80 °C overnight and used for high selective oxidation of benzylic C–H bonds. The resulting material was denoted as Fe3O4@SiO2-APTES-MnLGDC.General procedure in the C–H bond oxidation
Under atmosphere of argon in a 5 mL glass flask were placed catalyst (0.04 g, 2 mol%) and substrate (1 mmol) in solvent free condition, TBHP as oxidant was added. The reaction mixture was continuously stirred at 60 °C for the desired time and the reaction was monitored by TLC, after completion of the reaction, anisole (1 mmol, 1/1 substrate and anisole) as internal standard was added. In continue the mixture was extracted with ethyl acetate and then the catalyst was magnetically recovered by placing a permanent magnet in the reactor wall. Products were collected with a syringe and analyzed by gas chromatography (GC). The catalyst was washed several times with ethanol and acetone and then dried at 80 °C overnight before being used again for the next reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Institute for Advanced Studies in Basic Sciences (IASBS), the chemistry department of Shiraz University and Bahman Farnoudi for proofreading of the manuscript.Notes and references
- (a) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed; (b) K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS PubMed.
- (a) M. Chen, Y. Pan, H.-K. Kwong, R. J. Zeng, K.-C. Lau and T.-C. Lau, Chem. Commun., 2015, 51, 13686–13689 RSC; (b) R. Banerjee, Y. Proshlyakov, J. D. Lipscomb and D. A. Proshlyakov, Nature, 2015, 518, 431–434 CrossRef CAS PubMed; (c) R. Balasubramanian, S. M. Smith, S. Rawat, L. A. Yatsunyk, T. L. Stemmler and A. C. Rosenzweig, Nature, 2010, 465, 115–119 CrossRef CAS PubMed; (d) G. Xue, D. Wang, R. De Hont, A. T. Fiedler, X. Shan, E. Münck and L. Que, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20713–20718 CrossRef CAS PubMed; (e) C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 CrossRef CAS PubMed; (f) D. Wang, E. R. Farquhar, A. Stubna, E. Münck and L. Que, Nat. Chem., 2009, 1, 145–150 CrossRef CAS PubMed.
- V. C. C. Wang, S. Maji, P. P. Y. Chen, H. K. Lee, S. S. F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574–8621 CrossRef CAS.
- (a) J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS PubMed; (b) B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed.
- (a) J.-M. Li, Y.-H. Wang, Y. Yu, R.-B. Wu, J. Weng and G. Lu, ACS Catal., 2017, 7, 2661–2667 CrossRef CAS; (b) S. Tang, P. Wang, H. Li and A. Lei, Nat. Commun., 2016, 7, 11676 CrossRef PubMed.
- (a) S. Rana, A. Dey and D. Maiti, Chem. Commun., 2015, 51, 14469–14472 RSC; (b) J. Serrano-Plana, W. N. Oloo, L. Acosta-Rueda, K. K. Meier, B. Verdejo, E. García-España, M. G. Basallote, E. Münck, L. Que, A. Company and M. Costas, J. Am. Chem. Soc., 2015, 137, 15833–15842 CrossRef CAS PubMed.
- (a) A. Conde, G. Sabenya, M. Rodríguez, V. Postils, J. M. Luis, M. M. Díaz-Requejo, M. Costas and P. J. Pérez, Angew. Chem., Int. Ed., 2016, 55, 6530–6534 CrossRef CAS PubMed; (b) L. Carroll, H. L. Evans, A. C. Spivey and E. O. Aboagye, Chem. Commun., 2015, 51, 8439–8441 RSC.
- (a) H. Ren, Y.-P. Zhou, Y. Bai, C. Cui and M. Driess, Chem.–Eur. J., 2017, 23, 5663–5667 CrossRef CAS PubMed; (b) F. Yang, J. Yu, Y. Liu and J. Zhu, Org. Lett., 2017, 19, 2885–2888 CrossRef CAS PubMed.
- (a) Z. Ruan, D. Ghorai, G. Zanoni and L. Ackermann, Chem. Commun., 2017, 53, 9113–9116 RSC; (b) Y. He, Y. Cai and S. Zhu, J. Am. Chem. Soc., 2017, 139, 1061–1064 CrossRef CAS PubMed.
- (a) M. D. K. Boele, G. P. F. van Strijdonck, A. H. M. de Vries, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2002, 124, 1586–1587 CrossRef CAS PubMed; (b) R. Shi, H. Niu, L. Lu and A. Lei, Chem. Commun., 2017, 53, 1908–1911 RSC; (c) J. L. Nallasivam and R. A. Fernandes, J. Am. Chem. Soc., 2016, 138, 13238–13245 CrossRef CAS PubMed.
- C.-Q. Wang, L. Ye, C. Feng and T.-P. Loh, J. Am. Chem. Soc., 2017, 139, 1762–1765 CrossRef CAS PubMed.
- X. Wu, B. Wang, S. Zhou, Y. Zhou and H. Liu, ACS Catal., 2017, 7, 2494–2499 CrossRef CAS.
- B. C. Bailey, H. Fan, J. C. Huffman, M.-H. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2007, 129, 8781–8793 CrossRef CAS PubMed.
- (a) A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed; (b) S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed.
