
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of pyrano[3,2-e]indole alkaloid fontanesine B by a double cyclization strategy†
Tomoki Itoh,
Yuusuke Chiba,
Shunsuke Kawaguchi,
Yuki Koitaya,
Yuuma Yoneta,
Koji Yamada and
Takumi Abe *
*
Faculty of Pharmaceutical Sciences, Health Sciences University of Hokkaido, Ishikari-tobetsu, Hokkaido 0610293, Japan. E-mail: abe-t@hoku-iryo-u.ac.jp
First published on 3rd April 2019
Abstract
The regioselective synthesis of pyrano[3,2-e]indole alkaloid fontanesine B by two different cyclizations is described. The complete regioselectivity is controlled by the C4 Pictet–Spengler cyclization, in which an iminium ion acts as a transient directing (TDG) group. Furthermore, carbolines were constructed by a new Bischler–Napieralski-type cyclization, in which an unprecedented trichloromethyl carbamate serves as a reactive group.
Fontanesines A (1), B (2), and C (3) were isolated from the stem bark and leaf fractions of Conchocarpus fontanesianus by Queiroz and co-workers in 2016 (Fig. 1).1 These compounds have a characteristic pyrano[3,2-e]indole moiety fused with quinazolinone. A crucial challenge in the synthesis of fontanesines is the regioselective formation of the pyrano[3,2-e]indole core. Although the structures were unique and unprecedented, there are no reports on their partial preparation or total synthesis.
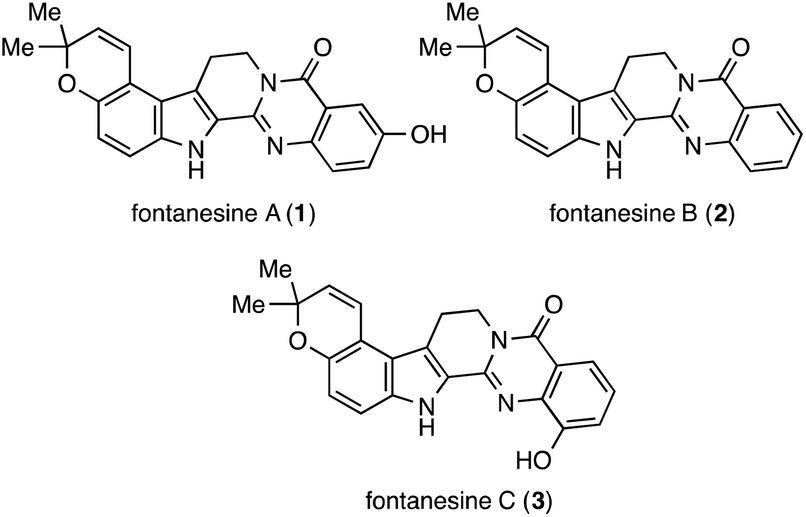 | ||
| Fig. 1 Fontanesines A (1), B (2), and C (3). | ||
The importance of a pyrano[3,2-e]indole framework in medicinal chemistry had encouraged Macor,2 Pandit,3 May,4 and Conforti5 to develop efficient methods for the regioselective construction of this framework. The majority of these methods relied on the thermal Claisen rearrangement,2–4 and Pt-mediated cyclization.5 To keep the pyran intact from earlier stage of total synthesis is difficult due to its instability.6
In our continuing efforts in the synthesis of indole alkaloids,7 we developed a novel strategy for the synthesis of azepinoindoles by C4 Pictet–Spengler reaction of serotonins8 or 5-hydroxytryptophans9 and aldehydes. This approach proved useful in the one-pot regioselective synthesis of pyrano[3,2-e]indoles.10 We considered the above facts and envisioned that the synthesis of pyrano[3,2-e]indoles by C4 Pictet–Spengler reaction would allow a rapid and regioselective formation of fontanesines, keeping the pyran intact. Herein, we report the results of our efforts to synthesize 2.
The retrosynthetic analysis of fontanesine B (2) is shown in Scheme 1. The quinazolinone moiety in 2 might be forged by a deprotection followed by condensation of anthranilic acid (11a) with carboline 9. One of the key steps in the synthetic route involved the carbonylative cyclization of pyrano[3,2-e]indole 8 to afford carboline 9. The pyrano[3,2-e]indole 8 could be accessible from aldehyde 5 and benzyl protected 5-hydroxytryptamine 4 using our developed C4 Pictet–Spengler/allylic transposition via the iminium intermediate 6 and azepinoindole 7.