- (a) S. Vásquez-Céspedes, K. M. Chepiga, N. Möller, A. H. Schäfer and F. Glorius, ACS Catal., 2016, 6, 5954–5961 CrossRef; (b) M. Kaur, S. Pramanik, M. Kumar and V. Bhalla, ACS Catal., 2017, 7, 2007–2021 CrossRef CAS; (c) S. Warratz, D. J. Burns, C. Zhu, K. Korvorapun, T. Rogge, J. Scholz, C. Jooss, D. Gelman and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 1557–1560 CrossRef CAS PubMed; (d) Y. Gao, P. Tang, H. Zhou, W. Zhang, H. Yang, N. Yan, G. Hu, D. Mei, J. Wang and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 3124–3128 CrossRef CAS PubMed; (e) T. V. Tran, H. T. N. Le, H. Q. Ha, X. N. T. Duong, L. H. T. Nguyen, T. L. H. Doan, H. L. Nguyen and T. Truong, Catal. Sci. Technol., 2017, 7, 3453–3458 RSC; (f) X. Tian, F. Yang, D. Rasina, M. Bauer, S. Warratz, F. Ferlin, L. Vaccaro and L. Ackermann, Chem. Commun., 2016, 52, 9777–9780 RSC; (g) S. Korwar, M. Burkholder, S. E. Gilliland, K. Brinkley, B. F. Gupton and K. C. Ellis, Chem. Commun., 2017, 53, 7022–7025 RSC; (h) B. Dong, Z. Han, Y. Zhang, Y. Yu, A. Kong and Y. Shan, Chem.–Eur. J., 2016, 22, 2046–2050 CrossRef CAS PubMed; (i) V. Pascanu, F. Carson, M. V. Solano, J. Su, X. Zou, M. J. Johansson and B. Martín-Matute, Chem.–Eur. J., 2016, 22, 3729–3737 CrossRef PubMed; (j) S. Zhang, H. Wang, M. Li, J. Han, X. Liu and J. Gong, Chem. Sci., 2017, 8, 4489–4496 RSC; (k) X. Wang, M. Liu, Y. Wang, H. Fan, J. Wu, C. Huang and H. Hou, Inorg. Chem., 2017, 56, 13329–13336 CrossRef CAS PubMed; (l) D.-T. D. Tang, K. D. Collins and F. Glorius, J. Am. Chem. Soc., 2013, 135, 7450–7453 CrossRef CAS PubMed; (m) H. Fei and S. M. Cohen, J. Am. Chem. Soc., 2015, 137, 2191–2194 CrossRef CAS PubMed.
- (a) C. Rajendran and G. Satishkumar, ChemCatChem, 2017, 9, 1284–1291 CrossRef CAS; (b) S. Samanta and R. Srivastava, Appl. Catal., B, 2017, 218, 621–636 CrossRef CAS.
- (a) Y. Kuwahara, Y. Yoshimura and H. Yamashita, Catal. Sci. Technol., 2016, 6, 442–448 RSC; (b) S. Singha and K. M. Parida, Catal. Sci. Technol., 2011, 1, 1496–1505 RSC; (c) L. Fu, S. Zhao, Y. Chen and Z. Liu, Chem. Commun., 2016, 52, 5577–5580 RSC; (d) Y. Aratani, Y. Yamada and S. Fukuzumi, Chem. Commun., 2015, 51, 4662–4665 RSC; (e) L. Wang, G. Wang, J. Zhang, C. Bian, X. Meng and F.-S. Xiao, Nat. Commun., 2017, 8, 15240 CrossRef PubMed; (f) Y. Li, C. Liu and W. Yang, New J. Chem., 2017, 41, 8214–8221 RSC.
- T. Karimpour, E. Safaei, B. Karimi and Y. Lee, ChemCatchem, 2018, 10, 1889–1899 CrossRef CAS.
- (a) S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS PubMed; (b) M. Shao, F. Ning, J. Zhao, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2012, 134, 1071–1077 CrossRef CAS PubMed.
- Y. Liu, W. Zhang, X. Li, X. Le and J. Ma, New J. Chem., 2015, 39, 6474–6481 RSC.
- (a) X. Dong, X. Zhang, P. Wu, Y. Zhang, B. Liu, H. Hu and G. Xue, ChemCatChem, 2016, 8, 3680–3687 CrossRef CAS; (b) R. K. Sharma, M. Yadav, Y. Monga, R. Gaur, A. Adholeya, R. Zboril, R. S. Varma and M. B. Gawande, ACS Sustainable Chem. Eng., 2016, 4, 1123–1130 CrossRef CAS.
- (a) Y. Liu, J.-F. Chen, J. Bao and Y. Zhang, ACS Catal., 2015, 5, 3905–3909 CrossRef CAS; (b) L. Wang, X. Wang, J. Luo, B. N. Wanjala, C. Wang, N. A. Chernova, M. H. Engelhard, Y. Liu, I.-T. Bae and C.-J. Zhong, J. Am. Chem. Soc., 2010, 132, 17686–17689 CrossRef CAS PubMed.
- X. Xu, M. Tang, M. Li, H. Li and Y. Wang, ACS Catal., 2014, 4, 3132–3135 CrossRef CAS.
- (a) M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS PubMed; (b) X. Wu, M. S. Seo, K. M. Davis, Y.-M. Lee, J. Chen, K.-B. Cho, Y. N. Pushkar and W. Nam, J. Am. Chem. Soc., 2011, 133, 20088–20091 CrossRef CAS PubMed; (c) A. Gunay and K. H. Theopold, Chem. Rev., 2010, 110, 1060–1081 CrossRef CAS PubMed; (d) R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02284h |
| This journal is © The Royal Society of Chemistry 2019 |