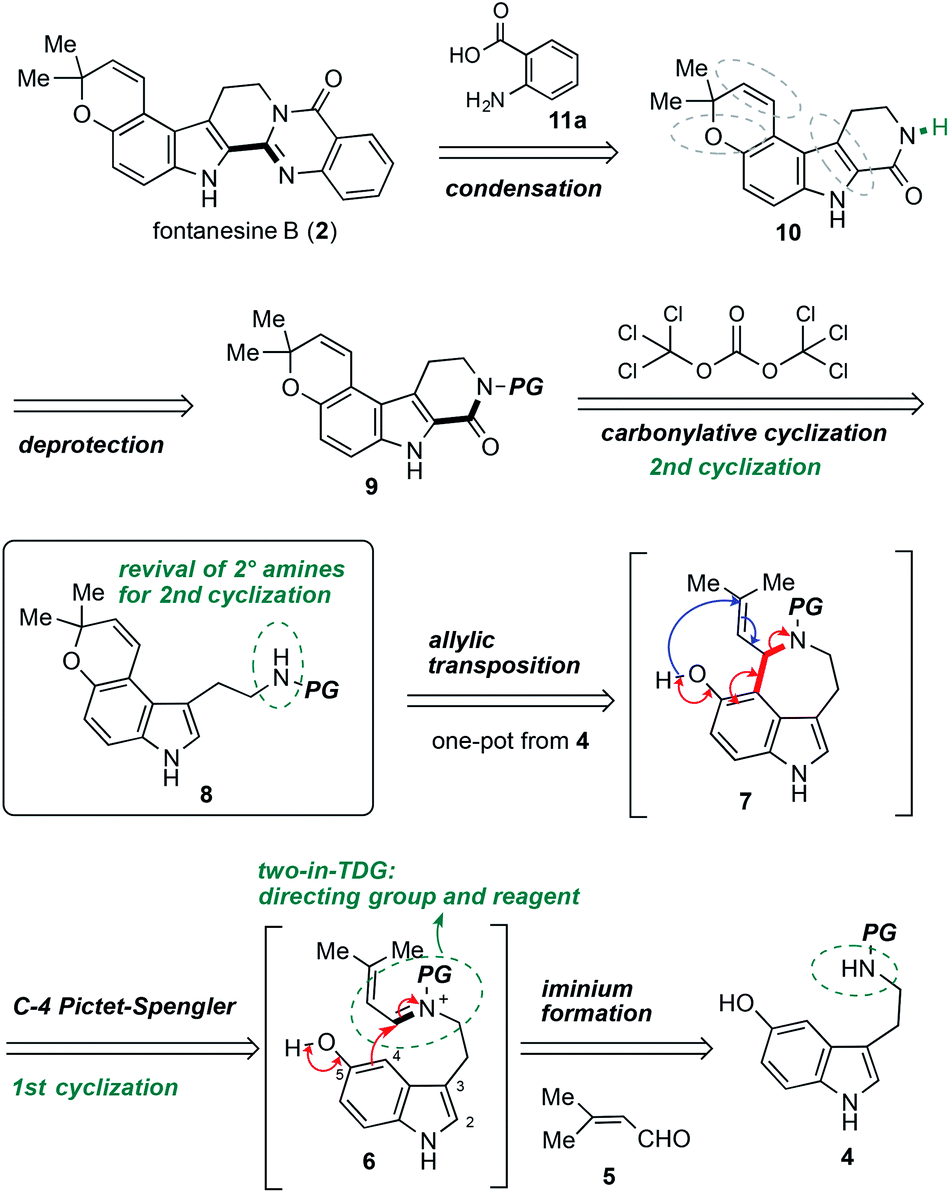 | ||
| Scheme 1 Retrosynthetic analysis of fontanesine B (2). | ||
Before synthetic studies, we could predict the difficulty of removing the protecting group on the nitrogen atom at the late stage. Therefore, we decided to prepare the several tryptamines 4 with different protecting groups. The synthesis was started from the benzyl protected 5-hydroxytryptamine 4 (Scheme 2). It was reacted with 3-methyl-2-butenal (5) in 2-propanol/Et3N under reflux to produce the desired pyrano[3,2-e]indole 8 in a one-pot reaction. Normal Pictet–Spengler reaction occurs at the C2 position of the indole ring under the acidic conditions. All steps of this one-pot sequence take place under basic conditions, which is presumably key to its success.
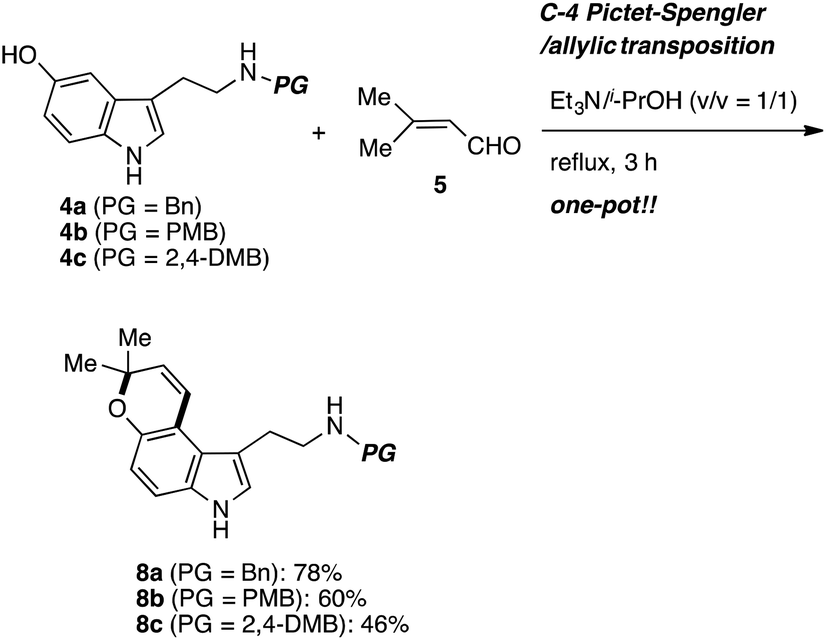 | ||
| Scheme 2 Synthesis of substrates 8. | ||
To test the feasibility of our approach, we resorted to the carbonylative cyclization of 8. According to the previous report on the reaction using triphosgene,11–13 which is a bench-stable solid and easy to handle,14 we investigated the conversion of 8 into 9 through intermediate 13 (ref. 15) (Scheme 3). Numerous attempts including screening of bases to achieve this have resulted in the polymerization and halogenation13 of 8 over the carbonylative cyclization.12 Upon exposure of 8c to triphosgene in the presence of Et3N followed by addition of HBr,12 the desired product 9c was obtained in low yield along with unstable brominated product 12. The acid lability of a pyrano[3,2-e]indole afforded troublesome, with polymerized materials being the major spot observed. As this polymerization presumably arises from activated urea intermediate 16, which was generated from less electrophilic acid chloride 14 (ref. 14 and 16b) or more electrophilic intermediate 15 by addition/elimination process by HBr and Et3N,16 it was clear that the Et3N14b and HBr would require to be dismissed at the cyclization step in our synthetic route.
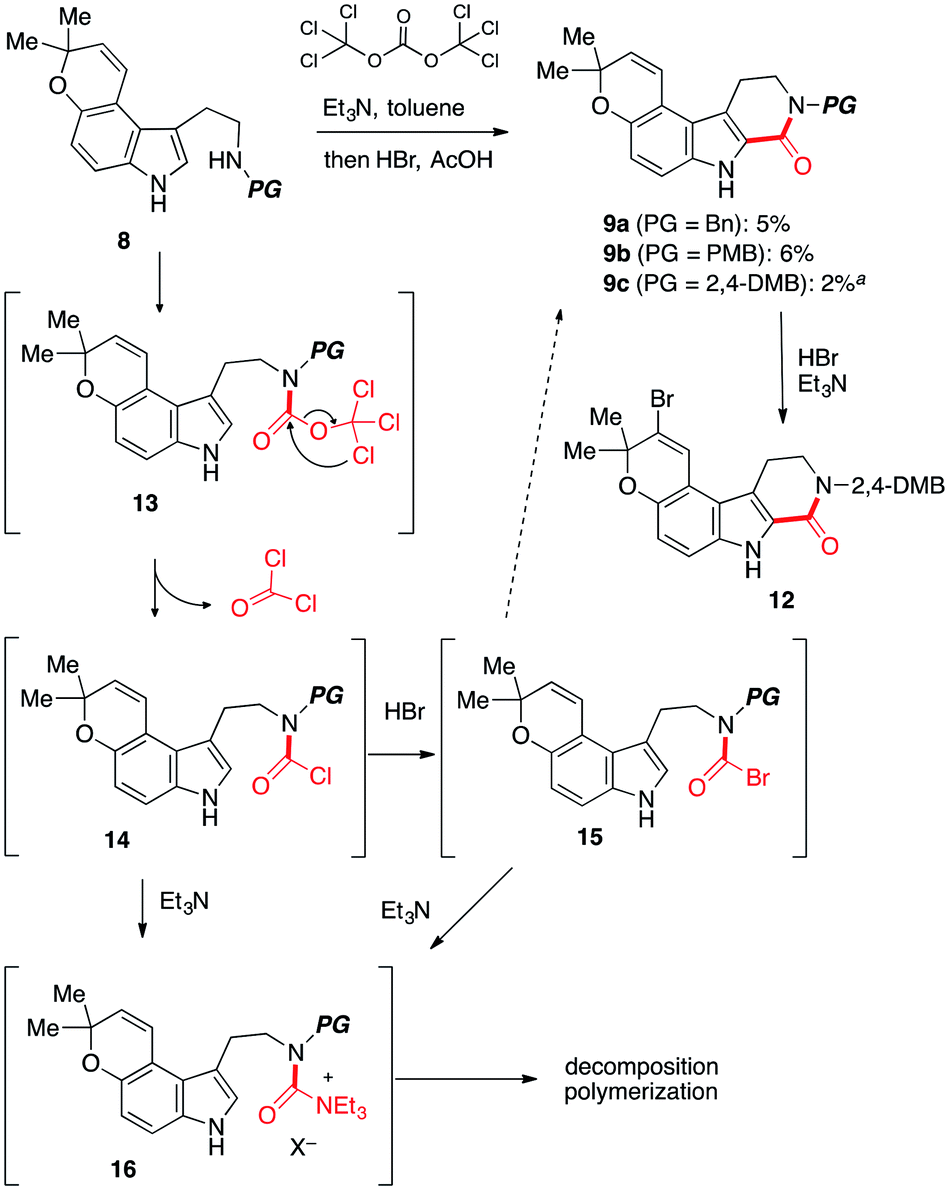 | ||
| Scheme 3 Attempted synthesis of 9. a12 was obtained in 14% yield. | ||
Because the product yield was not sufficient (up to 6% yield), further investigations were carried out. After intensive investigations, it was serendipity that we found that the treatment of 8 with triphosgene in the presence of Et3N at room temperature afforded a trichloromethyl carbamate intermediate 13b in 88% yield (Scheme 4).17 Then, after aqueous work-up to remove Et3N in the reaction media, 13b was heated in DMSO to afford 9b in 86% yield. Furthermore, by employing a stepwise method, we obtained 9 from 8 in good yield through the carbamoyl ion 17 (ref. 18) using a single column chromatography. To the best of our knowledge, this is the first time that an unstable trichloromethyl carbamate intermediate has been applied to the C–C bond formations.11–17 In contrast to the mild Bischler–Napieralski-type cyclization developed by Saikawa and Nakata,19 and Clayden,20 our protocol does not require additives to promote the cyclization.
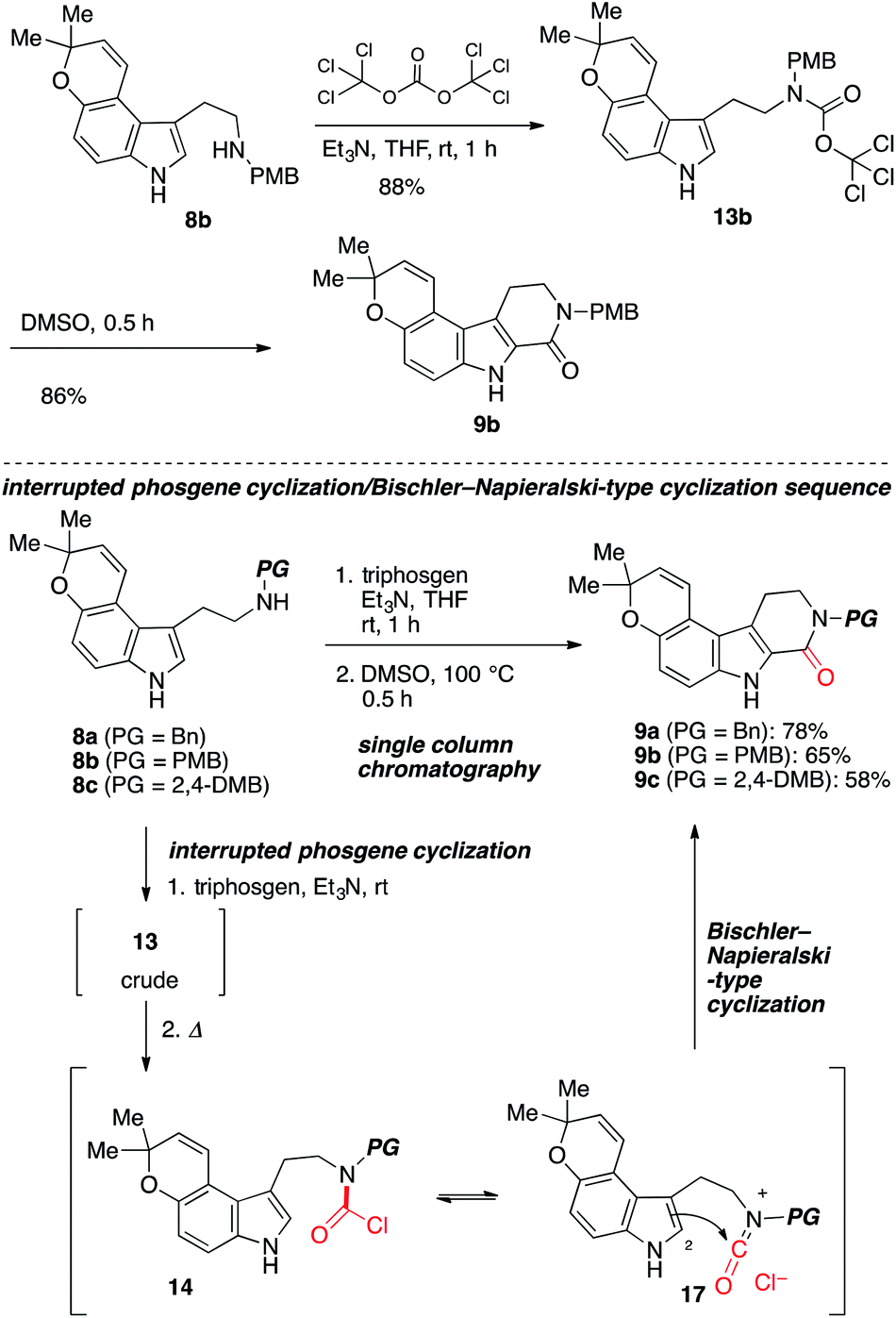 | ||
| Scheme 4 Improved synthesis of β-carbolines 9 from 8 via interrupted phosgene cyclization and Bischler–Napieralski-type cyclization. | ||
Numerous attempts were made in case of benzyl-substituted lactam 9a; however, all of them led to rapid decomposition (Scheme 5). On the other hand, treatment of 9b with p-toluenesulfonic acid (p-TsOH)21 afforded the deprotected lactam 10 in 17% yield. As expected, lactam 9c could also be deprotected under the same conditions to afford 10 in 67% yield. In general, 2,4-DMB group is more easily removed than PMB group.21
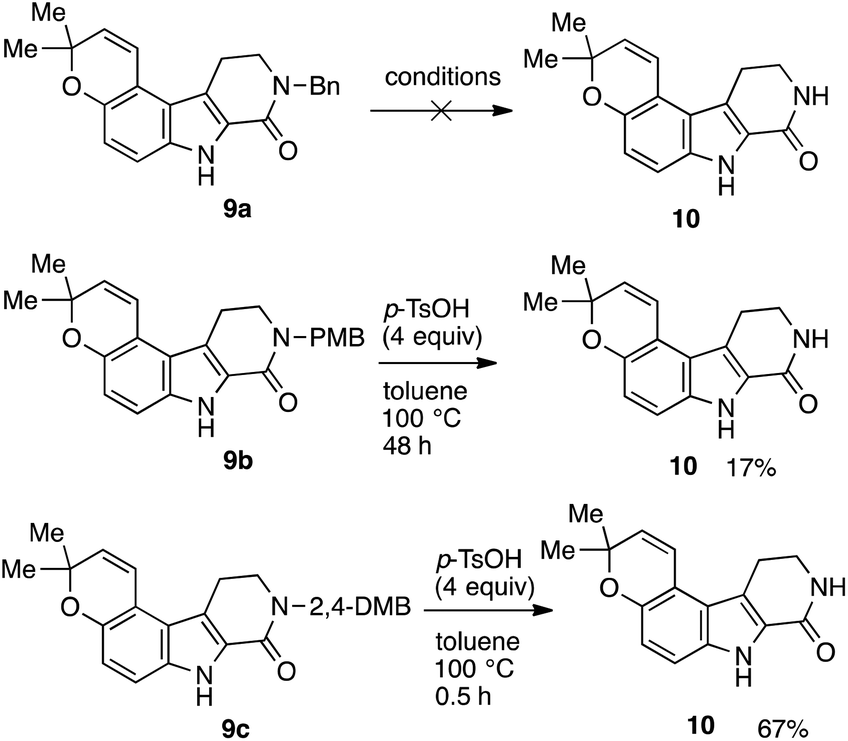 | ||
| Scheme 5 Removal of benzyl substituents on the nitrogen atoms in 9. | ||
With the synthetic access to 9, we were set to answer whether 9 could be deprotected keeping the alkene, pyran, and indole intact. Finally, the condensation of 10 and anthranilic acid (11a) in the presence of POCl3 (ref. 22) generated the final product 2 (Scheme 6), whose structure was determined by spectroscopic experiments. All the physical data of synthetic 2 were in good agreement with those reported for the natural product (Table 1).1
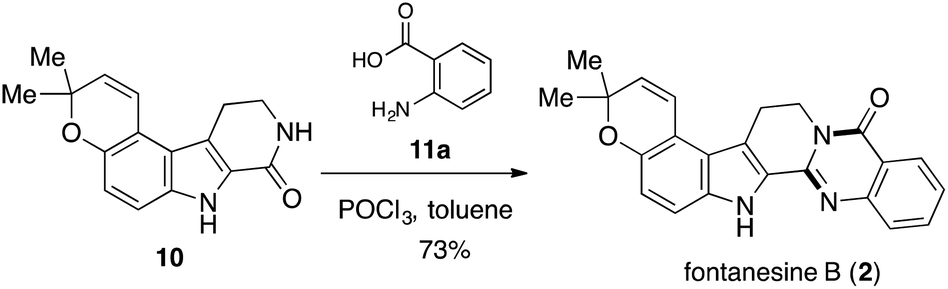 | ||
| Scheme 6 Completion of total synthesis of fontanesine B. | ||
|
|
|---|---|
| 1H NMR (DMSO-d6, 500 MHz, δ in ppm) | |
| Natural fontanesine B | Synthetic compound 2 |
| 1.40 (6H, s, CH3-25, 26) | 1.34 (6H, s, CH3-25, 26) |
| 3.33 (2H, t, J = 6.9 Hz, H-6) | 3.06 (2H, t, J = 7.5 Hz, H-6) |
| 4.43 (2H, t, J = 6.9 Hz, H-5) | 4.38 (2H, t, J = 6.9 Hz, H-5) |
| 5.77 (1H, d, J = 9.8 Hz, H-23) | 5.81 (1H, d, J = 9.8 Hz, H-23) |
| 6.77 (1H, d, J = 8.7 Hz, H-11) | 6.53 (1H, d, J = 9.7 Hz, H-11) |
| 6.88 (1H, d, J = 9.8 Hz, H-22) | 6.93 (1H, s, H-22) |
| 7.25 (1H, d, J = 8.7 Hz, H-12) | 7.12 (1H, s, H-12) |
| 7.47 (1H, ddd, J = 8.0, 7.1, 1.2 Hz, H-18) | 7.43 (1H, td, J = 7.4, 1.2 Hz, H-18) |
| 7.67 (1H, dd, J = 8.3, 1.2 Hz, H-16) | 7.64 (1H, d, J = 8.1 Hz, H-16) |
| 7.81 (1H, ddd, J = 8.3, 7.1, 1.5 Hz, H-17) | 7.77 (1H, td, J = 6.5, 1.2 Hz, H-17) |
| 8.16 (1H, dd, J = 8.0, 1.5 Hz, H-19) | 8.12 (1H, d, J = 8.0 Hz, H-19) |
| 11.72 (1H, s, H-1) | 11.71 (1H, s, H-1) |
| 13H NMR (DMSO-d6, 126 MHz, δ in ppm) | |
|---|---|
| Natural fontanesine B | Synthetic compound 2 |
| 20.7 (C-6) | 21.3 (C-6) |
| 27.0 (C-25, 26) | 27.5 (C-25, 26) |
| 40.6 (C-5) | 41.1 (C-5) |
| 75.0 (C-24) | 75.6 (C-24) |
| 112.6 (C-9) | 113.2 (C-9) |
| 112.7 (C-12) | 113.3 (C-12) |
| 115.6 (C-11) | 116.1 (C-11) |
| 116.5 (C-7) | 117.1 (C-7) |
| 119.2 (C-22) | 119.8 (C-22) |
| 120.6 (C-20) | 121.2 (C-20) |
| 120.9 (C-8) | 121.5 (C-8) |
| 125.9 (C-18) | 126.5 (C-18) |
| 126.4 (C-16) | 127.0 (C-16) |
| 126.5 (C-19) | 127.1 (C-19) |
| 127.9 (C-2) | 128.5 (C-2) |
| 130.1 (C-23) | 130.7 (C-23) |
| 134.3 (C-17) | 135.0 (C-17) |
| 134.4 (C-13) | 135.0 (C-13) |
| 145.2 (C-3) | 145.8 (C-3) |
| 146.2 (C-10) | 146.8 (C-10) |
| 147.4 (C-15) | 148.0 (C-15) |
| 160.5 (C-21) | 161.1 (C-21) |
In conclusion, we have successfully accomplished the total synthesis of fontanesine B using C4 Pictet–Spengler/allylic transposition as the key step to construct the pyrano[3,2-e]indole core using the transient directing group (TDG). In this cyclization, the TDG played the dual important role of directing group and reagent.23 In addition, the unprecedented carbamate intermediate produced in the carbonylative cyclization could be converted into pyrano[3,2-e]pyrido[3,4-b]indoles only by heating through the Bischler–Napieralski-type cyclization. Further investigations including application of the C2 and C4 cyclization strategy24 to the syntheses of other indole alkaloids is ongoing in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JSPS (KAKENHI Grant Number 16K18849 for T. A.) as a Grant-in-Aid for Young Scientists (B).Notes and references
- R. S. Cabral, P.-M. Allard, L. Marcourt, M. C. M. Young, E. F. Queiroz and J.-L. Wolfender, J. Nat. Prod., 2016, 79, 2270 CrossRef CAS PubMed.
- (a) J. E. Macor, C. B. Fox, C. J. Johnson, B. K. Koe, L. A. Lebel and S. H. Zorn, J. Med. Chem., 1992, 35, 3625 CrossRef CAS PubMed; (b) J. E. Macor, K. Ryan and M. E. Newman, Tetrahedron, 1992, 48, 1039 CrossRef; (c) J. E. Macor, D. H. Blank and R. J. Post, Tetrahedron Lett., 1994, 35, 45 CrossRef CAS; (d) J. E. Macor, Tetrahedron Lett., 1995, 36, 7019 CrossRef CAS; (e) J. E. Macor, O. D. Langer, J. Z. Gougoutas, M. F. Malley and L. A. M. Cornelius, Tetrahedron Lett., 2000, 41, 3541 CrossRef CAS.
- J. P. M. Plung, G.-J. Koomen and U. K. Pandit, Tetrahedron Lett., 1992, 33, 2179 CrossRef.
- J. A. May, H.-H. Chen, A. Rusinko, V. M. Lynch, N. A. Sharif and M. A. McLaughlin, J. Med. Chem., 2003, 46, 4188 CrossRef CAS PubMed.
- M. S. Sinicropi, A. Caruso, F. Conforti, M. Marrelli, H. EI Kashef, J.-C. Lancelot, S. Rault, G. A. Statti and F. Menichini, J. Enzyme Inhib. Med. Chem., 2009, 24, 1148 CrossRef CAS PubMed.
- In alkaloids synthesis, pyran formation had been conducted at a late stage. For reviews, see: (a) H.-J. Knölker and K. R. Reddy, Chem. Rev., 2002, 112, 3193 Search PubMed; (b) M. Ishikura, K. Yamada and T. Abe, Nat. Prod. Rep., 2010, 27, 1630 RSC; (c) A. W. Schmidt, K. R. Reddy and H.-J. Knölker, Chem. Rev., 2012, 102, 4303 Search PubMed; (d) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694 RSC; (e) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2015, 32, 1389 RSC.
- (a) T. Abe, T. Ikeda, R. Yanada and M. Ishikura, Org. Lett., 2011, 13, 3356 CrossRef CAS PubMed; (b) T. Abe, T. Ikeda, R. Yanada and M. Ishikura, Org. Lett., 2013, 15, 3622 CrossRef CAS PubMed; (c) T. Abe, T. Itoh, S. Hibino, T. Choshi and M. Ishikura, Tetrahedron Lett., 2014, 55, 5268 CrossRef CAS; (d) T. Itoh, T. Abe, S. Nakamura and M. Ishikura, Heterocycles, 2015, 91, 1423 CrossRef CAS; (e) T. Itoh, T. Abe, T. Choshi, T. Nishiyama and M. Ishikura, Heterocycles, 2016, 92, 1132 CrossRef CAS; (f) T. Abe and K. Yamada, Org. Lett., 2016, 18, 6504 CrossRef CAS PubMed; (g) T. Abe, K. Kida and K. Yamada, Chem. Commun., 2017, 53, 4362 RSC; (h) T. Itoh, T. Abe, T. Choshi, T. Nishiyama and M. Ishikura, Heterocycles, 2017, 95, 507 CrossRef CAS; (i) T. Abe, T. Suzuki, M. Anada, S. Matsunaga and K. Yamada, Org. Lett., 2017, 19, 4275 CrossRef CAS PubMed; (j) T. Abe and K. Yamada, Org. Lett., 2018, 20, 1469 CrossRef CAS PubMed; (k) T. Abe and M. Terasaki, Helv. Chim. Acta, 2018, 101, e1700284 CrossRef; (l) T. Abe, Heterocycles, 2018, 96, 490 CrossRef CAS.
- (a) K. Yamada, Y. Namerikawa, T. Abe and M. Ishikura, Heterocycles, 2009, 77, 825 CrossRef CAS; (b) K. Yamada, Y. Namerikawa, T. Haruyama, Y. Miwa, R. Yanada and M. Ishikura, Eur. J. Org. Chem., 2009, 5752 CrossRef CAS.
- (a) T. Abe and K. Yamada, J. Nat. Prod., 2017, 80, 241 CrossRef CAS PubMed; (b) T. Abe, T. Haruyama and K. Yamada, Synthesis, 2017, 49, 4141 CrossRef CAS.
- K. Yamada, S. Yamaguchi, N. Hatae, T. Abe, T. Iwamura and M. Ishikura, Heterocycles, 2011, 83, 815 CrossRef CAS.
- Phosgene cyclization of N-substituted tryptamines has not been reported. For examples of phosgene cyclization of primary tryptamines via isocyanates, see: (a) F. Bracher and D. Hilderband, Liebigs Ann. Chem., 1992, 1315 CrossRef CAS; (b) K. C. Nicolaou, J. L. Kiappes, W. Tian, V. B. Gondi and J. Becker, Org. Lett., 2011, 13, 3924 CrossRef CAS PubMed; (c) J. Zhang, S. Da, X. Feng, X. Chen, J. Jiang and Y. Li, Chin. J. Chem., 2013, 31, 123 CrossRef CAS.
- For examples of triphosgene mediated halogenations, see: (a) C. E. Ayala, A. Villalpando, A. L. Nguyen, G. T. McCandless and R. Kartika, Org. Lett., 2012, 14, 3676 CrossRef CAS PubMed; (b) A. Villalpando, C. E. Ayala, C. B. Watson and R. Kartika, J. Org. Chem., 2013, 78, 3989 CrossRef CAS PubMed; (c) M. A. Sapotra, L. Ngo and R. Kartika, J. Org. Chem., 2015, 80, 8815 CrossRef PubMed; (d) A. Villalpando, M. A. Sapotra, T. H. Tugwell and R. Kartika, Chem. Commun., 2015, 51, 15075 RSC; (e) A. H. Cleveland, F. R. Fronczek and R. Kartika, J. Org. Chem., 2018, 83, 3367 CrossRef CAS PubMed.
- For selected examples of coupling reactions using triphosgene, see: (a) A. Armstrong, I. D. Edmonds and M. E. Swarbrick, Tetrahedron Lett., 2003, 44, 5335 CrossRef CAS; (b) O. P. Gulin, F. Rabanal and E. Giralt, Org. Lett., 2006, 8, 5385 CrossRef CAS PubMed; (c) W. Su, S. J. Gray, R. Dondi and G. A. Burley, Org. Lett., 2009, 11, 3910 CrossRef CAS PubMed; (d) S. T. Le Quement, T. Flagstad, R. J. T. Mikkelsen, M. R. Hansen, M. C. Givskov and T. E. Nielsen, Org. Lett., 2012, 14, 640 CrossRef CAS PubMed; (e) Z. Wu, T. Hu, L. He and B. Gong, Org. Lett., 2012, 14, 2504 CrossRef CAS PubMed; (f) L. Fang, G. Yao, Z. Pan, C. Wu, H.-S. Wang, G. A. Burley and W. Su, Org. Lett., 2015, 17, 158 CrossRef CAS PubMed; (g) K. Leczycka-Wilk, K. Dabrowa, P. Cmoch and S. Jarosz, Org. Lett., 2017, 19, 4596 CrossRef CAS PubMed; (h) S. E. Varjosaari, P. Suating and M. J. Adler, Synthesis, 2016, 48, 43 Search PubMed.
- For reviews, see: (a) L. Cotarca, P. Delogu, A. Nardelli and V. Sunjic, Synthesis, 1995, 553 Search PubMed; (b) L. Cotarca, T. Geller and J. Répási, Org. Process Res. Dev., 2017, 21, 1439 CrossRef CAS.
- (a) J. Bermudez, S. Dabbs, K. A. Joiner and F. D. King, J. Med. Chem., 1990, 33, 1929 CrossRef CAS PubMed; (b) P. A. Barsanti, Y. Xia, W. Wang, K. G. Mendenhall, L. M. Langniton, S. Ramurthy, M. C. Phillips, S. Subramanian, R. Boyce, N. M. Brammeier, R. Constantine, D. Duhl, A. O. Walter, T. J. Abrams and P. A. Renhowe, US Pat. Appl. US 20070037853, CAN 146:251846, 2007.
- (a) L. Contarca and H. Eckert, Phosgenations – A Handbook, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2003 CrossRef; (b) L. Pasquato, G. Modena, L. Cotarca, P. Delouge and S. Mantovani, J. Org. Chem., 2000, 65, 8224 CrossRef CAS PubMed; (c) V. K. Gumaste and A. R. A. S. Deshmukh, Tetrahedron Lett., 2004, 45, 6571 CrossRef CAS.
- Trichloromethyl carbamate easily decomposes to give the corresponding carbamic acid, see: A. C. Sosa, R. Conway, R. T. Williamson, J. P. Suchy, W. Edwards and T. Cleary, Org. Process Res. Dev., 2011, 15, 1458 CrossRef.
- S. Hwang, D. Kim and S. Kim, Chem.–Eur. J., 2012, 18, 9977 CrossRef CAS PubMed.
- For the pioneering examples of Bischler–Napieralski-type cyclization using isopropyl carbamates and P2O5, see: (a) S. Adachi, K. Watanabe, Y. Iwata, S. Kameda, Y. Miyaoka, M. Onozuka, R. Mitsui, Y. Saikawa and M. Nakata, Angew. Chem., Int. Ed., 2013, 52, 2087 CrossRef CAS PubMed; (b) S. Adachi, M. Onozuka, Y. Yoshida, M. Ide, Y. Saikawa and M. Nakata, Org. Lett., 2014, 16, 358 CrossRef CAS PubMed.
- For an example of a mild Bishler–Napieralski-style cyclization using KI as a promoter, see: M. M. Amer, A. C. Carrasco, D. J. Leonard, J. W. Ward and J. Clayden, Org. Lett., 2018, 20, 7977 CrossRef CAS PubMed.
- C.-Y. Chern, Y.-P. Huang and W. M. Kan, Tetrahedron Lett., 2003, 44, 1039 CrossRef CAS.
- M. Decker, Eur. J. Med. Chem., 2005, 40, 305 CrossRef CAS PubMed.
- During the preparation of this manuscript, Zeng and Li reported a two-in-one strategy using a transient directing group (TDG) for palladium-catalyzed functionalization. See: H. Zheng, Z. Wang and C.-J. Li, Angew. Chem., Int. Ed., 2019, 58, 2859 CrossRef PubMed.
- To the best of our knowledge, the synthetic approach involving both Pictet–Spengler and Bischler–Napieralski cyclization has not been explored. For reviews, see: (a) J. Stöckigt, A. P. Antonchick, F. Wu and H. Waldman, Angew. Chem., Int. Ed., 2011, 50, 8538 CrossRef PubMed; (b) M. Chrzanowska, A. Grajewska and M. D. Rozwadowska, Chem. Rev., 2016, 116, 12369 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectra data for all compounds, including scanned images of 1H and 13C NMR spectra. See DOI: 10.1039/c9ra02321f |
| This journal is © The Royal Society of Chemistry 2019 |